

Abstract
Rasmussen’s encephalitis (RE) is a rare neurological disorder of inflammatory aetiology characterised by encephalitis, intractable seizures, hemiparesis, variable motor deficits, and dementia. It is not commonly considered in the clinical differential diagnosis of a patient with a past history of a stroke-like episode presenting with status epilepticus. It is suspected mainly in children in whom the disease is more common. Here we report a case of an adult man presenting with a common symptom of seizure caused by Rasmussen’s encephalitis.
Keywords: Rasmussen’s encephalitis, viral encephalitis
Implications for Practice:
-
What is known about this subject?
Rasmussen’s encephalitis, a rare neurological disorder that has been reported in children, causes cerebral atrophy and intractable seizures.
-
What new information is offered in this case study?
Rasmussen’s encephalitis, while rare in children, is even rarer in adults. The adult male in this case presented with a common symptom (seizure) caused by Rasmussen’s encephalitis.
-
What are the implications for research, policy, or practice?
Rasmussen’s encephalitis should be considered as a differential diagnosis not only in children but also in adults that present with intractable seizures.
Background
Rasmussen’s encephalitis (RE) is an uncommon disease of the nervous system that is usually seen in children. It is characterised by unilateral hemispheric atrophy, focal intractable seizures, and worsening neurological deficits. Historically, the condition was first described by Rasmussen et al. who published a clinico-pathological report of three children with a longstanding illness causing focal seizures and worsening damage to one cerebral hemisphere.1 Though considered as an illness in children, adult and adolescent patients account for 10 per cent of all cases2 with 58 years being the oldest reported age.3 Onset of symptoms in the adult age group is associated with a more protracted and milder clinical course with less residual functional deficit, lower degree of hemiatrophy, and more frequent occipital lobe seizure onset.2,4
The average age of clinical presentation is six years.2,5,6 Three clinical stages have been proposed:
The prodromal stage, which has a mean duration of 7.1 months (range: 0 months to 8.1 years),2,5–7 has a low seizure occurrence, and mild hemiparesis.
The acute stage has a median duration of eight months2,5,6 and is characterised by frequent seizures. The neurological symptoms become apparent in the form of worsening hemiparesis, hemianopia, and cognitive deterioration and aphasia if the dominant hemisphere is involved.2,5,6
The residual stage is the last stage with permanent damage and seizures being less frequent than in the acute stage.2,5,6
This complex case describes an adult patient who presented with what was eventually diagnosed as Rasmussen’s encephalitis.
Case details
A 29-year-old water tank cleaner presented to the emergency department with sudden onset of abnormal movements of both upper and lower limbs, loss of consciousness, and frothing at the mouth. He was apparently well until 18 months prior when he had a similar episode. The prior episode also involved a sudden loss of consciousness. Upon regaining consciousness he had complete weakness of the left upper and lower limbs with deviation of the angle of the mouth to the right side and slurring of speech. He was taken to a healthcare facility where a CT of the brain was performed (reports of which are unavailable), and subsequently initiated on physiotherapy and seizure medications. He was compliant with his therapy and the seizures did not recur. There was also an improvement in functional capacity whereby he was able to go about his activities of daily living independently. His speech became more intelligible over time. This recovery occurred gradually over three months after which time he discontinued allopathic medication and changed to alternative medicine. He remained seizure free until he presented to the emergency department after this second seizure.
There was no history of joint pain, rash, tuberculosis, diabetes mellitus, jaundice, head injury, or a family history of epilepsy. He neither smoked nor consumed alcohol. He had no history of sexual promiscuity. His bladder and bowel function, appetite, weight, and sleep were all within normal limits. On physical examination he was in status epilepticus with generalised tonic clonic seizures. His pulse rate was 110 beats/minute, blood pressure was 150/100mmHg, and interictal respiratory rate was 20 cycles per minute. He had normal body temperature, oxygen saturation of 95 per cent on room air, and Glasgow coma scale of 3/15. He was intubated and started on infusion of diazepam (as seizures could not be controlled with lorazepam, phosphenytoin, and levetiracetam) with which his seizures were brought under control.
