

Abstract
Background:
Acquired resistance to molecularly targeted therapeutics is a key challenge in personalised cancer medicine, highlighting the need for identifying the underlying mechanisms and early biomarkers of relapse, in order to guide subsequent patient management.
Methods:
Here we use human head and neck squamous cell carcinoma (HNSCC) models and nuclear magnetic resonance (NMR) spectroscopy to assess the metabolic changes that follow acquired resistance to EGFR tyrosine kinase inhibitors (TKIs), and which could serve as potential metabolic biomarkers of drug resistance.
Results:
Comparison of NMR metabolite profiles obtained from control (CALS) and EGFR TKI-resistant (CALR) cells grown as 2D monolayers, 3D spheroids or xenograft tumours in athymic mice revealed a number of differences between the sensitive and drug-resistant models. In particular, we observed elevated levels of glycerophosphocholine (GPC) in CALR relative to CALS monolayers, spheroids and tumours, independent of the growth rate or environment. In addition, there was an increase in alanine, aspartate and creatine+phosphocreatine in resistant spheroids and xenografts, and increased levels of lactate, branched-chain amino acids and a fall in phosphoethanolamine only in xenografts. The xenograft lactate build-up was associated with an increased expression of the glucose transporter GLUT-1, whereas the rise in GPC was attributed to inhibition of GPC phosphodiesterase. Reduced glycerophosphocholine (GPC) and phosphocholine were observed in a second HNSCC model probably indicative of a different drug resistance mechanism.
Conclusions:
Our studies reveal metabolic signatures associated not only with acquired EGFR TKI resistance but also growth pattern, microenvironment and contributing mechanisms in HNSCC models. These findings warrant further investigation as metabolic biomarkers of disease relapse in the clinic.
Keywords: acquired drug resistance, biomarkers, EGFR tyrosine kinase inhibitors, head and neck cancer, metabolism
Epidermal growth factor receptor (EGFR/HER1) is over-expressed in 95% of head and neck squamous cell carcinomas (HNSCC) and high levels are correlated with poor patient prognosis and decreased overall survival (Grandis et al, 1998; Ang et al, 2002), suggesting that the disease is EGFR-driven and making a strong case for targeting EGFR in HNSCC. Disappointingly, however, clinical evidence to date shows that EGFR-targeted agents, including monoclonal antibodies and tyrosine kinase inhibitors (TKIs), have little efficacy in HNSCC as a monotherapy, raising the possibility that alternative survival pathways may be activated in this disease (Box et al, 2013a, 2013b). To address this, new strategies for targeting EGFR in HNSCC are being evaluated clinically including the use of pan-HER inhibitors and rational drug combinations that target several signalling pathways simultaneously (Agulnik, 2012).
A key challenge for TKIs (including those targeting EGFR) in cancer is the development of acquired resistance, with many patients showing disease progression not long after the initial response (Wagle et al, 2011; Lovly and Shaw, 2014). Thus, the elucidation of underlying molecular mechanisms, as well as early biomarkers of patient relapse, is key for identifying subsequent therapies that may delay or indeed prevent further disease progression, and ultimately improve long-term survival.
Studies have shown that treatment failure is, in many cases, due to cancer cells acquiring new genetic and molecular alterations that enable them to survive in the face of therapeutic stress (Holohan et al, 2013). Additionally, transformed tumour cell metabolism may be equally important in this regard since alterations in key processes such as glucose, glutamine, nucleotide, fatty acid and phospholipid metabolism allow cancer cells to adapt to their growth demands and thrive in a hostile microenvironment (Bailey et al, 2012; Ward and Thompson, 2012). Several oncoproteins and oncogenic signalling pathways directly regulate vital components of the metabolic network, thus providing a link between the molecular abnormalities of cancer and the transformed metabolic phenotype (Deberardinis et al, 2008; Vander Heiden et al, 2009; Vousden and Ryan, 2009).
Altered metabolism is a hallmark of cancer (Hanahan and Weinberg, 2011) and metabolic features of cancer are being explored for diagnosis and staging as well as for informing on disease response and resistance to therapy (Beloueche-Babari et al, 2010; Beloueche-Babari et al, 2011). In this context, metabolic imaging and/or profiling techniques such as positron emission tomography, nuclear magnetic resonance (NMR) spectroscopy and mass spectrometry play an important role in translating findings from pre-clinical studies to humans.
We have developed a model of acquired resistance to EGFR TKIs based on the human HNSCC cell line CAL 27, and shown that a resistant derivative (CALR) exhibits a distinct protein expression signature compared with its sensitive counterpart (CALS) (Box et al, 2013a). Because of the central role that metabolism plays in tumour biology (Hanahan and Weinberg, 2011), we explore here the metabolic features of EGFR TKI sensitivity in CALS and CALR cells grown as 2D monolayers, 3D spheroids and tumour xenografts by using NMR spectroscopy. Our aim is to assess the value of 3D cultures, compared with 2D or xenograft tumour models, for the discovery of potential metabolic signatures underlying resistance that could potentially be developed as minimally invasive metabolic biomarkers of EGFR TKI resistance, and to interrogate the influence of the microenvironment on metabolic readouts.
Materials and methods
Cell culture and in vitro experiments
CALS/CALR and PJS/PJR HNSCC cell lines were generated and maintained as previously described (Box et al, 2013a). CALR and PJR cells are stably resistant to EGFR TKIs and were thus routinely grown in gefitinib-free media. Cells were screened regularly for mycoplasma and cultured for no longer than ten passages. 2D cell counts and diameter measurements were performed on a Beckman Coulter Vi-Cell Cell Viability Analyser.
3D CALS and CALR tumour spheroids were generated by plating 4000 cells per well in normal growth medium in round bottom ultra-low attachment 96-well plates (Corning) as previously documented (Vinci et al, 2013). After 4 days, spheroids were fed by 50% medium replenishment and harvested for analysis after a further 3 days. Spheroid diameters were measured on a Celigo cytometer (Nexcelom Bioscience). Gefitinib was purchased from ChemieTek.
