

Abstract
Insulin and IGF-1 are recognized as powerful regulators of the epithelial Na+ channel (ENaC) in the aldosterone-sensitive distal nephron. As previously described, these hormones both acutely increase ENaC activity in freshly isolated split open tubules and cultured principal cortical collecting duct cells. The present study was aimed at differentiating the effects of insulin and IGF-1 on Na+ transport in immortalized mpkCCDcl4 cells and defining their interrelations. We have shown that both insulin and IGF-1 applied basolaterally, but not apically, enhanced transepithelial Na+ transport in the mpkCCDcl4 cell line with EC50 values of 8.8 and 14.5 nM, respectively. Insulin treatment evoked phosphorylation of both insulin and IGF-1 receptors, whereas the effects of IGF-1 were more profound on its own receptor rather than the insulin receptor. AG-1024 and PPP, inhibitors of IGF-1 and insulin receptor tyrosine kinase activity, diminished insulin- and IGF-1-stimulated Na+ transport in mpkCCDcl4 cells. The effects of insulin and IGF-1 on ENaC-mediated currents were found to be additive, with insulin likely stimulating both IGF-1 and insulin receptors. We hypothesize that insulin activates IGF-1 receptors in addition to its own receptors, making the effects of these hormones interconnected.
Keywords: insulin, insulin-like growth factor 1, cortical collecting duct, epithelial Na+ channel, Na+ transport, kidney, tyrosine kinase
insulin receptor (IR) signaling is crucial for renal function in various segments of the kidney, particularly in glomeruli and tubules (19, 20). Its compromised action leads to a number of renal complications, including but not limited to hypertension, altered nutrient reabsorption, and glomerular diseases (38). Several groups in the 1990s characterized IR expression in the kidney using autoradiography (31) and 125I-labeled insulin binding in microdissected tubules (10); these experiments allowed the localization of IR to glomeruli, medullary vascular bundles, proximal convoluted tubules, distal convoluted tubules, outer medullary and cortical collecting ducts (CCDs), and cortical thick ascending limbs (TAL) of the loop of Henle. Most recently, using highly specific IR antibodies, Tiwari et al. (37) demonstrated that the IR is expressed in the proximal tubule, TAL, and collecting duct. IGF-1 is structurally similar to insulin, and its own IGF-1 receptor (IGF-1R) shares a high degree of homology with the IR (25, 30). Both receptors are members of the receptor tyrosine kinase family and are encoded by genetic loci believed to have evolved from a common ancestral gene (25). Insulin and IGF-1 are known to enhance Na+ reabsorption in the nephron; however, the role of IGF-1 in this process in much less defined and understood.
Insulin has been shown to stimulate Na+ reabsorption in the proximal tubule, TAL, and distal tubules, including CCDs (1, 9, 14, 23, 27, 35). Previous studies (11, 12) in dogs and humans have suggested that the distal tubule is the major nephron segment characterized by insulin-dependent Na+ transport. The activity of epithelial Na+ channels (ENaC), which represent the rate-limiting step for Na+ reabsorption in the distal nephron, has been shown to be upregulated by insulin ex vivo in the collecting duct or in cell cultures (5, 6, 13, 24, 32, 41). Activation of ENaC by IGF-1 has also been reported (3, 4, 18, 33). IGF-1 mRNA-positive cells have been found in collecting ducts and distal tubules (34). Net Na+ flux in a classical model of the mammalian distal nephron, the toad urinary bladder, is stimulated by concentrations of IGF-1 as low as 0.1 nM, and the maximal response has been detected at 10 nM (3). In the Madin-Darby canine kidney line of collecting duct cells, IGF-1 increased amiloride-sensitive Na+ transport along with aldosterone and antidiuretic hormone (7). Blazer-Yost et al. (8) demonstrated in 1992 that the majority of IR were associated with the basolateral membrane, whereas IGF-1R were found on basolateral and apical membranes. Gonzales-Rodriguez et al. (18) compared the effects of insulin and IGF-1 in a mCCDcl1 cell model. Their data revealed that this cell line was highly responsive to IGF-1, but not insulin, and they observed that insulin and IGF-1 effects were mainly mediated through IGF-1Rs and not IR. We (33) have also demonstrated that IGF-1 has acute effects on ENaC activity in isolated rat split open tubules.
We have recently benefited from knowledge obtained from transgenic mice lacking IR in specific parts of the nephron (26, 39). It has been shown that knockout of IR from portions of the renal tubule, including the TAL through the collecting duct, using a Ksp-cadherin promoter-targeted Cre-recombinase in these animals resulted in an impaired ability to excrete NaCl load, 50% reduced urinary nitric oxide excretion, and surprisingly elevated blood pressure (39). A previous study (26) also revealed that ENaC expression and blood pressure are reduced in mice with genetic knockout of IR specifically in collecting duct principal cells. Using mice lacking IR in principal cells of the CCD, we (29) have shown that insulin increases ENaC activity via its own receptor, affecting the channel open probability (Po). Thus, there is ample evidence suggesting a fundamental role of insulin in the regulation of Na+ homeostasis in the kidney. However, there are studies that contradict these findings. For example, data of Furuya et al. (17) and Vehaskari et al. (40) obtained in isolated perfused rabbit CCDs showed either a modest increase or no effect of insulin on Na+ transport. This is largely consistent with the data of Frindt and Palmer (15), who did not observe any stimulation of Na+ conductance by insulin in rat collecting ducts.