The left upper and lower limb had hypertonia, a power of 3/5, hyperreflexia, and an extensor plantar response. There was no anisocoria, abnormality of pupillary reflexes, extraocular eye movements, papilledema, or neck stiffness. The rest of the central nervous system and systemic examination were within normal limits. The clinical differential diagnoses were those of intracranial space-occupying lesion like tuberculoma or neurocysticercosis, tuberculous or viral encephalitis, primary or secondary central nervous system (CNS) angitis, and intracranial hemorrhage with raised intracranial pressure.
Blood investigations revealed leucocytosis with neutrophilic predominance (12,000/cu.mm and 61 per cent neutrophils). CSF analysis showed cytoalbuminodissociation [total wbc count-2 (all lymphocytes) and protein of 172mg%], and normal sugar level. The rest of the blood counts, renal function tests, liver function tests, vasculitic markers, electrolytes, blood glucose level, chest radiograph, and electrocardiogram were all within normal limits. The urine, blood, and cerebrospinal fluid cultures were all sterile.
After the initial investigations, encephalitis and CNS angitis were placed lower in the list of differentials. To differentiate between the intracranial space-occupying lesions, an MRI of the brain with gadolinium enhancement was performed. This showed atrophy of the right cerebral and left cerebellar hemispheres with dilatation of the right lateral and third ventricles, and hyperintensity of the grey and white matter of the right cerebral hemisphere on FLAIR (Figure 1).
Figure 1: MRI of the brain with gadolinium enhancement.

MRI of the brain with gadolinium enhancement shows atrophy of right cerebral hemisphere (white arrow) with dilatation of the right lateral ventricle (broken white arrow).
There was no midline shift or contrast uptake in any part of the brain. The rest of the brain parenchyma was normal. EEG showed 10Hz alpha rhythm responding to arousal. The background activity was made up of slow waves in the theta range. There was asymmetric slowing, with lower amplitude waves over the right hemisphere. Also present were intermittent sharp waves in the right fronto-parietal region and left hemisphere Figure 2), implying bilateral dysfunction (more on the right hemisphere) with active epileptiform activity. Hyperventilation and photostimulation were unremarkable.
Figure 2: EEG showing intermittent sharp waves (red circled).
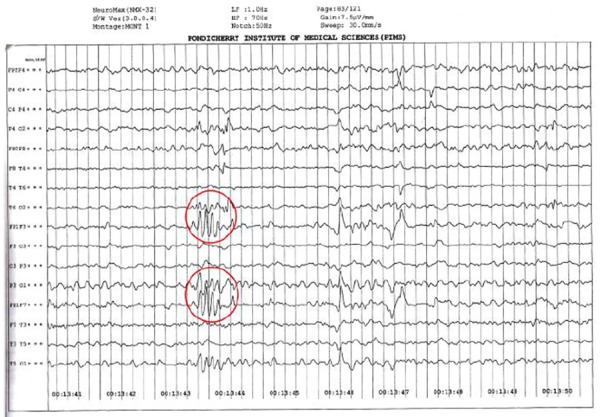
With the clinical findings of seizures, cortical deficit, and the aforementioned finding on the contrast-enhanced MRI of the brain and EEG, a diagnosis of Rasmussen’s encephalitis was made.
Discussion
The man in this case study most probably presented in the acute stage of Rasmussen’s encephalitis as he exhibited hemiparesis and other neurological symptoms, despite the large time span between the first and second seizures.
Monitoring of clinical improvement in these patients can be undertaken by follow-up of the hemiparesis,2,5,6 neuropsychological performance in patients without hemiparesis,2,8 and hemispheric ratio on neuroimaging. The hemispheric ratio is the ratio of affected/unaffected hemisphere on planimetry of axial and coronal slices, including the sylvian fissure. Most of the tissue loss occurs within the first year of onset of the acute stage.2,5,7
The aetiology and pathophysiology of RE remain elusive. Four groups have been described to assist in classifying the pathophysiology. Group 1 has inflammation with microglial nodules; group 2 reveals at least one gyral segment of complete necrosis; group 3 shows neuronal loss and gliosis; and group 4 displays gliosis and glial scarring.2,5,9,10 The microscopic picture may point toward a localised chronic encephalitis smouldering over a period of years. The localisation of the lesion along with its character is more consistent with viral encephalitis than with any other inflammatory disease of the central nervous system like post-infectious perivenous encephalitis or allergic encephalitis.1
There is no laboratory test that positively supports the diagnosis of RE. In a large series of CSF tests, in about one-half of the examinations cell counts and protein levels were within the normal range, and in the remainder, elevated cell counts (16–70 cells/ml, predominantly lymphocytes), and increased protein level (50–100mg/dl) were observed.2 Standard CSF tests are not useful in confirming or excluding the diagnosis.
