

Abstract
The endoplasmic reticulum (ER) responds to errors in protein folding or processing by induction of the unfolded protein response (UPR). During conditions of ER stress, unconventional splicing of an mRNA encoding the UPR-responsive transcription factor occurs at the ER surface, resulting in activation of the UPR. UPR activation is necessary for adaptation to ER stress and for the pathogenic fungus Cryptococcus neoformans is an absolute requirement for temperature adaptation and virulence. In this study, we have determined that C. neoformans has co-opted a conserved PUF RNA binding protein to regulate the posttranscriptional processing of the HXL1 mRNA encoding the UPR transcription factor. PUF elements were identified in both the 5′ and 3′ untranslated regions of the HXL1 transcript, and both elements bound Puf4. Deletion of PUF4 resulted in delayed unconventional splicing of HXL1 mRNA and delayed induction of Hxl1 target genes. In addition, the HXL1 transcript was stabilized in the absence of Puf4. The puf4Δ mutant exhibited temperature sensitivity but was as virulent as the wild type, despite a reduction in fungal burden in the brains of infected mice. Our results reveal a novel regulatory role in which a PUF protein influences the unconventional splicing of the mRNA encoding the UPR-responsive transcription factor. These data suggest a unique role for a PUF protein in controlling UPR kinetics via the posttranscriptional regulation of the mRNA encoding the UPR transcription factor Hxl1.
INTRODUCTION
Cryptococcus neoformans is one of the few fungal species that has evolved to successfully transition from its environmental niche to cause deep infections in humans, which requires adaptation to the mammalian core temperature. The mammalian host temperature exerts endoplasmic reticulum (ER) stress on C. neoformans, leading to a transient induction of the unfolded protein response (UPR) during temperature adaptation (1). The loss of UPR induction through deletion of the stress response mediators IRE1 and HXL1 impairs virulence and temperature adaptation (2). Likewise, prolonged ER stress resulting from the loss of mRNA degradation in a ccr4Δ mutant also impairs temperature adaptation (1). Together, this information suggests that a balance of UPR induction and resolution is critical for successful temperature adaptation in C. neoformans.
The UPR is induced in response to ER stress caused by aberrant protein folding or processing (3–5). A well-conserved feature of UPR activation is the spliceosome-independent removal of an unconventional intron from the mRNA encoding the major UPR transcription factor by the ER-resident transmembrane kinase/RNase Ire1 (5, 6). In Saccharomyces cerevisiae, this transcription factor is encoded by the HAC1 gene and the unconventional splicing event removes a 252-nucleotide intron from the mRNA, altering the reading frame and allowing production of a functional Hac1p transcription factor. In higher eukaryotes, the orthologous active protein Xbp1 is produced after removal of a 26-nucleotide intron from the XBP1 mRNA (7). This unconventional splicing event is an essential and rate-limiting step of UPR induction (5, 6).
The posttranscriptional events that influence splicing of the transcript encoding the UPR transcription factor have been well characterized in both yeast and mammals. In S. cerevisiae, the HAC1 transcript contains cis elements within both the 3′ untranslated region (UTR) and the unconventional intron which target HAC1 mRNA to Ire1p foci during ER stress (8). In higher eukaryotes, the unspliced XBP1 transcript is tethered to the ER surface in a translation-pausing reaction that results in the anchoring of a complex composed of the nascent Xbp1 peptide chain, ribosome, and unspliced XBP1 mRNA to the ER membrane. The anchoring of this complex to the ER membrane allows the proximal association of unspliced XBP1 mRNA to Ire1 and splicing by Ire1 upon UPR activation (9, 10). In addition to features encoded within the transcript, several protein partners have been implicated in the regulation of HAC1 mRNA. Of note are Ypt1p and Ada5p. Ypt1p is a ras GTPase which promotes HAC1 mRNA decay (11). Ada5p, a subunit of the SAGA transcriptional regulatory complex, interacts with Ire1 and is required for HAC1 splicing (12). However, neither Ypt1p nor Ada5p contains known RNA binding domains, suggesting that other protein partners may be facilitating their interaction with the HAC1 transcript.
The UPR transcription factor in C. neoformans is encoded by HXL1, named for HAC1, XBP1-like gene 1 (2). HXL1 displays limited homology to HAC1 and XBP1 and, consequently, contains neither the HAC1 cis Ire1 localization element previously described in S. cerevisiae nor the tethering domain of XBP1 (2, 13, 14). The limited homology of HXL1 to XBP1 and HAC1, combined with the absence of known posttranscriptional regulatory elements in HXL1, suggests that the unconventional splicing of HXL1 mRNA in C. neoformans is controlled through an alternative mechanism. Initial characterization of the UPR pathway in C. neoformans has shown the ER chaperone KAR2 to be essential for growth and both HXL1 and IRE1 to be critical for virulence and contribute to thermotolerance, cell wall integrity dynamics, and drug resistance (2, 15). Additionally, our previous work on the deadenylase Ccr4 demonstrated that posttranscriptional modulation of the ER stress response is required for resolution of the host temperature adaptation process in C. neoformans (1).
RNA binding proteins are the mediators of posttranscriptional regulation and exert their effects through interactions with specific sequences or structural motifs within their cognate mRNAs. Puf4 in C. neoformans is a member of the pumilio-FBF (PUF) family of mRNA binding proteins, which are found throughout the eukaryotic kingdom. PUF proteins regulate stability, translation, and the subcellular localization of functionally related target transcripts through interactions with conserved cis elements (16–24). In S. cerevisiae, Puf4p is bound to nuclear and nucleolar transcripts. However, the identification of putative Puf4-binding elements in the 3′ UTR of mitochondrial transcripts for the Pezizomycotina subdivision of fungi implies that Puf4 may control mitochondrial function in Pezizomycotina species (18, 21). These interspecies differences suggest a repurposing of the conserved Puf4 regulatory module to perform unique functions in different organisms. In this study, we report that in C. neoformans, Puf4 exerts a bimodal mechanism of action in the regulation of the pivotal UPR signaling transcript HXL1, regulating the rate of HXL1 splicing during UPR induction, as well as the attenuation of UPR signaling following adaptation to growth at 37°C through HXL1 mRNA decay. We postulate that by regulating the fate of HXL1 mRNA, Puf4 is able to modulate UPR kinetics in C. neoformans, controlling both the engagement and the resolution of this critical stress response. These data suggest that in C. neoformans, the convergence of two posttranscriptional regulatory mechanisms, the Puf4 module and the unconventional splicing of the UPR transcriptional activator, has occurred to balance ER stress response activation and resolution.
MATERIALS AND METHODS
Strains and constructs.
The clinical isolate Cryptococcus neoformans var. grubii strain H99 was the wild-type parent strain used to generate subsequent strains in this study. Strains were created using biolistic transformation of deletion and complementation cassettes (see Table S1 in the supplemental material for a list of the primers and oligonucleotides used in this study). Transformants were screened by PCR, Northern blot hybridization, and Southern blot hybridization to confirm integration.
Cloning and sequencing of PUF4 mRNA.
The PUF4 coding sequence and the 5′ and 3′ untranslated regions of PUF4 were identified using an Invitrogen rapid amplification of cDNA ends system. RNA isolated from wild-type cells was ligated to sequencing adapters, reverse transcribed, and amplified. The PCR product was inserted into a pCR4-TOPO vector and cloned, and plasmids from the resulting transformants were isolated and sequenced. The sequences were compared to those in the Cryptococcus neoformans var. grubii H99 database at the Broad Institute (www.broadinstitute.org).
Recombinant Puf4 protein.
A truncated PUF4 sequence containing the pumilio RNA binding domain was inserted into the pET14B vector and then transformed into Escherichia coli BL21(DE3)pLysS. The recombinant protein product was purified using nickel agarose column affinity purification (Qiagen). Protein lysate was dialyzed and stored in 50 mM PO4, pH 8, 250 mM NaCl, and 5% glycerol at 4°C.
EMSAs.
RNA binding assays were performed in a binding buffer containing 15 mM HEPES pH 7.4, 40 mM KCl, 5 mM MgCl2, 5% glycerol, 1 mM dithiothreitol (DTT), and 0.1 mg/ml yeast tRNA. TYE705-labeled RNA oligonucleotides, purchased from Integrated DNA Technologies, contained the PUF consensus sequence plus 7 to 8 bases of flanking sequence (see Table S1 in the supplemental material) (25). Competition was performed with 5-, 25-, or 50-fold excess competitor added. Reaction mixtures were incubated for 30 min, and native electrophoretic mobility shift assay (EMSA) reactions were run on a 6% DNA retardation gel. UV cross-linked EMSA reaction mixtures were cross-linked for 10 min in an HL-2000 Hybrilinker hybridization oven and electrophoresed on 4 to 12% Tris-glycine polyacrylamide gels (26, 27). EMSA gels were imaged using a LI-COR Odyssey infrared imaging system.
