

Abstract
Influenza viruses cause acute respiratory disease of great importance to public health. Alveolar type II (ATII) respiratory epithelial cells are central to normal lung function and are a site of influenza A virus replication in the distal lung. However, the consequences of infection for ATII cell function are poorly understood. To determine the impact of influenza infection on ATII cells we used C57BL/6-congenic SP-CGFP mice that express green fluorescent protein (GFP) under the control of the surfactant protein-C (SP-C) promoter, which is only active in ATII cells. Most cells isolated from the lungs of uninfected SP-CGFP mice were GFP+ but did not express the alveolar type I (ATI) antigen podoplanin (PODO). ATII cells were also EpCAM+ and α2,3-linked sialosaccharide+. Infection with influenza A/WSN/33 virus caused severe hypoxemia and pulmonary edema. This was accompanied by loss of whole lung GFP fluorescence, reduced ATII cell yields, increased ATII cell apoptosis, reduced SP-C gene and protein expression in ATII cell lysates, and increased PODO gene and protein levels. Flow cytometry indicated that infection decreased GFP+/PODO− cells and increased GFP−/PODO+ and GFP−/PODO− cells. Very few GFP+/PODO+ cells were detectable. Finally, infection resulted in a significant decline in EpCAM expression by PODO+ cells, but had limited effects on α2,3-linked sialosaccharides. Our findings indicate that influenza infection results in a progressive differentiation of ATII cells into ATI-like cells, possibly via an SP-C−/PODO− intermediate, to replace dying or dead ATI cells. However, impaired SP-C synthesis is likely to contribute significantly to reduced lung compliance in infected mice.
Keywords: surfactant protein-C, podoplanin, alveolar type I cell
infection with influenza viruses results in an acute respiratory disease that can range in severity from mild to fatal (45). In temperate climates, epidemics caused by seasonal influenza virus strains occur every fall and winter. Although limited information is available, it has been estimated that seasonal influenza infections result in 12 influenza-related deaths per 100,000 persons per year in the United States, the majority of which occur in the elderly (18, 41). Furthermore, the 2009–2010 H1N1 “swine flu” influenza A virus pandemic resulted in an estimated 14,800 excess deaths in the United States (9). Unfortunately, current vaccination uptake rates and efficacy are insufficient to provide “herd immunity” to either seasonal or pandemic influenza viruses, and the value of antiviral drugs has recently been called into question (11, 27, 39). It has been estimated that a mild pandemic similar to that of 1968 would kill 1.4 million people and cost approximately $330 billion [or 0.8% of global gross domestic product (GDP)] in lost economic output (38). Were a pandemic as severe as that of 1918–1919 to occur, more than 140 million people would die, and the world's GDP would suffer a loss of more than $4 trillion. Hence, there is an urgent need for better understanding of influenza pathogenesis to develop new influenza therapeutics.
The primary physiological function of the lungs is gas exchange. Gas exchange occurs within the respiratory zone of the lung, which is lined by a single layer of epithelial cells and an underlying plexus of pulmonary capillaries. The epithelial layer of this gas exchange surface is composed of two cell types [alveolar type I (ATI) and II (ATII) cells] that are covered with a thin layer of serous fluid and an overlying film of surfactant. Importantly, alveolar epithelial cells are the primary site of influenza A virus infection and replication in the distal lung and participate in the innate immune response to this virus (22, 47, 59).
Although ATI cells comprise only around 10% of the cells in the lung, they cover >98% of the alveolar surface area, with the remaining 2% being made up of ATII cells (46). ATI cells are large thin squamous epithelial cells that have been traditionally viewed as primarily serving in a structural capacity, although recent studies suggest that they may play an important role in alveolar physiology (15). Podoplanin (PODO) has been identified as an ATI epithelial cell-specific antigen in the lung, although its function in these cells is unknown (15). ATII cells are small cuboidal cells that synthesize, secrete, and recycle pulmonary surfactant lipids. They also produce four primary proteins that have been designated surfactant proteins (SP)-A, -B, -C, and -D (61). In addition, ATII cells regulate the depth of the alveolar lining fluid layer by active vectorial ion transport and serve as progenitors for ATI cells (13, 34). SP-C is only expressed by ATII cells and can therefore be used as an ATII cell-specific marker.
Despite being central to normal lung function and influenza A virus replication in the distal lung, the consequences of infection for ATII cell function and differentiation state are currently poorly understood. The goal of the current study was to determine the impact of influenza infection on ATII cells using C57BL/6-congenic SP-CGFP mice that express green fluorescent protein (GFP) under the control of the SP-C gene promoter, which is only active in ATII cells (17, 33).
MATERIALS AND METHODS
Animals.
Pathogen-free C57BL/6AnNCr mice (Mus musculus) were purchased from the National Cancer Institute (Frederick, MD). C57BL/6-congenic mice that express GFP under the control of the SP-C promoter (SP-CGFP mice) were bred in-house from founders that were kindly provided by the late Dr. Joe Rae Wright (Duke University Medical Center, Durham, NC) (33). Mice were maintained in standard ventilated racks and provided with food and water ad libitum, as well as appropriate environmental enrichment with Nestlets (Ancare, Bellmore, NY). All animal procedures were performed in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by The Ohio State University Institutional Animal Care and Use Committee. Ethical considerations precluded performance of survival studies, and every effort was made to minimize animal suffering.
Preparation of viral inoculum.
All studies used egg-grown mouse-adapted influenza A/WSN/33 (H1N1) virus. Absence of contamination with Mycoplasma pulmonis was confirmed by PCR (Charles River Research Animal Diagnostic Services, Wilmington, MA). Absence of endotoxin contamination was confirmed by a standard Limulus amebocyte assay (Lonza, Basel, Switzerland).
Mouse inoculation.
Pathogen-free, 8- to 12-wk-old SP-CGFP mice of either gender were anesthetized by intraperitoneal injection of ketamine (8.7 mg/kg)/xylazine (1.3 mg/kg), individually marked, and then inoculated intranasally with 10,000 plaque-forming units (pfu)/mouse of H1N1 virus in 50 μl PBS/0.1% BSA, as in our previous studies. In our hands, this inoculum induces hypoxemia and lung dysfunction in wild-type mice by 2 days postinfection (dpi), and results in 100% mortality by 8 dpi (median time to death: 7 days), but does not infect the brain (2, 3, 62).
Conscious mice were weighed every other day following infection, and carotid arterial O2 saturation was recorded by pulse oximetry, as in our previous studies (1, 2). Data for each experimental group were derived from at least three independent infections.
Lung wet-to-dry weight ratio.
Lung wet-to-dry weight ratio was measured as previously described (2). Briefly, mice were killed by intraperitoneal injection of ketamine (87 mg/kg)/xylazine (13 mg/kg) and then exsanguinated. Both lungs were removed, weighed, and dried in an oven at 55°C for 3 days. After being dried, the lungs were weighed again. Wet-to-dry weight ratio was then calculated as an index of intrapulmonary fluid accumulation, without correction for blood content.
Measurement of viral titers.
Viral titers were determined from serial dilutions of lung homogenates by a fluorescent-focus assay in NY3 fibroblasts (derived from STAT1−/− mice), as previously described (24).
Whole organ imaging.
Immediately before imaging, mice were killed as above. Both lungs were removed by careful dissection, and total lung GFP fluorescence was detected using the In Vivo Imaging System (IVIS)-200 bioluminescent imaging system (Perkin Elmer, Waltham, MA).
ATII cell isolation.
ATII cells were isolated from SP-CGFP mice by a standard lung digestion protocol (12). Briefly, mice were killed, the pulmonary artery was cannulated, and the lungs were perfused with normal saline in situ to flush out blood. The trachea was cannulated, and 2 ml dispase II (5 U/ml in PBS; Becton-Dickinson, San Jose, CA) were injected in the lungs followed by 0.3 ml warmed low-melting-point agarose (1% in PBS) to prevent the isolation of Clara cells and upper airway epithelial cells. The lungs were cooled on ice, dissected free, rinsed with saline, and placed in 5 ml dispase to digest at room temperature for 60 min with gentle rocking. Pancreatic DNase (0.01% in DMEM; Sigma-Aldrich) was added for the final 5 min of incubation. Lung tissue was teased apart, and the resulting cell suspension was filtered sequentially through 100-, 40-, and 21-μm sterile nylon meshes. Leukocytes were removed by panning with rat polyclonal anti-murine CD45 and anti-murine CD16/CD32 antibodies (both Becton-Dickinson) for 2 h at 37°C. Nonadherent cells were collected, pelleted by centrifugation, resuspended in normal saline, and counted using a hemocytometer. Purity of isolated ATII cell preparations was determined by visualization of lamellar bodies in modified Papanicolaou-stained cytospins.