Human tumour xenograft models
CALS and CALR subcutaneous tumours were established as described previously (Box et al, 2013a). Tumours were measured across two perpendicular diameters using callipers, and volumes were obtained from the following formula: V=4/3π [(d1+d2)/4]3. Once an appropriate tumour volume (∼350 mm3) was attained (∼2 weeks for CALR and ∼4 weeks for CALS), tumours were excised, carefully dissected clear of surrounding skin and fat, snap-frozen in liquid nitrogen then stored at −80 °C until further processing for western blotting or in vitro NMR spectroscopy. All experiments were performed in accordance with UK Home Office regulations under the Animals (Scientific Procedures) Act 1986 and UK National Cancer Research Institute (NCRI) Guidelines for the Welfare and Use of Animals in Cancer Research (Workman et al, 2010).
Metabolic evaluation of cells and tumours
For metabolic analysis by NMR spectroscopy, 2D subconfluent cell monolayers (∼5 × 107 cells) were extracted using a dual phase method in equal volumes of cold methanol, chloroform and water as previously described (Beloueche-Babari et al, 2012). For the 3D experiments, individual 7-day old spheroids (from six 96-well ULA plates/cell line) were pooled and extracted using a similar approach. Xenograft tumour material (∼150 mg) was ground under liquid nitrogen in a pestle and mortar then a dual phase extraction was used, as described above. After phase separation, the aqueous fraction from each sample was lyophilised and reconstituted in 540 μl of D2O containing 3-(trimethylsilyl) propionic-2,2,3,3-d4 acid (TSP) as an internal 1H NMR quantitation and chemical shift reference. For 31P NMR measurements, 60 μl of a D2O solution containing EDTA and methylenediphosphonic acid (MDPA, internal standard) to a final concentration of 10 and 0.43 mM respectively (pH 8.2) was added.
NMR spectroscopy data acquisition and processing
1H and 31P NMR spectroscopy data were acquired on a Bruker 500 MHz spectrometer at 25 °C. Acquisition and processing of spectra were performed as previously described (Beloueche-Babari et al, 2012). Metabolite content was measured by peak integration relative to TSP (1H NMR) or MDPA (31P NMR) and metabolite content normalised to cell number and volume for 2D cultures, and wet tissue weight for tumour samples. Spheroid data were expressed as ratios of metabolites.
Multivariate analysis of NMR spectroscopy data
1H NMR data from CALR and CALS tumours were subjected to unbiased metabolic profiling using principal component analysis (PCA), a method that uses the original variables (metabolite peaks) to derive a new smaller set of orthogonal (uncorrelated) variables, or principal components, that explain the variance in the original data set. For this, spectra were phased and baseline corrected then integrated in spectral regions (bins) of 0.04 p.p.m. within the 0.8–4.38 p.p.m. excluding the residual methanol peak at 3.36 ppm. Integrals from individual spectral bins were normalised to the sum of total integrals obtained and processed in SIMCA v13.0 (Umetrics – Umeå, Sweden) using a PCA model.
Western blotting
Cells were lysed as described previously (Rogers et al, 2009) except that tumour spheroids were stored at −80 °C overnight (to aid lysis) prior to centrifugation. Tumour samples were processed as previously documented (Beloueche-Babari et al, 2013). For analysis of GLUT-1 expression, proteins were electrophoresed under non-reducing conditions then transferred to PVDF membranes. Blots were probed with antibodies to GLUT-1 (Merck Millipore) or β-actin as a loading control (Abcam).
Glycerophosphocholine-phosphodiesterase (GPC-pd) assay
GPC-pd enzymatic activity was monitored by 1H NMR by using a modification of a previously described method (Iorio et al, 2010). 3 × 107 cells were pelleted, washed and homogenised then sonicated at 4 °C in 0.5 ml 100 mM Tris-HCl (pH 7.2) buffer containing 10 mM DTT and 1 mM EDTA in D2O. Following a 30 min centrifugation at 12 000 r.p.m., supernatants containing the cytosolic fractions were transferred to a 5 mm NMR tube. GPC-pd was assayed at 30 °C by monitoring the 1H NMR-observed build up of choline over time (35 min) following the addition of exogenous GPC and MgCl2 at a final concentration of 1.2 mM and 10 mM, respectively.
Statistical analysis
For comparison of metabolite levels, Student's t-test was used with P-values of ⩽0.05 considered to be statistically significant. Data represent the mean±s.e.
Results
Acquired resistance to EGFR TKI in HNSCC alters tumour metabolism
We have previously reported that CALR cells exhibit a faster growth rate compared with CALS when grown as subcutaneous xenograft tumours but not when cultured as monolayers in vitro (Box et al, 2013a). Interestingly, when grown as 3D spheroids, CALR grew more rapidly than CALS cells with average spheroid volumes at day 7 of 31±3 and 17±4 × 106 μm3 (n=5 biological repeats, P=0.02), respectively, indicative of a more aggressive growth phenotype as observed in vivo.
We hypothesised that our cell line models with distinct growth properties would possess different metabolic characteristics. Thus, prior to assessing metabolic differences associated with the resistance phenotype we explored the extent of any baseline metabolic variations between the three growth conditions: 2D, 3D and xenograft tumours, within each cell line.
As shown in Figure 1A, unbiased multivariate analysis with PCA of the 1H NMR spectral data indicated that the 2D cells, 3D spheroids and tumours exhibit separate clustering within each cell line, consistent with a distinct metabolic phenotype. The clustering was maintained even when data from CALS and CALR were merged, suggesting strong model-dependent patterns. The score scatter plots indicate that the variation along the PC1 axis is driven by differences between the 2D and tumour data vs the spheroid data while the variation along the PC2 axis is driven by differences between the 2D vs tumour data with spheroid data overlapping between the two. Thus, despite arising from the same cells of origin, the three experimental models used in this study have unique metabolic features which are likely to be a reflection of their growth phenotype and microenvironment.
Figure 1.

