

Abstract
Prolonged mechanical ventilation (MV) leads to rapid diaphragmatic atrophy and contractile dysfunction, which is collectively termed “ventilator-induced diaphragm dysfunction” (VIDD). Interestingly, endurance exercise training prior to MV has been shown to protect against VIDD. Further, recent evidence reveals that sedentary animals selectively bred to possess a high aerobic capacity possess a similar skeletal muscle phenotype to muscles from endurance trained animals. Therefore, we tested the hypothesis that animals with a high intrinsic aerobic capacity would naturally be afforded protection against VIDD. To this end, animals were selectively bred over 33 generations to create two divergent strains, differing in aerobic capacity: high-capacity runners (HCR) and low-capacity runners (LCR). Both groups of animals were subjected to 12 h of MV and compared with nonventilated control animals within the same strains. As expected, contrasted to LCR animals, the diaphragm muscle from the HCR animals contained higher levels of oxidative enzymes (e.g., citrate synthase) and antioxidant enzymes (e.g., superoxide dismutase and catalase). Nonetheless, compared with nonventilated controls, prolonged MV resulted in significant diaphragmatic atrophy and impaired diaphragm contractile function in both the HCR and LCR animals, and the magnitude of VIDD did not differ between strains. In conclusion, these data demonstrate that possession of a high intrinsic aerobic capacity alone does not afford protection against VIDD. Importantly, these results suggest that endurance exercise training differentially alters the diaphragm phenotype to resist VIDD. Interestingly, levels of heat shock protein 72 did not differ between strains, thus potentially representing an important area of difference between animals with intrinsically high aerobic capacity and exercise-trained animals.
Keywords: muscle atrophy, oxidative stress, mitochondria, reactive oxygen species
mechanical ventilation (mv) is a vital clinical intervention for critically ill patients that are unable to maintain adequate alveolar ventilation. Although MV can be lifesaving in patients suffering from respiratory failure, prolonged time on the ventilator results in rapid diaphragmatic atrophy and contractile dysfunction; this phenomenon is often termed ventilator-induced diaphragm dysfunction (VIDD) (44). VIDD is important because it can lead to problems in weaning patients from the ventilator (5). Prolonged time on the ventilator is a significant clinical problem because it increases the risk of patient morbidity and mortality due to infection and other complications (3, 4). Thus developing clinical interventions to prevent VIDD and reduce the risk of weaning problems is important.
Endurance exercise training is well-known to increase the oxidative and antioxidant capacity of the diaphragm (25, 26, 45, 46). Moreover, a recent study reveals that exercise training (i.e., exercise preconditioning) performed prior to MV exposure can protect against VIDD (39). This important finding indicates that exercise preconditioning results in a diaphragmatic phenotype that resists VIDD. Presently, the putative cytoprotective mediators of this effect are various members of the endogenous antioxidant system and heat shock proteins (39).
Interestingly, sedentary animals that have been selectively bred to have a high aerobic capacity (9) possess a skeletal muscle phenotype with numerous similarities to skeletal muscles from endurance trained animals (e.g., high oxidative capacity and elevated endogenous antioxidant capacity) (34, 35, 40, 43, 48). This observation raises the intriguing question, are the diaphragms from animals selectively bred to possess a high aerobic capacity protected against VIDD? Given the biochemical similarities of the diaphragm muscle between exercise-trained animals and animals that are selectively bred to possess a high aerobic capacity, we hypothesized that sedentary animals, with a high intrinsic aerobic capacity, would be naturally afforded protection against VIDD. We tested this hypothesis in a clinically relevant animal model of MV using rats selectively bred for a high aerobic capacity.
METHODS
Experimental Animals
All animal experiments were conducted with strict adherence to the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Moreover, the University of Florida (Gainesville) Institutional Animal Care and Use Committee approved these experiments.
Two strains of young adult rats (∼7–8 mo of age) that differ markedly in aerobic capacity, as a result of selective breeding, were used in the following experiments. The breeding paradigm has been described in detail previously (9). In brief, artificial selection for intrinsic aerobic endurance running capacity was started with the genetically heterogeneous N:NIH stock of rats as a founder population. From this founder population, selection for low and high capacity was determined based on distance run to exhaustion on a motorized treadmill by a velocity-ramped protocol (11). At each generation, 13 breeding pairs with the highest and lowest times to exhaustion were appropriately bred, which led to divergent strains of animals: one strain had a high aerobic capacity (high-capacity runners: HCR) and the other had a low aerobic capacity (low-capacity runners: LCR). At ∼3 mo of age, animals were phenotyped by assessing their running capacity during three separate trials on a treadmill running test. Running capacity was determined based on distance run to exhaustion. Each test was performed at a 15° incline by a velocity-ramped protocol that started at 10 m/min and was increased by 1 m/min every 2 min until failure. These tests were completed at the University of Michigan as previously described (9)(Table 1). For the current experiments, 40 female rats (20 HCR and 20 LCR animals) from generation 33, were transferred from the University of Michigan to the University of Florida. Animals were housed in customary caging maintained on a 12-h light-dark cycle with no access to wheel running. Animals were fed ad libitum standard rodent chow and water. Importantly, these animals were tested on the treadmill at ∼3 mo of age and were not subjected to the experimental protocol listed below until ∼7–8 mo of age. Thus these animals had no training intervention and can be classified as being sedentary.
Table 1.
Exercise capacity and body mass of experimental animals
| CON-LCR | MV-LCR | CON-HCR | MV-HCR | |
|---|---|---|---|---|
| Running capacity at 11 wk of age, m* | 242 ± 17 | 246 ± 21 | 2,322 ± 95 | 2,201 ± 43 |
| Running capacity at 11 wk of age, min* | 17.4 ± 0.9 | 17.5 ± 1.2 | 79.1 ± 1.9 | 76.7 ± 0.9 |
| Body mass at time of experiments, g*† | 239 ± 5 | 278 ± 7 | 226 ± 5 | 263 ± 3 |
Values are expressed as means ± SE. CON, control group; LCR, low-capacity runners; MV, mechanical ventilation; HCR, high-capacity runners.
Main effect of strain (LCR vs. HCR), P < 0.05.
Main effect of treatment (CON vs. MV), P < 0.05.
Experimental Design
LCR and HCR animals were assigned to one of four experimental groups (n = 10/group): 1) acutely anesthetized LCR (CON-LCR), 2) acutely anesthetized HCR (CON-HCR), 3) 12-h mechanically ventilated LCR (MV-LCR), and 4) 12-h mechanically ventilated HCR (MV-HCR). The 12-h duration of MV was selected based on previous findings from our lab and others demonstrating significant and repeatable levels of atrophy and contractile dysfunction (31, 32). Moreover, while difficult to directly compare, human investigations have also demonstrated marked atrophy in as few as 18 h (15, 32).
