

Abstract
In inflammatory bowel disease (IBD), obesity is associated with worsening of the course of disease. Here, we examined the role of obesity in the development of colitis and studied mesenteric fat-epithelial cell interactions in patients with IBD. We combined the diet-induce obesity with the trinitrobenzene sulfonic acid (TNBS) colitis mouse model to create groups with obesity, colitis, and their combination. Changes in the mesenteric fat and intestine were assessed by histology, myeloperoxidase assay, and cytokine mRNA expression by real-time PCR. Medium from human mesenteric fat and cultured preadipocytes was obtained from obese patients and those with IBD. Histological analysis showed inflammatory cell infiltrate and increased histological damage in the intestine and mesenteric fat of obese mice with colitis compared with all other groups. Obesity also increased the expression of proinflammatory cytokines including IL-1β, TNF-α, monocyte chemoattractant protein 1, and keratinocyte-derived chemokine, while it decreased the TNBS-induced increases in IL-2 and IFN-γ in mesenteric adipose and intestinal tissues. Human mesenteric fat isolated from obese patients and those with and IBD demonstrated differential release of adipokines and growth factors compared with controls. Fat-conditioned media reduced adiponectin receptor 1 (AdipoR1) expression in human NCM460 colonic epithelial cells. AdipoR1 intracolonic silencing in mice exacerbated TNBS-induced colitis. In conclusion, obesity worsens the outcome of experimental colitis, and obesity- and IBD-associated changes in adipose tissue promote differential mediator release in mesenteric fat that modulates colonocyte responses and may affect the course of colitis. Our results also suggest an important role for AdipoR1 for the fat-intestinal axis in the regulation of inflammation during colitis.
Keywords: obesity, adipose tissue, adipokines, colitis
obesity is an epidemic affecting one out of three Americans (8, 38) and a major risk factor for chronic diseases such as diabetes, cardiovascular diseases, and cancer (17, 30). Moreover, obesity-associated metabolic syndrome affects approximately one-fourth of the US population, with resulting comorbidities burdening the healthcare system (8, 38). Obesity involves a low-grade inflammatory state, mostly attributed to altered function of hypertrophic adipocytes. Adipose tissue is an active endocrine organ (1) and a source of cytokines, such as TNF-α, interleukins, and the adipokines adiponectin, leptin, and ghrelin (7, 9, 25, 44, 47). These mediators play proinflammatory, anti-inflammatory, or appetite-controlling roles depending on the conditions during their release (34, 42). Circulating levels of adipokines are also deregulated in obese patients (10), and this response may contribute to the pathophysiology of obesity-related diseases.
Frequency of inflammatory bowel disease (IBD), which includes ulcerative colitis (UC) and Crohn's Disease (CD), is elevated in the developed world and associated with increasing morbidity (36). Anatomically, the affected intestine is in immediate proximity to the intra-abdominal mesenteric and omental fat depots, which contain lymph nodes and are well vascularized. Although poorly understood, the presence of adipose tissue wrapping around intestinal lesions in patients with CD (creeping fat) has been well documented during surgery (15), whereas fat wrapping in UC has not been reported. Moreover, patients with higher body mass index (BMI) at diagnosis demonstrate an increased need for hospitalization during the course of the disease and a shorter time span between diagnosis and surgical intervention (6, 20). Recent studies demonstrated similarities in the expression patterns between adipocytes isolated from whole mesenteric fat depots obtained from obese patients and those with CD, with inflammation- and lipid metabolism-associated pathways showing the highest degree of convergence between the two groups (51). A recent report failed to establish a causative relationship between obesity and IBD (12). This investigation (12), however, was focused on BMI as a risk factor for developing IBD without assessing directly the effect of obesity on IBD outcome. Despite all the indications favoring a link between obesity and IBD outcome, evidence for this association is still limited.
Using intracolonic administration of 2,4,6-trinitrobenzenesulfonic acid (TNBS), we previously demonstrated histological changes in the mesenteric and epididymal fat depots that resemble changes described in CD and showed increased expression of proinflammatory mediators in these fat depots (21). TNF-α, leptin, and adiponectin have also been implicated in the induction of morphological changes in creeping fat adipocytes (11, 15, 18). Moreover, circulating levels of adiponectin, ghrelin, and resistin are increased, whereas those of leptin are decreased in patients with IBD (24), suggesting that adipose tissue-derived mediators may affect IBD pathophysiology. Another study showed that high-fat diet (HFD) exacerbates the outcome of colitis and is associated with increased fat mass (31). Mice on HFD also exhibited an increased number of natural killer T cells, which produced higher amounts of TNF-α and IFN-γ and a decreased number of regulatory T cells (31). HFD-adoptive transfer of regulatory T cells rescued colitis and lowered cytokine levels observed in the HFD group (31). These observations underline the potential involvement of adipose tissue-derived responses in IBD pathophysiology.
In the current study, we examined the effects of HFD-induced obesity on colitis outcomes by employing the well-characterized model of chronic TNBS-induced colitis in either lean or obese mice. We found striking differences in colitis-associated animal morbidity and mortality after TNBS administration between lean and obese mouse groups. More severe inflammatory changes and dramatically higher cytokine levels were also observed in the colon and the mesenteric adipose tissues in obese mice compared with lean mice. Adiponectin expression in the fat and adiponectin receptor (AdipoR) expression in the intestine were also different in obese vs. lean mice with colitis. Moreover, media obtained from mesenteric fat and cultured preadipocytes isolated from controls, obese patients, and patients with IBD exhibited significant differences in inflammatory mediator release and elicited condition-dependent changes on AdipoR1 expression in NCM460 human colonic epithelial cells, whereas silencing of AdopoR1 in mice showed increased inflammatory changes following experimental colitis.
MATERIALS AND METHODS
Human subjects.
Mesenteric fat tissues from patients with IBD (13 UC, 12 CD) and without it (12 control, 9 obese) were used. For the individuals without IBD, fat tissue was resected during gastric bypass (for the management of obesity), gynecological or adenocarcinoma surgery, other gastrointestinal complications, or vascular surgery. Human study protocols have been approved by the UCLA Institutional Review Board for Human Research (IRB no. 11-001527-AM-00003). All participants gave informed consent before taking part. All subjects were fasted for at least 10 h before surgery. Subjects with malignancies were not excluded because they may constitute an important subpopulation that could yield significant information for our intergroup comparisons. Tissues from Cedars-Sinai Medical Center were obtained after informed consent in accordance with procedures established by the Cedars-Sinai Institutional Review Board (IRBs 3358 and 23705).
Isolation of human preadipocytes.
Mesenteric fat tissue (2–5 g) was obtained from each patient. The tissue was placed into sterile 50-ml polypropylene tubes containing 15 ml of collagenase solution (1 mg of collagenase/1 ml of PBS, 3 ml of solution/1 g of tissue) and minced to a fine consistency. The solution was then vortexed for 20 s, and the tubes were placed in a 37°C shaking water bath (100 revolution/min) for 40 min. The solution was vortexed and filtered through a double-gauze-containing funnel. The homogenates were centrifuged (1,000 revolution/min, 10 min), and the top fatty layer was collected and washed three times with PBS. The pellet was then resuspended in 10 ml of erythrocyte lysis buffer (154 mM NH4Cl, 10 mM KHCO3, 1 mM EDTA), placed in a 37°C shaking water bath for 5 min at 100 revolution/min, and centrifuged at 1,000 revolution/min for 10 min. The pellet was resuspended in 10 ml of plating medium (DMEM, 0.1 mM penicillin, 0.06 mM streptomycin, 10% HI-FBS, pH 7.4), vortexed, plated onto 100-mm dishes, and incubated at 37°C.
Cell culture of human preadipocytes.