Unbiased metabolomic profiling of CALS and CALR tumour models. (A) 2D PCA score scatter plots showing a separate clustering for 1H NMR data from cells grown as 2D monolayers, 3D spheroids and xenograft tumours within the CALS and CALR cell lines separately and when the data are merged. (B) 2D PCA score scatter plots showing separate clustering for CALS and CALR 1H NMR data points within the 2D cell model, 3D spheroids and tumours. PC1 and PC2 are the two most important principal components explaining the variation in the data (shown as percentages in the x and y axes).
The metabolic characteristics of acquired EGFR TKI resistance were assessed with PCA of the 1H NMR data derived from CALS and CALR cells within each model. The separate clustering of the data points corresponding to CALS and CALR on the score scatter plots in Figure 1B indicates a distinct metabolic profile for the sensitive and the EGFR TKI-resistant cells in every model.
The clearest separation was obtained in the tumours which showed that variability in the data could be described according to three main principal components, PC1, PC2 and PC3 (Figure 1B and 2A), that between them explain ∼68% of the total variance (PC1: 34.8%, PC2: 18.4%, PC3: 15.1%). The resonances that appeared to be key in the separation between the CALS and CALR profiles include lactate, branched-chain amino acids (BCAAs), choline metabolites, acetate, myo-inositol, glutamine/glutamate and creatine (Cr)+phosphocreatine (PCr), as shown in Figure 2B.
Figure 2.

NMR profiling of CALS and CALR tumours. (A) Three-dimensional PCA score scatter plot showing separate clustering for 1H NMR data from CALS and CALR. (B) Score contribution plot showing changes in the 1H NMR peaks (and related metabolites) accounting for the differences between CALR and CALS tumours (plot obtained using the group-to-group comparison option in SIMCA). Positive scores represent increased levels, while negative scores indicate decreased levels in CALR relative to CALS. (C) Representative 31P NMR spectra showing the differences in 31P-containing metabolites between CALS and CALR tumours. Abbreviations: Asp=aspartate; BCAA=branched-chain amino acids; Cr=creatine; PCr=phosphocreatine; PC=phosphocholine; PE=phosphoethanolamine; GPC=glycerophosphocholine; GPE=glycerophosphoethanolamine; Pi=inorganic phosphate; Gln=glutamine; Glut=glutamate; Glx=glutathione; Myo-Ins=myo-inositol; ?=unidentified peak.
To validate the metabolite changes identified in the PCA, we performed a targeted analysis of the data by integrating the individual peaks in the 1H NMR spectra. As shown in Table 1, and in agreement with the PCA method, univariate 1H NMR revealed a number of metabolic alterations in CALR xenograft tumours compared with their CALS counterpart. Specifically, the levels of GPC, lactate, BCAAs, alanine and aspartate were significantly elevated in CALR relative to CALS tumours. Total choline, which is predominantly comprised of GPC, phosphocholine (PC) and free choline, was also increased in CALR compared with CALS. The levels of Cr/PCr, acetate and glutamate showed a trend towards an increase, while myo-inositol showed a trend towards a decrease in CALR compared with CALS tumours but these effects did not reach statistical significance.
Table 1. Metabolite content of CALR and CALS HNSCC human tumour xenografts as detected by 1H and 31P NMR spectroscopy analysis of excised tumour tissue.
| CALS | CALR | Pa | |
|---|---|---|---|
|
1H Metabolites (au g−1 wet tissue) | |||
| BCAA | 59±6 | 108±11 | 0.007 |
| Lactate | 40±4 | 67±8 | 0.02 |
| Alanine | 36±4 | 77±9 | 0.006 |
| Acetate | 12±2 | 9±1 | 0.16 |
| Glutamate | 65±6 | 83±9 | 0.14 |
| Glutamine | 39±4 | 41±5 | 0.74 |
| Glutathione | 25±4 | 24±4 | 0.86 |
| Aspartate | 1.8±0.5 | 2.7±0.2 | 0.01 |
| Cr+PCr | 46±3 | 62±7 | 0.07 |
| PC | 45±5 | 45±5 | 0.96 |
| GPC | 27±4 | 58±9 | 0.02 |
| Total choline | 75±7 | 118±16 | 0.05 |
| Myo-inositol | 10±1 | 8±1 | 0.25 |
|
31P Metabolites (μmol g−1 wet tissue) | |||
| PCr | 0.36±0.08 | 0.94±0.16 | 0.02 |
| Pi | 2.14±0.44 | 2.44±0.22 | 0.57 |
| GPC | 0.11±0.03 | 0.53±0.06 | 0.001 |
| GPE | 0.05±0.02 | 0.19±0.03 | 0.009 |
| PC | 0.33±0.03 | 0.34±0.03 | 0.84 |
| PE | 0.55±0.07 | 0.31±0.04 | 0.02 |
Two-tailed unpaired Student's t-test, n=5. Bold denotes statistically significant P values.
31P NMR analysis of CALS and CALR tumour extracts corroborated the elevation in GPC and PCr levels and additionally revealed a rise in glycerophosphoethanolamine (GPE) and a fall in phosphoethanolamine (PE) levels in CALR relative to CALS tumours (Table 1 and Figure 2C). NTP levels were not reproducibly detectable in the 31P NMR spectra of tumour extracts, but the consistent and significant increase in CALR PCr levels suggests that the drug-resistant tumours have improved bioenergetics.
Relationship between metabolism, drug resistance, growth environment and phenotype
To investigate the significance of the metabolic changes observed in relation to the acquired drug resistance phenotype and how this is impacted on by the microenvironment and the differential growth rates of the tumours, we assessed whether the metabolic signature of drug resistance identified in the xenograft tumours could also be present in cells grown in vitro. For these experiments we compared cells cultured as (a) 2D monolayers with comparable CALS/CALR growth rates and (b) 3D spheroids with a faster CALR growth rate, reminiscent of the in vivo xenograft model.