Western blot.
ATII cell preparations were homogenized in Cell Lysis buffer 9803 (Cell Signaling Technology, Danvers, MA), using a 30-gauge needle and syringe. Lysates were centrifuged to remove debris, and 30 μg of protein per sample were loaded on a 4–12% gradient polyacrylamide gel. Western blotting was performed by a standard protocol. Active caspase-3 was detected using a rabbit polyclonal antibody (ab2302; Abcam, San Francisco, CA), as were SP-C (AB3786; Millipore, Billerica, MA) and GAPDH (sc-25778; Santa Cruz Biotechnology, Santa Cruz, CA). PODO was detected using a rat monoclonal antibody (Novus Biologicals, Littleton, CO). H1N1 influenza nucleoprotein (NP) was detected using a rabbit polyclonal antibody (GenScript, Piscataway, NJ). Bound primary antibodies were detected using horseradish peroxidase-conjugated anti-rabbit IgG (Cell Signaling Technology) or anti-rat IgG (Novus Biologicals), as appropriate, and developed using ECL Western Blotting Substrate (Thermo Scientific, Rockford, IL). Densitometry of immunoreactive bands was performed on scanned film images using ImageJ software. Data were normalized to GAPDH.
qRT-PCR.
Freshly isolated ATII cells were lysed, and total RNA was extracted using the RNeasy system (Qiagen, Alameda, CA). Total RNA was then reverse transcribed into cDNA using the high-capacity cDNA reverse transcription kit (Applied Biosystems, Grand Island, NY). qRT-PCR amplification of cDNA was performed using the TaqMan Gene Expression system (Applied Biosystems). Expression of pro-sp-c and podo genes was determined by the ΔΔCt method and normalized to the endogenous control 18s rrna.
Flow cytometric analysis of ATII cells.
Isolated ATII cells were fixed in suspension in 4% paraformaldehyde and then immunostained using rabbit polyclonal antibodies to PODO (as above) and epithelial cell adhesion molecule (EpCAM/CD326; BioLegend, San Diego, CA), which were fluorochrome conjugated to allophycocyanin and phycoerythrin (PE), respectively, using LYNX rapid antibody conjugation kits (AbD Serotec, Raleigh, NC). In other studies, cells were stained with biotinylated Sambucus nigra lectin (SNA; Vector Laboratories, Burlingame, CA) and FITC-conjugated Maackia amurensis lectin-II (MAL-II; EY Laboratories, San Mateo, CA), followed by streptavidin-PE (BioLegend), as in previous studies (6). ATII cells immunostained with fluorochrome-labeled isotype control antibodies were used to define the threshold for positive fluorescence. Ten thousand fluorescence events were collected per sample using a FACSCalibur flow cytometer (Becton-Dickinson). Data were analyzed using FlowJo (version 10.0) software (TreeStar, Ashland, OR).
Statistical analyses.
Descriptive statistics were calculated using Instat 3.05 (GraphPad Software, San Diego, CA). Gaussian data distribution was verified by the method of Kolmogorov and Smirnov. Differences between group means were analyzed by ANOVA, with Tukey-Kramer multiple-comparison posttests. P < 0.05 was considered statistically significant. All data are presented as means ± SE.
RESULTS
Infection of C57BL/6-congenic SP-CGFP mice with influenza A/WSN/33 resulted in severe cachexia, hypoxemia, and pulmonary edema. Uninfected C57BL/6-congenic SP-CGFP mice were phenotypically normal. Following intranasal infection with H1N1 influenza A/WSN/33 (10,000 pfu/mouse), body weight decreased significantly by 2 dpi, and SP-CGFP mice lost >30% of starting body weight by 6 dpi (Fig. 1A). Mice were severely hypoxemic at 6 dpi (Fig. 1B), and significant pulmonary edema was found at both 2 and 6 dpi (Fig. 1C). Lung homogenate influenza viral titers did not differ significantly between 2 and 6 dpi (Fig. 1D). All data, which are consistent with development of severe acute lung injury (ALI), were comparable to those we reported previously in C57BL/6 mice infected with the same inoculum (1–3).
Fig. 1.

Infection of SP-CGFP mice with influenza A/WSN/33 (H1N1) resulted in severe cachexia, hypoxemia, and pulmonary edema. Effect of intranasal infection of C57BL/6-congenic SP-CGFP mice with H1N1 influenza A/WSN/33 (10,000 FFU/mouse) on: body weight (BWT; %change from day 0; n > 10 mice/group, A); carotid arterial oxygen saturation (SaO2; n > 10/group, B); lung water content (wet-to-dry weight ratio; n = 5–6/group, C); and lung homogenate viral titers (log FFU/g; n = 5/group, D). SP-C, surfactant protein-C; GFP, green fluorescent protein; FFU, focus-forming units. *P < 0.05 and #P < 0.001 vs. uninfected mice. ¶P < 0.001 vs. 2 days postinfection (dpi). Data are presented as means ± SE.
Influenza A virus infection reduced GFP fluorescence in lungs of SP-CGFP mice.
C57BL/6-congenic SP-CGFP mice express GFP under the control of the SP-C gene promoter, which is only active in ATII cells (17, 33). Hence, GFP serves as a specific ATII cell marker in these mice, and changes in GFP expression correlate to alterations in SP-C production. Unlike lungs from uninfected C57BL/6 mice, isolated whole lungs from uninfected SP-CGFP mice exhibited high GFP fluorescence when imaged by IVIS (Fig. 2A). However, no fluorescence was detected in the trachea or primary bronchi. GFP fluorescence intensity decreased modestly (but not significantly) at 2 dpi and was reduced by ∼50% at 6 dpi (Fig. 2B).
Fig. 2.
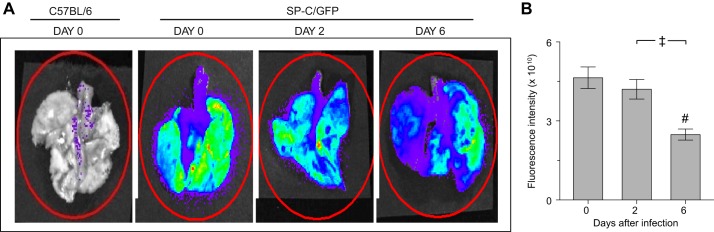
Influenza A virus infection reduced GFP fluorescence in lungs of SP-CGFP mice. Representative false-colored In Vivo Imaging System images of lungs from one uninfected C57BL/6 mouse, one uninfected SP-CGFP mouse, and one SP-CGFP mouse each at 2 and 6 dpi (A) and effect of infection with influenza A/WSN/33 virus on GFP fluorescence intensity in isolated lungs from SP-CGFP mice (n = 10/group, B). Red oval, area over which fluorescence was quantified. #P < 0.001 vs. uninfected mice. ‡P < 0.005 vs. 2 dpi. Data are presented as means ± SE.
ATII cell numbers decreased and ATII cell caspase-3 activation increased in SP-CGFP mice following influenza A virus infection.
To verify that infection-induced reductions in GFP expression at the whole lung level specifically resulted from changes in ATII cell SP-C expression or ATII cell loss by apoptosis, ATII cells were isolated from lungs of SP-CGFP mice by a standard digestion protocol. Characteristic Papanicolau-positive lamellar bodies (refractile inclusions containing stored surfactant lipids) were visible in cytospins of isolated ATII cells (Fig. 3A) (14). As in previous studies, 90–95% of cells in preparations from uninfected SP-CGFP mice were Papanicolau-positive (data not shown).
Fig. 3.

