

Abstract
To investigate the frequency and the prognostic impact of fibroblast growth factor receptor 1 (FGFR1) gene amplification in 526 curatively resected esophageal squamous cell carcinoma (ESCC). Using fluorescent in situ hybridization, high amplification was defined by an FGFR1/centromer 8 ratio is ≥ 2.0, or average number of FGFR1 signals/tumor cell nucleus ≥ 6.0, or percentage of tumor cells containing ≥ 15 FGFR1 signals or large cluster in ≥ 10%. Low amplification was defined by ≥ 5 FGFR1 signals in ≥ 50%. FGFR2 and FGFR3 mutations were assessed by direct sequencing in 388 cases and no mutation was detected. High and low amplification were detected in 8.6% and 1.1%, respectively. High FGFR1 amplification had significantly shorter disease-free survival (34.0 vs 158.5 months P=0.019) and overall survival (52.2 vs not reached P=0.022) than low/no amplification group. After adjusting for sex, smoking, stage, histology, and adjuvant treatment, high FGFR1 amplification had a greater risk of recurrence (adjusted hazard ratio [AHR], 1.6; P=0.029) and death (AHR, 1.53; P=0.050). High amplification was significantly higher in current smokers than former and never-smokers (Ptrend<0.001) and increased proportional to smoking dosage. High FGFR1 amplification is a frequent oncogenic alteration and an independent poor prognostic factor in resected ESCC.
Keywords: Fibroblast growth factor receptor 1, esophageal squamous cell carcinoma, gene amplification, fluorescent in situ hybridization, prognostic factor
INTRODUCTION
Esophageal cancer (EC) is the sixth most common cause of cancer death worldwide [1]. Despite the improvement of surgical technique and medical treatment, prognosis remains poor, with 5-year survival rate of less than 40%.[2] EC consists of two major histologic subtypes; esophageal squamous cell carcinoma (ESCC) and adenocarcinoma (EAC). Both histologic subtypes have different risk factors and epidemiology. ESCC continues to be the major subtype of EC in Asia, and the main risk factors include smoking and alcohol abuse.[3] In contrast, EAC predominately affects the Caucasian, and is closely associated with gastroesophageal reflux disease, Barrett's esophagus, and obesity. [4-7] Compared with ESCC, EAC shows younger age onset, male predominance and frequent occurrence in distal third of the esophagus.[8] Given widely different epidemiological and clinical features, EAC and ESCC may represent distinct disease entities which may benefit from different therapeutic approaches.
Amplification and overexpression of human epidermal growth factor receptor 2 (HER2; also known as ERBB2) was more frequent in EAC than ESCC and there was a strong concordance of HER2 status in primary and metastatic cancer.[9] Therefore, various clinical trials evaluating the efficacy of HER2 targeting agents were reported for the patients with advanced esophageal and esophagogastric junction (EGJ) adenocarcinoma.8-10 In the phase II trials for HER2 amplified EGJ adenocarcinoma, lapatinib, a dual tyrosine kinase inhibitor of ERBB1 and HER2 demonstrated modest response.[10, 11] The therapeutic implication of HER2 protein overexpression or gene amplification has been demonstrated in the Trastuzumab for Gastric Cancer (ToGA) trial [12]. In this trial, addition of trastuzumab (anti-HER2 antibody) to standard chemotherapy significantly improved response rate, progression-free survival and overall survival compared with chemotherapy alone in HER2-positive advanced gastric and EGJ adenocarcinoma. Based on the results of the ToGA trial, HER2 testing is routinely recommended for all patients with metastatic gastric or EGJ adenocarcinoma, and trastuzumab should be considered for HER2-positive cases.
Despite a rapid and enthusiastic development of targeted therapies in gastric or EGJ adenocarcinoma, no therapeutically tractable target has been identified for ESCC. Recently, comparative genomic analysis showed different DNA copy number alterations between EAC and ESCC.[13, 14] Among those, high copy number gains of cancer-associated genes, such as SOX2, PIK3CA, CCND1, and FGFR1, were more frequently observed in ESCC than in EAC, suggesting that genomic gain of these oncogenes may be therapeutic targets for ESCC.
Fibroblast growth factor receptor 1 (FGFR1) is a member of family of receptor tyrosine kinases (FGFR1-4), and its activation by amplification, mutation, or translocation leads to tumor cell proliferation and survival in many cancers.[15, 16] Potentially actionable FGFR rearrangements were identified in diverse solid tumors including lung cancer, oral cancer, and breast cancer. [17-19] FGFR1 amplification has been frequently reported in lung squamous cell carcinoma (SqCC), small cell lung cancer, and SqCC of head and neck (SCCHN), for which smoking is a clear and dominant risk factor.[15, 16, 20-28] Overall, the frequency of FGFR1 amplification was reported to be 5.6%-24.8% in lung SqCC and 15%-17.4% in SCCHN, suggesting that this genetic alteration mainly target squamous cell histology.[16, 20-25, 27, 28] Furthermore, FGFR1 amplification has been associated with poor prognosis or unfavorable clinicopathologic parameters in lung SqCC and SCCHN. Because ESCC has risk factors in common with lung SqCC and SCCHN, we hypothesized that FGFR1 amplifications is associated with pathogenesis and poor prognosis in ESCC.
In this study, we sought to determine the frequency, prognostic impact and association with smoking dosage of FGFR1 amplification in surgically resected ESCC. Furthermore, we also evaluated the frequency of FGFR2 and FGFR3 mutations in ESCC.