There is evidence that insulin can act through the IGF-1R and, likewise, that IGF-1 can signal via the IR, although the affinities are different (20). In general, accumulated findings suggest that physiological functions of IR and IGF-1R can be overlapping. For instance, recent data from our laboratory suggest that insulin and IGF-1 have a common stimulatory effect on ENaC mediated by the production of ROS (22). When studied in cultured cells, the two receptors are capable of producing similar cellular responses (25). However, some data support a concept that the actions of these hormones can be significantly different depending on the tissue where they are expressed (18). The present study was designed to examine the cross-talk between insulin, IGF-1, and their receptors in a model of mouse CCD cells (mpkCCDcl4 cells).
MATERIALS AND METHODS
Cell culture.
Immortalized mouse CCD principal cells (mpkCCDcl4 cells) were kindly provided by Dr. A. Vandewalle (Institut National de la Santé et de la Recherche Médicale, Paris, France) and grown in defined medium in cell culture flasks as previously described (2, 22, 33). Cells were then seeded onto permeable supports (Costar Transwells, 0.4 μm pore, 24-mm diameter) at a density of 0.2–0.3 × 106 cells/filter. mpkCCDcl4 cells were kept on filter supports for at least 7 days, and the medium was changed every second day. Cells were maintained with FBS and dexamethasone, allowing them to polarize and form a monolayer with high resistance and avid Na reabsorption. Growth medium was composed of equal volumes of DMEM and Ham's F-12, 60 nM Na+ selenate, 5 μg/ml transferrin, 50 nM dexamethasone, 1 nM triiodothyronine, 10 ng/ml EGF, 5 μg/ml insulin, 2% FCS, and 100 μg/ml penicillin-streptomycin. Cells were grown in a 5% CO2-95% air atmosphere incubator at 37°C. Typically, after 7 days, a confluent transporting cell monolayer developed, which was subsequently assessed by recording voltage and transepithelial resistance. Eighteen hours before use in any experiment, the medium of the cells incubated on filter supports was replaced with minimal medium that contained only DMEM, Ham's F-12, and antibiotics.
Equivalent open-circuit current experiments.
Transepithelial Na+ current across the mpkCCDcl4 cell monolayer was calculated according to Ohm's law as the quotient of transepithelial voltage to transepithelial resistance using a Millicel Electrical Resistance System (Millipore, Billerica, MA) to measure voltage and resistance as previously described (21, 22). To determine the net Na+ transport through ENaC, 10 μM amiloride was added to the apical cell surface at the end of each experiment.
Western blot analysis.
mpkCCDcl4 cells were washed twice in PBS and lysed in buffer as previously described (28). Equal amounts of proteins were separated using 10–20% SDS-PAGE, electrophoretically transferred onto nitrocellulose membranes (Millipore), immunoblotted with the appropriated antibody, and visualized by enhanced chemiluminescence (Amersham Biosciences, Piscataway, NJ).
Chemicals and antibodies.
Insulin (catalog no. I6634) and IGF-1 (catalog no. 200-05) were purchased from Sigma-Aldrich (St. Louis, MO) and Shenandoah Biotechnology (Warwick, PA), respectivley. Anti-phosphorylated (p)IR antibody (ab60946) was from Abcam (Cambridge, MA); anti-IR (sc-710), anti-pIGR-1R (sc-101704), and anti-IGF-1R (sc-713) antibodies as well as PPP (sc-204008) were from Santa Cruz Biotechnology. AG-1024 (catalog no. 121767) was purchased from EMD Millipore (Darmstadt, Germany).
Statistics.
Figure preparation and statistical analyses were carried out using MicroCal Origin software 9.0 (OriginLab). Dose responses were calculated using Pharmacology/DoseResp analyses in Origin 9.0 software. All summarized data are reported as means ± SE. Statistical differences were tested with either Student's (two-tailed) t-test or one-way ANOVA. Significance was defined at P < 0.05.
RESULTS
Insulin and IGF-1 enhance amiloride-sensitive currents across monolayers of mpkCCDcl4 cells.
The effects of insulin and IGF-1 were tested on polarized monolayers of mpkCCDcl4 cells, which develop robust amiloride-sensitive transepithelial Na+ transport. Various concentrations of insulin (1–200 nM) and IGF-1 (1–500 ng/ml) were applied basolaterally at the beginning of the experiment, and changes in transepithelial ENaC-mediated current were monitored in response to these drugs. Figures 1 and 2 show changes in amiloride-sensitive currents across mpkCCDcl4 monolayers in response to different concentrations of insulin (Fig. 1) and IGF-1 (Fig. 2). EC50 values for insulin and IGF-1 were 8.8 ± 2.4 and 14.5 ± 0.5 nM (110.5 ± 4.2 ng/ml), respectively. EC50 values were calculated at the time points corresponding to peaks of the response (2 and 4 h for insulin and 6 h of treatment with IGF-1). Summarized dose-response curves are shown in Figs. 1B and 2B, respectively. We also tested apical applications of both insulin and IGF-1 (see Fig. 3, A and B, for insulin and IGF-1 applications, respectively), which had no effect on recorded currents, as distinct from the basolateral application.