Alveolar type II (ATII) cell numbers decreased and ATII cell caspase-3 activation increased in SP-CGFP mice following influenza A virus infection. A: representative photomicrograph of Papanicolau-stained ATII cells isolated from an uninfected SP-CGFP mouse. Arrows indicate characteristic Papanicolau-positive lamellar bodies within the cell cytoplasm (no adjustments were made to original image other than conversion from color to grayscale). B: effect of infection with influenza A/WSN/33 virus on ATII cell yield/mouse (n = 7–11/group). C: Western blot for activated caspase-3 (A-CASP-3) protein in homogenates of ATII cells isolated from uninfected and influenza A/WSN/33 virus-infected SP-CGFP mice (no adjustments were made to original images other than conversion from color to grayscale). D: effect of infection with influenza A/WSN/33 virus on relative expression of A-CASP-3 in ATII cell homogenates (n = 3/group; normalized to GAPDH). ND, not detected. #P < 0.001 vs. uninfected mice. ¶P < 0.001 vs. 2 dpi. Data are presented as means ± SE.
The ATII cell isolation procedure yielded ∼8 × 106 ATII cells per uninfected SP-CGFP mouse (Fig. 3B). A modest but nonsignificant decrease in ATII cell yield per mouse was found at 2 dpi. However, ATII cell yields per mouse were decreased by >50% at 6 dpi. These data confirm results of our IVIS studies that showed a modest decrease in whole lung fluorescence at 2 dpi and a 50% reduction in fluorescence at 6 dpi.
To determine whether loss of ATII cells at 6 dpi might be a consequence of increased ATII cell apoptosis, levels of activated caspase-3 were measured by Western blot. No caspase-3 activation was detectable in ATII cells from uninfected mice or at 2 dpi (Fig. 3, C and D). However, high levels of active caspase-3 were present in ATII cell lysates at 6 dpi.
Infection with influenza A virus reduced expression of SP-C protein and increased expression of PODO protein by ATII cells isolated from SP-CGFP mice.
During epithelial repair following lung injury, SP-C-positive ATII cells can differentiate into ATI cells, which express PODO (5, 15). To establish whether influenza A virus infection had similar consequences, ATII cells isolated from SP-CGFP mice were subjected to Western blot analysis for both proteins (Fig. 4A). ATII cell expression of SP-C protein decreased significantly at 2 dpi, and SP-C was almost undetectable at 6 dpi (Fig. 4, A and B). This decrease in SP-C levels was accompanied by progressive reductions in expression of SP-A and SP-D, which are synthesized by ATII cells in the lung (Fig. 4A). Interestingly, levels of the supposedly ATI cell-specific protein PODO increased significantly at 2 dpi, and a further significant increase was observed at 6 dpi (Fig. 4, A and C). Influenza A virus NP protein could not be detected in freshly isolated ATII cells from uninfected mice but was present at 2 dpi (Fig. 4, A and D). NP expression levels were significantly higher at 6 dpi.
Fig. 4.

Infection with influenza A virus reduced expression of SP-C protein and increased expression of podoplanin (PODO) protein by ATII cells isolated from SP-CGFP mice. A: composite Western blot for SP-C, surfactant protein-A (SP-A), surfactant protein-D (SP-D), and PODO proteins and H1N1 influenza A virus nucleoprotein (NP) in homogenates of ATII cells isolated from uninfected and influenza A/WSN/33 virus-infected SP-CGFP mice (no adjustments were made to original images other than conversion from color to grayscale). B: effect of infection with influenza A/WSN/33 virus on relative expression of SP-C protein in ATII cell homogenates. C: effect of infection with influenza A/WSN/33 virus on relative expression of PODO protein in ATII cell homogenates. D: effect of infection with influenza A/WSN/33 virus on relative NP expression in ATII cell homogenates; n = 3/group; normalized to GAPDH. *P < 0.05 and #P < 0.001 vs. uninfected mice. †P < 0.05 and ¶P < 0.001 vs. 2 dpi. Data are presented as means ± SE.
Influenza A virus infection resulted in decreased expression of pro-sp-c and increased expression of podo genes by ATII cells isolated from SP-CGFP mice.
To determine whether alterations in SP-C and PODO protein expression were a consequence of reduced gene expression, mRNA was extracted from isolated SP-CGFP mouse ATII cells and subjected to qRT-PCR analysis. At 2 dpi, ATII cell pro-sp-c mRNA was reduced approximately fourfold relative to uninfected mice (Fig. 5A). At 6 dpi, pro-sp-c gene expression was reduced a further 40-fold. In contrast, podo mRNA only modestly increased at 2 dpi but was upregulated by approximately fourfold at 6 dpi relative to ATII cells isolated from uninfected mice (Fig. 5B). ATII cell 18S ribosomal RNA gene expression did not alter over the course of infection, indicating that changes in pro-sp-c and podo gene expression were not a consequence of alterations in expression of the gene to which they were normalized (Fig. 5C).
Fig. 5.
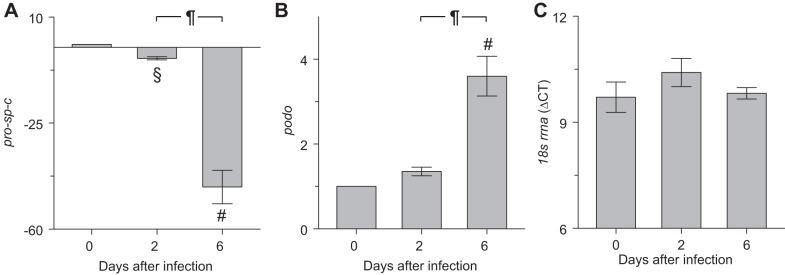
Influenza A virus infection resulted in decreased expression of pro-sp-c and increased expression of podo genes by ATII cells isolated from SP-CGFP mice. Effect of infection with influenza A/WSN/33 virus for 6 days on ATII cell expression of genes for: SP-C (pro-sp-C, A), podo (B), and 18S ribosomal RNA (18s rrna, C); n = 3–5/group; pro-sp-c and podo expression normalized to expression of the same gene in ATII cells from uninfected mice. §P < 0.005 and #P < 0.001 vs. uninfected mice. ¶P < 0.001 vs. 2 dpi. Data are presented as means ± SE.
Influenza A virus infection led to mutually exclusive changes in expression of SP-C and PODO by individual ATII cells isolated from SP-CGFP mice.
To determine whether ATII cell expression of SP-C and PODO was mutually exclusive, freshly isolated ATII cells from SP-CGFP mice were subjected to analysis by flow cytometry. ATII cells were identified by their large size (high forward scatter) and granularity (high side scatter). An ATII cell gate was established based on these characteristics in uninfected mice (Fig. 6A). This same gate was used to analyze ATII cells in all subsequent flow cytometric studies of cells isolated from influenza A virus-infected mice, to ensure that we would capture infection-induced changes in PODO, EpCAM, and sialic acid expression by isolated “ATII-like” cells even as SP-C expression declined.
Fig. 6.

Influenza A virus infection led to mutually exclusive changes in expression of SP-C and PODO by individual ATII cells isolated from SP-CGFP mice. A: representative flow cytometric quadrant plot showing the gating strategy for ATII cell flow cytometry studies, based on the forward- and side-scatter characteristics of ATII cells isolated from uninfected SP-CGFP mice. B: effect of infection with influenza A/WSN/33 virus on percentage of ATII cells that express either GFP or PODO, but not both. C: effect of infection with influenza A/WSN/33 virus on percentage of ATII cells that do not express either GFP or PODO and percentage that coexpress both proteins. D: effect of infection with influenza A/WSN/33 virus on ATII cell mean channel fluorescence (MCF) for GFP and PODO; n = 3 uninfected mice, 4 mice at 2 dpi, and 7 mice at 6 dpi. §P < 0.005 and #P < 0.001 vs. uninfected mice for each group. Data are presented as means ± SE.