RNA isolation and Northern blot analysis.
Overnight starter cultures were used to inoculate 30 ml yeast extract-peptone-dextrose (YPD) in baffled 250-ml Erlenmeyer flasks. Cells were grown to mid-log phase, and then either the cells were resuspended in prewarmed 37°C YPD or 1 μg/ml tunicamycin was added and the cells were harvested at the time points indicated. For RNA stability assays, 250 μg/ml 1,10-phenanthroline was added to halt transcription. Cell pellets were lysed using mechanical bead disruption, and RNA was isolated using a Qiagen RNeasy kit. RNA was electrophoresed on a 1% denaturing agarose gel and transferred to a nylon membrane by capillary action. Hybridization was performed as described previously (28). The hybridization signal of the transcript of interest was normalized to the intensity of the ribosomal bands using Quantity One software. For stability assays, data were analyzed by nonlinear regression using GraphPad software.
HXL1 mRNA splicing assays.
RNA was isolated and treated with DNase, and then cDNA was generated using an iScript cDNA synthesis kit. The cDNA was used in a semiquantitative PCR. The PCR cycle used the following steps: 95°C for 2 min; 35 cycles of 95°C for 30 s, 45°C for 30 s, and an extension phase of 72°C for 30 s; and a final extension of 72°C for 5 min. The PCR product was electrophoresed on a 1% agarose gel. Unspliced HXL1 cDNA generated a 505-bp PCR product, and spliced HXL1 cDNA amplified a 449-bp DNA fragment. Quantity One software was used for quantification of the pixel density of the DNA bands. The band intensity was plotted against the cycle number to verify that the intensity of both amplicons was within the linear range of the PCR. Reaction mixtures lacking reverse transcriptase were included as a control for DNA contamination.
Spot plate assays.
Overnight cultures of wild-type, puf4Δ, and puf4Δ::PUF4 strains were pelleted and resuspended in sterile water to an optical density at 600 nm of 1.0, and then 5-μl aliquots of six 10-fold serial dilutions were spotted onto YPD plates. YPD plates containing 15 mM DTT or 250 ng/ml tunicamycin were used for the drug sensitivity spot plate assays. The plates were incubated for 3 days at the temperatures indicated below and then photographed.
Virulence studies.
For intravenous inoculation, 6- to 8-week old NIH Swiss mice were given lateral tail vein injections of 106 CFU of wild-type, puf4Δ, or puf4Δ::PUF4 cells in a 100-μl volume. Intranasal inoculation used A/Jr mice given an intranasal bolus of 50 μl phosphate-buffered saline containing 105 CFU of wild-type, puf4Δ, or puf4Δ::PUF4 cells. Mice were monitored in accordance with the University at Buffalo IACUC guidelines for signs of morbidity and humanely euthanized. Statistical analysis utilized Kruskal-Wallis analysis (analysis of variance on ranks). For the brain accumulation assay, 9 NIH Swiss mice were given tail vein injections of 106 CFU of a mixture of the wild-type and puf4Δ strains at an approximately 1:1.48 ratio. Three mice were sacrificed per day for 3 days postinoculation. The brains were excised, and the brain homogenate was plated on YPD plus 100 μg/ml chloramphenicol or YPD plus 100 μg/ml chloramphenicol and 200 μg/ml nourseothricin. The numbers of organisms from these cultures were used to extrapolate the number of CFU per gram brain material for each strain. The significance of the difference between wild-type cell levels and puf4Δ cell levels for input samples and samples collected on days 1, 2, and 3 was assessed by Student's t test.
RESULTS
The HXL1 transcript, encoding a HAC1/XBP1 ortholog, contains a putative PUF consensus element in each UTR.
In the initial characterization of HXL1 in C. neoformans, it was noted that the Ire1 localization element previously identified in the homologous HAC1 transcripts for S. cerevisiae and other ascomycetes was completely absent from the HXL1 transcript, suggesting a divergent method of regulating HXL1 splicing in C. neoformans (2). To identify possible regulators of the fate of HXL1 mRNA, we screened the HXL1 transcript for potential RNA binding protein consensus motifs and identified two putative PUF elements within the 5′ and 3′ untranslated regions (UTRs) of HXL1. Consensus elements for Puf3p-Puf5p in S. cerevisiae are characterized by an invariant UGUA, followed by a 2- to 4-nucleotide variable region and an invariant UA at the terminus. The element identified in the 5′ UTR of HXL1 fits the known Puf5p consensus element in S. cerevisiae, whereas the sequence of the 3′ element deviates from the consensus sequence in the first and third positions of the variable region, encoding a C in the first position and a G in the third position that are either U or A in the published Puf5p consensus sequence (Table 1) (18).
TABLE 1.
Identification of putative PUF elements in UTRs of known HAC1/HXL1 orthologs in fungia
| Species | Strain | Locus identifier | Sequence |
Annotated UTR | |
|---|---|---|---|---|---|
| 5′ UTR | 3′ UTR | ||||
| Cryptococcus neoformans var. grubii | H99 | CNAG_06134 | UGUAACAGUA | UGUACCGAUA | Y |
| Cryptococcus neoformans var. neoformans | Jec21 | CNM01380 | UGUAACAUUA | UGUAUCGAUA | N |
| Cryptococcus neoformans var. neoformans | B-3501A | CNBM1240 | UGUAACAUUA | UGUAUCGAUA | N |
| Cryptococcus gattii | R265 | CNBG_4842.2 | UGUAACAUUA | Y | |
| Cryptococcus gattii | WM276 | CGB_M1590W | N | ||
| Coprinopsis cinerea | Okayama7#130 | CC1G_02249 | UGUAGCAGUA | N | |
| Ustilago maydis | 521 | UM03509.1 | UGUAAAUGUA | N | |
| Yarrowia lipolytica | CLIB122 | XP_500811 | N | ||
| Candida albicans | WO-1 | CAWG_00794.1 | N | ||
| Candida albicans | SC5314 | C1_06130C | N | ||
| Aspergillus fumigatus | H237 | ACJ61678 | Y | ||
| Aspergillus nidulans | FGSC 26 | Q8TFU8 | Y | ||
We screened the previously identified HAC1 orthologs (2) for putative PUF elements that match the PUF element in C. neoformans var. grubii HXL1. The putative PUF elements in the HXL1 homolog for each species are listed. Included in the table are both sequences with annotated UTRs (yes [Y]) and sequences without annotated UTRs (no [N]). For genes without annotated UTRs, sequences 500 bp upstream and downstream from the coding sequence were scanned for putative PUF elements. For putative PUF elements with variable regions that do not match the known Puf5p consensus sequence, the nonmatching nucleotide is shown in bold and underlined (18).
To determine if the presence of this putative PUF element is unique to C. neoformans, we analyzed the 5′ and 3′ UTRs of HAC1/HXL1 homologs in other species. Our analysis of the HAC1 transcript in yeast and the XBP1 transcript in humans revealed no PUF or PUM elements in either transcript (7, 18, 29–31). To ascertain whether the putative PUF element appears in the UTR of other fungi, we screened available HAC1 homologs for potential PUF elements (2). Interestingly, we found as we moved farther away from C. neoformans var. grubii on the phylogenetic tree that there is a loss of potential PUF elements in the putative UTR of HXL1 homologs (Table 1). The HXL1 gene in the closely related variant C. neoformans var. neoformans also encodes two putative PUF elements, one in the 5′ UTR and one in the 3′ UTR of HXL1. Cryptococcus gattii, a closely related species, has only one putative PUF element in the 5′ UTR of its HXL1 gene that is an exact match to the 5′ UTR element in the C. neoformans gene, suggesting that this element may function in both species. The two distantly related basidiomycetes Coprinopsis cinerea and Ustilago maydis each possess only one putative PUF element in the 3′ UTR. These elements lacked identity to the putative PUF elements found in C. neoformans and did not fit the consensus element for Puf5p in S. cerevisiae (Table 1) (18).
Puf4 binds the HXL1 PUF consensus elements.