RESULTS
Patient Characteristics
A total of 526 Korean patients who underwent curative esophagectomy were analyzed. The clinical characteristics of the enrolled patients are presented in Table 1. There were 489 male and 37 female with a median age of 66 years (range 35-98). Median tumor size was 3 cm and approximately half of tumors had pT3 or pN0. Pathologic stages were I in 22.4%, II in 42.8%, and III in 34.8%. Two thirds (60.3%) were located in the lower esophagus, and more than half of patients had moderately differentiated SqCCs. The majority of patients were current (39.0%) or former smokers (38.6%), and median smoking dosage was 30.0 pack-years (range 0-150). Adjuvant treatment was given in 140 patients (26.6%), and 87 of those (62.1%) were treated by concurrent chemoradiotherapy. The proportions of adjuvant therapy according to stage were 5.9% in stage I, 21.8% in stage II, and 45.9% in stage III.
Table 1. Patient characteristics according to FGFR1 amplification.
| Characteristics | All patients | High amplification* | Low amplification* | No amplification | |||||
|---|---|---|---|---|---|---|---|---|---|
| No. | % | No. | % | No. | % | No. | % | P† | |
| No of patients | 526 | 100 | 45 | 8.6 | 6 | 1.1 | 475 | 90.3 | |
| Age, years | 0.697 | ||||||||
| Median (range) | 66 (35-98) | 67 (52-77) | 68 (65-77) | 66 (35-98) | |||||
| Sex | 0.608 | ||||||||
| Male | 489 | 93.0 | 43 | 95.6 | 6 | 100 | 440 | 92.6 | |
| Female | 37 | 7.0 | 2 | 4.4 | 0 | 0 | 35 | 7.4 | |
| Tumor size, cm | 0.310 | ||||||||
| Median, range | 3.0 (0.2-10.5) | 3.0 (2.0-9.0) | 2.5 (2.0-4.5) | 3.0 (0.2-10.5) | |||||
| pT stage‡ | 0.618 | ||||||||
| T1 | 167 | 31.7 | 12 | 26.7 | 2 | 33.3 | 153 | 32.2 | |
| T2 | 106 | 20.2 | 12 | 26.7 | 1 | 16.7 | 93 | 19.6 | |
| T3 | 238 | 45.2 | 18 | 40.0 | 3 | 50.0 | 217 | 45.7 | |
| T4 | 15 | 2.9 | 3 | 6.7 | 0 | 0 | 12 | 2.5 | |
| pN stage‡ | 0.619 | ||||||||
| N0 | 259 | 49.2 | 22 | 48.9 | 3 | 50.0 | 234 | 49.3 | |
| N1 | 234 | 44.5 | 20 | 44.4 | 2 | 33.3 | 212 | 44.6 | |
| N2 | 22 | 4.2 | 3 | 6.7 | 1 | 16.7 | 18 | 3.8 | |
| N3 | 11 | 2.1 | 0 | 0 | 0 | 0 | 11 | 2.3 | |
| pTMN stage‡ | 0.822 | ||||||||
| I | 118 | 22.4 | 8 | 17.8 | 1 | 16.7 | 109 | 22.9 | |
| II | 225 | 42.8 | 19 | 42.2 | 2 | 33.3 | 204 | 42.9 | |
| III | 183 | 34.8 | 18 | 40.0 | 3 | 50.0 | 162 | 34.1 | |
| Location | 0.158 | ||||||||
| Upper | 66 | 12.5 | 5 | 11.1 | 0 | 0 | 61 | 12.8 | |
| Middle | 143 | 27.2 | 19 | 42.2 | 2 | 33.3 | 122 | 25.7 | |
| Lower | 317 | 60.3 | 21 | 46.7 | 4 | 66.7 | 292 | 61.5 | |
| Tumor grade | 0.424 | ||||||||
| Well | 128 | 24.3 | 12 | 26.7 | 2 | 33.3 | 114 | 24.0 | |
| Moderate | 314 | 59.7 | 29 | 64.4 | 4 | 66.7 | 281 | 59.2 | |
| Poorly | 84 | 16.0 | 4 | 8.9 | 0 | 0 | 80 | 16.8 | |
| Smoking status¶ | <0.001 | ||||||||
| Never-smoker | 118 | 22.4 | 1 | 2.2 | 0 | 0 | 117 | 24.6 | |
| Former smoker | 203 | 38.6 | 5 | 11.1 | 2 | 33.3 | 196 | 41.3 | |
| Current smoker | 205 | 39.0 | 39 | 86.7 | 4 | 66.7 | 162 | 34.1 | |
| Smoking dosage, pack-years | |||||||||
| Median | 30 | 39 | 35 | 30 | 0.002 | ||||
| Range | 0-150 | 0-99 | 13-60 | 0-150 | |||||
| Adjuvant therapy | 0.870 | ||||||||
| Yes | 140 | 26.6 | 13 | 28.9 | 2 | 33.3 | 125 | 26.3 | |
| No | 386 | 73.4 | 32 | 71.1 | 4 | 66.7 | 350 | 73.7 | |
| FGFR1 FISH§ | |||||||||
| Number (median, range) |
2.2 (0-15.5) | 6.4 (4.1-15.5) | 5.1 (5.0-5.6) | 2.2 (0-5.7) | <0.001 | ||||
| Ratio (mean, range) |
1.5 (0-7.8) | 2.9 (1.1-7.8) | 1.5(1.0-1.9) | 1.3 (0-1.6) | 0.003 | ||||
Abbreviations:
High FGFR1 amplification was defined as if one of the following criteria is fulfilled: (1) FGFR1/CEN8 ratio is ≥ 2.0, (2) average number of FGFR1 signal per nucleus ≥ 6.0, and (3) percentage of tumor cells containing ≥ 15 FGFR1 signals or large clusters in ≥ 10% cells. Low FGFR1 amplification was defined when the percentage of tumor cells containing ≥ 5 FGFR1 signals is ≥ 50 %.