In uninfected mice, the majority of cells in the ATII cell gate expressed GFP but not PODO, although ∼10% expressed PODO but not GFP, and a further 10% expressed neither antigen (Fig. 6, B and C). Importantly, almost no ATII cells coexpressed GFP and PODO. Following infection, the percentage of GFP+/PODO− cells within the ATII cell gate progressively declined, whereas GFP−/PODO+ cells increased. The percentage of cells that were GFP−/PODO− also increased over the course of infection, whereas the percentage of GFP+/PODO+ cells significantly decreased at 6 dpi. However, given the small size of the latter population, this effect was unlikely to be of biological significance. Finally, we found that GFP expression per GFP+ cells [GFP mean channel fluorescence (MCF)] declined significantly over the course of infection (Fig. 6D). In contrast, PODO MCF did not change.
EpCAM expression by ATII cells isolated from SP-CGFP mice decreased following influenza A virus infection.
Because we had found that influenza A/WSN/33 virus infection significantly altered expression of GFP (hence, SP-C) and PODO, which are generally viewed as markers of ATII and ATI cells, respectively, we wished to characterize the impact of infection on expression of EpCAM (CD326), which has been described as a non-cell-type-specific epithelial cell antigen (40). Approximately 90% of total gated cells from uninfected SP-CGFP mice expressed EpCAM (Fig. 7A). However, at 2 dpi, when total cell yields had not declined, both the percentage of EpCAM+ cells and EpCAM MCF was significantly reduced (Fig. 7, A and B). At 6 dpi, <20% of cells within the ATII cell gate expressed EpCAM, although EpCAM MCF was no longer reduced. Interestingly, while all GFP+ cells from uninfected mice were also EpCAM+, only ∼50% of PODO+ cells carried this marker before infection (Fig. 7C). Moreover, the overall decline in the percentage of EpCAM+ cells in the ATII cell gate following infection resulted primarily from a reduction in EpCAM expression by PODO+ cells.
Fig. 7.
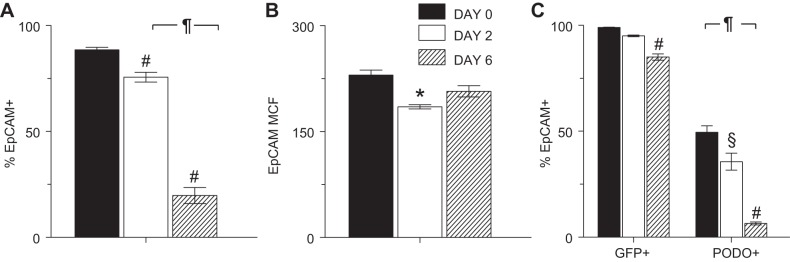
EpCAM expression by ATII cells isolated from SP-CGFP mice decreased following influenza A virus infection. Effect of infection with influenza A/WSN/33 virus on: percentage expression of EpCAM in all ATII cells (A), EpCAM MCF in all ATII cells (B), and percentage expression of EpCAM in GFP+ and PODO+ cells within the ATII cell gate (C); n = 3 uninfected mice, 4 mice at 2 dpi, and 7 mice at 6 dpi. Samples gated as in Fig. 6A. *P < 0.05, §P < 0.005, and #P < 0.001 vs. uninfected mice for each group. ¶P < 0.001 vs. 2 dpi. Data are presented as means ± SE.
Influenza A virus infection altered expression of α2,3-linked sialic acid moieties by ATII cells isolated from SP-CGFP mice.
ATI cells generally express α2,6-linked sialosaccharides, whereas ATII cells predominately express α2,3-linked sialosaccharides (6, 22, 65). Because we found significant alterations in expression of SP-C and PODO following infection, we wished to evaluate the impact of changes in these ATII and ATI cell markers on sialosaccharide expression. Before infection, we were unable to detect any staining with PE-conjugated SNA (which binds preferentially to α2,6-linked sialosaccharides) to cells within the ATII cell gate (data not shown). Likewise, despite increased PODO expression following infection, no binding to SNA was found. In uninfected SP-CGFP mice, ∼80% of cells within the ATII cell gate were labeled by MAL-II, which binds preferentially to α2,3-linked sialosaccharides (Fig. 8A). At 2 dpi, the percentage of MAL-II+ ATII cells did not change. However, MAL-II MCF decreased by 45% (Fig. 8B). At 6 dpi, the percentage of MAL-II+ cells declined, although this change was not statistically significant. In contrast, MAL-II MCF returned to preinfection levels.
Fig. 8.
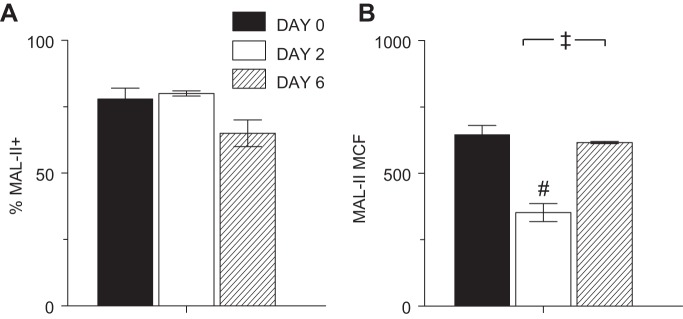
Influenza A virus infection altered expression of α2,3-linked sialic acid moieties by ATII cells isolated from SP-CGFP mice. Effect of infection with influenza A/WSN/33 virus on: percentage of Maackia amurensis lectin II+ (MAL-II+) ATII cells (A) and MCF of MAL-II+ ATII cells (B); n = 4 uninfected mice, 4 mice at 2 dpi, and 3 mice at 6 dpi. Samples gated as in Fig. 6A. #P < 0.001 vs. uninfected mice. ‡P < 0.005 vs. 2 dpi. Data are presented as means ± SE.
DISCUSSION
ATII cells are small cuboidal cells that are known to be essential to normal lung function. ATII cells synthesize, secrete, and recycle pulmonary surfactant lipids and proteins, which contribute to the maintenance of low intra-alveolar surface tension and thereby facilitate ventilation (61). They also regulate the depth of the alveolar lining fluid layer by active ion transport, participate in lung immune responses (10), and serve as progenitors for ATI cells (13, 34, 52). In addition, ATII cells are a primary site of influenza A virus replication in the distal lung and a central component of the innate antiviral immune response to infection (23). We have shown previously that infection of mice with an acutely lethal dose of mouse-adapted H1N1 induces ALI as early as 2 dpi without infecting brain tissue (3, 49). It also results in significant inhibition of alveolar fluid clearance, which is primarily mediated by ATII cell active ion transport (35, 62). This reduction in alveolar fluid clearance contributes significantly to the development of pulmonary edema and ALI in influenza A virus-infected mice (1–3). However, the effects of infection on other aspects of ATII cell function and their contributions to influenza pathogenesis have not been defined in detail.
In the current study, we found that the majority of ATII cells isolated from the lungs of uninfected SP-CGFP mice by a standard lung digestion protocol were GFP+/PODO− and expressed high levels of EpCAM and α2,3-linked sialosaccharides. As in our prior studies using C57BL/6 mice (1–3), infection of C57BL/6-congenic SP-CGFP mice with A/WSN/33 resulted in severe hypoxemia and pulmonary edema by 6 dpi, both of which are consistent with development of ALI. This was accompanied by loss of whole lung GFP fluorescence and a very significant reduction in the total number of ATII cells that could be isolated from the lung. Infection also resulted in increased ATII cell apoptosis, reduced sp-c gene and SP-A, SP-C, and SP-D protein expression in ATII cell lysates, and increased PODO gene and protein levels. These effects were also most pronounced at 6 dpi. At the single cell level, we found a progressive decrease in GFP+/PODO− cells, increased GFP−/PODO+ cells, and increased GFP−/PODO− cells by flow cytometry. Very few GFP+/PODO+ cells were present either before or after infection. Finally, infection resulted in a very significant decline in EpCAM expression by PODO+ cells at 6 dpi, but had limited effects on α2,3-linked sialosaccharide levels.