Puf4 in C. neoformans is homologous to both Puf4p and Puf5p in S. cerevisiae, as the latter arose from a gene duplication event (32). Despite the homology between Puf4p and Puf5p, each protein binds to a distinct element, with Puf4p binding elements with 3-nucleotide variant regions and Puf5p binding elements with 4-nucleotide variant regions (18). C. neoformans Puf4 bears a higher homology to Puf4p of S. cerevisiae; however, there is no Puf5p homolog in the C. neoformans genome (32). Therefore, we tested the pumilio domain of the C. neoformans Puf4 protein for the ability to bind the putative PUF elements identified in the 5′ UTR and 3′ UTR of the HXL1 transcript, using an RNA electrophoretic mobility shift assay (EMSA). Addition of the recombinant Puf4 pumilio domain to the TYE705-labeled oligonucleotide corresponding to either the 5′ or 3′ consensus element resulted in retardation of the oligonucleotide in the gel, indicating binding (Fig. 1A and B). Addition of an unlabeled competitor oligonucleotide efficiently competed binding, with the 3′ element that deviates from the Puf5p consensus sequence being competed at a lower concentration of competitor than the consensus 5′ element. Addition of an unlabeled competitor in which the element was mutated at the UGUA invariant region resulted in very little competition of the 5′ element. However, a moderate level of competition was seen with a mutated 3′ element oligonucleotide, again suggesting that the interaction of the 3′ element with Puf4 is less stringent than that of the 5′ element (Fig. 1A and B). We further verified the interaction between Puf4 and the HXL1 consensus elements by repeating our binding reactions using a whole-cell protein extract from wild-type C. neoformans, a puf4Δ mutant, or a puf4Δ::PUF4-complemented strain followed by UV cross-linking. We observed an ∼100-kDa band (the approximate size of Puf4) in the UV cross-linked reactions, indicating binding of a protein to the oligonucleotide (Fig. 2A and B). The protein band was present in the wild-type and PUF4-complemented cell lysates but absent in the puf4Δ mutant cell lysate, suggesting that the bound protein is Puf4 (Fig. 2A and B). Once again, we observed competition by an unlabeled competitor containing the respective PUF consensus element, while mutation of the consensus element restricted the ability of the unlabeled oligonucleotide to compete for Puf4 binding (Fig. 2A and B). These results indicate that Puf4 is able to bind to the PUF elements in both the 5′ and 3′ UTRs of HXL1 mRNA.
FIG 1.
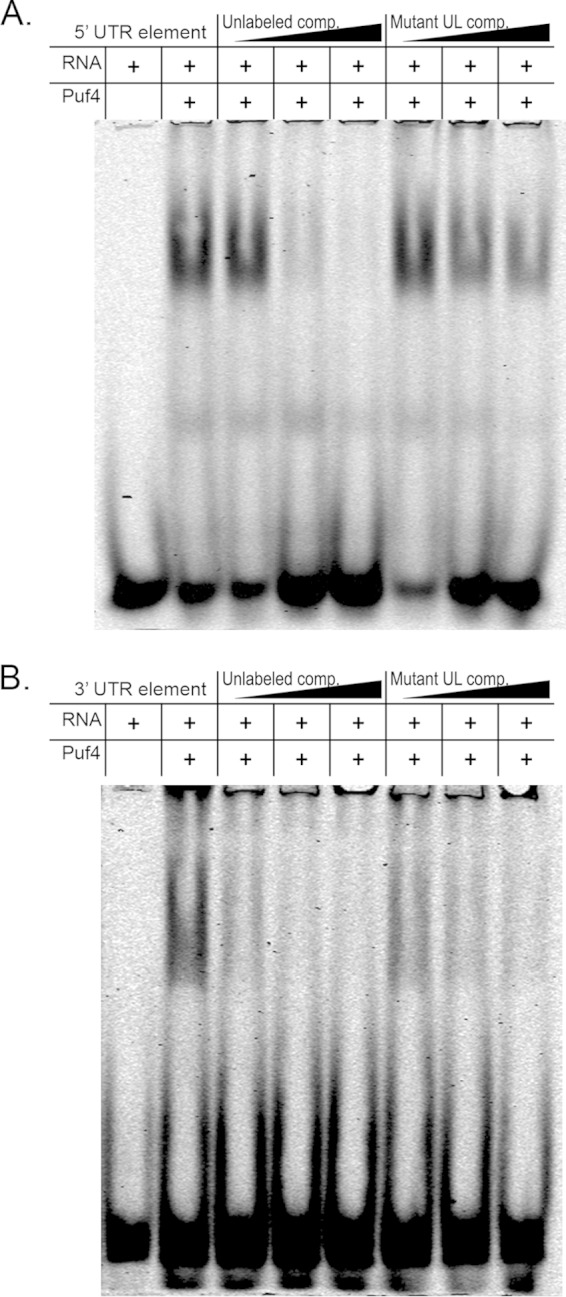
The recombinant Puf4 pumilio domain (Puf4-PD) binds to PUF consensus elements in HXL1 mRNA. Native electrophoretic mobility shift assays demonstrate recombinant Puf4 pumilio domain binding to either the 5′ UTR HXL1 oligonucleotide (A) or the 3′ UTR HXL1 oligonucleotide (B). Reaction mixtures to which labeled oligonucleotide probe was added are marked with a plus sign in the RNA row. Reaction mixtures to which the pumilio domain of Puf4 was added are indicated by a plus sign in the Puf4 row. Increasing concentrations of an unlabeled 5′ UTR (A) or 3′ UTR (B) HXL1 oligonucleotide competitor (Unlabeled comp.) were also added to the reaction mixtures to confirm competition. Unlabeled 5′ UTR (A) or 3′ UTR (B) HXL1 oligonucleotides with a mutated PUF competitor element (Mutant UL comp.) were also used in a competition assay to confirm the specificity of the interaction.
FIG 2.
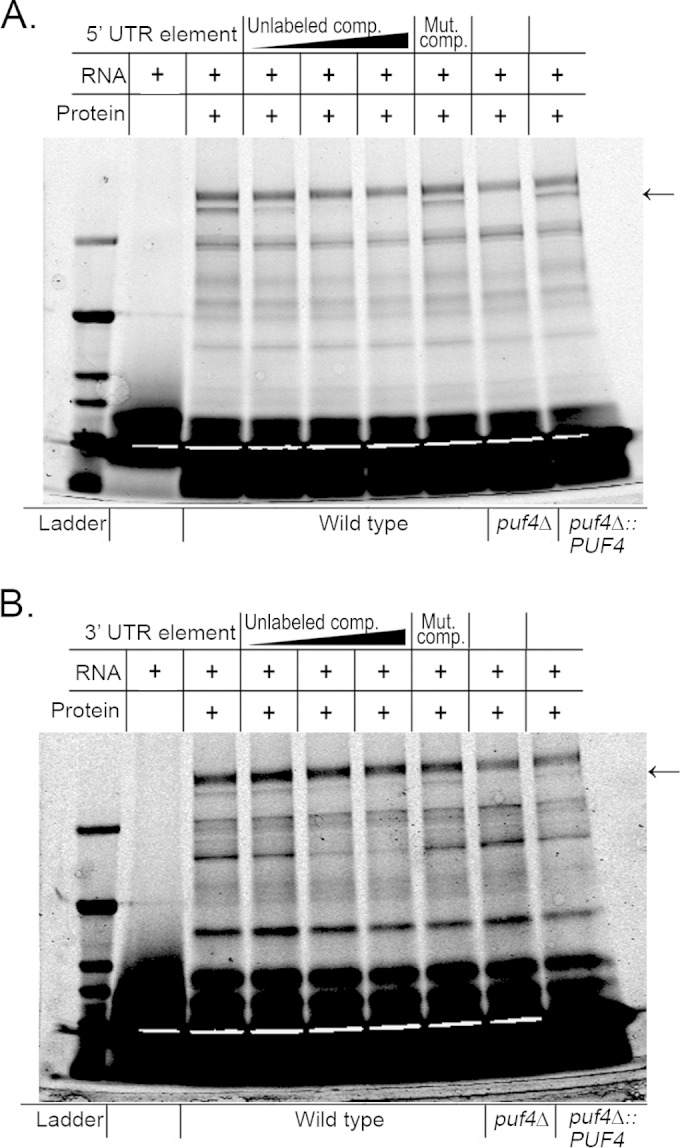
Cross-linked binding assays of the HXL1 putative PUF consensus elements with C. neoformans whole-cell extract. We performed UV cross-linked SDS-PAGE on reaction mixtures containing either the 5′ UTR HXL1 oligonucleotide (A) or the 3′ UTR HXL1 oligonucleotide (B) mixed with whole-cell lysate from wild-type, puf4Δ, or puf4Δ::PUF4 cells. Arrows, the band that is the size of Puf4. The fourth to sixth lanes contain increasing amounts of unlabeled oligonucleotide competitor corresponding to the 5′ UTR or the 3′ UTR of HXL1. The seventh lane contains a 50-fold excess unlabeled competitor with a mutated PUF element corresponding to the 5′ UTR (A) or the 3′ UTR (B) of HXL1. Mut. comp., mutant competitor.
Puf4 facilitates the unconventional splicing of HXL1 mRNA during periods of ER stress.