Fig. 1.
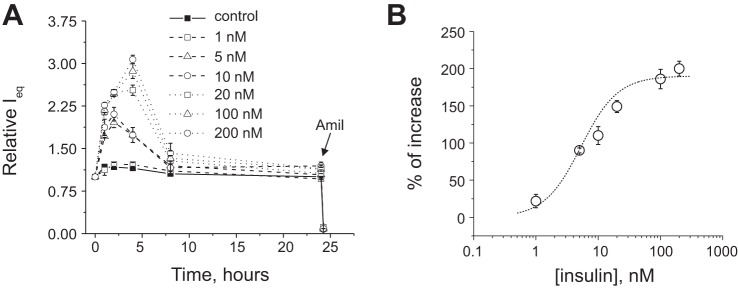
Insulin stimulates amiloride (Amil)-sensitive Na+ currents in mpkCCDcl4 cells as shown by equivalent transepithelial current (Ieq) measurements. A: time course of the action of insulin. Amiloride (10 μM) was added at the end of each experiment to ensure the epithelial Na+ channel (ENaC)-mediated nature of the current. B: summarized insulin dose-response curve approximated by Hill's equation. Each summarized point on the representative graph stands for at least n = 6.
Fig. 2.
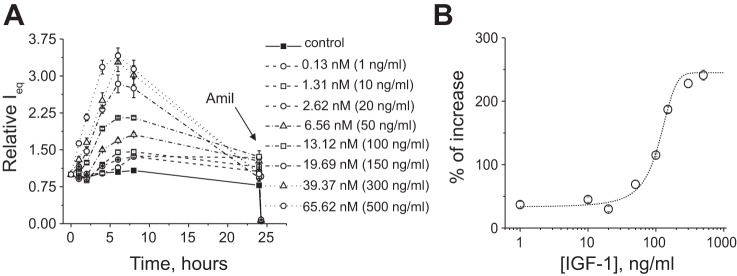
IGF-1 stimulates amiloride-sensitive Na+ currents in mpkCCDcl4 cells. A: time course of the action of IGF-1. B: summarized IGF-1 dose-response curve approximated by Hill's equation. Each summarized point on the representative graph stands for at least n = 6.
Fig. 3.
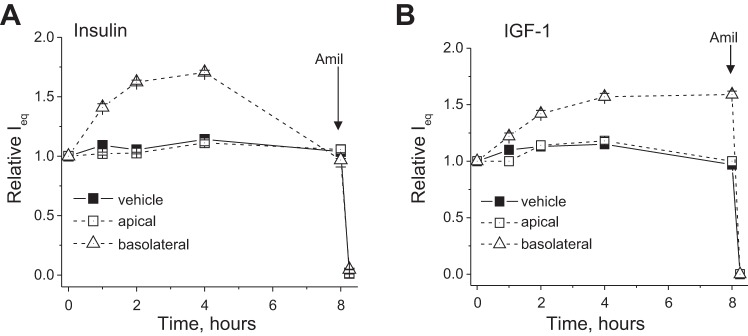
Time course of the actions of 20 nM insulin and 100 ng/ml (12 nM) IGF-1 on ENaC-mediated currents across mpkCCDcl4 monolayers. Insulin and IGF-1 were added apically or basolaterally. Each summarized point on the representative graph stands for at least n = 6.
Effects of IGF-1 and insulin on expression levels of total IGF-1, pIGF-1, and IR in mpkCCDcl4 cells.
We tested expression levels of IGF-1R and IR in response to insulin and IGF-1. After stimulation, mpkCCDcl4 cell lysates were obtained, and Western blot analysis was conducted using IR, IGF-1R, and their respective phospho-specific antibodies. As shown in Fig. 4, A and B, expression levels of total IGF-1R and IR were not significantly altered by incubation with saturating concentrations of insulin or IGF-1 separately or together for 4 h [100 nM insulin and 200 ng/ml (∼25 nM) IGF-1]. Moreover, we tested the effects of PPP and AG-1024 on levels of total IGF-1R and IR. These compounds are cell permeable and act as competitive, reversible, and potent inhibitors of IGF-1R and IR and exhibit little effect toward other members of the receptor tyrosine kinase family, such as the FGF receptor and EGF receptor. As shown in Fig. 4, A and B, neither incubation with 500 nM PPP (overnight) nor 500 nM AG-1024 (right before application of the hormones) exhibited any effect on total levels of IGF-1R or IR within cells in the absence of insulin or IGF-1 stimuli. Representative Western blot images and summarized data normalized to β-actin are shown in Fig. 4, top and bottom, respectively.