Currently, little information is available regarding the specific impact of acutely lethal influenza A virus infection on ATI and ATII cell phenotypes. Earlier in vitro studies showed that influenza A/PR/8 (H1N1) infection of differentiated human ATII cell cultures did not alter ATII cell differentiation and SP-C gene and protein expression (59). Histopathological evidence of ATII cell hyperplasia was found in mice infected with both influenza A/PR/8 and A/WSN/33 (32, 37), but no reduction in SP-C and PODO gene and protein expression occurred in A/PR/8-infected mice unless the alveolar macrophage response to infection was blocked by treatment with a neutralizing antibody to macrophage chemoattractant protein-1 (37). In other studies, sublethal A/PR/8 infection was shown to result in histopathological evidence of ATII cell loss by 3 dpi, which increased in severity until 7 dpi and then recovered (66). This was accompanied by loss of sp-c gene and SP-C and PODO protein expression in areas of alveolar destruction (19, 30, 68). Interestingly, downregulation of sp-c gene expression may have been restricted to infected ATII cells (19). We found that infection with A/WSN/33 for 6 days not only reduced total cell yields from the ATII cell isolation procedure by >50% but also induced a progressive shift from the ATII cell phenotype (GFP+/PODO−) toward an ATI-like (GFP−/PODO+) phenotype. This was accompanied by a progressive increase in GFP−/PODO− but not GFP+/PODO+ cells. To our knowledge, these effects of influenza infection on coexpression patterns of SP-C and PODO at the single cell level have not previously been reported.
Although PODO is known to be necessary for normal lung development and ATI cell morphogenesis (42), its function on ATI cells in the mature lung remains poorly understood (15). ATI and ATII cells have been shown to differentially express a variety of additional “terminal” markers. For example, expression of receptor for advanced glycosylation end products (RAGE), caveolin-1/-2, and aquaporin-5 is believed to be ATI cell-specific (36), whereas ATII cells have been shown to express ATP-binding cassette A3 protein (ABCA3), which is associated with lamellar bodies (64). Influenza A/PR/8 infection can induce de novo expression of RAGE on bronchial epithelial cells, and this phenomenon was associated with increased viral replication and disease severity (56). However, effects of influenza infection on ATI cell RAGE have not been reported. Likewise, with the exception of a report describing downregulation of aquaporin-5 following adenovirus infection (48), there is a distinct paucity of information regarding effects of any respiratory viral infections on caveolin-1/-2, aquaporin-5, and ABCA3 expression. Although some studies suggest that aquaporins are not required for normal alveolar fluid clearance (58), they may be important in resolution of pulmonary edema in the injured lung (63). It is therefore possible that altered aquaporin expression contributes to inhibition of alveolar fluid clearance by influenza A virus that we reported previously (62). However, we have yet to investigate this possibility.
Murine ATII cells have been shown to be capable of self-renewal and differentiation to ATI cells following bleomycin-induced ALI (4). Our findings indicate that a similar differentiation process may occur following the development of ALI in influenza-infected mice in an attempt to repair damaged bronchoalveolar epithelium. In addition, our results suggest that differentiation involves downregulation of SP-C followed by upregulation of PODO, rather than via a GFP+/PODO+ intermediate. Our qRT-PCR data showing that downregulation of pro-sp-c gene expression precedes upregulation of podo gene expression support this hypothesis, as does a prior report showing that an SP-C− progenitor cell population in the mouse lung may be important to ATII cell replenishment during recovery from injury (8). Given its known role in lung development (42), this raises the intriguing possibility that an increase in PODO expression may be necessary for lung regeneration. Unfortunately, however, our gating strategy for FACS analysis does not allow us to make this conclusion definitively. For example, it is also possible that the observed increase in GFP−/PODO− ATII cells at 6 dpi reflected expansion of a pool of distal airway stem cells. One recent study has shown that p63+/keratin 5+ cells in the terminal bronchioles can migrate into damaged lung parenchyma where they differentiate into PODO+ but not SP-C+ cells during the repair phase after sublethal infection with influenza A/PR/8 virus (30). These cells do not appear to be either ATII cell or upper airway basal cell derived (57). However, it is unclear whether induction of this stem cell population has sufficient time to occur in our model of acutely lethal infection. Moreover, the role of particular cell types as progenitors for ATI and ATII cells during repair may be dependent upon both the type of lung injury and the extent to which it is specifically directed at ATII cells. Hence, the tropism of influenza viruses for ATII cells may significantly impact their ability to serve as ATI cell progenitors.
In vitro infection with influenza A/PR/8 was shown to induce caspase-3 activation in differentiated human ATI-like and ATII cell cultures (28, 59). Likewise, we found that influenza A/WSN/33 infection resulted in caspase-3 activation at 6 dpi, which was temporally associated with a 55% reduction in total cell yields. Herold et al. reported that infection of mice with influenza A/PR/8 caused increased TRAIL receptor (DR5) expression and high levels of apoptosis in PODO+ alveolar epithelial cells isolated from lungs later in infection (20). Interestingly, they did not find significant numbers of apoptotic PODO− cells in their study. This suggests that the increased caspase-3 activation that we detected by Western blot in isolated ATII cells at 6 dpi in our study may be associated with gain of PODO expression.
Increased influenza severity has been correlated with spread of the virus to the lower respiratory tract and alveoli (21, 55). However, the cellular site of influenza virus replication in the distal lung remains unclear. In vitro, seasonal H1N1, 2009 pandemic H1N1, and H5N1 influenza A viruses were shown to productively replicate in primary cultures of polarized human ATI-like and ATII cells (7, 44, 67). Likewise, the mouse-adapted A/PR/8 (H1N1) influenza strain can infect primary human ATI-like and ATII cells in vitro (26, 28). In contrast, Weinheimer et al. reported that, while both seasonal and pandemic influenza A strains actively replicated in ATII cells in ex vivo human lung explants infected in vitro, replication within ATI cells did not occur, although they did find viral antigens on the surface of ATI cells (60). Because replication was higher in H5N1-infected ATII cells than those infected with seasonal H1N1, seasonal H3N2, and pandemic H1N1 viruses, these authors hypothesized that the extent of viral replication within ATII cells may be an important determinant of pathogenicity. In vivo, influenza NP antigen could be detected in both ATI and ATII cells from mice infected with the H1N1 influenza strain used in the current study (A/WSN/33) (32). Likewise, active viral replication could be detected in both SP-C+ cells and Clara cells in mice infected with a sublethal dose of influenza A/PR/8 virus (19). Importantly, both ATI and ATII cells in autopsy lung tissue from naturally infected humans and experimentally infected Cynomolgus macaques (Macaca fascicularis) were found to contain H5N1 influenza A NP (29, 51). Finally, experimental studies by van Riel et al. showed that seasonal and pandemic influenza viruses preferentially infect ATI cells, and these authors propose that increased ATII cell tropism underlies the increased pathogenicity of H5N1 viruses (53, 55). In our model, we found that development of ALI (similar to that seen following infection with H5N1 viruses) was associated with a progressive increase in NP expression in isolated ATII cells, which is consistent with this hypothesis.
When viral replication was measured in whole lung homogenates by a standard plaque assay, we did not find biologically significant changes in viral titers over the course of infection. In contrast, Western blot analysis indicated that NP expression was significantly higher at 6 dpi than 2 dpi in freshly isolated ATII cells. This suggests that whole lung homogenate data may not provide an accurate indication of the magnitude and kinetics of viral replication in individual respiratory epithelial cell types. Increased NP expression in ATII cell preparations was accompanied by a progressive loss of SP-A, SP-C, and SP-D protein expression, which was accompanied by an increase in PODO protein levels. This indicates that, in addition to caspase-3 activation, the extent of differentiation of ATII cells correlated with viral replication. It also suggests that intermediate (SP-C−/PODO−) or ATI-like (SP-C−/PODO+) cells may be a site of active viral replication in A/WSN/33-infected mice. However, it remains possible that increased viral replication at 6 dpi reflects persistence of virus within infected ATII cells even as they differentiate into SP-C−/PODO+ ATI-like cells and that murine ATI cells are not susceptible to direct infection by A/WSN/33. Finally, the high level of NP expression at 6 dpi may reflect viral replication within a subpopulation of SP-C−/PODO− Clara cells that appear to be resistant to influenza-induced cell death (19).