After 20 h, cells were washed three times with 10 ml PBS, and 1 ml trypsin solution (Invitrogen, Carlsbad, CA) was added. Trypsin was inactivated with 5 ml of plating medium, and cells were centrifuged at 1,000 revolution/min for 10 min. The supernatant was aspirated, cells were re-suspended in 10 ml of plating medium, and plated at 5 × 104 cells/cm2 in plating medium. Cells were incubated at 37°C until they reached confluence, and medium was changed with fresh medium every 48 h. This isolation procedure yields >99% pure preadipocyte populations (as determined by cloning of individual cells and counting of colonies derived from them that were able to accumulate lipid) (45). Cells were then subcultured three or four times to ensure removal of macrophages (46). No ADAM8, F4/80, or macrophage inflammatory protein-1a mRNA, markers of macrophages, were detected by Affymetrix array analysis of human mesenteric or omental preadipocytes prepared using this protocol, as we previously described (23).
Exposure of NCM460 human colonocytes.
For human mesenteric fat tissue media, after surgery, 100–200 mg of mesenteric fat tissue was removed with sterile scissors, placed in a 15-ml sterile polypropylene tube with 2 ml of FBS-free medium (MEM, 0.1 mM penicillin, 0.06 mM streptomycin, obtained as shown above), and kept in a 37°C shaking water bath for 24 h at 100 revolution/min. The conditioned media (200 μl) were then placed over confluent NCM460 colonocytes in 24-well plates for 24 h, and RNA was isolated in Trizol reagent.
For human preadipocyte media, preadipocyte medium was removed from confluent preadipocyte cultures of control patients and those with UC and CD during the third passage after isolation and exposure to confluent plates to fresh medium (MEM, 10% FBS, 1% penicillin/streptomycin,) for 24 h. NCM460 cells were grown to confluence in six-well plates, and 1 ml of preadipocyte media was added for 24 h. The wells were then washed once, and RNA was isolated using Trizol reagent.
For adiponectin, NCM460 cells were grown to confluence with culturing medium (M3D, 1% penicillin/streptomycin, and 10% FBS) in 24-well plates and were exposed to 10 μg/ml reconstituted adiponectin (Sigma-Aldrich, St. Louis, MO) or vehicle (PBS, 0.1% BSA) in treatment medium (M3D with 1% penicillin/streptomycin, FBS-free) for 24 h. The wells were then washed once with PBS, and RNA was isolated using Trizol reagent.
Animal groups.
Male C57BL/6 mice (18–20 g), 8–12 wk old (n = 8–16 per group) were purchased from Jackson Laboratories (Bar Harbor, ME). Mice were maintained on a normal light-dark cycle and provided with food and water ad libitum. Two groups of C57BL/6 mice were kept on HFD (Research Diets, New Brunswick, NJ) for 6–8 wk, while two additional groups were fed low-fat diet (LFD) for the same period (or until groups separated by 10 g). After the feeding, TNBS colitis was induced intracolonically to one of the HFD-fed and one of the LFD-fed groups while the other two groups received intracolonic ethanol (control) injections. Collectively, we produced the following four groups: 1) LFD-fed, non-TNBS (LFD-C); 2) LFD-fed, TNBS (LFD-TNBS); 3) HFD-fed, non-TNBS (HFD-C); and 4) HFD-fed, TNBS (HFD-TNBS).
All animal protocols were approved by the Institutional Animal Care and Use Committee at the David Geffen School of Medicine at UCLA, and studies were carried out in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80 23, revised 1978).
TNBS colitis.
Age- and weight-matched mice were lightly anesthetized with isofluorane, and a polyethylene cannula (Intramedic PE-20 tubing; Becton Dickinson, Parsippany, NJ) was inserted intracolonically (at a length of 4 cm). A solution of 40% ethanol (vehicle) or ethanol-containing TNBS (Sigma) was instilled into the colon (3–4 cm from the anus) using a syringe, whereas control animals were treated with vehicle alone. TNBS or vehicle injections were performed once a week for 6 wk of 1.0, 1.5, and 2.0 mg per 20 g (2 wk/dose). Mice were then left untreated for two more weeks. At the end of the study, body weight was assessed, and mice were then euthanized with isofluorane overdose. Pieces of mesenteric fat and intestine were either placed in formalin for immunohistochemistry or frozen for protein and RNA extraction. Colitis score was assessed as described previously (28).
Real-time PCR.
RNA was isolated from mouse and human mesenteric whole-fat tissue and human mesenteric preadipocytes using the Trizol method. RNA (1 μg) was reverse-transcribed into cDNA as previously described (22) and incubated with dual fluorogenic probes (Applied Biosystems, Foster City, CA). GAPDH and 18S were used as endogenous controls and were also detected using a dual-labeled fluorogenic probe (5′-FAM/3′-MGB probe, Applied Biosystems). Target mRNA (all from Applied Biosystems) levels were quantified using a fluorogenic 5′-nuclease PCR assay, as previously described (30) using a 7500 Fast Real-Time PCR sequence detection system (Applied Biosystems).
mRNA multiplex analysis.
Total RNA was isolated as described above, and inflammation-related gene expression was analyzed using the 42-plex FlexScript LDA inflammatory panel 3 (Luminex, Austin, TX). A sample of total RNA (20 ng) was loaded in each well following treatments described in the company manual (FlexScript LDA). The plate was run using Bio-plex 3D suspension array system (Bio-Rad, Hercules, CA). In addition to total RNA concentration, data were normalized to endogenous controls (GAPDH, B2M, β-actin) included within the gene panels.
Intracolonic AdipoR1 knockdown via siRNA.
C57BL/6 mice (10–12 wk old) were placed into three groups (sham-EtOH, scrambled, and siAdipoR1; n = 5 mice per group). At day 0, mice in the sham-EtOH group received intracolonically 100 μl of Lipofectamine 2000 (1:50/total volume). Mice in the scrambled group received intracolonically 4 nmol of scrambled nucleotides (SR30004; Origene Technologies, Rockville, MD) in Lipofectamine 2000, and mice in the siAdipoR1 group received 1.33 nmol of each of three anti-AdipoR1 siRNA duplexes (SR412651A, B, and C, Origene Technologies) in Lipofectamine 2000. The same injections were repeated on day 2, whereas, on day 3, mice were injected intracolonically with 5 mg TNBS. Mice were killed for analysis on day 5. Figure 9A includes a schematic representation of the design of these studies.
Fig. 9.

Intracolonic AdipoR1 knockdown exacerbates colitis in mice. A: schematic representation of AdipoR1 knockdown followed by induction of TNBS colitis in mice. Mice that received intracolonic injections of anti-AdipoR1 duplexes showed increased weight loss (B), increased macroscopic damage (C), as well as elevated colitis severity (D), mucosal damage (E), and crypt formation (F) compared with mice treated with scrambled control nucleotides. *P < 0.05, **P < 0.01 for scrambled-TNBS vs. sham; #P < 0.05 for scrambled-TNBS vs. AdipoR1-TNBS.
Immunohistochemistry.
Paraffin-embedded colon sections from patients with UC and CD and control patients (n = 4 per group) were mounted on slides. AdipoR1 staining was detected using an anti-AdipoR1 rabbit monoclonal antibody (1:100 dilution, ab126611; Abcam, Cambridge, MA) and the EnVision+ System HRP Labeled Polymer Anti-Rabbit kit (DAKO, Carpinteria, CA). Staining was performed at the Translational Pathology Core, UCLA following a standard procedure described in Millipore's manual for the primary antibody treatment (Billerica, MA).
Multiplex cytokine and phospho-protein immunoassays.
Human mesenteric fat tissue was isolated and plated as described above, and media were collected at the end of the 24-h period. Cytokine concentrations in conditioned media were determined using the Bio-Plex Pro Human Adipokine Magnetic Bead Panel 1 (Bio-Rad), and the final data were obtained and analyzed via the Bio-plex 3D Suspension array system (Bio-Rad). In addition to loading volume, results were normalized for total protein as well as tissue weight.
Statistical analysis.
Results were analyzed using the Prism professional statistics software program (Graphpad Software, San Diego, CA). One-way ANOVA and Mann-Whitney U-test (for comparisons between two groups) were used for intergroup comparisons. A P value of <0.05 was considered statistically significant.
RESULTS
HFD-induced obesity exacerbates the effects of TNBS colitis on mesenteric fat depot mass in C57BL/6 mice.