Fig. 4.

Expression levels of the insulin receptor (IR; A) and IGF-1 receptor (IGF-1R; B) after stimulation with IGF-1 or insulin (ins) and in the presence or absence of PPP and AG-1024 (AG). Representative Western blot analyses (n is shown on the summarizing graphs at the bottom) demonstrate the effects of 100 nM insulin and 200 ng/ml (25 nM) IGF-1 added separately and together, as well as the inhibitors PPP and AG-1024, on the expression of total levels of IR and IGF-1R in the mpkCCDcl4 cell line. β-Actin was used to verify equal loading. Cells were incubated with PPP (500 nM) overnight and acutely (right before the addition of insulin or IGF-1) with 500 nM AG-1024. veh, Vehicle; ns, not significant.
Figure 5, A–C, shows the effects of insulin and IGF-1 on levels of their phosphorylated receptors. Both insulin and IGF-1 increased phosphorylation of IR and IGF-1R. However, the overall increase in pIGF-1R phosphorylation in response to both insulin and IGF-1 was much more profound compared with IR phosphorylation. Neither PPP nor AG-1024 exhibited any influence on expression levels of pIR and pIGF-1R in the absence of hormonal stimuli. As shown in Fig. 5C, AG-1024 exhibited rather specific action on the phosphorylated form of IGF-1R; pIR levels were not altered by AG-1024 in the presence of either of the hormones. Interestingly, as shown in Fig. 5, A and B, application of both insulin and IGF-1 produced an additive effect on pIGF-1R expression, which was not observed for pIR.
Fig. 5.

Expression levels of phosphorylated (p)IR (A) and pIGF-1R (B) after stimulation with IGF-1 and insulin and in the presence of AG-1024 or PPP. C: expression levels of pIR and pIGF-1 stimulated by insulin or IGF-1 in the presence and absence of AG-1024. Representative Western blot analysis (n is shown on the summarizing graphs at the bottom) demonstrate the effects of 100 nM insulin and 200 ng/ml (25 nM) IGF-1 added separately and together, as well as AG-1024, on the expression of pIR and pIGF-1R in the mpkCCDcl4 cell line. β-actin was used to verify equal loading. Cells were incubated with PPP (500 nM) overnight and acutely (right before the addition of insulin or IGF-1) with 500 nM AG-1024. *P < 0.01 compared with control.
Inhibitors of IGF-1R and IR (PPP and AG-1024) decrease amiloride-sensitive insulin- or IGF-1-stimulated currents in mpkCCDcl4 cell monolayers.
Further experiments tested the effects of IGF-1R and IR inhibitors PPP and AG-1024 on insulin- and IGF-1-stimulated currents. mpkCCDcl4 cell monolayers grown on permeable supports were treated overnight with 500 nM PPP or vehicle and then stimulated with vehicle (control) or 12 nM IGF-1 (100 ng/ml). As shown in Fig. 6A, over the 24-h recording period, PPP significantly reduced the stimulatory effect of IGF-1, whereas overnight treatment with the compound itself did not have any significant effect on basal levels of the amiloride-sensitive current.
Fig. 6.
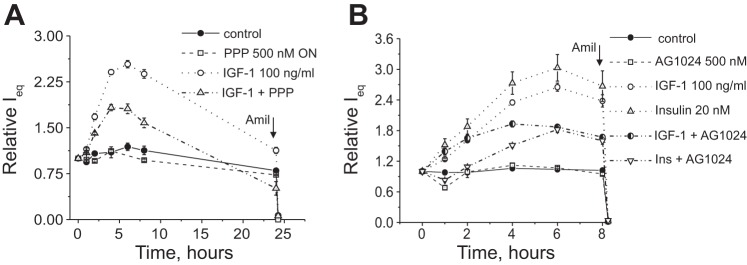
Effects of IR and IGF-1R inhibitors PPP and AG-1024 on ENaC-mediated currents stimulated by insulin and IGF-1. A: summarized time course of the action of the IGF-1 inhibitor PPP (500 nM, overnight treatment) on 100 ng/ml IGF-1-stimulated ENaC-mediated current. Amiloride (10 μM) was added at the end of experiments to ensure that the currents recorded were ENaC driven. Each summarized point on the representative graph stands for at least n = 6. B: AG-1024 inhibited both IGF-1- and insulin-simulated currents in the mpkCCDcl4 cell line. Shown is a summarized time course of the action of the IGF-1 inhibitor AG-1024 (500 nM, applied before the experiment) on IGF-1-stimulated (12 nM) and insulin-stimulated (20 nM) ENaC-mediated currents. Each summarized point on the representative graph stands for at least n = 6.
AG-1024 is known to be a cell-permeable, reversible, and specific inhibitor of IGF-1R and IR tyrosine kinase activity but exhibits lower IC50 values for IGF-1 (0.4 μM) than for IR. AG-1024 (500 nM) was applied immediately before transepithelial current measurements were started. As shown in Fig. 6B, AG-1024 itself did not cause any changes in basal levels of ENaC-mediated currents; however, treatment with this drug partially prevented the development of IGF-1 and insulin-stimulated responses (compared with treatment with IGF-1 or insulin only).