EpCAM is a transmembrane epithelial-specific intercellular cell adhesion molecule that is involved in cellular signaling, cell migration, proliferation, and differentiation (40, 50). Both ATI and ATII cells in adult human lung have been shown to express EpCAM by immunohistochemistry (25). However, EpCAM expression may be higher in human ATII cells than ATI cells (16). We found that EpCAM expression differed very significantly between ATI and ATII cells. In uninfected mice, all ATII cells but only one-half of ATI cells were EpCAM+. These findings agree with human cell studies (16) and indicate that, even in uninfected mice, EpCAM is neither a pan-epithelial antigen nor a reliable marker of ATI cells. Moreover, infection with influenza A virus only modestly reduced ATII cell EpCAM expression but decreased ATI cell EpCAM levels by almost 90%. Because we found almost no SP-C+/PODO+ ATII cells, this indicates that the overall reduction in EpCAM expression at 6 dpi was primarily correlated with the relative shift from SP-C+/PODO− to SP-C−/PODO+ ATII cells. Hence, these data further support our hypothesis that acutely lethal influenza A virus infection results in differentiation of ATII cells into ATI cells in an attempt to repair damaged bronchoalveolar epithelium. They also suggest that, by weakening intercellular adhesion, a reduction in EpCAM expression may contribute to the progressive loss of alveolar epithelial barrier function during the progression of influenza-induced ALI, although we have not yet confirmed this possibility. Finally, these results indicate that, over the course of infection, influenza can differentially impact an antigen that is expressed by both epithelial cell types. Infection of respiratory epithelial cells by influenza viruses requires binding of viral hemagglutinin to sialic acid moieties expressed by cell surface glycoproteins. Human influenza viruses have been shown to preferentially bind to α2,6-linked sialic acids, whereas avian viruses may preferentially bind to cells expressing α2,3-linked sialic acids (22, 43). We found that ATII cells from either uninfected or A/WSN/33-infected mice did not bind to the SNA lectin, indicating that they do not express α2,6-linked sialic acids. In contrast, the majority of cells isolated from uninfected SP-CGFP mice were labeled by MAL-II, which binds preferentially to α2,3-linked sialosaccharides. This is in agreement with previous studies (6, 65). Following infection, the percentage of isolated cells that were MAL-II+ did not change, although the extent of MAL-II binding per cell was reduced at 2 but not 6 dpi. Interestingly, van Riel et al. reported a similar reduction in MAL-II binding to both human and macaque ATII cells 24 h after H5N1 influenza A virus infection, although they did not evaluate later time points (54).
The primary physiological role of surfactant is to reduce alveolar surface tension and thereby facilitate alveolar inflation during inspiration. SP-C, which is the smallest surfactant-associated protein, enhances the movement of lipids between sheets and vesicles of surfactant, which is essential to stabilization of the surfactant film and surfactant recycling. Surfactant dysfunction will result in decreased lung compliance and increased work of breathing. We have shown previously that severe hypoxemia in influenza A/WSN/33-infected C57BL6 mice correlated with reductions in lung compliance (1–3). In the current study, pro-sp-c gene expression was reduced >40-fold at 6 dpi, and ATII cell lysate SP-C content was reduced by >95%. Finally, loss of ATII cells is also likely to result in reduced surfactant lipid levels, although these were not measured in the current study. Hence, while multiple factors are likely to underlie decreased lung compliance following influenza A virus infection, reduced SP-C and surfactant lipid synthesis is likely to contribute significantly to this effect.
We should note that there are certain limitations to our studies. First, although Papanicolau staining of cytospins of cells isolated from the lungs of uninfected SP-CGFP mice indicated that these preparations almost exclusively contained ATII cells, only around 75% of isolated cells were GFP+ by flow cytometry. Other investigators have reported similar findings and propose that SP-CGFP mice exhibit increasing heterogeneity of GFP expression as they get older, possibly as a result of differences in chromatin environment between mice of different ages (31). However, approximately one-half of all GFP− cells isolated were also PODO−, which argues that flow cytometry data may be an underestimate of the percentage of ATII cells present in isolated ATII cell preparations. An additional limitation of our studies is that we may be losing ATII cells after infection as a result of the isolation process itself: if GFP+ ATII cells become more fragile following infection, they may no longer be able to withstand lung digestion and filtration procedures. However, our whole lung fluorescence data do not support this possibility, at least with respect to the impact of influenza A virus infection on SP-C expression. Finally, we have not yet determined the mechanism underlying our observations. In our model, induction of severe ALI is associated with infiltration of neutrophils and macrophages and increased levels of interferon-γ, IL-6, interferon-γ -induced protein-10, and monocyte chemoattractant protein-1 in bronchoalveolar lavage fluid at 6 dpi (1, 3). Additional cell depletion and cytokine neutralization studies will be necessary to determine whether any of these factors contribute to changes in SP-C, PODO, and EpCAM expression that follow influenza A/WSN/33 infection. It is also possible that changes in bronchoalveolar epithelial cell phenotype result from direct viral infection. However, given the magnitude of the change relative to the likely numbers of infected cells, we believe that this possibility is unlikely.
In conclusion, we found that infection of SP-CGFP mice with an acutely lethal dose of H1N1 virus resulted in productive infection of ATII cells, loss of GFP+ ATII cells due to apoptosis, increased expression of the ATI cell marker PODO, and reduced expression of EpCAM. Infection also reduced pro-sp-c gene expression and SP-C protein synthesis. Our findings indicate that infection with an acutely lethal dose of influenza A virus results in a progressive differentiation of ATII cells into ATI-like cells, most likely to replace dying or dead ATI cells. Given the central role of ATII cells in maintaining normal alveolar fluid balance and surfactant function, we propose that this phenomenon contributes significantly to the pathogenesis of influenza-induced lung dysfunction and injury. New therapeutics that can promote ATII cell survival and increase surfactant lipid and protein synthesis are therefore likely to be of benefit in patients with influenza-induced ALI.
GRANTS
This work was supported by the United States Army Medical Department Long-term Health and Education Training (LTHET) Program, The Ohio State University Howard Hughes Medical Institute Med-to-Grad Training Program, and National Heart, Lung, and Blood Institute Grant R01-HL-102469.
DISCLOSURES
The authors have no associations that might pose conflicts of interest.
AUTHOR CONTRIBUTIONS
Author contributions: C.C.H., P.S.W., and I.C.D. conception and design of research; C.C.H. and P.S.W. performed experiments; C.C.H., P.S.W., and I.C.D. analyzed data; C.C.H., P.S.W., and I.C.D. interpreted results of experiments; C.C.H., P.S.W., and I.C.D. prepared figures; C.C.H., P.S.W., and I.C.D. edited and revised manuscript; I.C.D. drafted manuscript; I.C.D. approved final version of manuscript.
ACKNOWLEDGMENTS
We acknowledge Lisa Joseph and Jacqueline Nolting for technical support.
Present address for C. Hofer: Veterinary Medical Division, U.S. Army Medical Research Institute of Infectious Diseases, Fort Dietrick, MD 21702.