An MRI of the brain in most RE patients shows unilateral enlargement of the CSF compartment with maximum accentuation in the insular and peri-insular region, and increased signal intensity in the cortical, subcortical, or both regions. EEG shows unihemispherical impairment of background activity and sleep spindles, focal slow activity, multifocal ictal discharges, and subclinical ictal discharges. Our patient had MRI and EEG findings that suggested bilateral involvement, a relatively rare entity in patients with RE. However, the caudate nucleus, insular, and peri-insular regions were normal. Other investigations that can be performed to help confirm the unilateral nature of suspected RE include positron emission tomography (PET), single photon emission computed tomography (SPECT), and magnetic resonance spectroscopy (MRS).2 Brain biopsy is not needed for all patients as diagnosis can be made without it as well. The goals of therapy are to decrease inflammation, restore functional capacity, and control seizures. The various modalities of treatment available to achieve these goals include pharmacological, immunotherapeutic, surgical, and rehabilitative techniques.
The man in this case, based on clinical, EEG, and MRI brain findings, fulfilled the European consensus diagnostic criteria for the diagnosis of RE.2 He had a reasonable quality of life before this episode of status epilepticus, hence although surgical intervention would result in amelioration of seizure symptoms, it would also cause a disabling post-operative functional deterioration. The early institution of long-term immunotherapy to prevent functional decline was the recommended line of therapy.2,11–13
In a recent, first-ever randomised prospective treatment trial in RE, 16 patients with recent onset RE were randomised into either tacrolimus or intravenous immunoglobulin (IVIG). Though neither treatment was more efficacious than the other, immunotreated patients had a longer survival than the controls.14 Another study compared immunotherapy and surgery, finding that while nine out of 10 patients treated by surgery achieved seizure freedom, only one out of 11 patients who received immunotherapy did were seizure free.15 However, due to financial constraints (cost of immunotherapy), and after a detailed discussion with the family about the potential benefits and side effects, it was decided to manage him with antiepileptic drugs and physiotherapy.
Conclusion
RE is an uncommon cause of seizure and bilateral cortical involvement, such as that seen in this case, is a rare entity. Although rare in adults, RE should be considered as a possible diagnosis in patients who present with intractable seizures. As the aetiology is largely uncertain, the treatment is mainly symptomatic with other modalities being far from definite, and in India, being available only to those who can afford it.
ACKNOWLEDGEMENTS
Thank you to Dr Kurien Thomas for reviewing the manuscript for grammatical errors and for his constant encouragement and support.
Footnotes
PEER REVIEW
Not commissioned. Externally peer reviewed.
PATIENT CONSENT
The authors, Vimal J, Nagarajan R, Sriram D, declare that:
- They have obtained written, informed consent for the publication of the details relating to the patient(s) in this report.
- All possible steps have been taken to safeguard the identity of the patient(s).
- This submission is compliant with the requirements of local research ethics committees.