Univariate 1H NMR spectroscopy analysis of 2D cell extracts showed that, consistent with the tumour data, CALR cells exhibited higher levels of GPC relative to CALS cells while BCAAs lactate, Cr+PCr, aspartate and alanine, which were increased in the CALR tumours, were not significantly changed in the 2D cell cultures (Table 2).
Table 2. 1H NMR spectroscopy-detectable metabolite levels in CALR and CALS cells grown as 2D monolayers or as 3D spheroids.
| CALS | CALR | Pa | |
|---|---|---|---|
|
2D cell metabolites (au per cell volume) | |||
| BCAA | 13±1 | 15±1 | 0.16 |
| Lactate | 5.20±0.60 | 5.40±0.40 | 0.66 |
| Alanine | 9.50±1.20 | 9.20±0.80 | 0.84 |
| Glutamate | 5.50±0.50 | 6.00±0.50 | 0.54 |
| Aspartate | 0.49±0.06 | 0.34±0.04 | 0.08 |
| Cr+PCr | 2.20±0.20 | 1.9±0.20 | 0.44 |
| PC | 10±2 | 10±1 | 0.82 |
| GPC | 1.90±0.3 | 3.90±0.6 | 0.02 |
| PE | 0.61±0.06 | 0.54±0.09 | 0.53 |
|
3D spheroid metabolites (ratio to PC) | |||
| BCAA | 7.30±1.30 | 4.50±0.60 | 0.1 |
| Lactate | 1.52±0.16 | 1.34±0.28 | 0.58 |
| Alanine | 1.30±0.30 | 2.70±0.50 | 0.038 |
| Glutamate | 0.54±0.17 | 1.27±0.55 | 0.15 |
| Aspartate | 0.05±0.03 | 0.15±0.02 | 0.025 |
| Cr+PCr | 0.56±0.07 | 0.87±0.04 | 0.003 |
| GPC | 0.32±0.02 | 0.63±0.07 | 0.007 |
| PE | 0.36±0.10 | 0.19±0.07 | 0.29 |
Unpaired 2-tailed Student's t-test, n⩾3. Bold denotes statistically significant P values.
For the analysis of 3D spheroids, as it was not possible to obtain an accurate estimate of cell number, we normalised the metabolite content relative to the PC signal since this was found to be comparable in the tumours as well as the 2D cultured cells.
The data showed that, similar to the tumour profiles, the ratios of GPC/PC, PCr+Cr/PC, alanine/PC and aspartate/PC were significantly elevated. However, in contrast to the observations made in solid tumours, the ratio of BCAA/PC appeared to decrease in CALR compared with CALS spheroids, although this was not statistically significant, while lactate/PC remained constant within experimental uncertainly (Table 2).
The increased lactate observed in tumours but not 2D or 3D in vitro cultures could suggest a shift towards glycolytic metabolism (also known as the Warburg effect) in vivo. To investigate this possibility we assessed the protein expression levels of the glucose transporter GLUT-1. Our data indicate that CALR have upregulated GLUT-1 expression compared with CALS tumours, consistent with an increased Warburg effect. Interestingly, this difference was not observed in cells grown as 2D monolayers or 3D spheroids (Figure 3), where lactate levels were unchanged.
Figure 3.
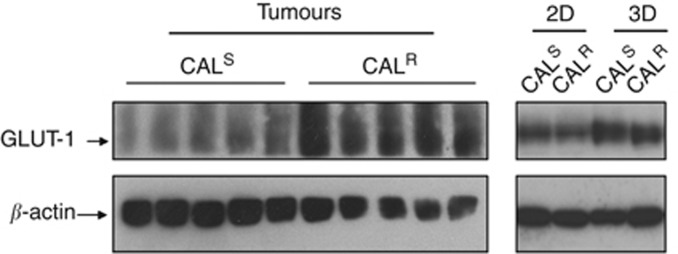
GLUT-1 expression in CALS and CALR tumour models. Western blots showing an elevated GLUT-1 protein expression in CALR relative to CALS xenograft tumours but not in 2D monolayers or 3D spheroids. The individual lanes in the tumour blot represent samples from individual xenografts.
Comparison of the data obtained from 2D cultures, 3D spheroids and xenograft tumours indicate that, relative to CALS, CALR cells displayed increased levels of lactate, BCAA and decreased PE in vivo only, elevated PCr, alanine and aspartate levels in 3D spheroids and tumours, and increased GPC in all three experimental models (Figure 4A and B). Thus the rises in lactate and GLUT-1 expression, together with increased BCAA and decreased PE are associated with the in vivo growth environment while the increase in alanine, aspartate and Cr+PCr is associated with the aggressive growth phenotype seen in 3D and in vivo. The build up of GPC is associated with EGFR TKI resistance independently of differences in growth rates or environment.
Figure 4.

Comparison of metabolite changes induced in EGFR TKI resistance models. (A) Diagram summarising the metabolite signature observed in CALR relative to CALS tumours and its presence in 2D and 3D in vitro cultures. (B) 1H NMR spectra of the choline region showing an increased GPC in CALR relative to CALS models. (C) Changes in GPC following an acute exposure to gefitinib (24 h, 1 μM) in CALS and CALR cell lines grown in 2D. Φ: not measured in spheroids or 2D monolayers, *P=0.04, NS: P=0.58, n=3 at least.
To assess the dynamics of the change in GPC, we monitored the levels of this metabolite following acute exposure to drug. 1H NMR spectroscopy of cells treated with 1μM gefitinib (EGFR TKI) for 24 h showed that GPC levels increased in CALS (to 157±21% relative to controls, P=0.04) but not in CALR cells (95±13% relative to controls, P=0.36), which were unresponsive to drug as determined by the effects of treatment on cell counts: 94±5% of controls (P=0.25) in CALR compared with 70±5% (P=0.001) in CALS (Figure 4C). Thus, the rise in GPC appears to be an early metabolic consequence of exposure to gefitinib, which is sustained with chronic exposure when cells acquire long-term drug resistance.