REFERENCES
- 1.Aeffner F, Abdulrahman B, Hickman-Davis JM, Janssen PM, Amer A, Bedwell DM, Sorscher EJ, Davis IC. Heterozygosity for the F508del mutation in the cystic fibrosis transmembrane conductance regulator anion channel attenuates influenza severity. J Infect Dis 208: 780–789, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aeffner F, Bratasz A, Flaño E, Powell KA, Davis IC. Post-infection A77–1726 treatment improves cardiopulmonary function in H1N1 influenza-infected mice. Am J Respir Cell Mol Biol 47: 543–551, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aeffner F, Woods PS, Davis IC. Activation of A1-adenosine receptors promotes leukocyte recruitment to the lung and attenuates acute lung injury in mice infected with influenza A/WSN/33 (H1N1) virus. J Virol 88: 10214–10227, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, Randell SH, Noble PW, Hogan BLM. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest 123: 3025–3036, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borok Z, Whitsett JA, Bitterman PB, Thannickal VJ, Kotton DN, Reynolds SD, Krasnow MA, Bianchi DW, Morrisey EE, Hogan BL, Kurie JM, Walker DC, Radisky DC, Nishimura SL, Violette SM, Noble PW, Shapiro SD, Blaisdell CJ, Chapman HA, Kiley J, Gail D, Hoshizaki D. Cell plasticity in lung injury and repair: report from an NHLBI workshop, April 19–20, 2010. Proc Am Thorac Soc 8: 215–222, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bowman AS, Nelson SW, Edwards JL, Hofer CC, Nolting JM, Davis IC, Slemons RD. Comparative effectiveness of isolation techniques for contemporary influenza A virus strains circulating in exhibition swine. J Vet Diagnostic Invest 25: 82–90, 2013. [DOI] [PubMed] [Google Scholar]
- 7.Chan M, Chan R, Yu W, Ho C, Chui WH, Lo CK, Yuen K, Guan Y, Nicholls J, Peiris JM. Influenza H5N1 virus infection of polarized human alveolar epithelial cells and lung microvascular endothelial cells (Abstract). Respir Res 10: 102, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chapman HA, Li X, Alexander JP, Brumwell A, Lorizio W, Tan K, Sonnenberg A, Wei Y, Vu TH. Integrin α6β4 identifies an adult distal lung epithelial population with regenerative potential in mice. J Clin Invest 121: 2855–2862, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Charu V, Simonsen L, Lustig R, Steiner C, Viboud C. Mortality burden of the 2009–10 influenza pandemic in the United States: improving the timeliness of influenza severity estimates using inpatient mortality records. Influenza Other Respir Viruses 7: 863–871, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chuquimia OD, Petursdottir DH, Rahman MJ, Hartl K, Singh M, Fernández C. The role of alveolar epithelial cells in initiating and shaping pulmonary immune responses: communication between innate and adaptive immune systems. PLoS ONE 7: e32125, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cochrane. Neuraminidase Inhibitors Review Team. Does oseltamivir really reduce complications of influenza? Clin Infect Dis 53: 1302–1303, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corti M, Brody AR, Harrison JH. Isolation and primary culture of murine alveolar type II cells. Am J Respir Cell Mol Biol 14: 309–315, 1996. [DOI] [PubMed] [Google Scholar]
- 13.Davis IC, Matalon S. Epithelial sodium channels in the adult lung–important modulators of pulmonary health and disease. Adv Exp Med Biol 618: 127–140, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dobbs LG. Isolation and culture of alveolar type II cells. Am J Physiol Lung Cell Mol Physiol 258: L134–L147, 1990. [DOI] [PubMed] [Google Scholar]
- 15.Dobbs LG, Johnson MD, Vanderbilt J, Allen L, Gonzalez R. The great big alveolar TI cell: evolving concepts and paradigms. Cell Physiol Biochem 25: 55–62, 2010. [DOI] [PubMed] [Google Scholar]
- 16.Fujino N, Kubo H, Ota C, Suzuki T, Suzuki S, Yamada M, Takahashi T, He M, Suzuki T, Kondo T, Yamaya M. A novel method for isolating individual cellular components from the adult human distal lung. Am J Respir Cell Mol Biol 46: 422–430, 2012. [DOI] [PubMed] [Google Scholar]
- 17.Glasser SW, Korfhagen TR, Wert SE, Bruno MD, McWilliams KM, Vorbroker DK, Whitsett JA. Genetic element from human surfactant protein SP-C gene confers bronchiolar-alveolar cell specificity in transgenic mice. Am J Physiol Lung Cell Mol Physiol 261: L349–L356, 1991. [DOI] [PubMed] [Google Scholar]
- 18.Goldstein E, Viboud C, Charu V, Lipsitch M. Improving the estimation of influenza-related mortality over a seasonal baseline. Epidemiology 23: 829–838, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heaton NS, Langlois RA, Sachs D, Lim JK, Palese P, tenOever BR. Long-term survival of influenza virus infected club cells drives immunopathology. J Exp Med 211: 1707–1714, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herold S, Steinmueller M, von Wulffen W, Cakarova L, Pinto R, Pleschka S, Mack M, Kuziel WA, Corazza N, Brunner T, Seeger W, Lohmeyer J. Lung epithelial apoptosis in influenza virus pneumonia: the role of macrophage-expressed TNF-related apoptosis-inducing ligand. J Exp Med 205: 3065–3077, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hrincius ER, Hennecke AK, Gensler L, Nordhoff C, Anhlan D, Vogel P, McCullers JA, Ludwig S, Ehrhardt C. A single point mutation (Y89F) within the non-structural protein 1 of influenza A viruses limits epithelial cell tropism and virulence in mice. Am J Pathol 180: 2361–2374, 2012. [DOI] [PubMed] [Google Scholar]
- 22.Ibricevic A, Pekosz A, Walter MJ, Newby C, Battaile JT, Brown EG, Holtzman MJ, Brody SL. Influenza virus receptor specificity and cell tropism in mouse and human airway epithelial cells. J Virol 80: 7469–7480, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol 14: 315–328, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jewell NA, Vaghefi N, Mertz SE, Akter P, Peebles RS Jr, Bakaletz LO, Durbin RK, Flaño E, Durbin JE. Differential type I interferon induction by respiratory syncytial virus and influenza A virus in vivo. J Virol 81: 9790–9800, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kasper M, Behrens J, Schuh D, Muller M. Distribution of E-cadherin and Ep-CAM in the human lung during development and after injury. Histochem Cell Biol 103: 281–286, 1995. [DOI] [PubMed] [Google Scholar]
- 26.Kebaabetswe LP, Haick AK, Miura TA. Differentiated phenotypes of primary murine alveolar epithelial cells and their susceptibility to infection by respiratory viruses. Virus Res 175: 110–119, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim TH. Seasonal influenza and vaccine herd effect. Clin Exp Vaccine Res 3: 128–132, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kosmider B, Messier E, Janssen W, Nahreini P, Wang J, Hartshorn K, Mason R. Nrf2 protects human alveolar epithelial cells against injury induced by influenza A virus (Abstract). Respir Res 13: 43, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuiken T, Rimmelzwaan GF, van Amerongen G, Osterhaus AD. Pathology of human influenza A (H5N1) virus infection in cynomolgus macaques (Macaca fascicularis). Vet Pathol 40: 304–310, 2003. [DOI] [PubMed] [Google Scholar]
- 30.Kumar PA, Hu Y, Yamamoto Y, Hoe NB, Wei TS, Mu D, Sun Y, Joo LS, Dagher R, Zielonka EM, Wang DY, Lim B, Chow VT, Crum CP, Xian W, McKeon F. Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell 147: 525–538, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee JH, Kim J, Gludish D, Roach RR, Saunders AH, Barrios J, Woo AJ, Chen H, Conner DA, Fujiwara Y, Stripp BR, Kim CF. Surfactant protein-C chromatin-bound green fluorescence protein reporter mice reveal heterogeneity of surfactant protein C-expressing lung cells. Am J Respir Cell Mol Biol 48: 288–298, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee YH, Lai CL, Hsieh SH, Shieh CC, Huang LM, Wu-Hsieh BA. Influenza A virus induction of oxidative stress and MMP-9 is associated with severe lung pathology in a mouse model. Virus Res 178: 411–422, 2013. [DOI] [PubMed] [Google Scholar]
- 33.Lo B, Hansen S, Evans K, Heath JK, Wright JR. Alveolar epithelial type II cells induce T cell tolerance to specific antigen. J Immunol 180: 881–888, 2008. [DOI] [PubMed] [Google Scholar]