Unconventional splicing of HXL1 is a crucial and rate-limiting step in UPR activation and is required for the production of a functional Hxl1 protein. As described above, C. neoformans appears to lack both the S. cerevisiae and mammalian features that regulate the splicing of HAC1 and XBP1 transcripts. Given the role of PUF proteins in posttranscriptional processes, we went on to test if deletion of Puf4 would result in splicing aberrancy. The rate of HXL1 mRNA splicing was compared for wild-type and puf4Δ cells during treatment with tunicamycin, a severe ER stressor, or growth at 37°C, a moderate ER stress event to which the cell can adapt. The unconventionally spliced intron of HXL1 is 56 nucleotides, and therefore, the spliced and unspliced variants of the HXL1 transcript are indiscernible by Northern blot analysis (2). Instead, we utilized semiquantitative reverse transcription-PCRs with primers that spanned the splice site to amplify a small section of HXL1 cDNA (Fig. 3). This allowed us to distinguish between and quantify the spliced and the unspliced HXL1 PCR products, which we used to calculate the percentage of the spliced form compared to the total amount of HXL1 mRNA within each strain under each condition (2). During growth in mid-log phase at 30°C, there was a similar, basal level of HXL1 splicing in both the wild type and the puf4Δ mutant (Fig. 3A). When cells were shifted abruptly to 37°C, there was an accumulation of the spliced form in the wild type that maximized by 12 min after the shift and persisted for the remainder of the time course. In contrast, there was little accumulation of the spliced form in the puf4Δ mutant, suggesting a defect in HXL1 splicing (Fig. 3A).
FIG 3.
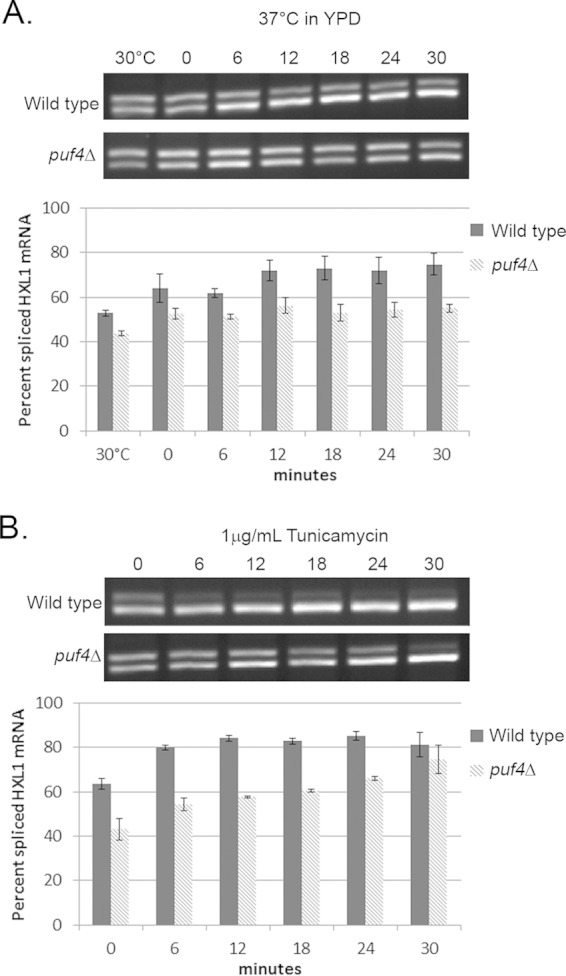
The rate of HXL1 mRNA splicing is delayed in the puf4Δ strain compared to that in the wild type. (A, B) The amount of unspliced and spliced HXL1 mRNA was compared between wild-type and puf4Δ cells. RNA was isolated from wild-type cells or puf4Δ cells during growth at 37°C (A) or treatment with tunicamycin (B). Afterward, the RNA was converted to cDNA and then amplified in a semiquantitative PCR using primers that span the HXL1 splice site and electrophoresed on an agarose gel. Unless indicated otherwise, the numbers above the gels are times (in minutes). The higher-molecular-mass band corresponds to unspliced HXL1 transcript. The lower-molecular-mass band corresponds to spliced HXL1 mRNA. The graphs depict the percentage of spliced HXL1 mRNA. Data are representative of those from at least three biological replicates.
Our previous work demonstrated that ER stress is transiently induced during the shift to 37°C. To evaluate a more intense ER stress induction, we added the ER stress-inducing agent tunicamycin to cultures grown at 30°C and followed HXL1 splicing over a time course of 30 min in the wild type and the puf4Δ mutant (Fig. 3B). In this case, time zero represents the time immediately after the addition of tunicamycin. As demonstrated in Fig. 3B, accumulation of spliced HXL1 was visible in the wild type immediately after tunicamycin addition, with maximal HXL1 splicing being seen by 12 min after tunicamycin addition. In contrast, there was a slow accumulation of spliced HXL1 in the puf4Δ mutant over the course of the 30 min, with splicing approaching wild-type levels by the 30-min time point. These data suggest that in the absence of Puf4, HXL1 splicing is delayed during induction of ER stress by either a shift in temperature or tunicamycin treatment.
A delay in UPR activation hinders UPR transcript induction in the puf4Δ mutant.
To determine whether the defect in HXL1 splicing seen in the puf4Δ mutant impedes the downstream effector function of Hxl1 and UPR induction, we measured the levels of ER stress response transcripts in response to either a shift to 37°C or treatment with tunicamycin. Using the ER chaperone KAR2 as a representative transcript of UPR transcriptional activation (1, 33), we performed a time course experiment measuring KAR2 and HXL1 mRNA levels following a shift to 37°C (Fig. 4A and B). As we have demonstrated previously (1), KAR2 mRNA accumulated in response to temperature stress, reaching maximum induction by 1 h after the shift to 37°C, after which the KAR2 abundance was reduced (Fig. 4A). In the puf4Δ mutant, the accumulation of KAR2 mRNA was slowed, with maximal induction occurring at 2 h after the shift to 37°C. Interestingly, KAR2 abundance was subsequently reduced to wild-type levels by 4 h after the shift to 37°C (Fig. 4A). A similar delay was seen for HXL1 during temperature adaptation, with maximal induction occurring after 30 min of temperature stress in the wild type but not until 1 h in the puf4Δ mutant (Fig. 4B).
FIG 4.
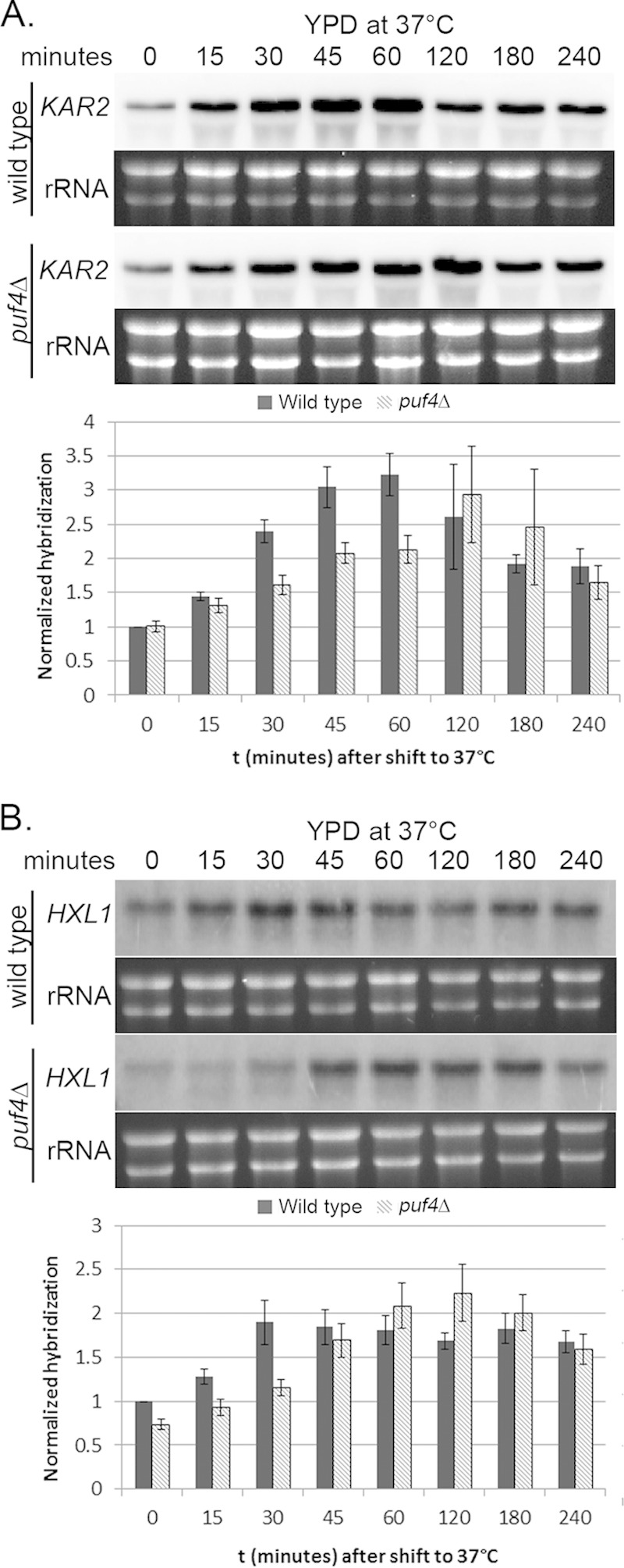
Delayed yet persistent UPR transcript induction is observed in the puf4Δ mutant during growth at 37°C. KAR2 (A) and HXL1 (B) mRNA levels in the wild-type strain or the puf4Δ mutant during growth at 37°C were analyzed by Northern blotting. Histograms were generated by normalizing the Northern blot hybridization levels of the given transcript to the rRNA levels to determine the change in transcript levels over time (t). Error bars are representative of at least three biological replicates.