Experimental Protocol
Acutely anesthetized controls.
Animals in the control groups (CON-LCR and CON-HCR) were anesthetized with an intraperitoneal (IP) injection of sodium pentobarbital (60 mg/kg body wt). Upon reaching a deep anesthetic state, the diaphragm muscle was rapidly dissected and processed as follows. One section of the medial costal diaphragm was used for in vitro contractile measurements, one section was stored for histochemical analyses, one section was used for mitochondrial isolation, and the remaining section was frozen in liquid nitrogen and stored at −80°C for subsequent biochemical analyses.
Mechanical ventilation.
All surgical procedures were performed by aseptic techniques. Following induction of anesthesia with an IP injection of sodium pentobarbital (60 mg/kg body wt), animals were subjected to a tracheostomy and mechanically ventilated with a pressure-controlled ventilator (Servo Ventilator 300; Siemens, Munich, Germany) in the control-MV mode. The use of this setting allows the ventilator to deliver the entire breath, thus completely unloading the diaphragm. The MV protocol lasted for 12 h with the following settings: upper airway pressure limit: 20 cmH2O; respiratory rate: 80 bpm; positive end-expiratory pressure: 1 cmH2O.
Following the tracheostomy, the carotid artery was cannulated to permit continuous measurement of blood pressure and to obtain periodic blood samples (∼100 μl) that were analyzed for arterial pCO2, (PaCO2), pO2 (PaO2), and pH with an electronic blood gas analyzer (GEM Premier 3000; Instrumentation Laboratory, Lexington, MA). Moreover, a venous catheter was inserted into the jugular vein for continuous infusion of anesthesia (sodium pentobarbital, ∼10 mg·kg−1·h−1) throughout the 12-h MV protocol. Of note, this anesthetic protocol has been shown not to adversely affect diaphragmatic contractile function, myofiber cross-sectional area (CSA), or mitochondrial function (8, 31).
Animals received constant care during the MV protocol, which included lubricating the eyes, expressing the bladder, removing airway mucus, and rotating the animal to prevent blood pooling. Body temperature was maintained ∼37°C by use of a recirculating heating blanket, and heart rate was monitored via lead II of an electrocardiograph. Lastly, animals received an intramuscular injection of glycopyrrolate (0.04–0.08 mg/kg) every 2 h during MV to reduce airway secretions. After 12 h of MV, the diaphragm was quickly removed and processed identically as described above.
Functional Measures
Measurement of in vitro diaphragmatic contractile properties.
A muscle strip (∼3 mm wide) was suspended vertically between two lightweight Plexiglas clamps (Altuglas International, Philadelphia, PA) with one end connected to an isometric force transducer (FT-03, Grass Instruments, Quincy, MA) within a jacketed tissue bath containing a Krebs-Hensleit solution equilibrated with 95% oxygen and 5% carbon dioxide gas. The muscle was electrically stimulated to contract, and the force output was recorded via a computerized data acquisition system as previously described (31). To compare diaphragmatic force across groups, force production was normalized to CSA to obtain muscle “specific force” production.
Histological Measures
Myofiber cross-sectional area.
Sections from frozen diaphragm samples were cut at 10 microns with a cryostat (HM 550 Cryostat, Thermo Scientific, Waltham, MA) and subsequently stained for dystrophin (Thermo Scientific), myosin heavy chain (MHC) I, and MHC type IIa proteins (Developmental Studies Hybridoma Bank; University of Iowa, Iowa City, IA). Thereafter, muscle fiber CSA was determined with Scion software (National Institutes of Health, Bethesda, MD) as described previously (20). Note that diaphragm CSA data was normalized to body mass to allow for comparisons between groups where weight differences were present.
Biochemical Measures
Isolation of mitochondria.
Diaphragmatic mitochondria were isolated from ∼250 mg of costal diaphragm muscle by the methods of Makinen and Lee (18) with minor modifications as previously described (8). Briefly, after the diaphragm was trimmed to remove fat and connective tissues, it was weighed and placed in 10 volumes of solution I [100 mM KCl, 40 mM Tris-HCl, 10 mM Tris base, 1 mM MgSO4, 0.1 mM EDTA, 0.2 mM ATP, and 2% (wt/vol) free fatty acid bovine serum albumin (BSA) (pH 7.40)]. Diaphragms were then minced with scissors, and the mince was homogenized with an Ultra-Turrax (Cincinnati, OH) blender for 15 s at 40% of full power. Protease (trypsin; Sigma Chemical, St. Louis, MO) was added (5 mg/g wet muscle), and the digested mince was mixed continually for 7 min. Digestion was terminated through the addition of an equal volume of solution I. The homogenate was centrifuged at 500 g for 10 min at 4°C to pellet down contractile protein and cellular debris. The supernatant was rapidly decanted through a double layer of cheesecloth and centrifuged at 3,500 g for 10 min to pellet down the mitochondrial fraction. The supernatant was discarded, and the mitochondrial pellet was resuspended in solution I. The suspension was centrifuged at 3500 g for 10 min. The supernatant was again discarded, and the pellet was resuspended in 10 volumes of solution II (similar to solution I, but without BSA). This resuspended pellet was subsequently centrifuged at 3500 g for 10 min. The final mitochondrial pellet was suspended in 250 μl of a solution containing (in mM) 220 mannitol, 70 sucrose, 10 Tris-HCl, and 1 EGTA (pH 7.40).
Mitochondrial respiration.
Mitochondrial oxygen consumption was measured by previously described techniques (8). Briefly, maximal oxygen consumption was measured polarographically in a respiration chamber maintained at 37°C (Hansatech Instruments, UK). Isolated mitochondria were incubated with 1 ml of respiration buffer [100 mM KCl, 50 mM Mops, 20 mM glucose, 10 mM K2PO4, 10 mM MgCl2, 1 mM EGTA, and 0.2% BSA (pH 7.00)] at 37°C in a water-jacketed respiratory chamber with continuous stirring (Hansatech Instruments). Maximal respiration (state 3) was obtained with complex I substrates 2 mM pyruvate and 2 mM malate in the presence of 0.25 mM ADP, and state 4 respiration was recorded following the phosphorylation of ADP. Thereafter, the respiratory control ratio (RCR) was calculated as the quotient of state 3 divided by state 4 respiration.
Western blot analysis.