We previously showed that mesenteric fat depots isolated from TNBS-treated mice express high levels of proinflammatory cytokines and increased inflammatory cell infiltrates (21). In this study, we separated mice in four groups (n = 8–16 per group) as described above. We observed that mesenteric fat depot expansion was evident in both lean and obese mice with colitis (Fig. 1). Obese mice with TNBS-induced colitis were the only group that had diarrhea and blood in the stool. Mice were killed 48 h after induction of colitis, and tissues were collected because of the high mortality of animals in the HFD-TNBS group. As expected, HFD-induced obesity alone was associated with increased mesenteric fat mass around the intestine compared with lean, LFD-fed mice (Fig. 1, C vs. A). In addition, obesity exacerbated this response in the mesenteric fat depots with more mesenteric fat attachment in the HFD-TNBS group compared with LFD-TNBS-exposed mice (Fig. 1, D vs. B). Indicative of the severity of colitis, mice in the obese group showed high mortality rates (50%) 48 h post-TNBS using a low TNBS dose commonly used to promote chronic disease (2 mg/20 g). In comparison, lean mice that received the same low TNBS dose were unaffected in terms of viability, weight loss, or diarrhea (data not shown). Thus conditions associated with increased fat mass during obesity contribute to a dramatic worsening of experimental colitis.
Fig. 1.

Macroscopic changes in mesenteric fat depots during experimental (trinitrobenzene sulfonic acid, TNBS) colitis resemble those observed in patients with Crohn's disease (CD). A: normal mouse intestine without obesity or colitis. B: TNBS colitis induces infiltration of fat and “wrapping” of the intestine around the affected area. LFD, low-fat diet. C: increased mesenteric fat mass proximal to the intestine without colitis. D: increased fat wrapping of the involved intestine during TNBS colitis in obese mice with strong attachment to multiple sites and signs of extensive angiogenesis. HFD, high-fat diet.
In separate experiments, we injected lower doses of TNBS (0.75, 1.00, and 1.25 mg/20 g) in groups of animals as described above in an attempt to reach the endpoint of 6 wk before death. Although the HFD-TNBS group did survive for the duration of the study (6 wk), at these low TNBS doses, we observed very low levels of inflammatory responses even in the obese group with minimal differences between groups [LFD-TNBS vs. HFD-TNBS, P = 0.02 for keratinocyte-derived chemokine (KC), n = 7]. Moreover, no signs of colitis were observed at the gross morphological level in any of the groups (data not shown).
HFD-induced obesity worsens TNBS-induced histological changes in mouse colon.
In the same experimental groups described above, colon was removed after the completion of the study, and histological sections were evaluated as described in materials and methods. There was significantly increased colonic inflammation in mice fed HFD and treated with TNBS (Fig. 2, D and E) compared with mice in the other three groups (LFD-C, LFD-TNBS, HFD-C, Fig. 2, A–C and E). Together, these data indicate that obesity may induce alterations in inflammatory mediator release that worsens colitis. Furthermore, mice in the HFD-TNBS group demonstrated increased weight loss compared with the LFD-TNBS group (Fig. 2F, P < 0.05) and had increased mortality with 50% of mice not surviving after the first 24–48 h (Fig. 2G). Tissues from mice that did not survive to the point of death were excluded from any analysis.
Fig. 2.

Obesity exacerbates experimental colitis in mice. We removed a 1-cm piece of colon from mice that included the visibly inflamed area located ∼3 cm from the anus and sectioned and stained with hematoxylin and eosin (H and E) stain. Histological sections (A–D) and clinical scoring (E) of mouse colons after treatment with TNBS reveals that colitis outcome is exacerbated in obese mice (HFD-TNBS) compared with lean mice with (LFD-TNBS) or without colitis (control) (LFD-C) and obese mice without colitis (HFD-C); ***P < 0.001. F: obese mice exhibited increased weight loss in response to TNBS colitis (HFD-TNBS) compared with their lean counterparts (LFD-TNBS); *P < 0.05. G: survival curve showing increased mortality in the HFD-TNBS compared with the LFD-TNBS group.
HFD-induced obesity increases proteinase 3 mRNA in the colon of TNBS-exposed mice.
To further evaluate increased obesity-associated increased immune cell infiltration in mice with colitis, we measured mRNA expression of neutrophil (proteinase 3) and macrophage (EMR1, human homologue of F4/80). Figure 3A (HFD-TNBS vs. LFD-TNBS) shows significant obesity-related increases in colonic proteinase 3 mRNA expression in TNBS-exposed mice, suggesting the increased presence of leukocytes in the intestine during colitis in HFD-fed mice. Obesity alone did not lead to any increases in proteinase 3 expression (Fig. 3A, HFD-C vs. LFD-C). In contrast, colonic EMR1 levels were decreased in HFD-TNBS group (Fig. 3B, P < 0.01, n = 6–8). Thus diet-induced obesity is associated with increased colonic neutrophil but not macrophage infiltration.
Fig. 3.
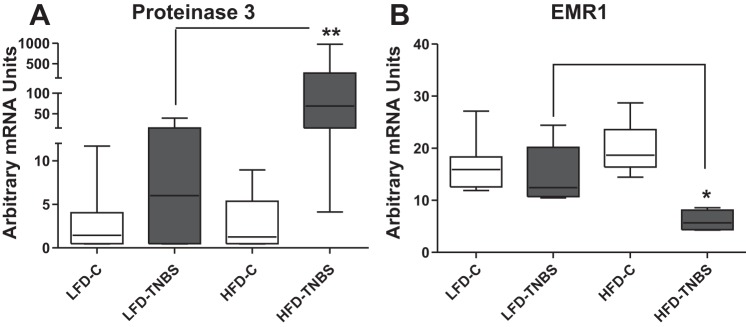
Obesity exacerbates inflammatory cell infiltrate in the colon of mice with TNBS-induced colitis. Real-time PCR on total intestinal RNA showed that mRNA levels of the neutrophil marker proteinase 3 increase in the intestine of obese mice 48 h after the induction of TNBS colitis (HFD-TNBS) compared with lean mice with (LFD-TNBS) or without (LFD-C) colitis and obese mice without colitis (HFD-C) (A), whereas those of the macrophage marker EMR1 decrease (B) (*P < 0.05, **P < 0.01).
HFD-induced obesity is associated with increased cytokine expression in mouse intestine during TNBS colitis.
We used real-time PCR to measure mRNA expression of inflammatory cytokines that may affect the development of colitis in mouse colon of all four mouse groups described above. We observed significant obesity-associated increases in the expression of IL-1β, IL-6, KC (Fig. 4, A–C, HFD-TNBS vs. LFD-TNBS, respectively, *P < 0.05, **P < 0.01, n = 6–8), and IL-10 (Fig. 4F, P < 0.001, n = 6–8/group) 48 h after TNBS treatment. In contrast, IFN-γ and IL-2 expression were lower in obese mice exposed to TNBS (Fig. 4, D and E, HFD-TNBS, *P < 0.05, **P < 0.01, n = 6–8/group), indicating an active mechanism that induces the observed changes rather than complete cytokine deregulation attributable to severe colitis. For some of these cytokines such as IL-1β, IL-6, IFN-γ, and IL-2, we observed increased RNA expression with obesity alone (Fig. 4, A, B, D, and E, LFD-C vs. HFD-C, ##P < 0.01, ###P < 0.001, n = 7–8). Such changes may be related to the exacerbated responses observed in the obese group during colitis (Fig. 4, A–F, HFD-TNBS).
Fig. 4.