χ2 test, Fisher's exact test, or Mann-Whitney U test.
Pathologic stage at the time of surgical resection was determined according to the American Joint Committee on Cancer (seventh edition) guidelines.
Never-smokers; a lifetime smoking dose of fewer than 100 cigarettes; former smokers, those who have stopped smoking for more than 1 year; current smokers, those who currently smoke or have quit for less than 1 year.
FGFR1 numbers are average numbers of FGFR1 signals per nucleus, and ratios are FGFR1/CEN8 ratios.
FGFR1 amplification status and clinicopathologic features
Among 526 patients, 45 (8.6%) were high FGFR1 amplification, 6 (1.1%) were low FGFR1 amplification, and 475 (90.3%) displayed no amplification (Table 1; Figure 1). The median FGFR1 gene copy number per nucleus and the mean FGFR1/CEN8 ratio in all patients were 2.2 (range, 0 to 15.5 copies per nucleus) and 1.5 (range, 0 to 7.8). The median FGFR1 gene copy number was 6.4 (range, 4.1 to 15.5) in high amplification, 5.1 (range, 5.0 to 5.6) in low amplification, and 2.2 (range 0 to 5.7) in no amplification group. The mean FGFR1/CEN8 ratio was 2.9 (range 1.1 to 7.8), 1.5 (range, 1.0 to 1.9), and 1.3 (range, 0 to 1.6) in high, low and no amplification group, respectively. Of 45 high FGFR1 amplified tumors, 12 cases (26.7%) only satisfied the criterion of an FGFR1:CEN8 ratio of ≥ 2.0, 7 cases (15.6%) only satisfied the criterion of an average number of FGFR1 signal per nucleus ≥6.0, and 4 cases (8.9%) only satisfied the criterion of percentage of tumor cells containing ≥ 15 FGFR1 signals or large clusters in ≥ 10% cells, respectively. 22 cases (48.9%) satisfied all three criteria for FGFR1 amplification simultaneously.
Figure 1. Fibroblast growth factor receptor 1 (FGFR1) amplification assessed by fluorescent in situ hybridization.
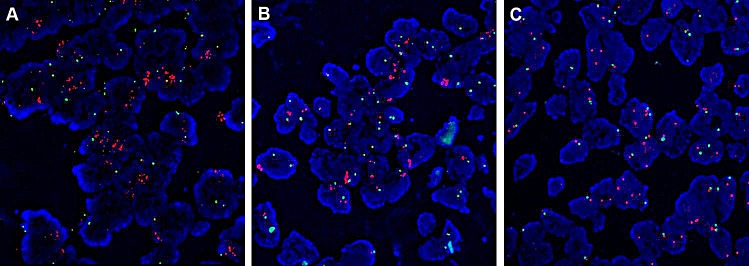
(A) High FGFR1 amplification; (B) Low FGFR1 amplification; (C) No amplification.
There was no significant difference among three FGFR groups regarding age, sex, tumor size, pathologic stage, location, histologic grade, and adjuvant therapy (Table 1). However, the incidence of high FGFR1 amplification was significantly higher in smokers than in never-smokers (P<0.001). Only 1 (2.2%) of 45 patients with high-level amplification was never smoker. The median smoking dosage was significantly higher for high amplification group than that for low or no amplification group(39 vs 35 vs 30 pack-years, P=0.002). Based on the pattern of high amplification (group fulfilling 3 criteria vs. 1 or 2 criteria), there was no significant difference in terms of age, sex, location, and pathologic stage.
Survival Outcomes According to FGFR1 Amplification
With a median follow-up time of 55.4 months, the 5 year DFS and OS rates for all patients were 52.8% and 59.8%, respectively. The 5-year DFS rate according to pTNM stages were 78.4% in stage I, 55.9% in stage II, and 33.2% in stage III patients. The 5-year OS rate according to pTNM stages were 86.8% for stage I, 65.3% for stage II, and 37.1% for stage III.
The median DFS for each of the three FGFR groups was 34.0 months in the high FGFR1 amplification, not reached (NR) in low amplification group, and 158.5 months in the no amplification group (Figure 2A). By using pair-wise comparison, patients with high FGFR1 amplification showed a significantly shorter DFS than those with no amplification (34.0 vs 158.5 months in no amplification, P=0.020). The median DFS of high amplification and low amplification were not significantly different (34.0 vs NR in low amplification, P=0.272), probably due to small sample size in the low amplification group. The median DFS was similar between low amplification and no amplification group in pair-wise comparison (P=0.639). The median OS for each of the three FGFR groups was 52.2 months in the high FGFR1 amplification, 72.0 months in the low amplification, and not reached in the no amplification (Figure 2B). By using pair-wise comparison, patients with high FGFR1 amplification showed a significantly shorter OS than those with no amplification (52.2 vs NR in no amplification, P=0.021). The OS times of high amplification and low amplification were not significantly different (52.2 vs 72.0 in low amplification, P=0.637), probably due to small sample size in the low amplification group. The median OS was similar between low amplification and no amplification group in pair-wise comparison (P=0.517).
Figure 2. Survival analysis on the basis of FGFR1 amplification (high, low, and no amplification).
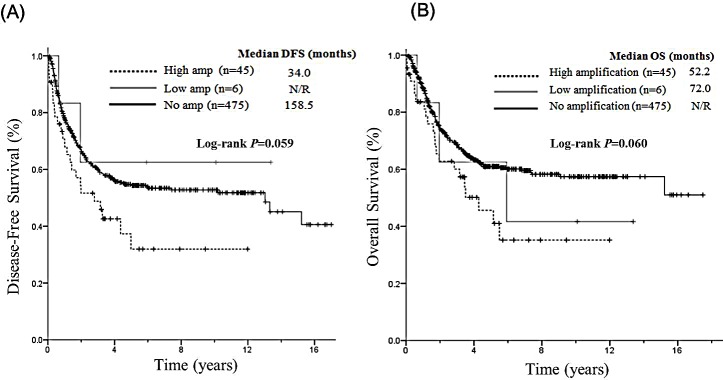
(A) Median DFS was 34.0 months in the high FGFR1 amplification group, not reached in low amplification group, and 158.5 months in the no amplification group. (B) The median OS was 52.2 months in the high FGFR1 amplification group, and 72.0 months in the low amplification group, and not reached in the no amplification group.