Preparation of muscle homogenates and Western blotting were performed according to standard procedures as described previously (27, 41) with modifications as outlined below. Briefly, diaphragm tissue samples were homogenized 1:10 (mg wt/μl buffer) in 5 mM Tris/5 mM EDTA (pH 7.5) with a protease inhibitor cocktail used 1:20 (vol/vol; Sigma-Aldrich, St. Louis, MO) and centrifuged at 30,000 g for 30 min at 4°C. After collection of the resulting supernatant, protein content was assessed by the method of Bradford (Sigma-Aldrich), and protein concentrations were normalized with Laemmli sample buffer (161-0737, Bio-Rad, Hercules, CA) containing 5% β-mercaptoethanol. Thereupon, proteins from the supernatant fraction of the diaphragm homogenates were separated on precast 4–20% gradient Criterion TGX stain-free gels (Bio-Rad), and then proteins were transferred onto nitrocellulose membrane. Following transfer, membranes were imaged to assess equal protein loading and transfer efficiency with the Gel Doc EZ System (Bio-Rad). Subsequently, membranes were blocked with Odyssey Blocking Buffer (LI-COR Biosciences, Lincoln, NE) diluted 1:1 with phosphate-buffered saline solution (PBS) for 1 h at room temperature, followed by incubation with primary antibodies directed against proteins of interest. Proteins of interest were diaphragmatic antioxidant levels, including copper-zinc superoxide dismutase (SOD1) (1:1,000; sc-11407; Santa Cruz Biotechnology, Santa Cruz, CA), manganese superoxide dismutase (SOD2) (1:1,000; sc-30080; Santa Cruz Biotechnology), glutathione peroxidase 1 (GPX1) (1:1,000; ab108427; Abcam, Cambridge, MA) and catalase (1:500; ab16731; Abcam). In addition, levels of citrate synthetase (CS) (1:2,000; ab96600; Abcam) and heat shock protein 72 (HSP72) (HSP01; 1:1,000; Calbiochem, San Diego, CA) were obtained. Following primary antibody incubation, membranes were washed in PBS containing Tween-20 (PBS-T) and then incubated for 1 h at room temperature with IRDye secondary antibodies (LI-COR Biosciences) diluted 1:20,000. Fluorescent images were collected on the Odyssey Infrared Imaging system (LI-COR Biosciences), and signals were quantified with Image Studio Lite software, Version 4.0 (LI-COR Biosciences). To control for protein loading and transfer differences, each protein was normalized to the level of α-tubulin (1:2,000; 12G10-c; Developmental Studies Hybrdioma Bank) detected on the same membrane as previously described (27, 41).
Statistical Analysis
Between-group comparisons for each dependent variable were made via two-way analysis of variance (ANOVA), with strain (LCR and HCR) and treatment (CON and MV) as fixed factors. Bonferroni post hoc tests were performed when significant main effects were present. Significance was established a priori at α < 0.05. Values are presented as means ± standard error.
RESULTS
Animal characteristics and phenotype.
Descriptive data for LCR and HCR rats are presented in Table 1. Importantly, running capacity assessed at 11 wk of age was over ninefold greater in the HCR rats, thus demonstrating the marked difference in aerobic phenotype between these strains due to the selective breeding paradigm (33). Note that the LCR animals were significantly heavier at the time of these experiments compared with the HCR animals (means ± SE, 245 ± 5 grams vs. 259 ± 6 grams; P < 0.05); similar findings have been reported previously by other investigators (10).
Systemic and biologic response to MV.
Because maintaining arterial blood gas and blood pressure homeostasis is important during prolonged MV, both parameters were carefully monitored throughout the protocol. Importantly, when PaCO2 began to exceed 40 mmHg and/or PaO2 approached 60 mmHg, ventilator adjustments were made, which included altering tidal volume and the fraction of inspired oxygen (FIO2; 22–26% oxygen) to maintain blood gas homeostasis as previously described (27). Evidence of our success in maintaining blood pressure and blood gas homeostasis in these animals during prolonged MV is illustrated in Table 2. Moreover, postmortem examination of the lungs and intestines did not reveal indications of infection or significant lung injury in the MV animals. This finding demonstrates that the aseptic surgical technique was successful and that prolonged MV did not result in major lung injury.
Table 2.
Systolic blood pressure, heart rate, arterial partial pressure of oxygen (PO2), arterial partial pressure of carbon dioxide (PCO2), and arterial pH were monitored at 3- to 6-h intervals during prolonged MV
| Physiological Variable | MV-LCR | MV-HCR |
|---|---|---|
| Systolic blood pressure, mmHg | 102 ± 4 | 103 ± 4 |
| Heart rate, beats/min | 380 ± 8 | 352 ± 7* |
| Arterial pO2, mmHg | 73 ± 3 | 73 ± 2 |
| Arterial pCO2, mmHg | 30 ± 1 | 33 ± 1 |
| Arterial pH | 7.46 ± 0.01 | 7.45 ± 0.01 |
Values are expressed as means ± SE.
Significant difference between groups.
MV-induced diaphragmatic contractile dysfunction.
To determine if having a high intrinsic aerobic capacity can protect the diaphragm against MV-induced contractile dysfunction, we measured the in vitro maximal isometric twitch force by using strips of diaphragm muscle and determined the force-frequency responses. First, focusing on animals with a high aerobic capacity, compared with control animals within the same strain (CON-HCR), 12 h of MV (MV-HCR) resulted in a significant reduction in diaphragm muscle force production at all stimulation frequencies (15–160 Hz; P < 0.05). Thus having a high intrinsic running capacity per se does not protect from contractile dysfunction during prolonged MV. Similarly, LCR animals responded to MV with significant reductions in diaphragm force production at all stimulation frequencies (15–160 Hz; P < 0.05). Importantly, no differences existed in diaphragm muscle contractile forces at any stimulation frequency between strains within a given treatment (CON-LCR vs. CON-HCR, P > 0.05; MV-LCR vs. MV-HCR, P > 0.05) (Fig. 1).
Fig. 1.