HFD-induced obesity affects cytokine expression in mouse intestine during TNBS colitis. Real-time PCR revealed that obese mice (HFD-TNBS) demonstrated increased IL-1β (A), IL-6 (B), keratinocyte-derived chemokine (KC) (C), and IL-10 mRNA (F) levels in the intestine 48 h after the induction of TNBS colitis compared with lean mice with (LFD-TNBS) or without (LFD-C) colitis and obese mice without colitis (HFD-C). Contrary to the aforementioned data showing dramatic upregulation of cytokines, mRNA expression of IFN-γ (D) and IL-2 (E) decreased significantly in obese mice 48 h after the induction of TNBS colitis (HFD-TNBS) compared with lean mice with (LFD-TNBS) or without (LFD-C) colitis and obese mice without colitis (HFD-C) in which group the mRNA levels of both cytokines increase. *P < 0.05, **P < 0.01, ***P < 0.001 for LFD-TNBS vs. HFD-TNBS; ##P < 0.01, ###P < 0.001 for LFD-C vs. HFD-C.
HFD-induced obesity is associated with increased inflammatory cell infiltrate in mesenteric fat depots during TNBS colitis.
CD-like increased infiltration of immune cells in adipose tissue can be replicated in the TNBS colitis model (21, 26). Mesenteric fat was removed from all mice groups at the end of the experiments and examined histologically. As in the intestine, hematoxylin and eosin (H and E)-stained histological sections showed that TNBS-associated inflammatory changes were dramatically exacerbated in the mesenteric fat depots by HFD-induced obesity, evidenced by increased inflammatory cell infiltrates in fat depots from obese mice with colitis (Fig. 5D), compared with mesenteric fat isolated from lean animals (Fig. 5B). No apparent inflammatory infiltration at the gross morphological level was observed in obese alone vs. lean controls in the absence of TNBS treatment.
Fig. 5.

Obesity exacerbates inflammatory cell infiltrate within mesenteric fat depots of mice with TNBS-induced colitis. H and E-stained histological sections showed that, after 48 h, TNBS colitis induced severe infiltration of inflammatory cells in mesenteric fat depots of obese mice (D) compared with lean mice with (LFD-TNBS) (B) or without (LFD-C) colitis (A) and obese mice without colitis (HFD-C) (C).
We also measured mRNA expression of inflammatory cell markers in the mesenteric fat depots in the different groups of mice. Using a myeloperoxidase (MPO) assay, we show that the levels of MPO, a marker of activated neutrophils, within fat depots were significantly higher during colitis only in the obese group (Fig. 6A, LFD-TNBS vs. HFD-TNBS, P < 0.01, n = 10–11). In agreement with our previous study (21), we observed increased proteinase mRNA levels in the mesenteric fat during colitis (Fig. 6B, LFD-C vs. LFD-TNBS, P < 0.05, n = 10–11), whereas obesity exacerbated this response (Fig. 6B, HFD-TNBS vs. LFD-TNBS, P < 0.01, n = 10–11). EMR1 mRNA is increased in obese mice without colitis vs. lean controls but not compared with lean TNBS and obese control mice (Fig. 6C). Thus obesity increases neutrophil infiltration in the mesenteric fat during colitis.
Fig. 6.

Total RNA was isolated from mesenteric fat depots and subjected to real-time PCR analysis. The protein levels of myeloperoxidase (MPO) (A) and the mRNA levels of proteinase 3 neutrophil markers (B) increased in the mesenteric adipose tissue of obese mice with colitis (HFD-TNBS) compared with their nonobese counterparts (LFD-TNBS) or obese (HFD-C) and lean (LFD-C) mice without colitis. C: whereas EMR1 mRNA levels increase with obesity alone (HFD-C), they are not affected by colitis either in lean (LFD-TNBS) or obese (HFD-TNBS) mice (**P < 0.01.).
HFD-induced obesity alters cytokine responses in mouse mesenteric fat depots during TNBS colitis.
We next isolated RNA from mesenteric fat of all experimental groups and analyzed them for the expression of proinflammatory cytokines that may be involved in the generation of adipocyte-specific effects in the intestine during colitis. Our results from mesenteric fat depots demonstrate dramatic increases with obesity in the mRNA expression of several proinflammatory cytokines such as IL-1β, IL-6, monocyte chemoattractant protein 1 (MCP-1), TNF-α, and KC (Fig. 7, A–E, HFD-TNBS, **P < 0.01, n = 5–8) 48 h after the induction of TNBS colitis compared with all other groups (n = 6–8). As expected for fat depots during obesity (HFD-C) (25, 44), the expression of several of these cytokines was increased (Fig. 7, TNF-α, MCP-1, IL-1β, and KC; LFD-C vs. HFD-C; #P < 0.01, n = 6–8). Again, the mRNA levels of INF-γ (but not IL-2) were dramatically reduced in the mesenteric fat of obese mice (HFD-TNBS) following TNBS compared with all other groups (Fig. 7F, *P < 0.05, n = 5–8). These results indicate that the effects of obesity in the mesenteric fat following colitis are specific and not attributable to complete dysregulation of the inflammatory response. Interestingly, the expression of the anti-inflammatory cytokine IL-10 was dramatically increased in the adipose tissues of obese mice after TNBS treatment (Fig. 7G, P < 0.01, n = 5–8).
Fig. 7.

HFD-induced obesity increases proinflammatory cytokine expression in mouse mesenteric adipose tissue during TNBS colitis. Total RNA was isolated from mouse mesenteric fat depots, and real-time PCR showed that obese mice (HFD-TNBS) demonstrated increased IL-1β (A), IL-6 (B), monocyte chemoattractant protein 1 (MCP-1) (C), TNF-α (D), KC (E), and IL-10 (G) mRNA levels in the mesenteric fat depot 48 h after the induction of TNBS colitis compared with lean mice with (LFD-TNBS) or without (LFD-C) colitis and obese mice without colitis (HFD-C). However, IFN-γ mRNA (F) levels decreased significantly in obese mice 48 h after the induction of TNBS colitis (HFD-TNBS) compared with lean mice with (LFD-TNBS) or without (LFD-C) colitis and obese mice without colitis (HFD-C). *P < 0.05, **P < 0.01 for LFD-TNBS vs. HFD-TNBS; #P < 0.05 for LFD-C vs. HFD-C.
HFD-induced obesity alters the expression of adiponectin and its receptors during TNBS colitis in mice.
Several adipokines have been implicated in IBD pathophysiology, including adiponectin. Adiponectin reduces inflammation by 1) inhibiting macrophage function (34, 48), 2) triggering proinflammatory cytokine secretion (34, 41), and 3) upregulating the protective cytokine IL-10. Its expression also decreases with obesity (2), whereas increased adiponectin during CD may facilitate mucosal healing. Here, we demonstrate that adiponectin mRNA expression was increased in the mesenteric depots following intracolonic TNBS (Fig. 8A, LFD-C vs. LFD-TNBS; #P < 0.01, n = 10–13) and decreased during obesity in the mesenteric fat depots of mice (Fig. 8A, LFD-C vs. HFD-C, n = 12–13). However, when we combined obesity with TNBS colitis (HFD-TNBS), the obesity-induced decrease in adiponectin expression was exacerbated (Fig. 8A, P < 0.001 compared with all groups). Moreover, the expression of its receptors AdipoR1 and AdipoR2 in the mesenteric fat was increased in response to TNBS (Fig. 8, B and C, LFD-C vs. LFD-TNBS; P < 0.01 and P < 0.05, respectively, n = 12–13) but remained unaffected by obesity (Fig. 8, B and C, HFD-C vs. LFD-C). In obese mice with colitis, both adiponectin receptors AdipoR1 and AdipoR2 remained at significantly lower levels compared with mice with TNBS alone (Fig. 8, B and C, LFD-TNBS vs. HFD-TNBS). In the intestine, both AdipoR1 and AdipoR2 mRNA levels decreased in obese mice with colitis (Fig. 8, D and E; P < 0.01 and P < 0.001, respectively; HFD-TNBS compared with all other groups). In contrast to our observations in mesenteric fat depots, TNBS colitis alone did not significantly alter colonic mRNA expression of these receptors (Fig. 8, D and E, LFD-C vs. LFD-TNBS; strong trend toward decrease), suggesting different roles for adiponectin in the colon and mesenteric fat during colitis.
Fig. 8.

