

Abstract
In pulmonary epithelia, β-adrenergic agonists regulate the membrane abundance of the epithelial sodium channel (ENaC) and, thereby, control the rate of transepithelial electrolyte absorption. This is a crucial regulatory mechanism for lung liquid clearance at birth and thereafter. This study investigated the influence of the gaseous signaling molecule hydrogen sulfide (H2S) on β-adrenergic agonist-regulated pulmonary sodium and liquid absorption. Application of the H2S-liberating molecule Na2S (50 μM) to the alveolar compartment of rat lungs in situ decreased baseline liquid absorption and abrogated the stimulation of liquid absorption by the β-adrenergic agonist terbutaline. There was no additional effect of Na2S over that of the ENaC inhibitor amiloride. In electrophysiological Ussing chamber experiments with native lung epithelia (Xenopus laevis), Na2S inhibited the stimulation of amiloride-sensitive current by terbutaline. β-adrenergic agonists generally increase ENaC abundance by cAMP formation and activation of PKA. Activation of this pathway by forskolin and 3-isobutyl-1-methylxanthine increased amiloride-sensitive currents in H441 pulmonary epithelial cells. This effect was inhibited by Na2S in a dose-dependent manner (5–50 μM). Na2S had no effect on cellular ATP concentration, cAMP formation, and activation of PKA. By contrast, Na2S prevented the cAMP-induced increase in ENaC activity in the apical membrane of H441 cells. H441 cells expressed the H2S-generating enzymes cystathionine-β-synthase, cystathionine-γ-lyase, and 3-mercaptopyruvate sulfurtransferase, and they produced H2S amounts within the employed concentration range. These data demonstrate that H2S prevents the stimulation of ENaC by cAMP/PKA and, thereby, inhibits the proabsorptive effect of β-adrenergic agonists on lung liquid clearance.
Keywords: H2S, hydrogen sulfide, ENaC, lung liquid clearance, gasotransmitter
hydrogen sulfide (h2s), a gas with the typical smell of rotten eggs, is well known as an environmental chemical threat (71). Despite its toxicity, H2S has recently been recognized as an interesting molecule for physiological research. Low, nontoxic concentrations of H2S seem to affect a variety of cellular processes specifically (86), suggesting that H2S might be a physiologically relevant molecule (85, 86).
H2S is endogenously generated by cells as a result of l-cysteine metabolism, which involves the enzymes cystathionine-β-synthase (CBS), cystathionine-γ-lyase (CSE), and 3-mercaptopyruvate sulfurtransferase (3-MST) (44, 76). H2S can also be generated by sulfur-metabolizing commensals (33) and pathogens (72). Therefore, epithelia of tissues, which are exposed to microbiota, such as the intestine or the respiratory tract, are predominantly exposed to H2S and possess a high capacity to metabolize excess and potentially toxic H2S (52). Furthermore, Diener and colleagues (67) demonstrated a secretory response to H2S in rat colon epithelia. In this tissue, H2S induces a secretion of chloride (37, 66), which would trigger transepithelial liquid secretion.
Aside from the secretion of chloride, the absorption of sodium is an important determinant of transepithelial liquid movement. Sodium ions enter epithelial cells at their apical membrane via sodium channels, such as the epithelial Na+ channel (ENaC) (14) and are actively pumped out of the cells by the Na+-K+-ATPase at the basolateral membrane (45). This vectorial net movement of sodium creates osmotic gradients, which consequently drive transepithelial liquid absorption. Transepithelial sodium transport is, thus, crucial for the maintenance of sodium and liquid homeostasis in various organs, such as sweat glands, kidneys, the intestine, or the lung (31).
The pulmonary epithelium is a particularly interesting epithelium, which evolves from a liquid-secreting epithelium during fetal life to a predominantly liquid-absorbing epithelium at birth and thereafter (1, 16, 62, 63). The switch to absorption of liquid during labor and the neonatal period is due to induction of the epithelial sodium-absorbing machinery by hormones (cortisol and thyroid hormones) and, particularly acutely, β-adrenergic agonists (11, 61, 83). The stimulation of sodium absorption by β-adrenergic agonists involves the adenylate cyclase (AC)/cAMP/PKA signaling axis (8, 17, 84). Activation of this pathway increases ENaC abundance in the apical membrane of the epithelial cells (8, 41, 79, 90) and facilitates enhanced sodium ion, and consequently, liquid absorption. This process is a crucial mechanism to clear the lung liquid during birth and the switch to air breathing. The importance of ENaC in this process was impressively demonstrated in studies by Hummler et al. (40) using transgenic mice with reduced ENaC expression in the lung. These mice died shortly after birth due to their inability to clear fetal lung liquid. More recent studies using phase-contrast X-ray imaging of aeration of newborn lungs propose that high transpulmonary pressure during inspiration plays a major role in lung liquid clearance (39, 73). The interaction between such a hydrostatic mechanism and ENaC-mediated sodium absorption needs to be elucidated.
In the adult lung, the sodium-absorbing machinery maintains a thin and tightly regulated liquid layer, which is important for normal lung physiology. In the airways, a dynamic equilibrium between chloride secretion and sodium transport determines the volume and composition of the airway surface liquid (ASL). A precise volume of this liquid is necessary for proper ciliary beating and, hence, mucociliary clearance (38). Hypoactivity of ENaC, as in type 1 pseudohypoaldosteronism, will lead to an increased volume of the ASL (42), whereas hyperactivity of ENaC leads to ASL volume depletion and, consequently, both situations lead to impaired mucociliary clearance (50). ENaC mutations or altered ENaC regulation in the airways is also associated with airway diseases, such as cystic fibrosis (3, 7, 50). In the distal lung, sodium and liquid absorption across the alveolar epithelium maintain a thin liquid layer, which is necessary for diffusion of O2/CO2. Impaired sodium absorption can lead to liquid accumulation in the lung and the formation of pulmonary edema (4). Understanding the physiological and pathophysiological regulation of ENaC-mediated sodium absorption is, thus, an important topic in lung physiology.