Additive effects of insulin and IGF-1 on ENaC-mediated currents in the mpkCCDcl4 cell line.
As shown in Fig. 5, there was an additive effect of insulin and IGF-1 treatment on levels of pIGF-1R, whereas pIR was not additively stimulated by IR and IGF-1. We further tested if there were similar effects on ENaC-mediated currents. To address this question, we performed stimulation of ENaC-mediated currents with saturating concentrations of insulin and IGF-1 [100 nM and 200 ng/ml (25 nM), respectively] together (Fig. 7A). As shown in Fig. 7A, stimulation of IR and IGF-1R together with insulin and IGF-1 resulted in an additive increase in the short-circuit current; this indicates that either one of the receptors, or both of them, can be cross-activated by the corresponding hormones or that there exist separate intracellular pathways mediated by IR and IGF-1 that eventually activate ENaC.
Fig. 7.
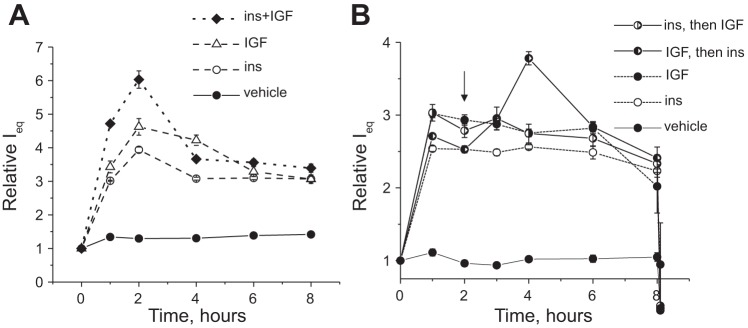
Additive effects of insulin and IGF-1 on ENaC-mediated currents in mpkCCDc14 cell monolayers. A: time courses of the action of saturating concentrations of insulin (100 nM) and IGF-1 (200 ng/ml or ∼25 nM) added separately or together at the 0-h time point. B: stimulation of amiloride-sensitive currents by 100 nM insulin or 200 ng/ml IGF-1 after an additional application of the saturating concentration of IGF-1 or insulin at the 2-h time point, respectively. Each summarized point on the representative graph stands for n = 6. The arrow denotes the addition of insulin or IGF-1 to cells pretreated with IGF-1 or insulin, respectively.
To further study the cross-reactivity of insulin and IGF-1, we stimulated ENaC-mediated currents in mpkCCDcl4 monolayers with saturating concentration of insulin and then added a saturating concentration of IGF-1, and vice versa. As shown in Fig. 7B, both insulin and IGF-1 produced increases ENaC-mediated current. However, addition of IGF-1 over insulin treatment did not produce any additional increase, whereas treatment with insulin over IGF-1 resulted in significant additive amiloride-sensitive current. These data clarify the results shown in Fig. 7A and are in accordance with the Western blot data shown in Fig. 5.
DISCUSSION
The present study was intended to define the interrelation between IR and IGF-1R in murine CCD cells. Our experiments demonstrated that both insulin and IGF-1 stimulate amiloride-sensitive ENaC-mediated currents in these cells. Insulin and IGF-1 demonstrated concentration-dependent effects on ENaC-mediated currents with EC50 values of 8.8 ± 2.4 and 14.5 ± 0.5 nM (110.5 ± 4.2 ng/ml), respectively. Theilig et al. (36) reported that increased ENaC expression was observed with insulin- and IGF-1-mediated apical stimulation of mpkCCDcl4 cells. To study the distribution of the receptors in cultured polarized mpkCCDcl4 cells, we tested insulin and IGF-1 applied to the apical side of the monolayer; neither insulin nor IGF-1 applied apically resulted in any induction of the amiloride-sensitive current (Fig. 3). This is consistent with the data of Gonzalez-Rodriguez et al. (18) indicating that the effects of IGF-1 and insulin are mediated by receptors located at the basolateral membrane.
Western blot analysis revealed the effects that insulin and IGF-1 have on the phosphorylation of the corresponding receptors. We assessed expression levels of IR and IGF-1R in their phosphorylated states (pIR and pIGF-1R). Incubation with insulin increased both pIR and pIGF-1R abundance, whereas IGF-1 influenced the phosphorylation of IR to a much lesser extent. Levels of total IGF-1R and IR were not affected by hormonal treatments.