Please cite this paper as: Vimal J, Nagarajan R, Sriram D. Rasmussen’s encephalitis: A case report. AMJ 2015;8(3):80–84. http//dx.doi.org/10.4066/AMJ.2015.2255
References
- 1.Rasmussen T, Olszewski J, Lloyd-Smith D. Focal seizures due to chronic localized encephalitis. Neurology. 1958;8:435–45. doi: 10.1212/wnl.8.6.435. [DOI] [PubMed] [Google Scholar]
- 2.Bien CG, Granata T, Antozzi C, Cross JH, Dulac O, Kurthen M. et al. Pathogenesis, diagnosis and treatment of Rasmussen encephalitis: a European consensus statement. Brain. 2005 Mar;128:454–71. doi: 10.1093/brain/awh415. [DOI] [PubMed] [Google Scholar]
- 3.Hunter GR, Donat J, Pryse-Phillips W, Harder S, Robinson CA. Rasmussen's encephalitis in a 58 year old female: still a variant? Can. J Neurol Sci. 2006;33:302–5. doi: 10.1017/s0317167100005175. [DOI] [PubMed] [Google Scholar]
- 4.Hart YM, Andermann F, Fish DR, Dubeau F, Robitaille Y, Rasmussen T. et al. Chronic encephalitis and epilepsy in adults and adolescents: a variant of Rasmussen's syndrome? Neurology. 1997;48:418–24. doi: 10.1212/wnl.48.2.418. [DOI] [PubMed] [Google Scholar]
- 5.Bien CG, Urbach H, Deckert M, Schramm J, Wiestler OD, Lassmann H. et al. Diagnosis and staging of Rasmussen's encephalitis by serial MRI and histopathology. Neurology. 2002;58:250–7. doi: 10.1212/wnl.58.2.250. [DOI] [PubMed] [Google Scholar]
- 6.Oguni H, Andermann F, Rasmussen TB. The natural history of the syndrome of chronic encephalitis and epilepsy: a study of the MRI series of forty eight cases. In: Chronic encephalitis and epilepsy: Rasmussen's syndrome. F. Andermann ed. Boston: Butterworth-Heinemann. 1991:7–35. [Google Scholar]
- 7.Bien CG, Widman G, Urbach H, Sassen R, Kuczaty S, Wiestler OD. et al. The natural history of Rasmussen's encephalitis. Brain. 2002;125:1751–9. doi: 10.1093/brain/awf176. [DOI] [PubMed] [Google Scholar]
- 8.Hennessy MJ, Koutroumanidis M, Dean AF, Jarosz J, Elwes RD, Binnie CD. et al. Chronic encephalitis and temporal lobe epilepsy: a variant of Rasmussen's syndrome? Neurology. 2001;56:678–81. doi: 10.1212/wnl.56.5.678. [DOI] [PubMed] [Google Scholar]
- 9.Bien CG, Elger CE, Wiendl H. Advances in pathogenic concepts and therapeutic agents in Rasmussen's encephalitis. Expert. Opin. Investig. Drugs. 2002;11:981–9. doi: 10.1517/13543784.11.7.981. [DOI] [PubMed] [Google Scholar]
- 10.Robitaille Y. Neuropathologic aspects of chronic encephalitis. In: Chronic encephalitis and epilepsy: Rasmussen's syndrome. F. Andermann ed. Boston: Butterworth-Heinemann. 1991:79–110. [Google Scholar]
- 11.Bien CG, Schramm J. Treatment of Rasmussen encephalitis half a century after its initial description: promising prospects and a dilemma. Epilepsy Res. 2009;86:101–2. doi: 10.1016/j.eplepsyres.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 12.Bien CG, Szinay M, Wagner J, Clusmann H, Becker AJ, Urbach H. Characteristics and surgical outcomes of patients with refractory magnetic resonance imaging-negative epilepsies. Arch. Neurol. 2009;66:1491–9. doi: 10.1001/archneurol.2009.283. [DOI] [PubMed] [Google Scholar]
- 13.Terra-Bustamante VC, Machado HR, Dos Santos OR, Serafini LN, Souza-Oliveira C, Escorsi-Rosset S. et al. Rasmussen encephalitis:long-term outcome after surgery. Childs Nerv. Syst. 2009;25:583–9. doi: 10.1007/s00381-008-0795-1. [DOI] [PubMed] [Google Scholar]
- 14.Bien CG, Tiemeier H, Sassen R, Kuczaty S, Urbach H, von Lehe M. et al. Rasmussen encephalitis: incidence and course under randomized therapy with tacrolimus or intravenous immunoglobulins. Epilepsia. 2013 Mar;54(3):543–0. doi: 10.1111/epi.12042. doi: 10.1111/epi.12042. [DOI] [PubMed] [Google Scholar]
- 15.Ramesha KN, Rajesh B, Ashalatha R, Kesavadas C, Abraham M. Radhakrishnan VV, et al. Rasmussen's encephalitis: experience from a developing country based on a group of medically and surgically treated patients. Seizure. 2009 Oct;18(8):567–72. doi: 10.1016/j.seizure.2009.05.010. doi: 10.1016/j.seizure.2009.05.010. [DOI] [PubMed] [Google Scholar]