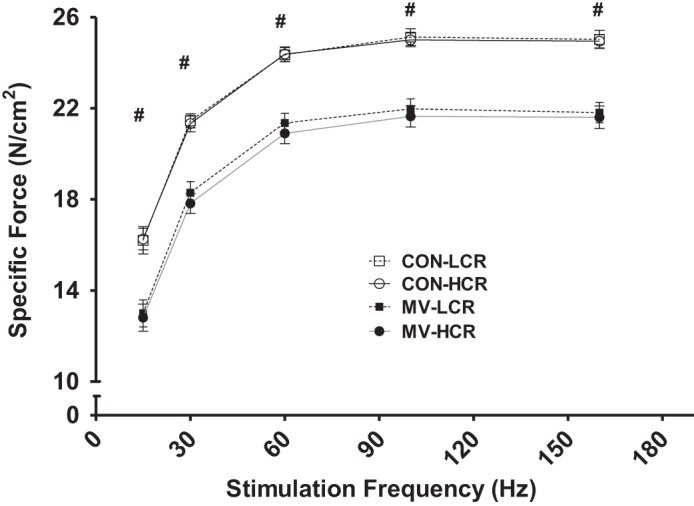
In vitro diaphragmatic force-frequency response. Values are means ± SE. MV, mechanical ventilation; LCR, low-capacity runner; HCR, high-capacity runner; CON, control group. #P < 0.05; MV-LCR and MV-HCR significantly different from CON-LCR and CON-HCR. No differences between CON-LCR and CON-HCR or MV-LCR and MV-HCR at any stimulation frequencies examined (n = 10/group). Note: because of similar values between control groups, symbols may be difficult to differentiate.
MV-induced diaphragm myofiber atrophy.
To determine if having a high intrinsic aerobic capacity can protect against MV-induced diaphragmatic atrophy, we measured diaphragm myofiber CSA in animals from all experimental groups. Diaphragm fiber CSA data were normalized to animal weight because greater body mass has been shown to increase diaphragmatic myofiber CSA (42). Importantly, prolonged MV resulted in atrophy (∼18% decrease in CSA) across all diaphragm myofiber types: type I (LCR, −19%; HCR, −18%), type IIa (LCR, −18%; HCR, −17%), and type IIb/x (LCR, −13%; HCR, −22%). Therefore, having an intrinsically high aerobic capacity alone does not afford protection against MV-induced diaphragmatic atrophy (Fig. 2).
Fig. 2.
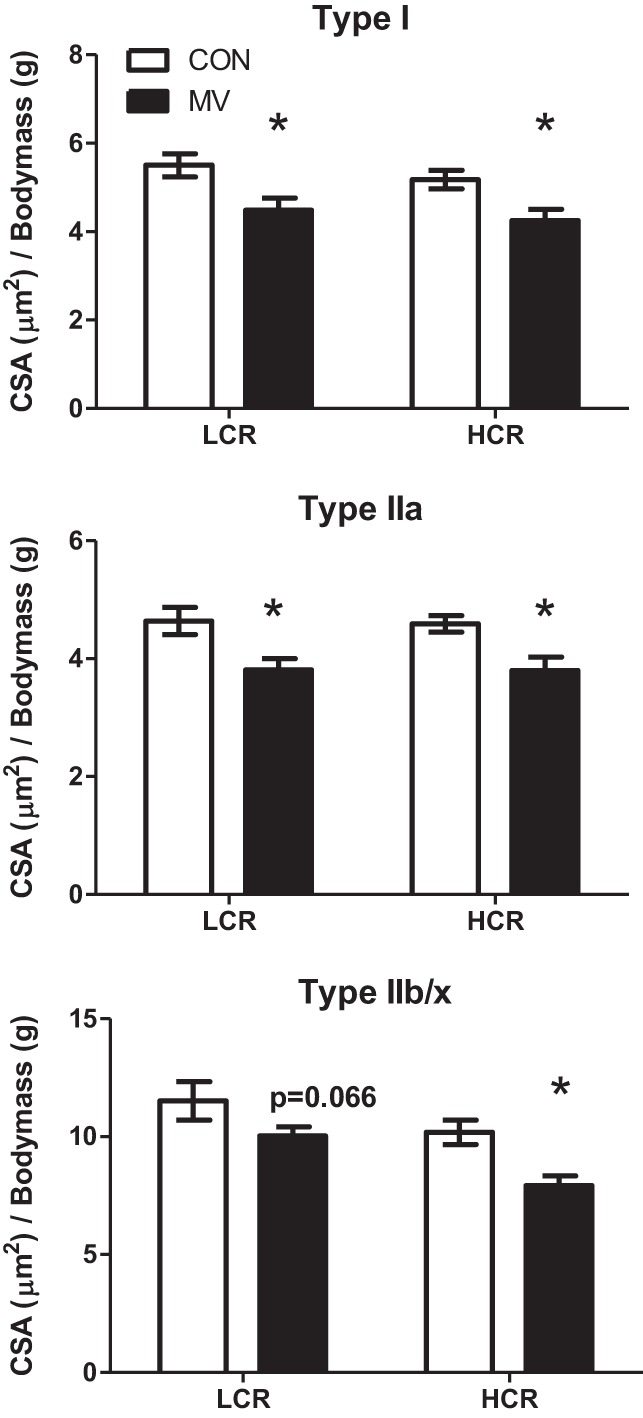
Fiber cross-sectional area (CSA) in diaphragm muscle myofibers from LCR and HCR CON or animals exposed to 12 h of MV. Values are normalized to body mass. Note that 12 h of MV caused significant atrophy of all fiber types for both strains. Values are means ± SE. *P < 0.05; MV significantly different from control within strain (HCR vs. LCR; n = 8–9/group).
MV-induced diaphragmatic mitochondrial dysfunction.
A hallmark of VIDD is mitochondrial dysfunction (8, 27). In this regard, our data indicate that 12 h of MV significantly reduces the RCR values in isolated diaphragmatic mitochondria from both HCR and LCR animals (Fig. 3). This MV-induced decrease in RCR was primarily a result of an increase in state 4 mitochondrial respiration in MV animals, thus recapitulating previous findings (8).
Fig. 3.