Because the DFS of patients with low and no amplification were similar but different from that of patients with high amplification, we categorized total patients into high amplification group and low/no amplification group and assessed the survival outcomes of these two groups. The median DFS of the high FGFR1 amplification group was significantly shorter compared with that of the low/no FGFR1 amplification group (34.0 vs 158.5 months P=0.019, Figure 3A). In addition, high FGFR1 amplification group demonstrated significantly shorter OS than low/no FGFR1 amplification group (52.2 vs NR, P=0.022, Figure 3B). In Cox proportional hazard model adjusted for sex, smoking history, pathologic stage, adjuvant treatment, and histologic grade, high FGFR1 amplification was significantly associated with a shorter DFS (AHR 1.61; 95% CI, 1.05-2.46; P=0.029, Table 2). There was a strong trend toward worse OS for high FGFR1 amplification compare to low/no FGFR1 amplification group in multivariate analysis (HR 1.53; 95% CI, 0.99-2.34, P=0.050). There was no significant difference in DFS and OS for sex, smoking status, and adjuvant treatment in multivariate analysis. There was no significant difference in DFS and OS between group fulfilling 3 criteria and group fulfilling 1 or 2 criteria.
Figure 3. Prognostic impact of high FGFR1 amplification and low/no amplification on DFS and OS.

(A) The median DFS of the high FGFR1 amplification group was significantly shorter compared with low/no FGFR1 amplification group (P=0.019) (B) The median OS of high FGFR1 amplification group demonstrated significantly shorter than low/no FGFR1 amplification group (P=0.022).
Table 2. Survival outcome in multivariate analysis.
| DFS | OS | ||||||
|---|---|---|---|---|---|---|---|
| Variable | Category | HR | 95% CI | P | HR | 95% CI | P |
| Sex | Male vs female (ref) | 0.91 | 0. 53-1.55 | 0.724 | 0.90 | 0.53-1.54 | 0.703 |
| Smoking status | Current smoker vs never/former-smoker (ref) | 1.08 | 0.82-1.43 | 0.595 | 1.02 | 0.77-1.35 | 0.898 |
| Pathologic stage* | III vs II/I (ref) | 2.56 | 1.97-3.34 | <0.001 | 2.53 | 1.94-3.29 | <0.001 |
| Histology | Poor vs well/moderate(ref) | 1.61 | 1.16-2.22 | 0.004 | 1.61 | 1.17-2.23 | 0.004 |
| Adjuvant treatment | Yes vs no (ref) | 1.11 | 0.82-1.50 | 0.497 | 1.05 | 0.78-1.43 | 0.731 |
| FGFR1 amplification | High vs Low /no (ref) | 1.61 | 1.05-2.46 | 0.029 | 1.53 | 0.99-2.34 | 0.050 |
Abbreviations: DFS, disease-free survival; OS, overall survival; ref, reference; amp, amplification;
Clinical stage at the time of initial diagnosis was determined according to the American Joint Committee on Cancer (seventh edition) guidelines.
FGFR2 and FGFR3 mutations
FGFR2 and FGFR3 mutations were assessed by direct sequencing in 388 cases with FFPE tissues available and no FGFR2 or FGFR3 mutation was detected.
Association between FGFR1 amplification and smoking
As shown in Figure 4A, the incidence of high-level FGFR1 amplification was significantly higher in current smokers than former or never-smokers (Ptrend <0.001). The incidences of high-level FGFR1 amplification in current, former and never-smokers were 19.0%, 2.5%, and 0.8%, respectively. With increment of total cigarette smoking dosage, the incidence of high-level FGFR1 amplification was significantly increased (Figure 4B, Ptrend = 0.001). High-level FGFR1 amplification were 0.8% for never-smokers, 5.8% in patients with 1-10 pack-years, 7.4% with 11-20 pack-years, 9.6% with 21-50 pack-years, and 19.3% with more than 50 pack-years.
Figure 4. Association of smoking status and high FGFR1 amplification.
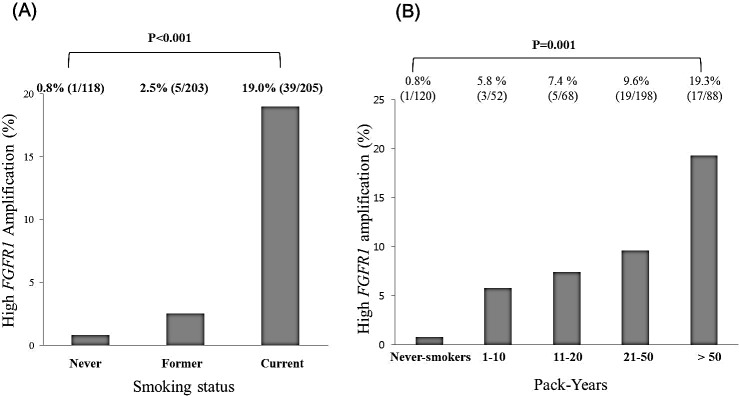
(A) Proportions of high FGFR1 amplification according to never-, former, and current smokers (B) Incidence of high FGFR1 amplification according to smoking dosage.