To specify the interrelation between actions of insulin and IGF-1 on IR and IGF-1R, we applied the IGF-1R and IR inhibitors PPP and AG-1024. Our results show that neither PPP nor AG-1024 had effects on the abundance of total IR and IGF-1R. AG-1024 (500 nM) decreased both insulin- and IGF-1-stimulated induction of the amiloride-sensitive current; treatment with 500 nM PPP also partially prevented the effects of IGF-1 on ENaC-mediated currents. This result, most likely, should be attributed to the fact that both PPP and AG-1024 exhibit nonspecific inhibitory effects toward phosphorylation of IR and IGF-1R. To briefly summarize, experiments using these pharmacological agents should be carefully considered since the autophosphorylation site, which most antibodies are designed for (Tyr1131/1135/1136 for pIGF-1R and Tyr1158/1162/1163 for pIR) is very conserved between IR and IGF-1R. Furthermore, PPP is actually inhibits phosphorylation at this site.
Nevertheless, it appears that there is an interrelation between the effects of IGF-1 and insulin on ENaC activity. These data add to our previous findings about the effects of insulin in IR knockout animals (29); we hypothesized there that insulin acutely increases ENaC activity via its own receptor, affecting channel Po. Interestingly, our patch-clamp single channel measurements in freshly isolated split-open mouse tubules showed that insulin treatment enhanced ENaC activity in tubules isolated from wild-type mice but did not have this effect in IR knockout mice (29). Similarly, we (33) previously demonstrated that IGF-I acutely increases the Po of ENaC in mpkCCDcl4 cells and native principal cells in a phosphatidylinositol 3-kinase-dependent manner.
Gonzales-Rodriguez et al. (18) studied the effects of IGF-1 and insulin in mCCDcl1 cells and suggested that this cell line is highly responsive to IGF-1, and not insulin, and that the observed insulin and IGF-1 responses were mainly signaled through IGF-1R and not IR. However, their observations regarding insulin responsiveness could be affected by the long exposure of cells to insulin under the culturing conditions, and this could explain the decreased insulin sensitivity. In our experiments, mpkCCDcl4 cells were responsive to both insulin and IGF-1 with similar EC50 values, although the time course of the response was different. The authors note that insulin circulating levels are low on fasting and increase after feeding, whereas free plasma IGF-1 does not fluctuate during the circadian cycle (16). Therefore, IGF-1 could be one of the hormones maintaining basal levels of Na+ reabsorption, whereas insulin could determine acute Na+ balance.
Taken together with previously published data, our results contribute to the understanding of the interdependence of insulin/IGF-1 signaling; as shown in Figs. 5 and 7, insulin has the ability to induce the phosphorylation of both IR and IGF-1R to the same extent, whereas IGF-1R has a lower affinity to IR compared with its own receptor. The electrophysiological data (Fig. 7) demonstrate that IGF-1 is unable to additionally stimulate ENaC-mediated currents once insulin has saturated IR and IGF-1R. However, saturating concentrations of IGF-1 allow room for the stimulation of ENaC currents by insulin. This led us to the hypothesis that insulin can stimulate two intracellular pathways (likely involving both IR and IGF-1R), whereas IGF-1 signaling only saturates one of these pathways. This conclusion further suggests that the action of insulin is essential for ENaC activity, whereas IGF-1 might be responsible for the activation of a pathway that could be a compensatory pathway for the maintenance of renal function related to Na+ reabsorption in CCD principal cells. However, this can get more complicated if hybrid receptors are formed, by combination of IR and IGF-1R, through which both hormones can transduce the signal with different affinities (20). Nevertheless, it is clear that insulin signaling is the predominant pathway responsible for the activation and maintenance of ENaC reabsorption in cells of the CCD.
In summary, manipulation of IR and IGF-1R is a promising means of treating kidney diseases associated with abnormalities in Na+ reabsorption mediated via insulin and IGF-1 signaling. Additional studies using transgenic models with knockouts of IR and IGF-1R in principal cells of the CCD are required. Double knockout of IR and IGF-1R should also provide important information about role of these receptors in the development of kidney disease and hypertension.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants R01-HL-108880 (to A. Staruschenko), R01-HL-056259 (to M. W. Brands), and K99-HL-116603 (to T. S. Pavlov), American Diabetes Association Grant 1-10-BS-168 (to A. Staruschenko), a Medical College of Wisconsin Research Affairs Committee Pilot Grant (to T. S. Pavlov), and a Ben J. Lipps Research Fellowship from the American Society of Nephrology (to D. V. Ilatovskaya).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: D.V.I. and A.S. conception and design of research; D.V.I., V.L., and T.S.P. performed experiments; D.V.I., V.L., and T.S.P. analyzed data; D.V.I., M.W.B., and A.S. interpreted results of experiments; D.V.I. and V.L. prepared figures; D.V.I. and A.S. drafted manuscript; D.V.I., T.S.P., and A.S. edited and revised manuscript; D.V.I., V.L., M.W.B., T.S.P., and A.S. approved final version of manuscript.