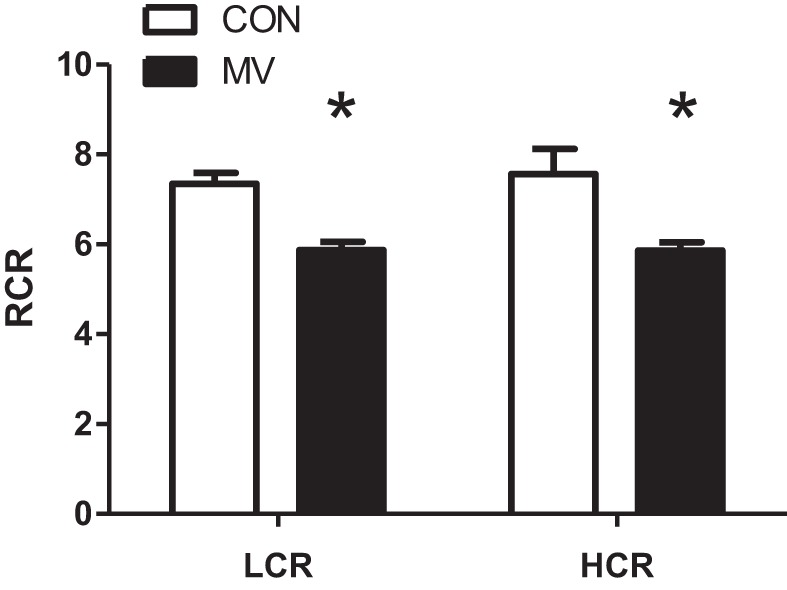
Isolated diaphragm mitochondria respiratory control ratio (RCR) from both LCR and HCR animals. Mitochondria were isolated from acutely anesthetized animals (CON) or animals exposed to 12 h of MV. RCR was obtained by diving state 3 and state 4 respiration. Data were obtained using pyruvate/malate as the substrate. Values are means ± SE. *P < 0.05; MV significantly different from control within strain (HCR vs. LCR; n = 7–9/group).
Antioxidant and oxidative enzyme levels in diaphragm muscle.
Because an increase in diaphragmatic production of reactive oxygen species (ROS) is required to develop VIDD (1, 27), we measured the relative protein abundance of key antioxidant enzymes in the diaphragm. Alike to previous reports, which examined limb muscles of HCR animals (35, 43), antioxidant levels in the diaphragm of HCRs were higher than LCRs. Specifically, compared with LCR animals, the diaphragms of HCR animals contained higher levels of SOD1, SOD2, and catalase. In contrast, regarding the relative protein abundance of GPX1, no differences were detected between groups (Fig. 4).
Fig. 4.

Relative protein abundance of primary antioxidants in diaphragm muscle. Values are mean percent change compared with LCR control ± SE and normalized to α-tubulin. Representative Western blots for the antioxidants are shown above the respective graphs. A: protein levels of SOD1 (CuZn-SOD). B: protein levels of SOD2 (MnSOD). C: protein levels of GPX1. D: protein levels of catalase. ¶P < 0.05; main effect of strain (HCR vs. LCR). #P < 0.05; main effect of treatment (CON vs. MV). †P < 0.05; difference between strains within CON (HCR vs. LCR). ŦP < 0.05; difference between strains within MV (HCR vs. LCR; n = 9–10).
Importantly, the diaphragm muscle from HCR animals contained significantly higher levels of the Krebs cycle enzyme citrate synthase, which has long been regarded as a proxy for mitochondrial content (Fig. 5). These data demonstrate an enhanced mitochondrial volume in the diaphragm muscle of HCR animals; similar findings have been reported in the limb muscles from these animals (40).
Fig. 5.

Assessment of citrate synthetase (CS) relative protein abundance in diaphragm muscle. Values are mean percent change compared with LCR control ± SE and normalized to α-tubulin. A representative Western blot is shown at top. ¶P < 0.05; main effect of strain (HCR vs. LCR). #P < 0.05; main effect of treatment (CON vs. MV). †P < 0.05; difference between strains within CON (HCR vs. LCR). ŦP < 0.05; difference between strains within MV (HCR vs. LCR). *P < 0.05; difference between treatment within HCR (CON vs. MV; n = 9–10).
Diaphragmatic HSP72 levels do not differ between HCR and LCR animals.
The 72-kDa heat shock protein (HSP72) is a putative mediator of exercise-induced cytoprotection in skeletal muscles and is increased in the diaphragm of exercise-trained animals (39). Therefore, we measured the relative protein abundance of HSP72 in the diaphragms of both HCR and LCR animals. Interestingly, no differences were present between strains regarding this protein (Fig. 6).
Fig. 6.

Relative protein abundance of heat shock protein 72 (HSP72). Values are mean percent change compared with LCR control ± SE and normalized to α-tubulin. A representative Western blot is shown at top graph. Note: no differences exist between any groups (n = 8–10/group).
DISCUSSION
Overview of Principle Findings
These experiments tested the hypothesis that animals selectively bred to possess a high intrinsic aerobic capacity (i.e., HCR animals) would be protected against VIDD. This hypothesis was formulated based on the recent evidence that endurance exercise training, performed prior to MV, results in a diaphragmatic phenotype that resists VIDD (39). However, contrary to our prediction, HCR animals were not protected against VIDD, as they experienced significant diaphragmatic atrophy and contractile dysfunction in response to prolonged MV. Therefore, our findings reveal that a high intrinsic aerobic capacity alone is not sufficient to protect against VIDD. A detailed discussion of these and other important findings follows.
HCRs and LCRs as an Experimental Model
Regular physical exercise is a well-established intervention used to reduce the risk of numerous diseases (e.g., heart disease, diabetes, etc.) (2), and it appears that this exercise-induced reduction in disease risk is linked to the oxidative capacity of the individual (22). Researchers have exploited this notion by selectively breeding animals to differ in intrinsic aerobic capacity, which has now become a powerful experimental model to investigate disease progression (9). Indeed, selective breeding over multiple generations has created two divergent strains of rats that differ dramatically in their intrinsic aerobic capacities and their susceptibility to various diseases (10).
Continued selective breeding over the generations has further increased the difference in running capacity (33), which has spurred numerous investigations to assess the differences in the skeletal muscle phenotype between animal strains. It is now well-established that HCR animals display a skeletal muscle phenotype with many similarities to exercise-trained muscle (e.g., increased antioxidants, uncoupling proteins, PGC-1α, etc.) (35, 40, 43, 48). These findings lead to the intriguing question, given that exercise training protects the diaphragm against VIDD (39), is the diaphragm of selectively bred HCR animals also protected against VIDD? This question forms the basis for the current experiments.
A High Intrinsic Aerobic Capacity Does Not Protect against VIDD
The present results clearly demonstrate that HCR animals experienced the same level of VIDD as LCR animals, therefore, raising a key question: what differences exist between an exercise-trained muscle and a muscle with an intrinsically high aerobic capacity? On the basis of the known cellular mechanisms that contribute to VIDD, we investigated the differences in the levels of primary antioxidant enzymes and an important member of the heat shock family of proteins, HSP72, in the diaphragm of these experimental animals. A brief discussion of these results follows.
Selective breeding for aerobic capacity and antioxidant capacity.