DISCUSSION
In this study, we investigated the frequency and the prognostic impact of FGFR1 amplification in resected ESCC. To our knowledge, this is the first report on the prognostic impact of FGFR1 amplification in the largest-ever cohort of resected ESCC patients from East Asian. Our study demonstrated high FGFR1 amplification is a common genetic alteration (8.6%) and an independent negative prognostic factor in resected ESCC. Interestingly, it was associated with cigarette smoking in a dose-dependent manner.
The frequency of FGFR1 amplification has been reported to range from 6% to 9.4%. In the DNA array hybridization study,[29] 2 out of 32 ESCC cases (6%) demonstrated amplified 8p11 locus which containing FGFR1 gene. A comparative genomic study revealed many focal DNA amplifications or losses such as SOX2, PIK3CA, CCND1, FGFR1, MYC, GATA4, and GATA6 in ESCC and EAC.[13] Of note, FGFR1 was amplified in 21% of ESCC samples compare with 8% in EAC. Recently, FGFR1 amplification was reported to be 9.4% in the cohort with Western Europe.[30] The frequency of high FGFR1 amplification (8.6%), as determined by FISH analysis in our study, was comparable to those reported in previous studies.[13, 29, 30] Given the relatively high frequency of FGFR1 amplification, it may represent an attractive therapeutic target for ESCC.
Given the histological similarity and common risk factors, it is not surprising that FGFR1 amplification has been reported in upper aerodigestive tract cancers such as lung SqCC, SCCHN, and small cell lung cancer.[16, 20-28] However, the prognostic significance of FGFR1 amplification in these cancers has shown controversial results. In resected lung SqCC, Kim et al[16] reported FGFR1 amplification as negative prognostic factor, whereas Heist et al [24] observed no significant difference in OS. In SCCHN, FGFR1 amplification was significantly associated with poor prognostic factors such as higher T stage, lymphovascular invasion, and higher numbers of visceral metastases.[22]Tumor heterogeneity, unstandardized FISH criteria for FGFR1 amplification, varying adjuvant treatment, and small sample size may contribute to the controversial results. In our study, by applying more sophisticated criteria for FGFR1 amplification, we were able to divide FGFR1 amplification into high and low amplification. Low FGFR1 amplification occurred only in 1.1% and survival outcome of low FGFR1 amplification group was similar to no amplification group. Compared with low/no amplification group, high FGFR1 amplification was significantly associated with shorter DFS regardless of sex, histology, and adjuvant therapy, implying that FGFR1 amplification as an independent negative prognostic factor in curatively resected ESCC.
The identification of FGFR alteration in human cancers led to rapid development of selective FGFR inhibitor such as BGJ398 and AZD4547. In phase I study with BGJ398,[31] 1 patient with FGFR1 amplified lung SqCC with FGFR1/CEN8 ratio of 2.6 by FISH analysis had partial response. In another phase I study with FGFR inhibitor AZD4547,[32] 1 partial response was also seen in lung SqCC patient with FGFR1 amplification (FGFR1/CEN8 ratio > 2.8). Various FGFR inhibitors are currently in clinical development for the patients with FGFR aberrations. Therefore, our study strongly suggests the therapeutic potential of FGFR inhibitor for ESCC, and future clinical trials for advanced high FGFR1 amplified ESCC are strongly warranted.
Unfortunately, FISH criteria for FGFR1 amplification has not yet been standardized. Indeed, definition of FGFR1 amplification by FISH technique has been highly variable in the previous studies in lung cancer and breast cancer.[16, 24, 25, 28, 33, 34] Unlike breast cancer, lung SqCC exhibits small-clusters and co-amplifications of FGFR1 and CEN8.[25] Therefore, FGFR1 FISH assay needs to differentiate between true amplification and polysomy. In a large cohort study, Schildhause et al [25] proposed a more sophisticated FGFR1 FISH criteria using average gene copy number per nucleus, FGFR1/CEN8 ratio, and percentage of gene clusters at the same time. By the addition of FGFR1/CEN8 ratio, 8 out of 47 cases (17.0%) were newly classified as high amplification in this study.(25) Similarly, in our study, 12 out of 45 high amplification group (26.7%) might have been misclassified as low/no amplification group if the criterion for FGFR1/CEN8 ratio was not included. Considering clinical benefit appeared in patients with high FGFR1/CEN8 ratio in clinical trials with FGFR inhibitors,[31, 32] this scoring system might serve as a standardized screening tool to select patients who gain greater benefit from treatment with FGFR inhibitors. Our criteria need further validation in the future clinical trials with FGFR inhibitors in ESCC patients.
Accumulating evidence has shown that FGFR1 amplification correlated with smoking status in squamous cell biology. FGFR1 amplification occurs significantly more often in the smokers of the lung SqCC, SCCHN, and small cell lung cancer.[16, 22, 23, 26] The proportion of FGFR1 amplification among current smokers were reported 15.8% to 28.9% in lung SqCC and 17.7% in SCCHN in a dose dependent manner.[16, 22, 23] In small cell lung cancer cohort, all FGFR1 amplified cases were current or former smokers.[26] In our study, FGFR1 amplification was significantly more likely to be smokers, and 44 out of 45 high FGFR1 amplification cases were current or former smokers. Similar to the findings in lung SqCC and SCCHN, acquisition of FGFR1 amplification in our study was also significantly increased proportional to smoking dosage. Therefore, FGFR1 amplification may be an oncogenic driver mutation in tobacco-associated cancers of the aerodigestive tract. An interesting question of the role of FGFR1 amplification on the smoking-associated carcinogenesis still remained to be solved.