EGFR TKI resistance in the CALS/CALR model is associated with inhibition of GPC-pd
GPC is produced from the breakdown of the membrane phospholipid phosphatidylcholine (PtdCho) in a 2-step reaction mediated via phospholipase A and lysophospholipase, which concomitantly leads to the formation of free fatty acids (FFAs). GPC is further metabolised to glycerol-3 phosphate and choline in a reaction catalysed by GPC phosphodiesterase (GPC-pd).
To determine the metabolic processes underpinning the elevation in GPC levels, we initially assessed the levels of FFA signals in the 1H NMR spectra of the organic phase of cells extracts. No significant differences were recorded between the profiles of CALS and CALR cells indicating that the rates of PtdCho breakdown were comparable in the two cell lines (Supplementary Table S1).
The breakdown of GPC via GPC-pd was next investigated using a 1H NMR-based assay that monitored the formation of choline over time as a measure of GPC-pd activity (Figure 5A and B). This showed that the rate of choline formation as a result of GPC hydrolysis was 97±19 pmol per cell per min in CALS monolayers, decreasing to 45±13 pmol per cell per min in CALR monolayers (P=0.03, n=4), consistent with lower GPC-pd activity in this cell line (Figure 5C). Taken together these findings show that the increase in GPC content in the EGFR TKI-resistant CALR cells is likely to be due to a reduction of its metabolism by GPC-pd.
Figure 5.

GPC-pd activity assay in CALR and CALS monolayers. (A) Sequential 1H NMR spectra from the GPC-pd assay showing the changes in choline and GPC resonances over time. (B) Build up of choline in 2D CALR and CALS cells over time (total of 35 min) following addition of GPC. (C) Quantification of GPC-pd activity shows a decrease in CALR relative to CALS cells. *P=0.03, n=4.
The metabolic signatures of acquired EGFR TKI resistance may be mechanism-dependent
Finally, to test the generalisability of our observations we assessed a second sensitive/resistant HNSCC model that we have previously described: the PJS and PJR cell lines (Box et al, 2013a). DNA staining and flow cytometry showed polyploidy in PJR cells, a phenomenon that was not observed in PJS, CALS or CALR cell lines (supplementary Figure S1). Polyploidy has previously been linked to drug resistance (Puig et al, 2008; Shen et al, 2008), suggesting a potential difference in the underlying mechanisms of acquired resistance to EGFR TKIs between PJR and CALR models. Our 1H NMR data show that, unlike the case for CALR vs CALS, drug-resistant PJR cells in standard 2D cultures exhibited reduced levels of GPC and PC compared with the sensitive PJS cells with no significant changes in the levels of lactate, alanine, glutamate or BCAA (Table 3). Thus, acquired resistance to EGFR TKIs in the PJR/PJS model is associated with a differential metabolic response compared with the CALR/CALS model, which is likely to be related to the differences in accompanying cellular processes and potentially the underlying drug resistance mechanisms.
Table 3. 1H NMR spectroscopy-detectable metabolite levels in PJR and PJS cells grown as 2D monolayers.
| 2D cell metabolites (au/cell volume) | PJS | PJR | Pa |
|---|---|---|---|
| BCAA | 4.80±0.25 | 5.4±0.35 | 0.23 |
| Lactate | 6.90±0.80 | 7.50±1.00 | 0.66 |
| Alanine | 2.20±0.30 | 2.20±0.38 | 0.96 |
| Glutamate | 2.10±0.20 | 2.40±0.30 | 0.46 |
| PC | 17±2 | 11±1 | 0.05 |
| GPC | 1.50±0.10 | 0.54±0.03 | 0.01 |
| PE | 0.50±0.14 | 0.20±0.05 | 0.17 |
Unpaired 2-tailed Student's t-test, n=3. Bold denotes statistically significant P values.
Discussion
Acquired resistance to TKIs is a major challenge in personalised cancer medicine and the identification of mechanisms, as well as early biomarkers of patient relapse, will enable a timely switch to alternative therapies prior to further disease progression.
In this study we used TKI sensitive (CALS) and resistant (CALR) human HNSCC cells grown as standard 2D monolayers, 3D spheroids or tumour xenografts alongside NMR spectroscopy to explore metabolic characteristics of acquired resistance to EGFR TKIs which, if independently validated, could have potential as minimally invasive biomarkers, and to assess the influence of the microenvironment on the metabolic readouts.
1H and 31P NMR spectroscopy combined with PCA and univariate metabolite analysis of tumour extracts revealed a number of metabolic differences between CALR and CALS tumours, in particular increased lactate, GPC, BCAA, alanine, aspartate, PCr and decreased PE. These changes are likely to reflect differences in metabolism between CALR and CALS tumour cells since the extent of stromal enrichment, as revealed by haematoxylin and eosin staining (Supplementary Figure S2), was comparable between the CALS and CALR tumours, indicating that any contributions from non-tumour cells to the metabolic readouts should be similar between the two models.
Assessment of this metabolic signature in in vitro models indicated that in 3D spheroids, where CALR had a more aggressive growth phenotype relative to CALS, increased GPC, Cr/PCr, aspartate as well as alanine were also observed alongside drug resistance. On the other hand, in 2D cultures, where CALR did not exhibit a faster growth rate than CALS cells, only an increase in GPC was detectable. Taken together, these data suggest that the increase in tumour lactate and BCAA and decreased PE, only detectable in vivo, could reflect contribution from the tumour microenvironment, while the increase in PCr, aspartate and alanine levels observed in the CALR tumours and 3D spheroids may be linked to the aggressive growth phenotype they exhibit in comparison with their CALS counterpart. In contrast, the rise in GPC, which was observed in all three growth conditions irrespective of their growth rate, is likely to be associated with acquired resistance to EGFR TKI in our HNSCC model. Our data also show that the metabolic phenotype exhibited by CALR and CALS xenograft tumours was reflected more accurately by the 3D spheroids than by the 2D monolayers, which reinforces the increased value of 3D cultures as biologically relevant experimental models of cancer (Vinci et al, 2012).