REFERENCES
- 1.Baum M. Insulin stimulates volume absorption in the rabbit proximal convoluted tubule. J Clin Invest 79: 1104–1109, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bens M, Vallet V, Cluzeaud F, Pascual-Letallec L, Kahn A, Rafestin-Oblin ME, Rossier BC, Vandewalle A. Corticosteroid-dependent sodium transport in a novel immortalized mouse collecting duct principal cell line. J Am Soc Nephrol 10: 923–934, 1999. [DOI] [PubMed] [Google Scholar]
- 3.Blazer-Yost BL, Cox M. Insulin-like growth factor 1 stimulates renal epithelial Na+ transport. Am J Physiol Cell Physiol 255: C413–C417, 1988. [DOI] [PubMed] [Google Scholar]
- 4.Blazer-Yost BL, Cox M, Furlanetto R. Insulin and IGF I receptor-mediated Na+ transport in toad urinary bladders. Am J Physiol Cell Physiol 257: C612–C620, 1989. [DOI] [PubMed] [Google Scholar]
- 5.Blazer-Yost BL, Esterman MA, Vlahos CJ. Insulin-stimulated trafficking of ENaC in renal cells requires PI 3-kinase activity. Am J Physiol Cell Physiol 284: C1645–C1653, 2003. [DOI] [PubMed] [Google Scholar]
- 6.Blazer-Yost BL, Liu X, Helman SI. Hormonal regulation of ENaCs: insulin and aldosterone. Am J Physiol Cell Physiol 274: C1373–C1379, 1998. [DOI] [PubMed] [Google Scholar]
- 7.Blazer-Yost BL, Record RD, Oberleithner H. Characterization of hormone-stimulated Na+ transport in a high-resistance clone of the MDCK cell line. Pflügers Arch 432: 685–691, 1996. [DOI] [PubMed] [Google Scholar]
- 8.Blazer-Yost BL, Shah N, Jarett L, Cox M, Smith RM. Insulin and IGF1 receptors in a model renal epithelium: receptor localization and characterization. Biochem Int 28: 143–153, 1992. [PubMed] [Google Scholar]
- 9.Brands MW, Manhiani MM. Sodium-retaining effect of insulin in diabetes. Am J Physiol Regul Integr Comp Physiol 303: R1101–R1109, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Butlen D, Vadrot S, Roseau S, Morel F. Insulin receptors along the rat nephron: [125I] insulin binding in microdissected glomeruli and tubules. Pflügers Arch 412: 604–612, 1988. [DOI] [PubMed] [Google Scholar]
- 11.DeFronzo RA, Cooke CR, Andres R, Faloona GR, Davis PJ. The effect of insulin on renal handling of sodium, potassium, calcium, and phosphate in man. J Clin Invest 55: 845–855, 1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeFronzo RA, Goldberg M, Agus ZS. The effects of glucose and insulin on renal electrolyte transport. J Clin Invest 58: 83–90, 1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Faletti CJ, Perrotti N, Taylor SI, Blazer-Yost BL. sgk: an essential convergence point for peptide and steroid hormone regulation of ENaC-mediated Na+ transport. Am J Physiol Cell Physiol 282: C494–C500, 2002. [DOI] [PubMed] [Google Scholar]
- 14.Feraille E, Rousselot M, Rajerison R, Favre H. Effect of insulin on Na+, K+-ATPase in rat collecting duct. J Physiol 488: 171–180, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frindt G, Palmer LG. Effects of insulin on Na and K transporters in the rat CCD. Am J Physiol Renal Physiol 302: F1227–F1233, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frystyk J. Free insulin-like growth factors–measurements and relationships to growth hormone secretion and glucose homeostasis. Growth Horm IGF Res 14: 337–375, 2004. [DOI] [PubMed] [Google Scholar]
- 17.Furuya H, Tabei K, Muto S, Asano Y. Effect of insulin on potassium secretion in rabbit cortical collecting ducts. Am J Physiol Renal Fluid Electrolyte Physiol 262: F30–F35, 1992. [DOI] [PubMed] [Google Scholar]
- 18.Gonzalez-Rodriguez E, Gaeggeler HP, Rossier BC. IGF-1 vs. insulin: respective roles in modulating sodium transport via the PI-3 kinase/Sgk1 pathway in a cortical collecting duct cell line. Kidney Int 71: 116–125, 2007. [DOI] [PubMed] [Google Scholar]
- 19.Hale LJ, Coward RJ. The insulin receptor and the kidney. Curr Opin Nephrol Hypertens 22: 100–106, 2013. [DOI] [PubMed] [Google Scholar]
- 20.Hale LJ, Coward RJ. Insulin signalling to the kidney in health and disease. Clin Sci (Lond) 124: 351–370, 2013. [DOI] [PubMed] [Google Scholar]
- 21.Ilatovskaya DV, Pavlov TS, Levchenko V, Negulyaev YA, Staruschenko A. Cortical actin binding protein cortactin mediates ENaC activity via Arp2/3 complex. FASEB J 25: 2688–2699, 2011. [DOI] [PubMed] [Google Scholar]