In response to the addition of tunicamycin, we again observed a delay in the induction of both KAR2 and HXL1 transcripts in the puf4Δ mutant compared to the rate of induction in the wild-type strain (Fig. 5A and B). For KAR2, the maximal induction in response to tunicamycin treatment was observed at 1 h in the wild type but not until 2 h in the puf4Δ mutant (Fig. 5A). For HXL1, maximal induction occurred by 2 h in the wild type and by 3 h in the puf4Δ mutant (Fig. 5B). The absence of a putative Puf4 consensus element in the KAR2 transcript suggests that the influence of Puf4 on KAR2 mRNA levels (Fig. 4A and 5A) is due to an upstream component. Given that splicing of HXL1 is essential for the activation of UPR gene expression, we hypothesize that the slower induction of UPR transcripts under both stress conditions is due to a delay in HXL1 splicing in the puf4Δ mutant. This delay in splicing then affects downstream components, including the transcription of UPR genes. Interestingly, induction of HXL1 expression is itself delayed in the puf4Δ mutant (Fig. 4B and 5B), suggesting a feed-forward autoregulation of HXL1 expression.
FIG 5.
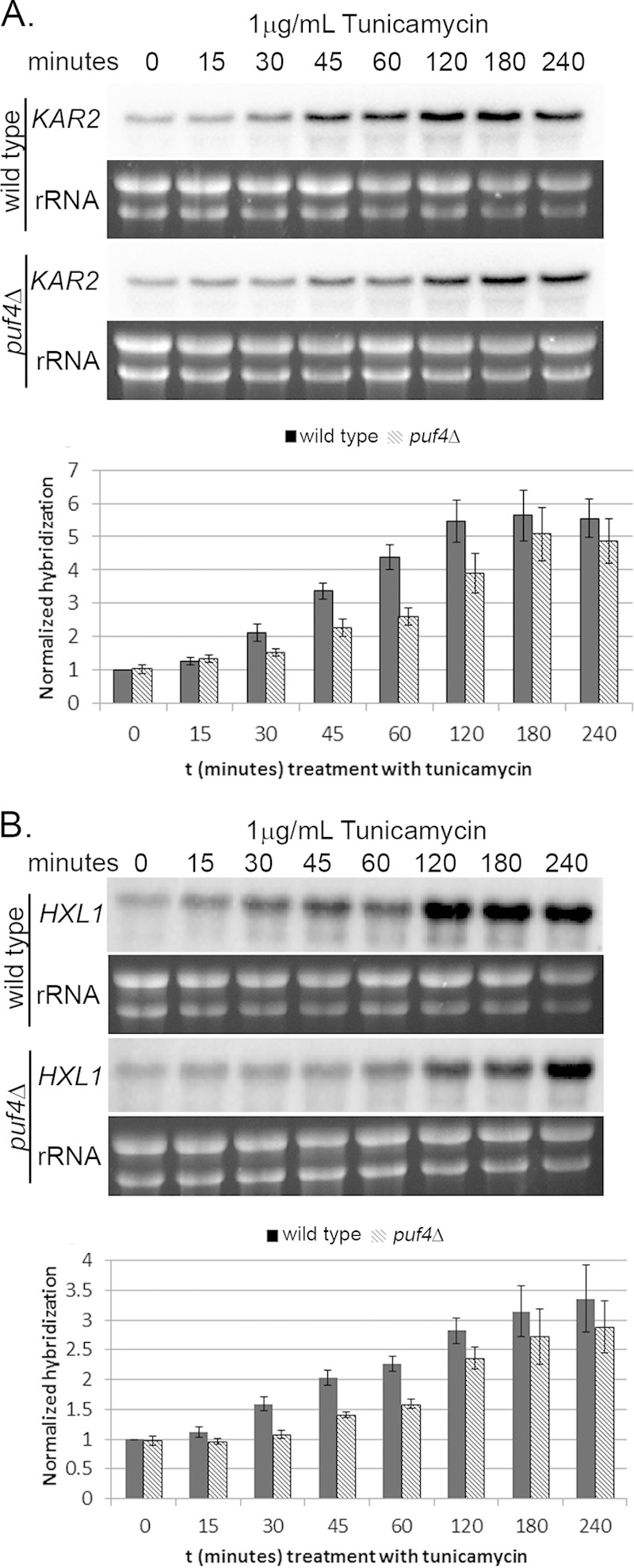
Delayed UPR transcript induction is observed in puf4Δ cells during tunicamycin treatment. KAR2 (A) and HXL1 (B) mRNA levels in the wild-type strain or the puf4Δ mutant during treatment with tunicamycin were assayed by Northern blotting. Histograms were generated by normalizing the Northern blot hybridization levels of the given transcript to the rRNA levels to determine the change in transcript levels over time. Error bars are representative of at least three biological replicates.
HXL1 mRNA decay is defective in the absence of Puf4.
The mRNA deadenylase Ccr4 facilitates the degradation of ER transcripts following their maximum induction after 1 h at 37°C (1). Because PUF proteins are known regulators of RNA stability, we tested whether Puf4 is mediating the decay of HXL1 at 1 h after the shift to 37°C. To assay transcript stability, we performed RNA time course assays using the transcriptional inhibitor 1,10-phenanthroline and compared HXL1 and KAR2 mRNA decay rates between the wild-type strain and the puf4Δ mutant (1, 28). Log2 plots graphing HXL1 mRNA decay between the strains show that the HXL1 transcript was significantly stabilized in the puf4Δ mutant compared to the stability in the wild-type strain after 1 h at 37°C (Fig. 6A). No difference in HXL1 stability was observed under unstressed conditions or after tunicamycin treatment (data not shown), suggesting that the influence of Puf4 on HXL1 mRNA stability is temporally controlled and specific to temperature stress. To test whether Puf4-mediated decay is transcript specific, we compared the stability of KAR2 mRNA in wild-type cells and the puf4Δ mutant. We observed similar decay patterns for the KAR2 transcript in puf4Δ mutant and wild-type cells (Fig. 6B), which was expected, given the lack of a Puf4 consensus element in the KAR2 transcript. These data suggest that, in addition to controlling the activation of the ER stress response through the splicing of HXL1 mRNA, Puf4 may also control the intensity and duration of the response by promoting the degradation of HXL1 mRNA during stress response attenuation.
FIG 6.