HFD-induced obesity lowers mRNA expression of adiponectin in the fat and of adiponectin receptors 1 and 2 in the intestine during TNBS colitis in mice. We performed real-time PCR on adipose tissue and intestine total RNA and observed increased levels adiponectin (AdipoQ) mRNA (A) in adipose tissue of lean mice with colitis (LFD-TNBS) compared with lean controls (LFD-C) and significantly decreased levels in obese mice 48 h after the induction of TNBS colitis (HFD-TNBS) compared with lean mice with (LFD-TNBS) or without (LFD-C) colitis and obese mice without colitis (HFD-C). Adiponectin receptor 1 (AdipoR1) (B) and AdipoR2 (C) mRNA levels decrease significantly in the adipose tissue of obese mice 48 h after the induction of TNBS colitis (HFD-TNBS) only compared with lean mice with colitis (LFD-TNBS). AdipoR1 (D) and AdipoR2 (E) decreased significantly in the intestine of obese mice 48 h after the induction of TNBS colitis (HFD-TNBS) compared with lean mice with (LFD-TNBS) or without (LFD-C) colitis and obese mice without colitis (HFD-C). #P < 0.05 vs. LFD-C; *P < 0.05, **P < 0.01, ***P < 0.001.
Intracolonic AdipoR1 knockdown worsens TNBS colitis in mice.
To highlight the potential role for AdipoR1 in intestinal inflammation, we knocked down this receptor via intracolonic administration of anti-AdipoR1 siRNAs before the induction of colitis (siAdipoR1, Fig. 9A). Expectedly, both the siAdipoR1 and scramble groups showed increased weight lost compared with sham treatment attributable to the TNBS induction (Fig. 9B, *P < 0.05, n = 5–6). However, the siAdipoR1 group lost more weight even compared with the scrambled group at day 5 of the study (Fig. 9B, #P < 0.05, n = 6). H and E staining of intestinal sections from the three groups showed worsening of colitis in the siAdipoR1 group compared with both the scrambled and sham (EtOH) groups (Fig. 9C). Colitis severity (Fig. 9D, P < 0.01 vs. sham, P < 0.05 vs. scrambled), mucosal damage (Fig. 9E, P < 0.01 vs. sham, P < 0.05 vs. scrambled), and crypt formation (Fig. 9F, P < 0.01 vs. sham, P < 0.05 vs. scrambled) were also exacerbated.
Differential AdipoR1 expression of human colonic epithelial cells following exposure to conditioned media from control, obese, and IBD preadipocytes.
To provide evidence of potential adipose tissue-derived effects during obesity or IBD on adiponectin-associated responses in colonocytes, we exposed NCM460 human colonic epithelial cells to conditioned media derived from 8–19 mesenteric fat tissues of control patients and those with UC and CD and from cultured human preadipocytes of 6–9 control patients, obese patients, and those with UC and CD. We then examined the mRNA levels of adiponectin receptors in these cells. We observed that conditioned media derived from mesenteric fat tissues of both patients with UC and CD reduced the expression levels of AdipoR1 in NCM460 colonocytes (Fig. 10A, P < 0.05, n = 8–19). In addition, conditioned media from preadipocytes from patients with UC induced a significant decrease in AdipoR1 mRNA levels in NCM460 colonocytes (Fig. 10B, P < 0.01, UC vs. C, n = 6–9) but not AdipoR2 (not shown). Strong trends toward decreased AdipoR1 levels were also observed in colonocytes exposed to conditioned media from preadipocytes obtained from obese patients and those with and CD (Fig. 10B, n = 6–9, P = 0.0667 and 0.0867 for obese patients and those with CD, respectively). Thus changes in adipose tissue-derived mediator secretion during obesity and IBD may alter the capacity of intestinal epithelial cells to respond to adiponectin. Phospho-protein multiplex analysis revealed that phospho-insulin-like growth factor 1 receptor (IGF-1R) levels of NCM460 cells decrease after exposure to conditioned media from preadipocytes isolated from patients with UC and CD compared with those from control patients (Fig. 10C, P < 0.01 for UC, P < 0.05 for CD, n = 6–9). Conditioned media from fat tissue isolated from patients with IBD decreased mRNA expression of the transcription factor peroxisome proliferator-activated receptor-γ (PPAR-γ) in NCM460 colonocytes (Fig. 10D, P < 0.01 for UC, trend for CD, n = 8–21). Finally, IGF-1 treatment increased mRNA expression of AdipoR1 in NCM460 colonocytes (Fig. 10E, P < 0.01, n = 6) suggesting that AdipoR1 regulation by preadipocytes and fat tissue conditioned media may be mediated via downregulation of IGF-1R signaling.
Fig. 10.

Conditioned media isolated from preadipocyte cultures from controls, obese patients, and patients with inflammatory bowel disease (IBD) (n = 6–9) induce differential mRNA expression responses of AdipoR1 in human colonic epithelial NCM460 cells. A: human colonic epithelial NCM460 cells were exposed to conditioned media from mesenteric fat depots of controls and patients with ulcerative colitis (UC) and CD, and AdipoR1 mRNA expression decreased significantly compared with controls. B: NCM460 colonocytes were treated with conditioned media from preadipocytes of controls, obese patients, and patients with IBD for 24 h, and total RNA was collected. Real-time PCR revealed that AdipoR1 mRNA levels are significantly decreased in human NCM460 colonocytes after exposure to media from preadipocytes from patients with UC, whereas there is also a strong trend toward decrease when media from preadipocytes from patients with CD and obese patients are employed. Conditioned media from IBD preadipocytes decrease phospho insulin-like growth factor (IGF)-1R levels (C), whereas conditioned media from fat tissues from patients with IBD (D) decrease peroxisome proliferator-activated receptor-γ (PPAR-γ) mRNA levels in NCM460 cells compared with media from control patients. E: treatment of NCM460 colonocytes with IGF-1 increased AdipoR1 mRNA levels. *P < 0.05, **P < 0.01.
AdipoR1 levels increase in colonic biopsies of patients with IBD vs. controls, and adiponectin reduces cytokine expression in human NCM460 colonocytes.
Immunohistochemistry for the detection of the levels of AdipoR1 revealed increased receptor-positive cells in the colonic mucosa of patients with UC and CD compared with control subjects (Fig. 11A). To investigate the effects of adiponectin in the intestine, we treated human NCM460 colonocytes and collected RNA for the measurement of mRNA expression levels of several cytokines. Among the 42 cytokines measured, we observed that adiponectin treatment led to decreased mRNA levels of IL-2, IL-5, IL-8, IL-17, IL-23, and transforming growth factor-β2 (Fig. 11, B–G, *P < 0.05, **P < 0.01, n = 6) and increased the mRNA levels of VEGFA (Fig. 11H, P < 0.01, n = 6). Thus obesity-associated regulation of AdipoR1 in the intestinal epithelium may affect the ability of these cells to respond to inflammatory regulation by adiponectin during colitis.
Fig. 11.

AdipoR1 levels increase in human colonic biopsies during IBD, and adiponectin reduces cytokine mRNA expression in human NCM460 colonocytes. A: immunohistochemistry of human colonic sections shows elevated levels of AdipoR1 during IBD compared with non-IBD controls. Human colonic epithelial NCM460 cells were exposed to 10 ng/ml adiponectin for 24 h in the medium, and the mRNA levels were determined using a multiplex assay (42-plex). B–G: IL-2, IL-5, IL-2, IL-17, IL-23, and transforming growth factor (TGF)-β2 mRNA levels were significantly reduced in adiponectin-treated NCM460 colonocytes compared with untreated controls. H: mRNA levels of VEGFA were significantly increased. *P < 0.05, **P < 0.01.
Human mesenteric fat depots demonstrate distinct mediator release when isolated from patients with UC and CD.