In reference to the mechanisms responsible for triggering VIDD, administration of select exogenous antioxidants can protect the diaphragm from both atrophy and contractile dysfunction during prolonged MV; these findings establish that increased oxidative stress is a requirement for VIDD (1, 27, 38, 47). Further, endurance exercise training increases the antioxidant capacity of the diaphragm muscle, which could potentially prevent MV-induced oxidative stress and thus protect against VIDD (30, 39, 45, 46). Similar to endurance-trained animals, diaphragm muscle in HCR animals also possesses higher levels of endogenous antioxidant enzymes (e.g., SOD1, SOD2, and catalase) compared with LCR animals (Fig. 4). Nonetheless, unlike exercise-trained animals, HCR animals are not protected against VIDD. A potential explanation for the failure of the HCR animals to possess innate protection against VIDD is that while the HCR animals possess higher levels of primary antioxidant enzymes in the diaphragm, this increase may not be sufficient to protect against MV-induced oxidative stress. Moreover, the measurement of primary antioxidant enzyme levels alone does not reflect the antioxidant capacity of specific cellular compartments or the total antioxidant capacity of the cell.
While the current results suggest that small increases in select diaphragmatic antioxidant enzymes levels alone is not sufficient to afford protection against VIDD, it is important to appreciate that this finding does not eliminate the possibility that exercise-induced increases in antioxidant molecules can provide protection against VIDD. For example, in addition to increases in primary antioxidant enzymes, exercise training can also increase the abundance of numerous molecules that contribute to the overall antioxidant capacity of muscle fibers (e.g., thioredoxins, perioxiredoxins, and/or glutathione) (28). It is also possible that exercise training results in posttranslational modifications to numerous antioxidant molecules resulting in an increased antioxidant capacity in the cell (6). Further, exercise training also increases the levels of HSP72 in skeletal muscles (21, 23); this exercise-induced increase in muscle HSP72 levels could complement the increase in cellular antioxidant capacity and might be an important contributor to exercise-induced protection against VIDD (39).
Selective breeding for aerobic capacity and heat shock protein 72.
As introduced above, increased levels of HSP72 in the diaphragm could play a protective role against VIDD. Indeed, heat stress, a major stimulator of HSP72 expression, has been shown to protect limb skeletal muscles against disuse atrophy (24, 36). More importantly, a recent study demonstrated that heat stress, prior to MV, significantly increased diaphragmatic levels of HSP72 and protected against MV-induced diaphragmatic atrophy (7). Theoretically, HSP72 can protect against disuse muscle atrophy in several potential ways: 1) HSP72 can maintain mitochondrial integrity by protecting mitochondria against apoptotic stimuli (17, 29, 49); 2) HSP72 can prevent proteolysis by binding to oxidized proteins and assisting in their refolding (16, 24); and/or 3) HSP72 can inhibit the activity of two transcriptional activators, FoxO3a and NF-κB, which play key roles in the atrophic process (37). Our previous work reveals that compared with sedentary controls, endurance exercise training results in a significant (e.g., 50%) increase in diaphragmatic levels of HSP72 (39). However, between strains, diaphragm muscle from HCR and LCR animals contain similar levels of HSP72, and both of these experimental groups were equally affected by VIDD. These findings raise the question of whether exercise-induced increases in diaphragmatic levels of HSP72 play an essential role in exercise-induced protection against VIDD; this is a testable hypothesis worthy of further investigation.
Areas for Future Investigation
The current results, in combination with previous findings in animals that display a divergent capacity to respond to exercise training, raise several interesting questions regarding the impact of exercise training on diaphragmatic preconditioning. For example, an intriguing area of inquiry relates to two new strains of rats that differ in their response to exercise training. Specifically, one of these strains, termed low-response trainers, displays a blunted response to endurance exercise training, whereas another strain (i.e., high-response trainers) are readily adaptable to exercise training (12). Following exercise training the low-responding animals do not improve their ability to run during a graded exercise test, yet these animals demonstrate increases in skeletal muscle mitochondrial volume and improved mitochondrial function (13, 19). Therefore, it would be interesting to determine if endurance exercise training results in diaphragmatic preconditioning in both these low-response and the high-response training animals. Importantly, the LCR animals used in the current study do respond to endurance exercise training as demonstrated by improvements in their skeletal muscle metabolic function after training (14); therefore, it would be interesting to determine if LCR animals can achieve the same level of exercise preconditioning in the diaphragm as observed in our previous study with outbred Sprague-Dawley rats (39). Ultimately, the most important translational area for future research would entail investigating the susceptibility of patients to VIDD based on their initial level of physical fitness.
SUMMARY AND CONCLUSIONS
Recent evidence reveals that exercise training (i.e., exercise preconditioning) alters the diaphragmatic phenotype to resist VIDD (39). This led to the hypothesis that animals possessing a high intrinsic aerobic capacity would also be protected against VIDD. Nonetheless, our results demonstrate that rats selectively bred to have a high aerobic capacity are not protected from VIDD. These novel findings disclose that endurance exercise training is a unique stimulus for diaphragmatic adaptation. This point highlights the fact that muscular exercise is a powerful experimental tool for identifying biological targets to prevent VIDD and this remains an exciting area for future research.
GRANTS
This work was supported by National Institutes of Health Grant RO1-AR-064189 (to S. Powers); the LCR-HCR rat model system was supported by National Institutes of Health Grants R24-OD-010950 and R01-DK-099034 (to L. Koch and S. Britton), R01-GM-104194 (to S. Britton), and R01-DK-077200 (to S. Britton).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
K.J.S., L.G.K., S.L.B., and S.K.P. conception and design of research; K.J.S., A.J.S., M.P.W., and A.B.M. performed experiments; K.J.S., A.J.S., M.P.W., and A.B.M. analyzed data; K.J.S., A.J.S., M.P.W., A.B.M., and S.K.P. interpreted results of experiments; K.J.S., A.J.S., M.P.W., and A.B.M. prepared figures; K.J.S. and S.K.P. drafted manuscript; K.J.S., A.J.S., M.P.W., A.B.M., L.G.K., S.L.B., and S.K.P. edited and revised manuscript; K.J.S., A.J.S., M.P.W., A.B.M., L.G.K., S.L.B., and S.K.P. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Stephanie Hall for providing valuable feedback on this manuscript. We also thank Dr. Jatin Burniston for helping us establish this collaborative effort with the University of Michigan. We acknowledge the expert care of the rat colony provided by Molly Kalahar and Lori Heckenkamp. Contact LK (lgkoch@med.umich.edu) or SB (brittons@umich.edu) for information on the LCR and HCR rats: these rat models are maintained as an international resource with support from the Department of Anesthesiology at the University of Michigan, Ann Arbor, Michigan.