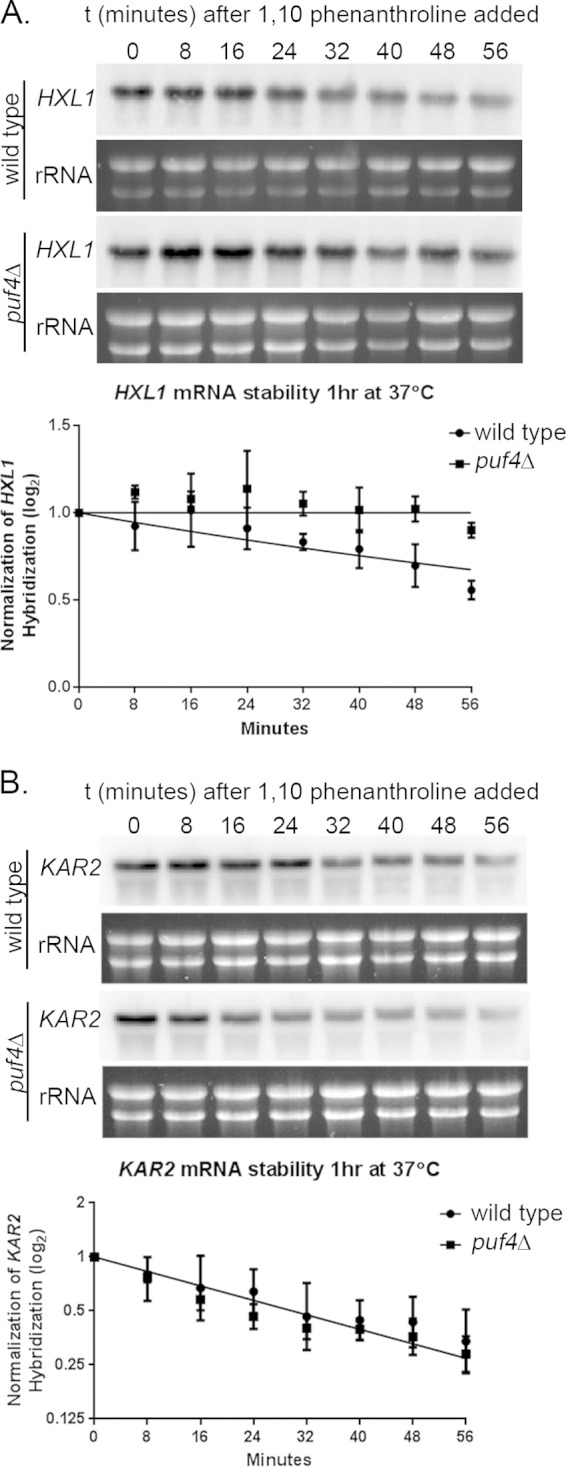
Puf4 regulates the stability of HXL1 mRNA but not KAR2 mRNA. Strains were grown for 1 h at 37°C, at which time 1,10-phenanthroline was added to halt transcription. RNA was harvested from cells at 8-min intervals and used for Northern blot analysis of HXL1 (A) and KAR2 (B) transcripts to measure mRNA stability over time. Graphs were generated by normalizing the Northern blot hybridization levels of the given transcript to the rRNA levels to determine the change in transcript stability over time. Graphs depict log2 plots of the mRNA half-life of pooled data from at least three biological replicates.
PUF4 deletion gives rise to a pleiotropic ER stress phenotype.
C. neoformans mutants that are defective in UPR activation are sensitive to UPR-inducing agents, such as dithiothreitol (DTT) and tunicamycin (2, 15). To determine if deletion of Puf4 also results in the same drug sensitivity phenotypes found in previous UPR mutants, we compared the sensitivity of the wild type, the puf4Δ mutant, and the complemented strain to DTT and tunicamycin using spot plate analysis. As expected, the puf4Δ mutant was sensitive to DTT in the spot plate assay (Fig. 7A). Surprisingly, however, the puf4Δ mutant was found to be resistant to tunicamycin at 250 ng/ml (Fig. 7A). Growth for all three strains was inhibited completely at 1 μg/ml, the concentration used in the splicing experiments described above. Complementation of the deletion strain with wild-type PUF4 restored wild-type sensitivity to both agents.
FIG 7.

Deletion of PUF4 results in pleiotropic ER stress phenotypes. (A) Serial dilutions of cells on spot plates comparing the growth of the wild-type, puf4Δ, and puf4Δ::PUF4 strains during treatment with DTT or tunicamycin (TM). Cells were spotted onto YPD agar plates containing either 15 mM DTT or 250 ng/ml tunicamycin and then incubated for 3 days and photographed. (B) Native electrophoretic mobility shift assay of the recombinant Puf4 pumilio domain incubated with an RNA oligonucleotide corresponding to a portion of the 3′ UTR of ALG7. Reaction mixtures to which labeled oligonucleotide probe was added are marked with a plus sign in the RNA row. Reaction mixtures to which the pumilio domain of Puf4 was added are indicated by a plus sign in the Puf4 row. Increasing amounts of unlabeled 3′ UTR ALG7 oligonucleotide competitor (Unlabeled comp.) were added to confirm competition. Unlabeled 3′ UTR ALG7 oligonucleotides with a mutated PUF competitor element (Mutant UL comp.) were added to confirm the specificity of the interaction. (C) UV cross-linked SDS-PAGE with the 3′ UTR ALG7 oligonucleotide and whole-cell lysate from the wild-type, puf4Δ, or puf4Δ::PUF4 strain. Arrow, the band that is the size of Puf4. The fourth to sixth lanes contain increasing amounts of the unlabeled 3′ UTR ALG7 oligonucleotide competitor. The seventh lane contains a 50-fold excess unlabeled competitor for the 3′ UTR of ALG7 with the PUF element mutated.
There is a fundamental difference in the mechanism of ER stress induction between DTT and tunicamycin. DTT acts to nonspecifically alter protein folding in the ER through the reduction of disulfide bonds, whereas tunicamycin is a specific inhibitor of Alg7, a UDP-N-acetylglucosamine-1-phosphate transferase required for N-linked protein glycosylation (34, 35). Because resistance to tunicamycin could be conferred by dysregulation of ALG7 mRNA (36, 37), we assessed the 5′ and 3′ UTRs of ALG7 for putative Puf4-binding elements. The 3′ UTR of ALG7 was found to contain a putative Puf4 consensus sequence. This consensus element bound the pumilio domain of Puf4 in vitro (Fig. 7B and C), indicating that the resistance to tunicamycin in the puf4Δ mutant may be a result of Puf4-dependent regulation of the ALG7 transcript. Analysis of ALG7 mRNA stability did not reveal any significant differences between the wild type and the puf4Δ mutant under any of the conditions tested (data not shown), suggesting that Puf4-mediated regulation may be at the level of translation. Further studies are needed to decipher the relationship between Puf4, ALG7, and the tunicamycin resistance phenotype of the puf4Δ mutant.
Loss of Puf4 reduces in vitro and in vivo growth but not virulence.
Previous studies have shown that components of the ER stress response in C. neoformans are critical for virulence, with deletion of either HXL1 or IRE1 resulting in attenuated virulence (2). Given the effect of Puf4 on HXL1 splicing and subsequent downstream signaling, we predicted that deletion of Puf4 would result in an attenuation of growth at 37°C and virulence. Growth was assayed by spot plate assays at 30°C, 37°C, and 39°C. As demonstrated in Fig. 8A, the puf4Δ mutant grew at the same rate as the wild type at 30°C, but growth was modestly attenuated at 37°C and robustly attenuated at 39°C, supporting a role for Puf4 in temperature adaptation. To assay pathogenesis, mouse models of both intranasal and intravenous inoculation were used. In both assays, there was no significant difference in virulence between wild-type cells, the puf4Δ mutant, and the puf4Δ::PUF4-complemented strain (Fig. 8B). To further examine the virulence potential of the puf4Δ mutant, we performed a competition assay in which wild-type cells and puf4Δ cells were injected into mice and then the brains of the mice were harvested on days 1, 2, and 3 following infection and the numbers of CFU (measured as the number of CFU per gram of brain tissue) of each strain were counted. The initial inoculum had slightly higher levels of viable puf4Δ mutant cells than wild-type cells, yet we observed significantly lower levels of puf4Δ cells in the brain on days 1 to 3 (Fig. 8C). Even though the puf4Δ mutant had lower cell counts than the wild type, the brains of the mice still had a high fungal burden of the puf4Δ mutant. Therefore, it is unsurprising that mice infected with the puf4Δ strain died at the same rate as those infected with the wild-type strain. The lower number of puf4Δ mutant cells in the brain is likely attributable to the slight defect in growth at 37°C. However, it is also possible that in the puf4Δ mutant, the increased stability of the HXL1 mRNA may compensate for the delay in ER stress induction, balancing the stress response and promoting host adaptation in the absence of Puf4.
FIG 8.

Deletion of PUF4 impairs in vitro and in vivo growth but not virulence. (A) Serial dilutions of cells on spot plates comparing the growth of the wild-type, puf4Δ, and puf4Δ::PUF4 strains during growth at 30°C, 37°C, or 39°C. Cells were spotted onto YPD agar plates and then incubated for 3 days and photographed. (B) (Top) Mice were given an intranasal inoculation of 105 CFU of the wild-type, puf4Δ, or puf4Δ::PUF4 strain or 50 μl of saline as a negative control, and then the mice were monitored for 60 days. The percent survival is displayed. (Bottom) Mice were given an intravenous tail vein injection of 106 CFU of the wild-type, puf4Δ, or puf4Δ::PUF4 strain or 100 μl of saline as a negative control, and morbidity was monitored for 25 days. The percent survival of the mice is plotted. (C) To assay strain competition in vivo, a suspension containing equal amounts of wild-type cells and puf4Δ cells was injected into mice. The mice were sacrificed, and brain CFU counts for each strain were obtained on days 1, 2, and 3. The levels of the wild-type strain and puf4Δ cells used in the injected suspension are also listed and are marked input. Error bars were generated with results from 3 mice. Error bars for input wild-type and puf4Δ cell counts were calculated using three separate plating CFU counts. *, P < 0.05.