Systemic inflammatory changes are considered a hallmark of obesity (5, 19, 44). Several of these responses were observed systemically and within fat depots during IBD (51). We have obtained mesenteric fat depots of 8 control, 14 patients with UC, and 14 patients with CD and collected conditioned media for multiplex adipokine analysis after 24 h in culture. Analysis of the 11 adipokines revealed IBD-associated changes in the release of mediators from human mesenteric fat compared with controls (Fig. 12, A–F, *P < 0.05, **P < 0.01, #P < 0.1, n = 8–14). We observed increased adiponectin, TNF-α, and IL-1β (Fig. 12, A, D, and F) release in mesenteric fat from both patients with UC and CD. Moreover, compared with controls, release of leptin was increased only in patients with UC (Fig. 12B), whereas IL-8 release was increased in patients with CD (Fig. 12E). Resistin release was also decreased in mesenteric fat depots from patients with CD compared with controls (Fig. 12F).
Fig. 12.

Mesenteric fat depots removed from patients with UC and CD demonstrate differential mediator release from each other and from depots isolated from healthy controls (n = 8–14). Multiplex analysis of an adipokine-containing panel from 11 humans revealed that mesenteric fat isolated from patients with UC and CD secretes higher levels of adiponectin, IL-1β, and TNF-α, (A, D, and F, respectively), compared with controls. Mesenteric fat depots from patients with UC secrete higher levels of leptin (B), and those of patients with CD secrete higher levels of IL-8 (E) compared with controls. G: mesenteric fat depots from patients with CD secrete lower levels of resistin compared with controls. *P < 0.05, #P < 0.1.
DISCUSSION
Our data strongly support the notion that obesity affects the outcome of experimental colitis and produces a dramatically altered inflammatory environment both in fat and the intestine. We demonstrate significant increases in inflammatory cell infiltration in both the intestine and mesenteric fat depots (Figs. 3 and 6) in obese mice during colitis, likely attributable to effects of increased adiposity in the production of inflammatory mediators in both tissues (Figs. 4 and 7). Mice were killed only 2 days after TNBS administration and not 6 wk as originally planned. This adjustment was necessary because of the detrimental response of the obese mice to TNBS even at low chronic doses, which failed to cause any inflammatory changes in lean mice.
It is striking that, for the mediators reported here, the responses in the obese group during colitis are almost identical for both the intestine and adipose tissue. However, not all mediators exhibit the same patterns of expression with obesity alone (HFD-C), with the adipose tissue showing a wider range of responses. Significant decreases in the expression of both IFN-γ and IL-2 (Fig. 4, D and E, respectively) demonstrate that the effect of obesity on cytokine expression during colitis is a result of specific and controlled transcriptional modulation. Both IFN-γ and IL-2 are involved in T cell maturation processes, suggesting an important role for obesity in the modulation of T cell responses during colitis. Interestingly, mRNA expression of these two mediators increases significantly during obesity (IL-2, IFN-γ; Fig. 4, D and E) in the colon but not in adipose tissue. In addition, several of the cytokines dramatically changed in the obese group with colitis (HFD-TNBS) have been shown to participate in the development of experimental colitis and IBD pathophysiology. Briefly, TNF-α antagonism represents one of the main treatment modalities for IBD (39). IL-1β has been shown to promote the accumulation and survival of pathogenic CD4(+) T cells in the T cell transfer mouse model of colitis (13), whereas the levels of this cytokine are elevated and are associated with increased disease activity in the colon of patients with IBD (29, 32). Similar associations were also demonstrated for the colonic levels of IL-8 in patients with IBD (14), whereas the IL-10 mouse knockout model provides one of the most common tools for the study of colitis (43).
As expected, obesity induces differential responses in the expression of proinflammatory mediators both in the adipose tissue and the intestine, suggesting interactions between these two tissues during obesity that may alter the course of colitis. Indeed, novel experimental evidence demonstrating that the intramesenteric adipose tissue mediator content could influence the availability of macrophage subtypes in creeping fat during CD (27) also supports this hypothesis. Collectively, our study is the first to demonstrate direct (and adverse) effects of obesity in the outcome of experimental colitis and elucidate that obesity-related mesenteric fat responses, although not identical, resemble a proinflammatory phenotype during colitis.
Adiponectin has anti-inflammatory properties, and its levels are adversely affected by obesity (33). A potential protective role of adiponectin in IBD is suggested by its significant increase in mesenteric fat depots (creeping fat) of patients with CD (49). Our data in Fig. 8A show an obesity-induced reversal in increased adiponectin expression during colitis. Abolishment of a potential protective role of adiponectin may be responsible for the dramatic exacerbation of inflammatory responses observed in the obese group during colitis in our study. We also observed differential expression of both adiponectin receptors in the intestine and adipose tissue during obesity and colitis (Fig. 8, B–E, HFD-TNBS vs. LFD-TNBS), with levels dropping significantly below those of control animals in the intestine (HFD-TNBS vs. LFD-C). The potential importance of the regulation of AdipoR1 levels during colitis is also highlighted by our data in Fig. 9 demonstrating increased TNBS colitis-associated weight loss and colonic damage after siRNA-induced AdipoR1 knockdown and by our human data in Fig. 11A showing increased colonic levels of AdipoR1 receptor protein in patients with IBD.
Considering the highly conflicting reports on the effects of adiponectin ablation on the course of experimental colitis stemming from studies employing null mice (16, 37, 40), our data, along with reports on the anti-inflammatory and healing roles of this adipokine in the intestine, suggest the need for additional studies employing more tissue-specific or tissue-limited approaches. Arsenescu et al. (4) showed that overexpression of adiponectin increased serum and colonic levels of IL-10, whereas Th1 cytokines were downregulated. A plant homolog of adiponectin had an identical effect on IL-10 production (4). Furthermore, increased adiponectin expression correlated with resistance to development of colitis, upregulation of Treg response, and downregulation of Th17 pathway mediators (3). Increased IL-10 expression in the colon and fat in our study (Figs. 4F and 7G) suggests a generalized anti-inflammatory damage-control mechanism potentially being activated in our colitis model with obesity.
Our results show altered levels of colonocyte adiponectin receptor in response to conditioned medium from preadipocytes from patients with IBD and whole-fat tissue (Fig. 10), suggesting that changes in mediator expression within fat depots with colitis could affect adiponectin colonocyte signaling. Colitis-associated downregulation of AdipoR1 in the colon may involve IGF-1R signaling-associated pathways, as suggested by our data (Fig. 10), showing that conditioned media from preadipocytes and fat tissue from patients with IBD reduce the levels of phosphor-IGF-1R (Tyr1131) and PPAR-γ mRNA. Interestingly, PPAR-γ has been shown to affect the transcription of both IGF-1R and AdipoR1 (35, 50). Furthermore, to establish a potential link between IGF-1R and AdipoR1, we treated NCM460 cells with IGF-1 and observed increased AdipoR1 expression. The potential importance of the reduction of colonocyte adiponectin receptor levels by adipose-derived products included in the conditioned media is highlighted by the anti-inflammatory effects of adiponectin treatment in the same cells (Fig. 11). Such effects may be diminished during colitis in obese patients, depriving these individuals of potentially beneficial effects of adiponectin (expression is increased in patients with IBD, Fig. 12A) and thus exacerbating colitis in these patients. It is thus conceivable that the intestine is exposed to intra-abdominal fat-derived products attributable to the close proximity of these tissues or possibly via the circulation during obesity. It is also likely that such exposure to differentially expressed mediators (Fig. 12) takes place during IBD or experimental colitis, especially in cases where the intestinal wall is compromised. This may lead to exacerbation of colitis such as in our case where obese mice show high mortality with a dose to which their lean counterparts remain unaffected. These results suggest that fat-promoted alterations in adiponectin-AdipoR1 signaling may affect the course of colitis during obesity.
In summary, our results implicate obesity-associated changes in the mesenteric fat depots as an important component of the severity of experimental colitis. Our data also provide the first link between altered adipose tissue function during obesity or IBD and intestinal epithelial cell responses and highlight that the adiponectin-adiponectin receptor axis may play a significant role in the regulation of colitis in obese patients.