Somatic mutations of FGFR2 and FGFR3 were reported in 12% and 30% of endometrial and urothelial cell carcinomas, respectively.[35, 36] Inhibitor–sensitive 5 mutation loci for FGFR2 and 6 mutation loci for FGFR3 were noticed in 3% of lung SqCC samples.[37]Here, we show no FGFR2 and FGFR3 mutations in our 388 ESCC patients. This may be explained by the limited sensitivity of the Sanger sequencing, tumor heterogeneity or molecular difference of ESCC from lung SqCC.
The main limitation of our study include its retrospective nature and patient selection. Therefore, our findings should be validated in an independent cohort and response data to FGFR-targeted therapies in the future clinical trials.
In conclusion, we demonstrated high FGFR1 amplification is an independent poor prognostic factor in resected ESCC. Patients with FGFR1 amplification were significantly more likely to be smokers and the frequency of FGFR1 amplification was also increased proportional to smoking dosage, suggesting FGFR1 amplification as an oncogenic aberration induced by smoking carcinogen. Our finding indicates FGFR1 amplification is a promising therapeutic target in ESCC.
PATIENTS AND METHODS
Patients and tissue samples
This study was conducted in a cohort of patients with ESCC who underwent radical esophagectomy at Severance Hospital and Samsung Medical Center, Seoul, Korea, between 2002 and 2010. The criteria used for patient selection included (1) surgically resected SqCC of the thoracic esophagus (R0 resection), (2) availability of primary tumor tissue, (3) no distant metastasis, and (4) no preoperative treatment. The tumor samples of 664 patients were available for examination of FGFR1 amplification. We excluded 115 cases (17.3%) who received neoadjuvant treatment. Twenty-three patients (3.5%) were excluded because of incomplete survival follow-up and/or smoking data. All diagnosis were reviewed by two experienced pathologist (Y.L.C. and H.K.K) and confirmed by hematoxylin and eosin staining. Three representative cores measuring in 2-mm-diameter for each paraffin-embedded primary tumor were assembled into tissue microarray blocks.
Patients' information was collected by reviewing the medical records for evaluation of clinicopathologic characteristics and survival outcome. Staging was determined using the 7th edition American Joint Committee on Cancer guideline of tumor, node, and metastasis (TNM) classification. Smoking status such as never-smoker, former smoker, and current smoker were defined as previous studies.[38] The study was approved by the institutional review board of Severance Hospital and Samsung Medical Center.
FGFR1 Fluorescence In Situ Hybridization
Fluorescence in situ hybridization (FISH) to detect FGFR1 amplification was performed at the chromosomal level of the tissue microarray. We performed fluorescence signal detection with two probes on chromosome 8. The target probe is located on the FGFR1 locus spanning 8p12 - 8p11.23 with spectrum orange (red) and CEN 8 with Spectrum Green (Abbott Molecular, Abbott Park, IL) following routine method. The evaluation was done independently by two experienced pathologists (Y.L.C. and H.K.) blinded to clinical information, and at least 100 nuclei per case were evaluated. FGFR1 amplification was defined based on the previous studies.[25, 26] High FGFR1 amplification was defined as if one of the following criteria is fulfilled: (1) FGFR1/CEN8 ratio is ≥ 2.0, (2) average number of FGFR1 signal per nucleus ≥ 6.0, and (3) percentage of tumor cells containing ≥ 15 FGFR1 signals or large clusters in ≥ 10% cells. Low FGFR1 amplification was defined when the percentage of tumor cells containing ≥ 5 FGFR1 signals is ≥ 50%.
Mutation analysis
Genomic DNA was extracted from 388 formalin-fixed paraffin-embedded (FFPE) tissue specimens using an QIAamp DNA Micro kit (Qiagen, CA, USA) according to the manufacturer's instruction. The extracted DNA was used in a PCR amplification reaction. Based on the previous study in lung SqCC,[37] we evaluated the W290C, S320C, and K660E/K660N mutations in FGFR2, and R248C and S249C mutations in FGFR3. PCR amplification primers were designed to amplify following regions; W290C; 5′-TCCACAGTGGTCGGAGGAG-3′; 5′- AAAGTCCTCACCTTGAGAACCTTG-3′, S320C; 5′-CCTGGTTGGCCGTTATATTG-3′; 5′- TGTTTTGGCAGGACAGTGAG-3′, K660E/K660N; 5′- ATTCATCGAGATTTAGCAGCCAG-3′; 5′- ACATTCTGAGCCTCACCCC-3′, R248C and S249C; 5′-TGGCGGTGGTGGTGAG-3′; 5′- ATTCACCTCCACGTGCTTGA-3′.
PCR was carried out with the following conditions : initial denaturation at 95°C for 10 min, followed by 45 cycles for 30 s, 54°C for 60 s, 72°C for 45 s and final polymerization step of 72°C for 5 min in a GeneAmp PCR system 2720 (Life technologies, CA, USA). The amplified DNA product was visualized by gel electrophoresis and PCR products were sequenced using the Big dye terminator sequencing kit (Life technologies, CA, USA) according to the manufactures' instruction. Sequence reactions were the subjected to electrophoresis on an Applied Biosystems 3130XL DNA Analyzer (Life technologies, CA, USA).
Statistical Analysis
We evaluated association between FGFR1 amplification status and clinical significance using the χ2test or Fisher's exact test and trend was analyzed using linear regression. We also assessed whether FGFR1 amplification influenced survival outcome using Kaplan-Meier curves with a log-rank test. Disease free survival (DFS) was defined from the time of surgery to initial relapse or death. Overall survival (OS) was measured from the time of surgery to death or last follow-up date, and 95% CIs were evaluated by survival analysis using the Kaplan-Meier method. Statistical significance was set at P < 0.05 for all analyses. Multivariate analysis was done using Cox regression analysis for following variables: sex, smoking status, stage, histology, adjuvant treatment, and FGFR1 status. All statistical analysis was performed using SPSS version 18.0 (SPSS, Chicago, IL).