- 34.Marconett CN, Zhou B, Rieger ME, Selamat SA, Dubourd M, Fang X, Lynch SK, Stueve TR, Siegmund KD, Berman BP, Borok Z, Laird-Offringa IA. Integrated transcriptomic and epigenomic analysis of primary human lung epithelial cell differentiation. PLoS Genet 9: e1003513, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matalon S, Davis IC. Vectorial sodium transport across the mammalian alveolar epithelium: it occurs but through which cells? Circ Res 92: 348–349, 2003. [DOI] [PubMed] [Google Scholar]
- 36.McElroy MC, Kasper M. The use of alveolar epithelial type I cell-selective markers to investigate lung injury and repair. Eur Respir J 24: 664–673, 2004. [DOI] [PubMed] [Google Scholar]
- 37.Narasaraju T, Ng HH, Phoon MC, Chow VTK. MCP-1 antibody treatment enhances damage and impedes repair of the alveolar epithelium in influenza pneumonitis. Am J Respir Cell Mol Biol 42: 732–743, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Osterholm MT. Pandemic influenza vaccine: the US government is not doing enough. Clin Pharmacol Ther 82: 635–637, 2007. [DOI] [PubMed] [Google Scholar]
- 39.Osterholm MT, Ballering KS, Kelley NS. Major challenges in providing an effective and timely pandemic vaccine for influenza A(H7N9). J Am Med Assoc 309: 2557–2558, 2013. [DOI] [PubMed] [Google Scholar]
- 40.Patriarca C, Macchi RM, Marschner AK, Mellstedt H. Epithelial cell adhesion molecule expression (CD326) in cancer: A short review. Cancer Treatment Rev 38: 68–75, 2012. [DOI] [PubMed] [Google Scholar]
- 41.Quandelacy TM, Viboud C, Charu V, Lipsitch M, Goldstein E. Age- and sex-related risk factors for influenza-associated mortality in the United States between 1997–2007. Am J Epidemiol 179: 156–167, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramirez MI, Millien G, Hinds A, Cao Y, Seldin DC, Williams MC. T1alpha, a lung type I cell differentiation gene, is required for normal lung cell proliferation and alveolus formation at birth. Dev Biol 256: 61–72, 2003. [DOI] [PubMed] [Google Scholar]
- 43.Scull MA, Gillim-Ross L, Santos C, Roberts KL, Bordonali E, Subbarao K, Barclay WS, Pickles RJ. Avian influenza virus glycoproteins restrict virus replication and spread through human airway epithelium at temperatures of the proximal airways. PLoS Pathog 5: e1000424, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shieh WJ, Blau DM, Denison AM, Deleon-Carnes M, Adem P, Bhatnagar J, Sumner J, Liu L, Patel M, Batten B, Greer P, Jones T, Smith C, Bartlett J, Montague J, White E, Rollin D, Gao R, Seales C, Jost H, Metcalfe M, Goldsmith CS, Humphrey C, Schmitz A, Drew C, Paddock C, Uyeki TM, Zaki SR. 2009 pandemic influenza A (H1N1): Pathology and pathogenesis of 100 fatal cases in the United States. Am J Pathol 177: 166–175, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Short KR, Kroeze EJBV, Fouchier RAM, Kuiken T. Pathogenesis of influenza-induced acute respiratory distress syndrome. Lancet Infect Dis 14: 57–69, 2014. [DOI] [PubMed] [Google Scholar]
- 46.Stone KC, Mercer RR, Gehr P, Stockstill B, Crapo JD. Allometric relationships of cell numbers and size in the mammalian lung. Am J Respir Cell Mol Biol 6: 235–243, 1992. [DOI] [PubMed] [Google Scholar]
- 47.Thompson CI, Barclay WS, Zambon MC, Pickles RJ. Infection of human airway epithelium by human and avian strains of influenza A virus. J Virol 80: 8060–8068, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Towne JE, Harrod KS, Krane CM, Menon AG. Decreased expression of aquaporin (AQP)1 and AQP5 in mouse lung after acute viral infection. Am J Respir Cell Mol Biol 22: 34–44, 2000. [DOI] [PubMed] [Google Scholar]
- 49.Traylor ZP, Aeffner F, Davis IC. Influenza A H1N1 induces declines in alveolar gas exchange in mice consistent with rapid post-infection progression from acute lung injury to ARDS. Influenza Other Respir Viruses 7: 472–479, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Trzpis M, McLaughlin PMJ, de Leij LMFH, Harmsen MC. Epithelial cell adhesion molecule: more than a carcinoma marker and adhesion molecule. Am J Pathol 171: 386–395, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Uiprasertkul M, Puthavathana P, Sangsiriwut K, Pooruk P, Srisook K, Peiris M, Nicholls JM, Chokephaibulkit K, Vanprapar N, Auewarakul P. Influenza A H5N1 replication sites in humans. Emerg Infect Dis 11: 1036–1041, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Van der Velden J, Bertoncello I, McQualter J. LysoTracker is a marker of differentiated alveolar type II cells (Abstract). Respir Res 14: 123, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Riel D. H5N1 virus attachment to lower respiratory tract (Absract). Science 312: 399, 2006. [DOI] [PubMed] [Google Scholar]
- 54.van Riel D, Leijten LM, Kochs G, Osterhaus ADME, Kuiken T. Decrease of virus receptors during highly pathogenic H5N1 virus infection in humans and other mammals. Am J Pathol 183: 1382–1389, 2013. [DOI] [PubMed] [Google Scholar]
- 55.van Riel D, Munster VJ, de Wit E, Rimmelzwaan GF, Fouchier RAM, Osterhaus ADME, Kuiken T. Human and avian influenza viruses target different cells in the lower respiratory tract of humans and other mammals. Am J Pathol 171: 1215–1223, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Zoelen MAD, van der Sluijs KF, Achouiti A, Florquin S, Braun-Pater JM, Yang H, Nawroth PP, Tracey KJ, Bierhaus A, van der Poll T. Receptor for advanced glycation end products is detrimental during influenza A virus pneumonia. Virology 391: 265–273, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vaughan AE, Brumwell AN, Xi Y, Gotts JE, Brownfield DG, Treutlein B, Tan K, Tan V, Liu FC, Looney MR, Matthay MA, Rock JR, Chapman HA. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nat Advance online publication2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Verkman AS. Role of aquaporins in lung liquid physiology. Respir Physiol Neurobiol 159: 324–330, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang J, Nikrad MP, Phang T, Gao B, Alford T, Ito Y, Edeen K, Travanty EA, Kosmider B, Hartshorn K, Mason RJ. Innate immune response to influenza A virus in differentiated human alveolar type II cells. Am J Respir Cell Mol Biol 45: 582–591, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weinheimer VK, Becher A, Tõnnies M, Holland G, Knepper J, Bauer TT, Schneider P, Neudecker J, Rückert JC, Szymanski K, Temmesfeld-Wollbrueck B, Gruber AD, Bannert N, Suttorp N, Hippenstiel S, Wolff T, Hocke AC. Influenza A viruses target type II pneumocytes in the human lung. J Infect Dis 206: 1685–1694, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Whitsett JA, Wert SE, Weaver TE. Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Ann Rev Med 61: 105–119, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wolk KE, Lazarowski ER, Traylor ZP, Yu EN, Jewell NA, Durbin RK, Durbin JE, Davis IC. Influenza A virus inhibits alveolar fluid clearance in BALB/c mice. Am J Respir Crit Care Med 178: 969–976, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu XM, Wang HY, Li GF, Zang B, Chen WM. Dobutamine enhances alveolar fluid clearance in a rat model of acute lung injury. Lung 187: 225–231, 2009. [DOI] [PubMed] [Google Scholar]
- 64.Yamano G, Funahashi H, Kawanami O, Zhao LX, Ban N, Uchida Y, Morohoshi T, Ogawa J, Shioda S, Inagaki N. ABCA3 is a lamellar body membrane protein in human lung alveolar type II cells. FEBS Lett 508: 221–225, 2001. [DOI] [PubMed] [Google Scholar]
- 65.Yao L, Korteweg C, Hsueh W, Gu J. Avian influenza receptor expression in H5N1-infected and noninfected human tissues. FASEB J 22: 733–740, 2008. [DOI] [PubMed] [Google Scholar]
- 66.Yin L, Xu S, Cheng J, Zheng D, Limmon GV, Leung NHN, Rajapakse JC, Chow VTK, Chen J, Yu H. Spatiotemporal quantification of cell dynamics in the lung following influenza virus infection (Abstract). J Biomed Optic 18: 046001, 2013. [DOI] [PubMed] [Google Scholar]
- 67.Yu WCL, Chan RWY, Wang J, Travanty EA, Nicholls JM, Peiris JSM, Mason RJ, Chan MCW. Viral replication and innate host responses in primary human alveolar epithelial cells and alveolar macrophages infected with influenza H5N1 and H1N1 viruses. J Virol 85: 6844–6855, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zuo W, Zhang T, Wu DZ, Guan SP, Liew AA, Yamamoto Y, Wang X, Lim SJ, Vincent M, Lessard M, Crum CP, Xian W, McKeon F. p63+Krt5+ distal airway stem cells are essential for lung regeneration. Nature doi: 10.1038/nature13903, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]