- 22.Ilatovskaya DV, Pavlov TS, Levchenko V, Staruschenko A. ROS production as a common mechanism of ENaC regulation by EGF, insulin and IGF-1. Am J Physiol Cell Physiol 304: C102–C111, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ito O, Kondo Y, Oba M, Takahashi N, Omata K, Abe K. Tyrosine kinase, phosphatidylinositol 3-kinase, and protein kinase C regulate insulin-stimulated NaCl absorption in the thick ascending limb. Kidney Int 51: 1037–1041, 1997. [DOI] [PubMed] [Google Scholar]
- 24.Kamynina E, Staub O. Concerted action of ENaC, Nedd4-2, and Sgk1 in transepithelial Na+ transport. Am J Physiol Renal Physiol 283: F377–F387, 2002. [DOI] [PubMed] [Google Scholar]
- 25.Kim JJ, Accili D. Signalling through IGF-I and insulin receptors: where is the specificity? Growth Horm IGF Res 12: 84–90, 2002. [DOI] [PubMed] [Google Scholar]
- 26.Li L, Garikepati RM, Tsukerman S, Kohan D, Wade JB, Tiwari S, Ecelbarger CM. Reduced ENaC activity and blood pressure in mice with genetic knockout of the insulin receptor in the renal collecting duct. Am J Physiol Renal Physiol 304: F279–F288, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mandon B, Siga E, Chabardes D, Firsov D, Roinel N, De Rouffignac C. Insulin stimulates Na+, Cl−, Ca2+, and Mg2+ transports in TAL of mouse nephron: cross-potentiation with AVP. Am J Physiol Renal Fluid Electrolyte Physiol 265: F361–F369, 1993. [DOI] [PubMed] [Google Scholar]
- 28.Pavlov TS, Chahdi A, Ilatovskaya DV, Levchenko V, Vandewalle A, Pochynyuk O, Sorokin A, Staruschenko A. Endothelin-1 inhibits the epithelial Na+ channel through βPix/14-3-3/Nedd4-2. J Am Soc Nephrol 21: 833–843, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pavlov TS, Ilatovskaya DV, Levchenko V, Li L, Ecelbarger CM, Staruschenko A. Regulation of ENaC in mice lacking renal insulin receptors in the collecting duct. FASEB J 27: 2723–2732, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Samani AA, Yakar S, LeRoith D, Brodt P. The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev 28: 20–47, 2007. [DOI] [PubMed] [Google Scholar]
- 31.Sechi LA, De Carli S, Bartoli E. In situ characterization of renal insulin receptors in the rat. J Recept Res 14: 347–356, 1994. [DOI] [PubMed] [Google Scholar]
- 32.Shimkets RA, Lifton R, Canessa CM. In vivo phosphorylation of the epithelial sodium channel. Proc Natl Acad Sci USA 95: 3301–3305, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Staruschenko A, Pochynyuk O, Vandewalle A, Bugaj V, Stockand JD. Acute regulation of the epithelial Na+ channel by phosphatidylinositide 3-OH kinase signaling in native collecting duct principal cells. J Am Soc Nephrol 18: 1652–1661, 2007. [DOI] [PubMed] [Google Scholar]
- 34.Sugimoto H, Shikata K, Makino H, Ota K, Ota Z. Increased gene expression of insulin-like growth factor-I receptor in experimental diabetic rat glomeruli. Nephron 72: 648–653, 1996. [DOI] [PubMed] [Google Scholar]
- 35.Takahashi N, Ito O, Abe K. Tubular effects of insulin. Hypertens Res 19, Suppl 1: S41–S45, 1996. [DOI] [PubMed] [Google Scholar]
- 36.Theilig F, Geers C, Spohr C, Enke A, Peters H. Proteinuria induces increased insulin- and IGF1-receptor signalling, a mechanism of ENaC-mediated volume retention. J Am Soc Nephrol 22: 526A, 2011.21258036 [Google Scholar]
- 37.Tiwari S, Halagappa VK, Riazi S, Hu X, Ecelbarger CA. Reduced expression of insulin receptors in the kidneys of insulin-resistant rats. J Am Soc Nephrol 18: 2661–2671, 2007. [DOI] [PubMed] [Google Scholar]
- 38.Tiwari S, Riazi S, Ecelbarger CA. Insulin's impact on renal sodium transport and blood pressure in health, obesity, and diabetes. Am J Physiol Renal Physiol 293: F974–F984, 2007. [DOI] [PubMed] [Google Scholar]
- 39.Tiwari S, Sharma N, Gill PS, Igarashi P, Kahn CR, Wade JB, Ecelbarger CM. Impaired sodium excretion and increased blood pressure in mice with targeted deletion of renal epithelial insulin receptor. Proc Natl Acad Sci USA 105: 6469–6474, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vehaskari VM, Hering-Smith KS, Moskowitz DW, Weiner ID, Hamm LL. Effect of epidermal growth factor on sodium transport in the cortical collecting tubule. Am J Physiol Renal Fluid Electrolyte Physiol 256: F803–F809, 1989. [DOI] [PubMed] [Google Scholar]
- 41.Wang J, Barbry P, Maiyar AC, Rozansky DJ, Bhargava A, Leong M, Firestone GL, Pearce D. SGK integrates insulin and mineralocorticoid regulation of epithelial sodium transport. Am J Physiol Renal Physiol 280: F303–F313, 2001. [DOI] [PubMed] [Google Scholar]