GRANTS
This work was supported by the Research Fellowship Awards from the Crohn's Colitis Foundation of America (I. Karagiannides), Research Grant from the Broad Medical Foundation (I. Karagiannides), the Neuroendocrine Assay Core and Project 2 supported by NIDDK P50 DK 64539 (I. Karagiannides and C. Pothoulakis), and NIH NIDDK grant RO-1 DK 47343 (C. Pothoulakis).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: A.S., D.S., C.B., D.Q.S., P.F., V.A., R.A., J.R.T., and I.K. performed experiments; A.S., D.S., C.B., and I.K. analyzed data; A.S., R.A., C.P., and I.K. interpreted results of experiments; A.S., C.B., and I.K. prepared figures; A.S., C.P., and I.K. drafted manuscript; D.Q.S., P.F., V.A., R.A., J.R.T., C.P., and I.K. edited and revised manuscript; C.P. and I.K. conception and design of research; C.P. and I.K. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Sarah Dry and the Translational Pathology Core Laboratory, Department of Pathology, University of California at Los Angeles, for providing human mesenteric fat tissue samples for our studies.
REFERENCES
- 1.Ahima RS, Flier JS. Adipose tissue as an endocrine organ. Trends Endocrinol Metab 11: 327–332, 2000. [DOI] [PubMed] [Google Scholar]
- 2.Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, Hotta K, Shimomura I, Nakamura T, Miyaoka K, Kuriyama H, Nishida M, Yamashita S, Okubo K, Matsubara K, Muraguchi M, Ohmoto Y, Funahashi T, Matsuzawa Y. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun 257: 79–83, 1999. [DOI] [PubMed] [Google Scholar]
- 3.Arsenescu R, Arsenescu V, Zhong J, Nasser M, Melinte R, Dingle RW, Swanson H, de Villiers WJ. Role of the xenobiotic receptor in inflammatory bowel disease. Inflamm Bowel Dis 17: 1149–1162, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arsenescu V, Narasimhan ML, Halide T, Bressan RA, Barisione C, Cohen DA, de Villiers WJ, Arsenescu R. Adiponectin and plant-derived mammalian adiponectin homolog exert a protective effect in murine colitis. Dig Dis Sci 56: 2818–2832, 2011. [DOI] [PubMed] [Google Scholar]
- 5.Batra A, Okur B, Glauben R, Erben U, Ihbe J, Stroh T, Fedke I, Chang HD, Zeitz M, Siegmund B. Leptin: a critical regulator of CD4+ T-cell polarization in vitro and in vivo. Endocrinology 151: 56–62, 2010. [DOI] [PubMed] [Google Scholar]
- 6.Blain A, Cattan S, Beaugerie L, Carbonnel F, Gendre JP, Cosnes J. Crohn's disease clinical course and severity in obese patients. Clin Nutr 21: 51–57, 2002. [DOI] [PubMed] [Google Scholar]
- 7.Bouloumie A, Curat CA, Sengenes C, Lolmede K, Miranville A, Busse R. Role of macrophage tissue infiltration in metabolic diseases. Curr Opin Clin Nutr Metab Care 8: 347–354, 2005. [DOI] [PubMed] [Google Scholar]
- 8.Bray GA, Bellanger T. Epidemiology, trends, and morbidities of obesity and the metabolic syndrome. Endocrine 29: 109–117, 2006. [DOI] [PubMed] [Google Scholar]
- 9.Bruun JM, Verdich C, Toubro S, Astrup A, Richelsen B. Association between measures of insulin sensitivity and circulating levels of interleukin-8, interleukin-6 and tumor necrosis factor-alpha. Effect of weight loss in obese men. Eur J Endocrinol 148: 535–542, 2003. [DOI] [PubMed] [Google Scholar]
- 10.Cao H. Adipocytokines in obesity and metabolic disease. J Endocrinol 220: T47–T59, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carey R, Jurickova I, Ballard E, Bonkowski E, Han X, Xu H, Denson LA. Activation of an IL-6:STAT3-dependent transcriptome in pediatric-onset inflammatory bowel disease. Inflamm Bowel Dis 14: 446–457, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan SSM, Luben R, Olsen A, Tjonneland A, Kaaks R, Teucher B, Lindgren S, Grip O, Key T, Crowe FL, Bergmann MM, Boeing H, Hallmans G, Karling P, Overvad K, Palli D, Masala G, Kennedy H, vanSchaik F, Bueno-de-Mesquita B, Oldenburg B, Khaw K-T, Riboli E, Hart AR. Body mass index and the risk for Crohn's disease and ulcerative colitis: data from a European Prospective Cohort Study (The IBD in EPIC Study). Am J Gastroenterol 108: 575–582, 2013. [DOI] [PubMed] [Google Scholar]
- 13.Coccia M, Harrison OJ, Schiering C, Asquith MJ, Becher B, Powrie F, Maloy KJ. IL-1beta mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4(+) Th17 cells. J Exp Med 209: 1595–1609, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daig R, Andus T, Aschenbrenner E, Falk W, Scholmerich J, Gross V. Increased interleukin 8 expression in the colon mucosa of patients with inflammatory bowel disease. Gut 38: 216–222, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Desreumaux P, Ernst O, Geboes K, Gambiez L, Berrebi D, Muller-Alouf H, Hafraoui S, Emilie D, Ectors N, Peuchmaur M, Cortot A, Capron M, Auwerx J, Colombel JF. Inflammatory alterations in mesenteric adipose tissue in Crohn's disease. Gastroenterology 117: 73–81, 1999. [DOI] [PubMed] [Google Scholar]
- 16.Fayad R, Pini M, Sennello JA, Cabay RJ, Chan L, Xu A, Fantuzzi G. Adiponectin deficiency protects mice from chemically induced colonic inflammation. Gastroenterology 132: 601–614, 2007. [DOI] [PubMed] [Google Scholar]
- 17.Fortuno A, Rodriguez A, Gomez-Ambrosi J, Fruhbeck G, Diez J. Adipose tissue as an endocrine organ: role of leptin and adiponectin in the pathogenesis of cardiovascular diseases. J Physiol Biochem 59: 51–60, 2003. [DOI] [PubMed] [Google Scholar]
- 18.Gambero A, Marostica M, Abdalla Saad MJ, Pedrazzoli J Jr.. Mesenteric adipose tissue alterations resulting from experimental reactivated colitis. Inflamm Bowel Dis 13: 1357–1364, 2007. [DOI] [PubMed] [Google Scholar]
- 19.Harman-Boehm I, Bluher M, Redel H, Sion-Vardy N, Ovadia S, Avinoach E, Shai I, Kloting N, Stumvoll M, Bashan N, Rudich A. Macrophage infiltration into omental versus subcutaneous fat across different populations: effect of regional adiposity and the comorbidities of obesity. J Clin Endocrinol Metab 92: 2240–2247, 2007. [DOI] [PubMed] [Google Scholar]
- 20.Hass DJ, Brensinger CM, Lewis JD, Lichtenstein GR. The impact of increased body mass index on the clinical course of Crohn's disease. Clin Gastroenterol Hepatol 4: 482–488, 2006. [DOI] [PubMed] [Google Scholar]
- 21.Karagiannides I, Kokkotou E, Tansky M, Tchkonia T, Giorgadze N, O'Brien M, Leeman SE, Kirkland JL, Pothoulakis C. Induction of colitis causes inflammatory responses in fat depots: evidence for substance P pathways in human mesenteric preadipocytes. Proc Natl Acad Sci USA 103: 5207–5212, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karagiannides I, Stavrakis D, Bakirtzi K, Kokkotou E, Pirtskhalava T, Nayeb-Hashemi H, Bowe C, Bugni JM, Nuno M, Lu B, Gerard NP, Leeman SE, Kirkland JL, Pothoulakis C. Substance P (SP)-neurokinin-1 receptor (NK-1R) alters adipose tissue responses to high-fat diet and insulin action. Endocrinology 152: 2197–2205, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karagiannides I, Tchkonia T, Dobson DE, Steppan CM, Cummins P, Chan G, Salvatori K, Hadzopoulou-Cladaras M, Kirkland JL. Altered expression of C/EBP family members results in decreased adipogenesis with aging. Am J Physiol Regul Integr Comp Physiol 280: R1772–R1780, 2001. [DOI] [PubMed] [Google Scholar]