Increased glycolytic metabolism has been reported in association with resistance to chemotherapeutic drugs (Zhou et al, 2012). Although we did not observe such effects in our in vitro models, lactate levels were increased in the drug-resistant xenografts and the concomitant upregulation of the glucose transporter GLUT-1 in CALR relative to CALS tumours is consistent with a switch to glycolytic metabolism. Levels of GLUT-1 were comparable between CALR and CALS in 2D monolayers and 3D spheroids, where lactate levels were unchanged, indicating that the Warburg effect observed in tumours is not an inherent feature of the parental cells rather it is a consequence of the in vivo microenvironment. Oxidative stress and hypoxia may be underlying causes of this glycolytic switch as they can induce the expression of hypoxia inducible factor-1 alpha, which can, in turn, upregulate the transcription of GLUT-1 (Verschoor et al, 2010; Brahimi-Horn et al, 2011). Hypoxia is also a major cause of treatment-resistance in HNSCC patients (Koukourakis et al, 2006; Gee et al, 2010). Further work is required to characterise the physiologic phenotype of CALR and CALS tumours and assess if any differences could account for the increase in GLUT-1 expression and lactate levels observed here.
High levels of cellular bioenergy metabolite levels including the phosphagen metabolite PCr have been reported to be linked to chemoresistance (Kaplan et al, 1991; Zhou et al, 2012) and could indicate an enhancement in energy metabolism required for sustaining increased cell proliferation (Cairns et al, 2011). We also observed increased aspartate and alanine in CALR relative to CALS tumours and spheroids. This is of interest as aspartate constitutes a pivotal component of the malate-aspartate shuttle, a process that enables efficient energy production in the cell (Wallace, 2012). A key role has recently emerged for glutamine-derived aspartate in the generation of reducing equivalents necessary for protection against oxidative stress (Son et al, 2013). Although such functions would fit with the demands for improved bioenergetics evident in the aggressive growth phenotype of CALR tumours and spheroids, further work is necessary to assess if the rise in aspartate (and alanine) observed here are linked to this process or other mechanisms.
Our data point to increased GPC as a metabolic biomarker of acquired EGFR TKI resistance in the CALR/CALS model that is independent of any growth environment or phenotype. This is of interest as choline metabolism is altered in cancer cells and both PC and GPC are increased with malignancy, not only as a result of increased cell proliferation but also because of a direct oncogenic protein regulation (Glunde et al, 2011). GPC levels were also increased in the sensitive CALS but not the resistant CALR cells following acute exposure to the EGFR TKI gefitinib, indicating that the processes leading to the rise in GPC are triggered by drug treatment and persist when the cells become chronically resistant to drug. An increase in GPC has been observed with various chemotherapeutic and targeted anti-cancer agents (Beloueche-Babari et al, 2010), thus it is unlikely to be specific to EGFR TKIs.
The activity of GPC-pd, the enzyme that cleaves GPC into choline and glycerol-3 phosphate, was significantly reduced in CALR relative to CALS cells. In the absence of significant differences in fatty acyl signals between the two cell lines, that is, no major changes in lipid breakdown, we conclude that inhibition of GPC-pd is likely to be the major cause for the elevation in GPC content in the drug-resistant CALR cells. Further work is necessary to establish the molecular and cellular mechanisms linking acquired resistance to EGFR TKIs in this model and the ensuing inhibition of GPC-pd.
Finally, to test if our metabolic signature of resistance is applicable to other HNSCC models, we tested a second sensitive/resistant cell line pair: PJS/PJR. We found that in this case acquired resistance to EGFR TKI was also associated with changes in choline metabolism (indicating altered membrane phospholipid turnover) but, unlike the CALR/CALS model, a reduction in GPC and PC levels was observed. The basis for this difference is unclear but may be related to differences in underlying drug resistance mechanisms, including polyploidy (previously linked to resistance to cytotoxic drugs (Puig et al, 2008; Shen et al, 2008)), which was observed in the PJ but not the CAL model. Differences in the anatomical site of origin of the cells (tongue for CALS/CALR and oral cavity (excluding tongue) for PJS/PJR) may also be involved. Alterations in GPC levels have previously been reported in human cancer cells exhibiting multidrug resistance but the direction of the change was cell line dependent and likely to reflect cellular processes accompanying the induction of the drug resistance phenotype (Kaplan et al, 1991), in agreement with our findings. Clearly, analysis of a larger number of isogenic sensitive/resistant cell line pairs is required to assess the general applicability of the metabolic signatures observed here, and unravel the mechanisms underlying them.
A key objective of our study was to identify metabolic characteristics of acquired resistance to EGFR TKIs that could be developed as non-invasive imaging biomarkers. The glycolytic phenotype observed in the tumours can be monitored with 18F-fluorodeoxyglucose (FDG)-PET, the gold-standard method for clinical metabolic imaging, as well as lactate-selective in vivo 1H NMR spectroscopic imaging, which have both been used clinically in HNSCC (Le et al, 2008; Jansen et al, 2012). The changes in GPC and total choline signal, as well as Cr+PCr observed with acquired resistance can also be monitored with standard in vivo 1H NMR spectroscopy techniques as shown in previous patient studies (including HNSCC) (King et al, 2010; Kim et al, 2011; Germuska et al, 2012; Jansen et al, 2012). Ex vivo analysis of tumour tissue may be particularly useful for monitoring metabolites not measurable by currently available imaging techniques, for example, aspartate and alanine as previously documented in clinical HNSCC samples (Somashekar et al, 2011). Longitudinal studies assessing the metabolic characteristics of human HNSCC and correlating them with patient outcome are, however, necessary to assess the utility of metabolic biomarkers for patient follow-up.
In summary, this study has revealed metabolic signatures associated with acquired EGFR TKI resistance that may be linked to the underlying drug resistance mechanisms as well as aggressive growth patterns in HNSCC models. The changes reported here can be monitored through in vivo as well as ex vivo examinations of patient tumours, highlighting their potential for translation to the clinic.