Disclosure of Potential Conflicts of Interest
The authors have no potential conflicts of interest to disclose.
Acknowledgments
The FGFR1 FISH probe used in this study was supported by Abbott molecular Inc. (Abbott Park, IL).
Footnotes
Disclosure of Potential Conflicts of Interest
The authors have no potential conflicts of interest to disclose.
Grant
This study was supported by a grant from the Korea Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI12C1440, B. C. Cho) and by a faculty research grant of Yonsei University College of Medicine for 2012(6-2012-0044, H. Kim) and by the R&D Program for Society of the National Research Foundation (NRF) funded by the Ministry of Science, ICT & Future Planning (NRF-2013M3C8A1078501, Y. L. Choi).
REFERENCES
- 1.Kamangar F, Dores GM, Anderson WF. Patterns of cancer incidence, mortality, and prevalence across five continents: Defining priorities to reduce cancer disparities in different geographic regions of the world. Journal of Clinical Oncology. 2006;24:2137–50. doi: 10.1200/JCO.2005.05.2308. [DOI] [PubMed] [Google Scholar]
- 2.Ando N, Ozawa S, Kitagawa Y, Shinozawa Y, Kitajima M. Improvement in the results of surgical treatment of advanced squamous esophageal carcinoma during 15 consecutive years. Ann Surg. 2000;232:225–32. doi: 10.1097/00000658-200008000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 4.Wong A, Fitzgerald RC. Epidemiologic risk factors for Barrett's esophagus and associated adenocarcinoma. Clin Gastroenterol Hepatol. 2005;3:1–10. doi: 10.1016/s1542-3565(04)00602-0. [DOI] [PubMed] [Google Scholar]
- 5.Sampliner RE. Epidemiology, pathophysiology, and treatment of Barrett's esophagus: reducing mortality from esophageal adenocarcinoma. Med Clin North Am. 2005;89:293–312. doi: 10.1016/j.mcna.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 6.Layke JC, Lopez PP. Esophageal cancer: a review and update. Am Fam Physician. 2006;73:2187–94. [PubMed] [Google Scholar]
- 7.Freeman HJ. Risk of gastrointestinal malignancies and mechanisms of cancer development with obesity and its treatment. Best Pract Res Clin Gastroenterol. 2004;18:1167–75. doi: 10.1016/j.bpg.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 8.Siewert JR, Stein HJ, Feith M, Bruecher BLDM, Bartels H, Fink U. Histologic tumor type is an independent prognostic parameter in esophageal cancer: Lessons from more than 1,000 consecutive resections at a single center in the Western world. Annals of Surgery. 2001;234:360–7. doi: 10.1097/00000658-200109000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reichelt U, Duesedau P, Tsourlakis M, Quaas A, Link BC, Schurr PG, et al. Frequent homogeneous HER-2 amplification in primary and metastatic adenocarcinoma of the esophagus. Mod Pathol. 2007;20:120–9. doi: 10.1038/modpathol.3800712. [DOI] [PubMed] [Google Scholar]
- 10.Galsky MD, Von Hoff DD, Neubauer M, Anderson T, Fleming M, Nagarwala Y, et al. Target-specific, histology-independent, randomized discontinuation study of lapatinib in patients with HER2-amplified solid tumors. Invest New Drugs. 2012;30:695–701. doi: 10.1007/s10637-010-9541-0. [DOI] [PubMed] [Google Scholar]
- 11.Hecht J, Urba S, Koehler M, Ellis C, Gagnon R, Kemner A, et al. Lapatinib monotherapy in recurrent upper gastrointestinal malignancy: phase II efficacy and biomarker analyses. Proc GI ASCO. 2008:43. [Google Scholar]
- 12.Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–97. doi: 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- 13.Bandla S, Pennathur A, Luketich JD, Beer DG, Lin L, Bass AJ, et al. Comparative Genomics of Esophageal Adenocarcinoma and Squamous Cell Carcinoma. Annals of Thoracic Surgery. 2012;93:1101–6. doi: 10.1016/j.athoracsur.2012.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rumiato E, Pasello G, Montagna M, Scaini MC, De Salvo GL, Parenti A, et al. DNA copy number profile discriminates between esophageal adenocarcinoma and squamous cell carcinoma and represents an independent prognostic parameter in esophageal adenocarcinoma. Cancer Letters. 2011;310:84–93. doi: 10.1016/j.canlet.2011.06.017. [DOI] [PubMed] [Google Scholar]
- 15.Kim HS, Mitsudomi T, Soo RA, Cho BC. Personalized therapy on the horizon for squamous cell carcinoma of the lung. Lung Cancer. 2013;80:249–55. doi: 10.1016/j.lungcan.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 16.Kim HR, Kim DJ, Kang DR, Lee JG, Lim SM, Lee CY, et al. Fibroblast Growth Factor Receptor 1 Gene Amplification Is Associated With Poor Survival and Cigarette Smoking Dosage in Patients With Resected Squamous Cell Lung Cancer. Journal of Clinical Oncology. 2013;31:731–7. doi: 10.1200/JCO.2012.43.8622. [DOI] [PubMed] [Google Scholar]
- 17.Majewski IJ, Mittempergher L, Davidson NM, Bosma A, Willems SM, Horlings HM, et al. Identification of recurrent FGFR3 fusion genes in lung cancer through kinome-centred RNA sequencing. J Pathol. 2013;230:270–6. doi: 10.1002/path.4209. [DOI] [PubMed] [Google Scholar]