DISCUSSION
The UPR is essential for recognizing ER perturbations and triggering the activation of the appropriate response machinery under conditions of ER stress. In C. neoformans, the UPR is also necessary for virulence and adaptation to growth at elevated temperatures (1, 2, 15). The unconventional splicing of the mRNA encoding the major ER stress-responsive transcription factor is a highly conserved process by which the UPR is engaged; however, the mechanisms that regulate this unconventional splicing reaction are highly divergent between yeast and mammals.
In this study, we report that an evolutionarily conserved posttranscriptional regulatory unit, a pumilio protein and its cognate element, has been co-opted by C. neoformans to regulate not only the unconventional splicing of the HXL1 mRNA encoding the UPR transcription factor but also HXL1 mRNA decay. The influence of Puf4 on HXL1 mRNA splicing represents a novel role for a PUF protein. We hypothesize that Puf4-mediated regulation of both HXL1 mRNA splicing and stability allows a single mRNA binding protein to coordinate the posttranscriptional coupling of UPR induction via splicing and UPR resolution by HXL1 mRNA decay. The importance of controlling the activation and attenuation of the UPR has previously been reported in S. cerevisiae, where Kar2p (also known as BiP), Ire1p, and Ptc2p (a phosphatase controlling Ire1p kinase activity) have been shown to control the kinetics of splicing, with a disruption in splicing dynamics resulting in defective responses to ER stress (38–41). Similarly, we observed that deletion of Puf4 resulted in delays in HXL1 splicing efficiency and, as a consequence, a change in the dynamics of the UPR. These effects likely contribute to the temperature sensitivity observed in the puf4Δ mutant.
Despite the conservation of PUF proteins and their cognate elements across evolution, there is a lack of conservation in the mRNA targets that are regulated by PUF proteins, even among closely related species. Previous studies in S. cerevisiae have shown that Puf4p and Puf5p bind nuclear and nucleolar transcripts (18). This is in contrast to our data describing Puf4 regulation of ER-related transcripts in C. neoformans, indicating an evolutionary divergence in the role of Puf4/Puf5 between the two species. Also of note, deletion of Pum1, a Puf3p homolog in C. neoformans, does not result in the characteristic mitochondrial phenotypes seen in the puf3Δ mutant of S. cerevisiae (42, 43; A. L. Bloom and J. C. Panepinto, unpublished data). Therefore, Pum1 in C. neoformans has also likely evolved to regulate a divergent subset of transcripts from Puf3p in S. cerevisiae. It will be of interest to see whether the remaining PUF proteins in C. neoformans have evolved to coordinate RNA regulons that are alternatives to their corresponding homologs in S. cerevisiae.
There are two Puf4-binding elements in the HXL1 transcript, one in the 5′ UTR that fits the published consensus sequence for S. cerevisiae Puf5p and one in the 3′ UTR that deviates in the variable region from this published consensus sequence (18). The PUF regulatory paradigm places the cognate cis elements in the 3′ UTR with little variation (44). However, this study identifies elements within both the 5′ UTR and the 3′ UTR of the HXL1 transcript that bind Puf4. The in vitro recombinant pumilio domain binding studies suggest that the 5′ element is more specific than the 3′ element in interacting with Puf4, though both elements interact with Puf4 in cell extracts. It is possible that one element may influence splicing, while the other element regulates HXL1 mRNA stability. Ongoing studies will determine the contributions of each element to the posttranscriptional regulation of the HXL1 transcript by Puf4.
While screening for ER-related phenotypes in the puf4Δ mutant, we identified the puf4Δ mutant to be sensitive to DTT, a reducing agent that influences protein folding in the ER, suggesting that Puf4 is required for a properly functioning ER. However, the puf4Δ strain also exhibited resistance to the ER stress-inducing drug tunicamycin. The dichotomy between these two phenotypes suggests that the influence of Puf4 on the ER is complex and multifaceted. DTT and tunicamycin each target different aspects of the ER to induce ER stress. Tunicamycin interferes with protein glycosylation by targeting Alg7. Investigation into a potential role for Puf4 in regulating Alg7 identified a Puf4 consensus element in the ALG7 transcript. We hypothesize that the tunicamycin resistance phenotype is the result of Puf4 posttranscriptionally regulating the ALG7 transcript, resulting in an increase in Alg7 protein levels that confers tunicamycin resistance. However, the mechanism by which Puf4 regulates ALG7 mRNA remains unclear. The identification of the ALG7 transcript as an additional target for Puf4 suggests that Puf4 has multiple roles in modulating the ER, aside from its role in the regulation of HXL1 and the UPR. Characterization of the Puf4 regulon will be necessary to determine the extent to which Puf4 is influencing different ER functions.
In C. neoformans, the puf4Δ mutant, despite exhibiting temperature sensitivity and cell wall integrity defects, was found to be equivalent to the wild-type strain in the ability to cause mortality in a mouse model of cryptococcosis (32). Further investigation of the in vivo growth rate within a mouse model revealed that the puf4Δ mutant had lower numbers of CFU in the brain than the wild-type cells, but not to an extent that would impact survival in our model. The unexpected virulence capacity of the puf4Δ mutant suggests that the increased stability of the HXL1 mRNA may compensate for the delay in ER stress induction, balancing stress induction and resolution and promoting adaptation. It is also possible that additional Puf4 targets influence the ER stress response and promote adaptation in vivo.
C. neoformans, as a soil-dwelling microorganism capable of causing disease in humans, must be highly adaptable to new environments. Our results demonstrate that C. neoformans has co-opted a conserved RNA regulatory module for the purpose of controlling the rates of both the engagement and the resolution of the UPR. The regulation of the ER is a novel function for a pumilio protein and is likely unique to the genus Cryptococcus. Because the ER stress response is fundamental to the adaptability of C. neoformans, additional work to define unique aspects of this response in C. neoformans and other fungi could lead to novel therapeutic targets.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded in part by NIAID grant R01-AI089920 to J.C.P. and an American Heart Association predoctoral fellowship to V.E.G.
We thank Amy Jacobs for use of the LI-COR Odyssey imaging technology and Laurie K. Read for her assistance with manuscript preparation.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00273-14.