REFERENCES
- 1.Betters JL, Criswell DS, Shanely RA, Van Gammeren D, Falk D, Deruisseau KC, Deering M, Yimlamai T, Powers SK. Trolox attenuates mechanical ventilation-induced diaphragmatic dysfunction and proteolysis. Am J Respir Crit Care Med 170: 1179–1184, 2004. [DOI] [PubMed] [Google Scholar]
- 2.Chodzko-Zajko WJ, Proctor DN, Fiatarone Singh MA, Minson CT, Nigg CR, Salem GJ, Skinner JS. American College of Sports Medicine position stand. Exercise and physical activity for older adults. Med Sci Sports Exerc 41: 1510–1530, 2009. [DOI] [PubMed] [Google Scholar]
- 3.Dasta JF, McLaughlin TP, Mody SH, Piech CT. Daily cost of an intensive care unit day: the contribution of mechanical ventilation. Crit Care Med 33: 1266–1271, 2005. [DOI] [PubMed] [Google Scholar]
- 4.Esteban A, et al. . Evolution of mechanical ventilation in response to clinical research. Am J Respir Crit Care Med 177: 170–177, 2008. [DOI] [PubMed] [Google Scholar]
- 5.Esteban A, Frutos F, Tobin MJ, Alia I, Solsona JF, Valverdu I, Fernandez R, de la Cal MA, Benito S, Tomas R, et al. . A comparison of four methods of weaning patients from mechanical ventilation. Spanish Lung Failure Collaborative Group. N Engl J Med 332: 345–350, 1995. [DOI] [PubMed] [Google Scholar]
- 6.Hollander J, Bejma J, Ookawara T, Ohno H, Ji LL. Superoxide dismutase gene expression in skeletal muscle: fiber-specific effect of age. Mech Ageing Dev 116: 33–45, 2000. [DOI] [PubMed] [Google Scholar]
- 7.Ichinoseki-Sekine N, Yoshihara T, Kakigi R, Sugiura T, Powers SK, Naito H. Heat stress protects against mechanical ventilation-induced diaphragmatic atrophy. J Appl Physiol 117: 518–524, 2014. [DOI] [PubMed] [Google Scholar]
- 8.Kavazis AN, Talbert EE, Smuder AJ, Hudson MB, Nelson WB, Powers SK. Mechanical ventilation induces diaphragmatic mitochondrial dysfunction and increased oxidant production. Free Radic Biol Med 46: 842–850, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koch LG, Britton SL. Artificial selection for intrinsic aerobic endurance running capacity in rats. Physiol Genomics 5: 45–52, 2001. [DOI] [PubMed] [Google Scholar]
- 10.Koch LG, Britton SL, Wisloff U. A rat model system to study complex disease risks, fitness, aging, and longevity. Trends Cardiovasc Med 22: 29–34, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koch LG, Meredith TA, Fraker TD, Metting PJ, Britton SL. Heritability of treadmill running endurance in rats. Am J Physiol Regul Integr Comp Physiol 275: R1455–R1460, 1998. [DOI] [PubMed] [Google Scholar]
- 12.Koch LG, Pollott GE, Britton SL. Selectively bred rat model system for low and high response to exercise training. Physiol Genomics 45: 606–614, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lessard SJ, Rivas DA, Alves-Wagner AB, Hirshman MF, Gallagher IJ, Constantin-Teodosiu D, Atkins R, Greenhaff PL, Qi NR, Gustafsson T, Fielding RA, Timmons JA, Britton SL, Koch LG, Goodyear LJ. Resistance to aerobic exercise training causes metabolic dysfunction and reveals novel exercise-regulated signaling networks. Diabetes 62: 2717–2727, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lessard SJ, Rivas DA, Stephenson EJ, Yaspelkis BB 3rd, Koch LG, Britton SL, Hawley JA. Exercise training reverses impaired skeletal muscle metabolism induced by artificial selection for low aerobic capacity. Am J Physiol Regul Integr Comp Physiol 300: R175–R182, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levine S, Nguyen T, Taylor N, Friscia ME, Budak MT, Rothenberg P, Zhu J, Sachdeva R, Sonnad S, Kaiser LR, Rubinstein NA, Powers SK, Shrager JB. Rapid disuse atrophy of diaphragm fibers in mechanically ventilated humans. N Engl J Med 358: 1327–1335, 2008. [DOI] [PubMed] [Google Scholar]
- 16.Locke M, Tanguay RM, Klabunde RE, Ianuzzo CD. Enhanced postischemic myocardial recovery following exercise induction of HSP 72. Am J Physiol Heart Circ Physiol 269: H320–H325, 1995. [DOI] [PubMed] [Google Scholar]
- 17.Maglara AA, Vasilaki A, Jackson MJ, McArdle A. Damage to developing mouse skeletal muscle myotubes in culture: protective effect of heat shock proteins. J Physiol 548: 837–846, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Makinen MW, Lee CP. Biochemical studies of skeletal muscle mitochondria. I. Microanalysis of cytochrome content, oxidative and phosphorylative activities of mammalian skeletal muscle mitochondria. Arch Biochem Biophys 126: 75–82, 1968. [DOI] [PubMed] [Google Scholar]
- 19.Marton O, Koltai E, Takeda M, Koch LG, Britton SL, Davies KJ, Boldogh I, Radak Z. Mitochondrial biogenesis-associated factors underlie the magnitude of response to aerobic endurance training in rats. Pflugers Arch. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McClung JM, Kavazis AN, Whidden MA, DeRuisseau KC, Falk DJ, Criswell DS, Powers SK. Antioxidant administration attenuates mechanical ventilation-induced rat diaphragm muscle atrophy independent of protein kinase B (PKB Akt) signalling. J Physiol 585: 203–215, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morton JP, Kayani AC, McArdle A, Drust B. The exercise-induced stress response of skeletal muscle, with specific emphasis on humans. Sports Med 39: 643–662, 2009. [DOI] [PubMed] [Google Scholar]
- 22.Myers J, Prakash M, Froelicher V, Do D, Partington S, Atwood JE. Exercise capacity and mortality among men referred for exercise testing. N Engl J Med 346: 793–801, 2002. [DOI] [PubMed] [Google Scholar]
- 23.Naito H, Powers SK, Demirel HA, Aoki J. Exercise training increases heat shock protein in skeletal muscles of old rats. Med Sci Sports Exerc 33: 729–734, 2001. [DOI] [PubMed] [Google Scholar]
- 24.Naito H, Powers SK, Demirel HA, Sugiura T, Dodd SL, Aoki J. Heat stress attenuates skeletal muscle atrophy in hindlimb-unweighted rats. J Appl Physiol 88: 359–363, 2000. [DOI] [PubMed] [Google Scholar]