Acknowledgments
We thank staff at the Biological Services Unit, ICR, for animal care and maintenance and Dr Ian Titley and Ms Gowri Vijayaraghavan for help with flow cytometry analyses. This work was funded by Cancer Research UK (grants C1090/A16464 and C309/A8274). In addition, we acknowledge the support received from the Cancer Research UK and EPSRC Cancer Imaging Centre in association with the MRC and Department of Health (England) grant C1060/A10334, NHS funding to the NIHR Biomedical Research Centre and the Clinical Research Facility in Imaging, and Oracle Cancer Trust. MOL is an NIHR Senior Investigator.
The author declares no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on British Journal of Cancer website (http://www.nature.com/bjc)
Supplementary Material
References
- Agulnik M. New approaches to EGFR inhibition for locally advanced or metastatic squamous cell carcinoma of the head and neck (SCCHN) Med Oncol. 2012;29 (4:2481–2491. doi: 10.1007/s12032-012-0159-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang KK, Berkey BA, Tu X, Zhang HZ, Katz R, Hammond EH, Fu KK, Milas L. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res. 2002;62 (24:7350–7356. [PubMed] [Google Scholar]
- Bailey KM, Wojtkowiak JW, Hashim AI, Gillies RJ. Targeting the metabolic microenvironment of tumors. Adv Pharmacol. 2012;65:63–107. doi: 10.1016/B978-0-12-397927-8.00004-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beloueche-Babari M, Arunan V, Troy H, te Poele RH, te Fong AC, Jackson LE, Payne GS, Griffiths JR, Judson IR, Workman P, Leach MO, Chung YL. Histone deacetylase inhibition increases levels of choline kinase alpha and phosphocholine facilitating noninvasive imaging in human cancers. Cancer Res. 2012;72 (4:990–1000. doi: 10.1158/0008-5472.CAN-11-2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beloueche-Babari M, Chung YL, Al-Saffar NM, Falck-Miniotis M, Leach MO. Metabolic assessment of the action of targeted cancer therapeutics using magnetic resonance spectroscopy. Br J Cancer. 2010;102 (1:1–7. doi: 10.1038/sj.bjc.6605457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beloueche-Babari M, Jamin Y, Arunan V, Walker-Samuel S, Revill M, Smith PD, Halliday J, Waterton JC, Barjat H, Workman P, Leach MO, Robinson SP. Acute tumour response to the MEK1/2 inhibitor selumetinib (AZD6244, ARRY-142886) evaluated by non-invasive diffusion-weighted MRI. Br J Cancer. 2013;109 (6:1562–1569. doi: 10.1038/bjc.2013.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beloueche-Babari M, Workman P, Leach MO. Exploiting tumor metabolism for non-invasive imaging of the therapeutic activity of molecularly targeted anticancer agents. Cell Cycle. 2011;10:17. doi: 10.4161/cc.10.17.17192. [DOI] [PubMed] [Google Scholar]
- Box C, Mendiola M, Gowan S, Box GM, Valenti M, Brandon AD, Al-Lazikani B, Rogers SJ, Wilkins A, Harrington KJ, Eccles SA. A novel serum protein signature associated with resistance to epidermal growth factor receptor tyrosine kinase inhibitors in head and neck squamous cell carcinoma. Eur J Cancer. 2013;49 (11:2512–2521. doi: 10.1016/j.ejca.2013.03.011. [DOI] [PubMed] [Google Scholar]
- Box C, Zimmermann M, Eccles S. Molecular markers of response and resistance to EGFR inhibitors in head and neck cancers. Front Biosci. 2013;18:520–542. doi: 10.2741/4118. [DOI] [PubMed] [Google Scholar]
- Brahimi-Horn MC, Bellot G, Pouyssegur J. Hypoxia and energetic tumour metabolism. Curr Opin Genet Dev. 2011;21 (1:67–72. doi: 10.1016/j.gde.2010.10.006. [DOI] [PubMed] [Google Scholar]
- Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11 (2:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- Deberardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7 (1:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Gee HE, Camps C, Buffa FM, Patiar S, Winter SC, Betts G, Homer J, Corbridge R, Cox G, West CM, Ragoussis J, Harris AL. hsa-mir-210 is a marker of tumor hypoxia and a prognostic factor in head and neck cancer. Cancer. 2010;116 (9:2148–2158. doi: 10.1002/cncr.25009. [DOI] [PubMed] [Google Scholar]
- Germuska M, Tunariu N, Leach MO, Xu J, Payne GS. An evaluation of motion compensation strategies and repeatability for abdominal (1)H MR spectroscopy measurements in volunteer studies and clinical trials. NMR Biomed. 2012;25 (6:859–865. doi: 10.1002/nbm.1802. [DOI] [PubMed] [Google Scholar]
- Glunde K, Bhujwalla ZM, Ronen SM. Choline metabolism in malignant transformation. Nat Rev Cancer. 2011;11 (12:835–848. doi: 10.1038/nrc3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandis JR, Melhem MF, Gooding WE, Day R, Holst VA, Wagener MM, Drenning SD, Tweardy DJ. Levels of TGF-alpha and EGFR protein in head and neck squamous cell carcinoma and patient survival. J Natl Cancer Inst. 1998;90 (11:824–832. doi: 10.1093/jnci/90.11.824. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144 (5:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13 (10:714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- Iorio E, Ricci A, Bagnoli M, Pisanu ME, Castellano G, Di VM, Venturini E, Glunde K, Bhujwalla ZM, Mezzanzanica D, Canevari S, Podo F. Activation of phosphatidylcholine cycle enzymes in human epithelial ovarian cancer cells. Cancer Res. 2010;70 (5:2126–2135. doi: 10.1158/0008-5472.CAN-09-3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen JF, Schoder H, Lee NY, Stambuk HE, Wang Y, Fury MG, Patel SG, Pfister DG, Shah JP, Koutcher JA, Shukla-Dave A. Tumor metabolism and perfusion in head and neck squamous cell carcinoma: pretreatment multimodality imaging with 1H magnetic resonance spectroscopy, dynamic contrast-enhanced MRI, and [18F]FDG-PET. Int J Radiat Oncol Biol Phys. 2012;82 (1:299–307. doi: 10.1016/j.ijrobp.2010.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan O, Jaroszewski JW, Clarke R, Fairchild CR, Schoenlein P, Goldenberg S, Gottesman MM, Cohen JS. The multidrug resistance phenotype: 31P nuclear magnetic resonance characterization and 2-deoxyglucose toxicity. Cancer Res. 1991;51 (6:1638–1644. [PubMed] [Google Scholar]