- 24.Karmiris K, Koutroubakis IE, Xidakis C, Polychronaki M, Voudouri T, Kouroumalis EA. Circulating levels of leptin, adiponectin, resistin, and ghrelin in inflammatory bowel disease. Inflamm Bowel Dis 12: 100–105, 2006. [DOI] [PubMed] [Google Scholar]
- 25.Kern PA, Saghizadeh M, Ong JM, Bosch RJ, Deem R, Simsolo RB. The expression of tumor necrosis factor in human adipose tissue. Regulation by obesity, weight loss, and relationship to lipoprotein lipase. J Clin Invest 95: 2111–2119, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koon HW, Kim YS, Xu H, Kumar A, Zhao D, Karagiannides I, Dobner PR, Pothoulakis C. Neurotensin induces IL-6 secretion in mouse preadipocytes and adipose tissues during 2,4,6,-trinitrobenzensulphonic acid-induced colitis. Proc Natl Acad Sci USA 106: 8766–8771, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kredel LI, Batra A, Stroh T, Kühl AA, Zeitz M, Erben U, Siegmund B. Adipokines from local fat cells shape the macrophage compartment of the creeping fat in Crohn's disease. Gut 62: 852–862, 2013. [DOI] [PubMed] [Google Scholar]
- 28.Kruschewski M, Foitzik T, Perez-Canto A, Hubotter A, Buhr HJ. Changes of colonic mucosal microcirculation and histology in two colitis models: an experimental study using intravital microscopy and a new histological scoring system. Dig Dis Sci 46: 2336–2343, 2001. [DOI] [PubMed] [Google Scholar]
- 29.Ludwiczek O, Vannier E, Borggraefe I, Kaser A, Siegmund B, Dinarello CA, Tilg H. Imbalance between interleukin-1 agonists and antagonists: relationship to severity of inflammatory bowel disease. Clin Exp Immunol 138: 323–329, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lyon CJ, Law RE, Hsueh WA. Minireview: adiposity, inflammation, and atherogenesis. Endocrinology 144: 2195–2200, 2003. [DOI] [PubMed] [Google Scholar]
- 31.Ma X, Torbenson M, Hamad AR, Soloski MJ, Li Z. High-fat diet modulates non-CD1d-restricted natural killer T cells and regulatory T cells in mouse colon and exacerbates experimental colitis. Clin Exp Immunol 151: 130–138, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mahida YR, Wu K, Jewell DP. Enhanced production of interleukin 1-beta by mononuclear cells isolated from mucosa with active ulcerative colitis of Crohn's disease. Gut 30: 835–838, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsuzawa Y. Adiponectin: a key player in obesity related disorders. Curr Pharm Des 16: 1896–1901, 2010. [DOI] [PubMed] [Google Scholar]
- 34.Neumeier M, Weigert J, Schaffler A, Wehrwein G, Muller-Ladner U, Scholmerich J, Wrede C, Buechler C. Different effects of adiponectin isoforms in human monocytic cells. J Leukoc Biol 79: 803–808, 2006. [DOI] [PubMed] [Google Scholar]
- 35.Neumeier M, Weigert J, Schaffler A, Weiss T, Kirchner S, Laberer S, Scholmerich J, Buechler C. Regulation of adiponectin receptor 1 in human hepatocytes by agonists of nuclear receptors. Biochem Biophys Res Commun 334: 924–929, 2005. [DOI] [PubMed] [Google Scholar]
- 36.Ng SC, Bernstein CN, Vatn MH, Lakatos PL, Loftus EV Jr, Tysk C, O'Morain C, Moum B, Colombel JF. Geographical variability and environmental risk factors in inflammatory bowel disease. Gut 62: 630–649, 2013. [DOI] [PubMed] [Google Scholar]
- 37.Nishihara T, Matsuda M, Araki H, Oshima K, Kihara S, Funahashi T, Shimomura I. Effect of adiponectin on murine colitis induced by dextran sulfate sodium. Gastroenterology 131: 853–861, 2006. [DOI] [PubMed] [Google Scholar]
- 38.Ogden CL, Lamb MM, Carroll MD, Flegal KM. Obesity and socioeconomic status in children and adolescents: United States, 2005–2008. NCHS Data Brief 51: 1–8, 2010. [PubMed] [Google Scholar]
- 39.Peyrin-Biroulet L. Anti-TNF therapy in inflammatory bowel diseases: a huge review. Minerva Gastroenterol Dietol 56: 233–243, 2010. [PubMed] [Google Scholar]
- 40.Pini M, Gove ME, Fayad R, Cabay RJ, Fantuzzi G. Adiponectin deficiency does not affect development and progression of spontaneous colitis in IL-10 knockout mice. Am J Physiol Gastrointest Liver Physiol 296: G382–G387, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saijo S, Nagata K, Nakano Y, Tobe T, Kobayashi Y. Inhibition by adiponectin of IL-8 production by human macrophages upon coculturing with late apoptotic cells. Biochem Biophys Res Commun 334: 1180–1183, 2005. [DOI] [PubMed] [Google Scholar]
- 42.Sennello JA, Fayad R, Pini M, Gove ME, Fantuzzi G. Transplantation of wild-type white adipose tissue normalizes metabolic, immune and inflammatory alterations in leptin-deficient ob/ob mice. Cytokine 36: 261–266, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singh UP, Singh S, Taub DD, Lillard JW Jr.. Inhibition of IFN-gamma-inducible protein-10 abrogates colitis in IL-10-/- mice. J Immunol 171: 1401–1406, 2003. [DOI] [PubMed] [Google Scholar]
- 44.Skurk T, Alberti-Huber C, Herder C, Hauner H. Relationship between adipocyte size and adipokine expression and secretion. J Clin Endocrinol Metab 92: 1023–1033, 2006. [DOI] [PubMed] [Google Scholar]
- 45.Tchkonia T, Giorgadze N, Pirtskhalava T, Tchoukalova Y, Karagiannides I, Forse RA, DePonte M, Stevenson M, Guo W, Han J, Waloga G, Lash TL, Jensen MD, Kirkland JL. Fat depot origin affects adipogenesis in primary cultured and cloned human preadipocytes. Am J Physiol Regul Integr Comp Physiol 282: R1286–R1296, 2002. [DOI] [PubMed] [Google Scholar]
- 46.van Furth R, Goud TJ, van der Meer JW, Blusse van Oud Alblas A, Diesselhoff-den Dulk MM, Schadewijk-Nieuwstad M. Comparison of the in vivo and in vitro proliferation of monoblasts, promonocytes, and the macrophage cell line J774. Adv Exp Med Biol 155: 175–187, 1982. [DOI] [PubMed] [Google Scholar]
- 47.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr.. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112: 1796–1808, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamaguchi N, Argueta JG, Masuhiro Y, Kagishita M, Nonaka K, Saito T, Hanazawa S, Yamashita Y. Adiponectin inhibits Toll-like receptor family-induced signaling. FEBS Lett 579: 6821–6826, 2005. [DOI] [PubMed] [Google Scholar]
- 49.Yamamoto K, Kiyohara T, Murayama Y, Kihara S, Okamoto Y, Funahashi T, Ito T, Nezu R, Tsutsui S, Miyagawa JI, Tamura S, Matsuzawa Y, Shimomura I, Shinomura Y. Production of adiponectin, an anti-inflammatory protein, in mesenteric adipose tissue in Crohn's disease. Gut 54: 789–796, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao P, Deng Y, Gu P, Wang Y, Zhou H, Hu Y, Chen P, Fan X. Insulin-like growth factor 1 promotes the proliferation and adipogenesis of orbital adipose-derived stromal cells in thyroid-associated ophthalmopathy. Exp Eye Res 107: 65–73. [DOI] [PubMed] [Google Scholar]
- 51.Zulian A, Cancello R, Micheletto G, Gentilini D, Gilardini L, Danelli P, Invitti C. Visceral adipocytes: old actors in obesity and new protagonists in Crohn's disease? Gut 61: 86–94, 2012. [DOI] [PubMed] [Google Scholar]