REFERENCES
- 1.Havel VE, Wool NK, Ayad D, Downey KM, Wilson CF, Larsen P, Djordjevic JT, Panepinto JC. 2011. Ccr4 promotes resolution of the endoplasmic reticulum stress response during host temperature adaptation in Cryptococcus neoformans. Eukaryot Cell 10:895–901. doi: 10.1128/EC.00006-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheon SA, Jung KW, Chen YL, Heitman J, Bahn YS, Kang HA. 2011. Unique evolution of the UPR pathway with a novel bZIP transcription factor, Hxl1, for controlling pathogenicity of Cryptococcus neoformans. PLoS Pathog 7:e1002177. doi: 10.1371/journal.ppat.1002177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. 2000. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell 11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaufman RJ. 1999. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev 13:1211–1233. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- 5.Schroder M, Kaufman RJ. 2005. ER stress and the unfolded protein response. Mutat Res 569:29–63. doi: 10.1016/j.mrfmmm.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 6.Cox JS, Shamu CE, Walter P. 1993. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73:1197–1206. doi: 10.1016/0092-8674(93)90648-A. [DOI] [PubMed] [Google Scholar]
- 7.Hooks KB, Griffiths-Jones S. 2011. Conserved RNA structures in the non-canonical Hac1/Xbp1 intron. RNA Biol 8:552–556. doi: 10.4161/rna.8.4.15396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aragon T, van Anken E, Pincus D, Serafimova IM, Korennykh AV, Rubio CA, Walter P. 2009. Messenger RNA targeting to endoplasmic reticulum stress signalling sites. Nature 457:736–740. doi: 10.1038/nature07641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yanagitani K, Imagawa Y, Iwawaki T, Hosoda A, Saito M, Kimata Y, Kohno K. 2009. Cotranslational targeting of XBP1 protein to the membrane promotes cytoplasmic splicing of its own mRNA. Mol Cell 34:191–200. doi: 10.1016/j.molcel.2009.02.033. [DOI] [PubMed] [Google Scholar]
- 10.Yanagitani K, Kimata Y, Kadokura H, Kohno K. 2011. Translational pausing ensures membrane targeting and cytoplasmic splicing of XBP1u mRNA. Science 331:586–589. doi: 10.1126/science.1197142. [DOI] [PubMed] [Google Scholar]
- 11.Tsvetanova NG, Riordan DP, Brown PO. 2012. The yeast Rab GTPase Ypt1 modulates unfolded protein response dynamics by regulating the stability of HAC1 RNA. PLoS Genet 8:e1002862. doi: 10.1371/journal.pgen.1002862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Welihinda AA, Tirasophon W, Kaufman RJ. 2000. The transcriptional co-activator ADA5 is required for HAC1 mRNA processing in vivo. J Biol Chem 275:3377–3381. doi: 10.1074/jbc.275.5.3377. [DOI] [PubMed] [Google Scholar]
- 13.Cheon SA, Jung KW, Bahn YS, Kang HA. 2014. The unfolded protein response (UPR) pathway in Cryptococcus. Virulence 5:341–350. doi: 10.4161/viru.26774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glazier VE, Panepinto JC. 2014. The ER stress response and host temperature adaptation in the human fungal pathogen Cryptococcus neoformans. Virulence 5:351–356. doi: 10.4161/viru.27187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jung KW, Kang HA, Bahn YS. 2013. Essential roles of the Kar2/BiP molecular chaperone downstream of the UPR pathway in Cryptococcus neoformans. PLoS One 8:e58956. doi: 10.1371/journal.pone.0058956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blewett NH, Goldstrohm AC. 2012. A eukaryotic translation initiation factor 4E-binding protein promotes mRNA decapping and is required for PUF repression. Mol Cell Biol 32:4181–4194. doi: 10.1128/MCB.00483-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garcia-Rodriguez LJ, Gay AC, Pon LA. 2007. Puf3p, a pumilio family RNA binding protein, localizes to mitochondria and regulates mitochondrial biogenesis and motility in budding yeast. J Cell Biol 176:197–207. doi: 10.1083/jcb.200606054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerber AP, Herschlag D, Brown PO. 2004. Extensive association of functionally and cytotopically related mRNAs with Puf family RNA-binding proteins in yeast. PLoS Biol 2:E79. doi: 10.1371/journal.pbio.0020079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu W, Deng Y, Zenklusen D, Singer RH. 2004. A new yeast PUF family protein, Puf6p, represses ASH1 mRNA translation and is required for its localization. Genes Dev 18:1452–1465. doi: 10.1101/gad.1189004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hook BA, Goldstrohm AC, Seay DJ, Wickens M. 2007. Two yeast PUF proteins negatively regulate a single mRNA. J Biol Chem 282:15430–15438. doi: 10.1074/jbc.M611253200. [DOI] [PubMed] [Google Scholar]
- 21.Jiang H, Guo X, Xu L, Gu Z. 2012. Rewiring of posttranscriptional RNA regulons: Puf4p in fungi as an example. Mol Biol Evol 29:2169–2176. doi: 10.1093/molbev/mss085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keene JD. 2007. RNA regulons: coordination of post-transcriptional events. Nat Rev Genet 8:533–543. doi: 10.1038/nrg2111. [DOI] [PubMed] [Google Scholar]
- 23.Quenault T, Lithgow T, Traven A. 2011. PUF proteins: repression, activation and mRNA localization. Trends Cell Biol 21:104–112. doi: 10.1016/j.tcb.2010.09.013. [DOI] [PubMed] [Google Scholar]
- 24.Saint-Georges Y, Garcia M, Delaveau T, Jourdren L, Le Crom S, Lemoine S, Tanty V, Devaux F, Jacq C. 2008. Yeast mitochondrial biogenesis: a role for the PUF RNA-binding protein Puf3p in mRNA localization. PLoS One 3:e2293. doi: 10.1371/journal.pone.0002293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bell-Pedersen D, Lewis ZA, Loros JJ, Dunlap JC. 2001. The Neurospora circadian clock regulates a transcription factor that controls rhythmic expression of the output eas(ccg-2) gene. Mol Microbiol 41:897–909. doi: 10.1046/j.1365-2958.2001.02558.x. [DOI] [PubMed] [Google Scholar]
- 26.Dean JL, Wait R, Mahtani KR, Sully G, Clark AR, Saklatvala J. 2001. The 3′ untranslated region of tumor necrosis factor alpha mRNA is a target of the mRNA-stabilizing factor HuR. Mol Cell Biol 21:721–730. doi: 10.1128/MCB.21.3.721-730.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chritton JJ, Wickens M. 2010. Translational repression by PUF proteins in vitro. RNA 16:1217–1225. doi: 10.1261/rna.2070110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bloom AL, Solomons JT, Havel VE, Panepinto JC. 2013. Uncoupling of mRNA synthesis and degradation impairs adaptation to host temperature in Cryptococcus neoformans. Mol Microbiol 89:65–83. doi: 10.1111/mmi.12258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cox JS, Walter P. 1996. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell 87:391–404. doi: 10.1016/S0092-8674(00)81360-4. [DOI] [PubMed] [Google Scholar]
- 30.Spassov DS, Jurecic R. 2003. The PUF family of RNA-binding proteins: does evolutionarily conserved structure equal conserved function? IUBMB Life 55:359–366. doi: 10.1080/15216540310001603093. [DOI] [PubMed] [Google Scholar]
- 31.Wang X, McLachlan J, Zamore PD, Hall TM. 2002. Modular recognition of RNA by a human pumilio-homology domain. Cell 110:501–512. doi: 10.1016/S0092-8674(02)00873-5. [DOI] [PubMed] [Google Scholar]
- 32.Gerik KJ, Donlin MJ, Soto CE, Banks AM, Banks IR, Maligie MA, Selitrennikoff CP, Lodge JK. 2005. Cell wall integrity is dependent on the PKC1 signal transduction pathway in Cryptococcus neoformans. Mol Microbiol 58:393–408. doi: 10.1111/j.1365-2958.2005.04843.x. [DOI] [PubMed] [Google Scholar]
- 33.Miyazaki T, Nakayama H, Nagayoshi Y, Kakeya H, Kohno S. 2013. Dissection of Ire1 functions reveals stress response mechanisms uniquely evolved in Candida glabrata. PLoS Pathog 9:e1003160. doi: 10.1371/journal.ppat.1003160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barnes G, Hansen WJ, Holcomb CL, Rine J. 1984. Asparagine-linked glycosylation in Saccharomyces cerevisiae: genetic analysis of an early step. Mol Cell Biol 4:2381–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Braakman I, Helenius J, Helenius A. 1992. Manipulating disulfide bond formation and protein folding in the endoplasmic reticulum. EMBO J 11:1717–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen Y, Feldman DE, Deng C, Brown JA, De Giacomo AF, Gaw AF, Shi G, Le QT, Brown JM, Koong AC. 2005. Identification of mitogen-activated protein kinase signaling pathways that confer resistance to endoplasmic reticulum stress in Saccharomyces cerevisiae. Mol Cancer Res 3:669–677. doi: 10.1158/1541-7786.MCR-05-0181. [DOI] [PubMed] [Google Scholar]
- 37.Scocca JR, Hartog KO, Krag SS. 1988. Evidence of gene amplification in tunicamycin-resistant Chinese hamster ovary cells. Biochem Biophys Res Commun 156:1063–1069. doi: 10.1016/S0006-291X(88)80740-X. [DOI] [PubMed] [Google Scholar]
- 38.Chawla A, Chakrabarti S, Ghosh G, Niwa M. 2011. Attenuation of yeast UPR is essential for survival and is mediated by IRE1 kinase. J Cell Biol 193:41–50. doi: 10.1083/jcb.201008071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pincus D, Chevalier MW, Aragon T, van Anken E, Vidal SE, El-Samad H, Walter P. 2010. BiP binding to the ER-stress sensor Ire1 tunes the homeostatic behavior of the unfolded protein response. PLoS Biol 8:e1000415. doi: 10.1371/journal.pbio.1000415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rubio C, Pincus D, Korennykh A, Schuck S, El-Samad H, Walter P. 2011. Homeostatic adaptation to endoplasmic reticulum stress depends on Ire1 kinase activity. J Cell Biol 193:171–184. doi: 10.1083/jcb.201007077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Welihinda AA, Tirasophon W, Green SR, Kaufman RJ. 1998. Protein serine/threonine phosphatase Ptc2p negatively regulates the unfolded-protein response by dephosphorylating Ire1p kinase. Mol Cell Biol 18:1967–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiang H, Guan W, Gu Z. 2010. Tinkering evolution of post-transcriptional RNA regulons: puf3p in fungi as an example. PLoS Genet 6:e1001030. doi: 10.1371/journal.pgen.1001030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang L, Tian X, Gyawali R, Upadhyay S, Foyle D, Wang G, Cai JJ, Lin X. 2014. Morphotype transition and sexual reproduction are genetically associated in a ubiquitous environmental pathogen. PLoS Pathog 10:e1004185. doi: 10.1371/journal.ppat.1004185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wickens M, Bernstein DS, Kimble J, Parker R. 2002. A PUF family portrait: 3′UTR regulation as a way of life. Trends Genet 18:150–157. doi: 10.1016/S0168-9525(01)02616-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.