- Kim H, Catana C, Ratai EM, Andronesi OC, Jennings DL, Batchelor TT, Jain RK, Sorensen AG. Serial magnetic resonance spectroscopy reveals a direct metabolic effect of cediranib in glioblastoma. Cancer Res. 2011;71 (11:3745–3752. doi: 10.1158/0008-5472.CAN-10-2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AD, Yeung DK, Yu KH, Mo FK, Hu CW, Bhatia KS, Tse GM, Vlantis AC, Wong JK, Ahuja AT. Monitoring of treatment response after chemoradiotherapy for head and neck cancer using in vivo 1H MR spectroscopy. Eur Radiol. 2010;20 (1:165–172. doi: 10.1007/s00330-009-1531-2. [DOI] [PubMed] [Google Scholar]
- Koukourakis MI, Bentzen SM, Giatromanolaki A, Wilson GD, Daley FM, Saunders MI, Dische S, Sivridis E, Harris AL. Endogenous markers of two separate hypoxia response pathways (hypoxia inducible factor 2 alpha and carbonic anhydrase 9) are associated with radiotherapy failure in head and neck cancer patients recruited in the CHART randomized trial. J Clin Oncol. 2006;24 (5:727–735. doi: 10.1200/JCO.2005.02.7474. [DOI] [PubMed] [Google Scholar]
- Le QT, Koong A, Lieskovsky YY, Narasimhan B, Graves E, Pinto H, Brown JM, Spielman D. In vivo 1H magnetic resonance spectroscopy of lactate in patients with stage IV head and neck squamous cell carcinoma. Int J Radiat Oncol Biol Phys. 2008;71 (4:1151–1157. doi: 10.1016/j.ijrobp.2007.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovly CM, Shaw AT. Molecular pathways: resistance to kinase inhibitors and implications for therapeutic strategies. Clin Cancer Res. 2014;20 (9:2249–2256. doi: 10.1158/1078-0432.CCR-13-1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig PE, Guilly MN, Bouchot A, Droin N, Cathelin D, Bouyer F, Favier L, Ghiringhelli F, Kroemer G, Solary E, Martin F, Chauffert B. Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy. Cell Biol Int. 2008;32 (9:1031–1043. doi: 10.1016/j.cellbi.2008.04.021. [DOI] [PubMed] [Google Scholar]
- Rogers SJ, Box C, Chambers P, Barbachano Y, Nutting CM, Rhys-Evans P, Workman P, Harrington KJ, Eccles SA. Determinants of response to epidermal growth factor receptor tyrosine kinase inhibition in squamous cell carcinoma of the head and neck. J Pathol. 2009;218 (1:122–130. doi: 10.1002/path.2515. [DOI] [PubMed] [Google Scholar]
- Shen H, Moran DM, Maki CG. Transient nutlin-3a treatment promotes endoreduplication and the generation of therapy-resistant tetraploid cells. Cancer Res. 2008;68 (20:8260–8268. doi: 10.1158/0008-5472.CAN-08-1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somashekar BS, Kamarajan P, Danciu T, Kapila YL, Chinnaiyan AM, Rajendiran TM, Ramamoorthy A. Magic angle spinning NMR-based metabolic profiling of head and neck squamous cell carcinoma tissues. J proteome Res. 2011;10 (11:5232–5241. doi: 10.1021/pr200800w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, Kang Y, Fleming JB, Bardeesy N, Asara JM, Haigis MC, DePinho RA, Cantley LC, Kimmelman AC. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496 (7443:101–105. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324 (5930:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verschoor ML, Wilson LA, Singh G. Mechanisms associated with mitochondrial-generated reactive oxygen species in cancer. Can J Physiol Pharmacol. 2010;88 (3:204–219. doi: 10.1139/Y09-135. [DOI] [PubMed] [Google Scholar]
- Vinci M, Box C, Zimmermann M, Eccles SA. Tumor spheroid-based migration assays for evaluation of therapeutic agents. Methods Mol Biol. 2013;986:253–266. doi: 10.1007/978-1-62703-311-4_16. [DOI] [PubMed] [Google Scholar]
- Vinci M, Gowan S, Boxall F, Patterson L, Zimmermann M, Court W, Lomas C, Mendiola M, Hardisson D, Eccles SA. Advances in establishment and analysis of three-dimensional tumor spheroid-based functional assays for target validation and drug evaluation. BMC Biol. 2012;10:29. doi: 10.1186/1741-7007-10-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer. 2009;9 (10:691–700. doi: 10.1038/nrc2715. [DOI] [PubMed] [Google Scholar]
- Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, Kehoe SM, Johannessen CM, Macconaill LE, Hahn WC, Meyerson M, Garraway LA. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29 (22:3085–3096. doi: 10.1200/JCO.2010.33.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12 (10:685–698. doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21 (3:297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman P, Aboagye EO, Balkwill F, Balmain A, Bruder G, Chaplin DJ, Double JA, Everitt J, Farningham DA, Glennie MJ, Kelland LR, Robinson V, Stratford IJ, Tozer GM, Watson S, Wedge SR, Eccles SA. Guidelines for the welfare and use of animals in cancer research. Br J Cancer. 2010;102 (11:1555–1577. doi: 10.1038/sj.bjc.6605642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Tozzi F, Chen J, Fan F, Xia L, Wang J, Gao G, Zhang A, Xia X, Brasher H, Widger W, Ellis LM, Weihua Z. Intracellular ATP levels are a pivotal determinant of chemoresistance in colon cancer cells. Cancer Res. 2012;72 (1:304–314. doi: 10.1158/0008-5472.CAN-11-1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.