- 25.Powers SK, Criswell D, Lawler J, Martin D, Ji LL, Herb RA, Dudley G. Regional training-induced alterations in diaphragmatic oxidative and antioxidant enzymes. Respir Physiol 95: 227–237, 1994. [DOI] [PubMed] [Google Scholar]
- 26.Powers SK, Criswell D, Lieu FK, Dodd S, Silverman H. Diaphragmatic fiber type specific adaptation to endurance exercise. Respir Physiol 89: 195–207, 1992. [DOI] [PubMed] [Google Scholar]
- 27.Powers SK, Hudson MB, Nelson WB, Talbert EE, Min K, Szeto HH, Kavazis AN, Smuder AJ. Mitochondria-targeted antioxidants protect against mechanical ventilation-induced diaphragm weakness. Crit Care Med 39: 1749–1759, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Powers SK, Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev 88: 1243–1276, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Powers SK, MLA, Demirel HA. Exercise, heat shock proteins, and myocardial protection from I-R injury. Med Sci Sports Exerc 33: 386–392, 2001. [DOI] [PubMed] [Google Scholar]
- 30.Powers SK, Shanely RA. Exercise-induced changes in diaphragmatic bioenergetic and antioxidant capacity. Exerc Sport Sci Rev 30: 69–74, 2002. [DOI] [PubMed] [Google Scholar]
- 31.Powers SK, Shanely RA, Coombes JS, Koesterer TJ, McKenzie M, Van Gammeren D, Cicale M, Dodd SL. Mechanical ventilation results in progressive contractile dysfunction in the diaphragm. J Appl Physiol 92: 1851–1858, 2002. [DOI] [PubMed] [Google Scholar]
- 32.Powers SK, Wiggs MP, Sollanek KJ, Smuder AJ. Ventilator-induced diaphragm dysfunction: cause and effect. Am J Physiol Regul Integr Comp Physiol 305: R464–R477, 2013. [DOI] [PubMed] [Google Scholar]
- 33.Ren YY, Overmyer KA, Qi NR, Treutelaar MK, Heckenkamp L, Kalahar M, Koch LG, Britton SL, Burant CF, Li JZ. Genetic analysis of a rat model of aerobic capacity and metabolic fitness. PLoS One 8: e77588, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rivas DA, Lessard SJ, Saito M, Friedhuber AM, Koch LG, Britton SL, Yaspelkis BB 3rd Hawley JA. Low intrinsic running capacity is associated with reduced skeletal muscle substrate oxidation and lower mitochondrial content in white skeletal muscle. Am J Physiol Regul Integr Comp Physiol 300: R835–R843, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seifert EL, Bastianelli M, Aguer C, Moffat C, Estey C, Koch LG, Britton SL, Harper ME. Intrinsic aerobic capacity correlates with greater inherent mitochondrial oxidative and H2O2 emission capacities without major shifts in myosin heavy chain isoform. J Appl Physiol 113: 1624–1634, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Selsby JT, Dodd SL. Heat treatment reduces oxidative stress and protects muscle mass during immobilization. Am J Physiol Regul Integr Comp Physiol 289: R134–R139, 2005. [DOI] [PubMed] [Google Scholar]
- 37.Senf SM, Dodd SL, McClung JM, Judge AR. Hsp70 overexpression inhibits NF-kappaB and Foxo3a transcriptional activities and prevents skeletal muscle atrophy. FASEB J 22: 3836–3845, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smuder AJ, Hudson MB, Nelson WB, Kavazis AN, Powers SK. Nuclear factor-kappaB signaling contributes to mechanical ventilation-induced diaphragm weakness. Crit Care Med 40: 927–934, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smuder AJ, Min K, Hudson MB, Kavazis AN, Kwon OS, Nelson WB, Powers SK. Endurance exercise attenuates ventilator-induced diaphragm dysfunction. J Appl Physiol 112: 501–510, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stephenson EJ, Stepto NK, Koch LG, Britton SL, Hawley JA. Divergent skeletal muscle respiratory capacities in rats artificially selected for high and low running ability: a role for Nor1? J Appl Physiol 113: 1403–1412, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Talbert EE, Smuder AJ, Min K, Kwon OS, Szeto HH, Powers SK. Immobilization-induced activation of key proteolytic systems in skeletal muscles is prevented by a mitochondria-targeted antioxidant. J Appl Physiol 115: 529–538, 2013. [DOI] [PubMed] [Google Scholar]
- 42.Tamaki N. Effect of growth on muscle capillarity and fiber type composition in rat diaphragm. Eur J Appl Physiol Occup Physiol 54: 24–29, 1985. [DOI] [PubMed] [Google Scholar]
- 43.Tweedie C, Romestaing C, Burelle Y, Safdar A, Tarnopolsky MA, Seadon S, Britton SL, Koch LG, Hepple RT. Lower oxidative DNA damage despite greater ROS production in muscles from rats selectively bred for high running capacity. Am J Physiol Regul Integr Comp Physiol 300: R544–R553, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vassilakopoulos T, Petrof BJ. Ventilator-induced diaphragmatic dysfunction. Am J Respir Crit Care Med 169: 336–341, 2004. [DOI] [PubMed] [Google Scholar]
- 45.Vincent HK, Powers SK, Demirel HA, Coombes JS, Naito H. Exercise training protects against contraction-induced lipid peroxidation in the diaphragm. Eur J Appl Physiol Occup Physiol 79: 268–273, 1999. [DOI] [PubMed] [Google Scholar]
- 46.Vincent HK, Powers SK, Stewart DJ, Demirel HA, Shanely RA, Naito H. Short-term exercise training improves diaphragm antioxidant capacity and endurance. Eur J Appl Physiol 81: 67–74, 2000. [DOI] [PubMed] [Google Scholar]
- 47.Whidden MA, Smuder AJ, Wu M, Hudson MB, Nelson WB, Powers SK. Oxidative stress is required for mechanical ventilation-induced protease activation in the diaphragm. J Appl Physiol 108: 1376–1382, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wisloff U, Najjar SM, Ellingsen O, Haram PM, Swoap S, Al-Share Q, Fernstrom M, Rezaei K, Lee SJ, Koch LG, Britton SL. Cardiovascular risk factors emerge after artificial selection for low aerobic capacity. Science 307: 418–420, 2005. [DOI] [PubMed] [Google Scholar]
- 49.Xiao R, Ferry AL, Dupont-Versteegden EE. Cell death-resistance of differentiated myotubes is associated with enhanced anti-apoptotic mechanisms compared to myoblasts. Apoptosis 16: 221–234, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]