- 18.Seo JS, Ju YS, Lee WC, Shin JY, Lee JK, Bleazard T, et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 2012;22:2109–19. doi: 10.1101/gr.145144.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu YM, Su F, Kalyana-Sundaram S, Khazanov N, Ateeq B, Cao X, et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013;3:636–47. doi: 10.1158/2159-8290.CD-13-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freier K, Schwaenen C, Sticht C, Flechtenmacher C, Muhling J, Hofele C, et al. Recurrent FGFR1 amplification and high FGFR1 protein expression in oral squamous cell carcinoma (OSCC) Oral Oncol. 2007;43:60–6. doi: 10.1016/j.oraloncology.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 21.Gadgeel SM, Chen W, Cote ML, Bollig-Fischer A, Land S, Schwartz AG, et al. Fibroblast growth factor receptor 1 amplification in non-small cell lung cancer by quantitative real-time PCR. PLoS One. 2013;8:e79820. doi: 10.1371/journal.pone.0079820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goke F, Bode M, Franzen A, Kirsten R, Goltz D, Goke A, et al. Fibroblast growth factor receptor 1 amplification is a common event in squamous cell carcinoma of the head and neck. Modern Pathology. 2013;26:1298–306. doi: 10.1038/modpathol.2013.58. [DOI] [PubMed] [Google Scholar]
- 23.Goke F, Franzen A, Menon R, Goltz D, Kirsten R, Boehm D, et al. Rationale for treatment of metastatic squamous cell carcinoma of the lung using fibroblast growth factor receptor inhibitors. Chest. 2012;142:1020–6. doi: 10.1378/chest.11-2943. [DOI] [PubMed] [Google Scholar]
- 24.Heist RS, Mino-Kenudson M, Sequist LV, Tammireddy S, Morrissey L, Christiani DC, et al. FGFR1 amplification in squamous cell carcinoma of the lung. J Thorac Oncol. 2012;7:1775–80. doi: 10.1097/JTO.0b013e31826aed28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schildhaus HU, Heukamp LC, Merkelbach-Bruse S, Riesner K, Schmitz K, Binot E, et al. Definition of a fluorescence in-situ hybridization score identifies high- and low-level FGFR1 amplification types in squamous cell lung cancer. Mod Pathol. 2012;25:1473–80. doi: 10.1038/modpathol.2012.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schultheis AM, Bos M, Schmitz K, Wilsberg L, Binot E, Wolf J, et al. Fibroblast growth factor receptor 1 (FGFR1) amplification is a potential therapeutic target in small-cell lung cancer. Mod Pathol. 2013 doi: 10.1038/modpathol.2013.141. [DOI] [PubMed] [Google Scholar]
- 27.Tran TN, Selinger CI, Kohonen-Corish MRJ, McCaughan BC, Kennedy CW, O'Toole SA, et al. Fibroblast growth factor receptor 1 (FGFR1) copy number is an independent prognostic factor in non-small cell lung cancer. Lung Cancer. 2013;81:462–7. doi: 10.1016/j.lungcan.2013.05.015. [DOI] [PubMed] [Google Scholar]
- 28.Weiss J, Sos ML, Seidel D, Peifer M, Zander T, Heuckmann JM, et al. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci Transl Med. 2010;2:62ra93. doi: 10.1126/scitranslmed.3001451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ishizuka T, Tanabe C, Sakamoto H, Aoyagi K, Maekawa M, Matsukura N, et al. Gene amplification profiling of esophageal squamous cell carcinomas by DNA array CGH. Biochem Biophys Res Commun. 2002;296:152–5. doi: 10.1016/s0006-291x(02)00836-7. [DOI] [PubMed] [Google Scholar]
- 30.Loga KV, Kohlhaussen J, Marx AH, Sauter G, Grob T, Quaas A. FGFR1 amplification is linked to the squamous cell carcinoma subtype in esophageal carcinoma. Cancer research. 2013;73 abstr 3024. [Google Scholar]
- 31.Phase I study of BGJ398, a selective pan-FGFR inhibitor in genetically preselected advanced solid tumors. In: Lowe SW, editor. AACR Annual Meeting 2014; 2014; San Diego. 2014. p. CT326. [Google Scholar]
- 32.Andre F, Ranson M, Dean E, Varga A, Noll Rvd, Stockman PK, et al. Results of a phase I study of AZD4547, an inhibitor of fibroblast growth factor receptor (FGFR), in patients with advanced solid tumors. Cancer Res. 2013;73 abstr LB-145. [Google Scholar]
- 33.Elbauomy Elsheikh S, Green AR, Lambros MB, Turner NC, Grainge MJ, Powe D, et al. FGFR1 amplification in breast carcinomas: a chromogenic in situ hybridisation analysis. Breast Cancer Res. 2007;9:R23. doi: 10.1186/bcr1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-Garcia MA, et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010;70:2085–94. doi: 10.1158/0008-5472.CAN-09-3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dutt A, Salvesen HB, Chen TH, Ramos AH, Onofrio RC, Hatton C, et al. Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc Natl Acad Sci U S A. 2008;105:8713–7. doi: 10.1073/pnas.0803379105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cappellen D, De Oliveira C, Ricol D, de Medina S, Bourdin J, Sastre-Garau X, et al. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet. 1999;23:18–20. doi: 10.1038/12615. [DOI] [PubMed] [Google Scholar]
- 37.Liao RG, Jung J, Tchaicha J, Wilkerson MD, Sivachenko A, Beauchamp EM, et al. Inhibitor-sensitive FGFR2 and FGFR3 mutations in lung squamous cell carcinoma. Cancer Res. 2013;73:5195–205. doi: 10.1158/0008-5472.CAN-12-3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Couraud S, Zalcman G, Milleron B, Morin F, Souquet PJ. Lung cancer in never smokers - A review. European Journal of Cancer. 2012;48:1299–311. doi: 10.1016/j.ejca.2012.03.007. [DOI] [PubMed] [Google Scholar]