We have recently shown that exogenously applied H2S decreases basal sodium fluxes across pulmonary epithelial cells (2, 6, 24). This antiabsorptive effect is consistent with the generally observed secretory epithelial response to this gas (67). However, despite the present studies on ion transport regulation by H2S, a direct effect of H2S on transepithelial lung liquid movement has not been demonstrated. Furthermore, there is no knowledge about the interference of H2S with proabsorptive regulators of lung liquid clearance, such as β-adrenergic agonists. Therefore, we investigated the effect of exogenous H2S on basal and β-adrenergic agonist-stimulated lung liquid clearance in situ and transepithelial sodium absorption in vitro. We demonstrate that H2S inhibits the proabsorptive effect of β-adrenergic agonists on ENaC-mediated sodium absorption.
MATERIALS AND METHODS
Application of H2S and used chemicals.
H2S was applied by using the sulfur salt Na2S (5 × 10−6 M to 2.5 × 10−4 M; Sigma, Taufkirchen, Germany) (Fig. 1). Generally, H2S will equilibrate with its concentration in the air and, therefore, evaporate from the buffer solutions over time (21). This loss of H2S from buffers was initially determined by measuring H2S concentrations at various time points after application of 5 × 10−5 M Na2S to H441 Ringer solution (for composition, see below). H2S was measured by the formation of methylene blue exactly as previously described (70). Methylene blue/H2S levels stayed constant for up to 5 min postapplication of Na2S and decreased afterward to baseline levels (Fig. 1). Therefore, Na2S was applied repetitively in experiments that required exposure times >5 min. For biochemical experiments, loss of H2S was compensated by increasing the concentration of Na2S by five-fold.
Fig. 1.
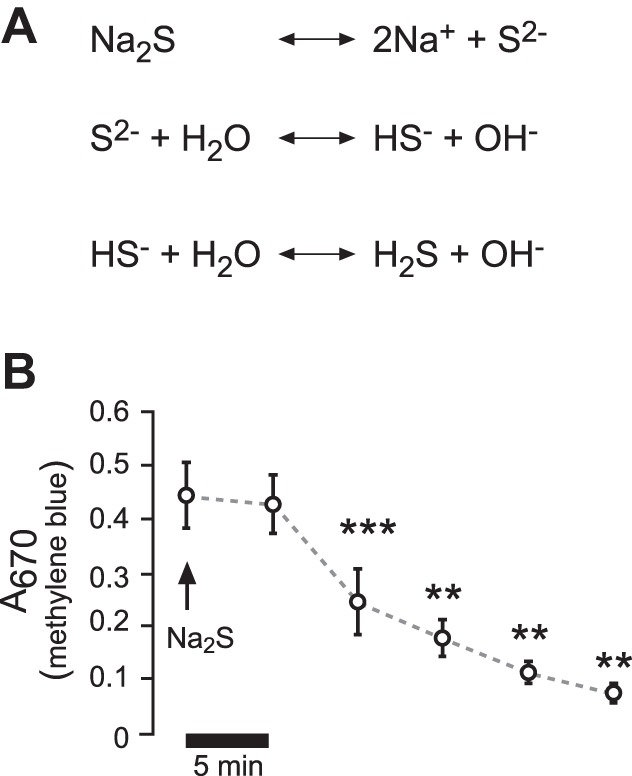
Administration of H2S and stability in buffer solutions. A: sulfur salt Na2S was employed to deliver H2S. According to the shown dissociation equations, a Na2S-containing solution would react basic. The employed buffers were adjusted, so that there were no pH changes with the employed Na2S concentrations (5 × 10−5 − 2.5 × 10−4 M). B: Na2S (5 × 10−5) was applied to buffers in Ussing chambers containing H441 Ringer solution, and aliquots were taken every 5 min. H2S was indirectly measured by the formation of methylene blue and its absorption at 670 nm. **P < 0.01, ***P < 0.001. Significant difference (Student's paired t-test) with respect to the initial value (n = 5).
Terbutaline (1–3 × 10−4 M; Sigma) was used as a β-adrenergic receptor agonist and amiloride (10−5 M; Sigma) as an inhibitor of ENaC. The adenylate cyclase activator forskolin (10−5 M; AG Scientific, distributed by MoBiTec, Göttingen, Germany) and the phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (IBMX; 10−4 M; Sigma) were employed to increase intracellular cAMP. l-Cysteine (10−3 M; Sigma) and pyridoxal phosphate (2 × 10−4 M; Sigma) were employed as precursors of endogenous H2S synthesis. In some cases, DMSO (Sigma) was used as a solvent for stock solutions, and control experiments were performed with this drug accordingly.
Measurement of in situ lung liquid clearance in rats.
The in situ perfused rat preparation used in these experiments has been described previously (75, 89). All animal experiments were approved by independent committees. Rat experiments were performed under license from the United Kingdom Home Office in accordance with the Animals (Scientific Procedures) Act 1986. Male Wistar rats (Charles River Laboratories, Kent, UK) were given terminal anesthesia with intraperitoneal injections of a mixture of 0.5 ml Hypnorm (fentanyl 0.316 mg/ml and fluanisone 10 mg/ml) and 0.5 ml Hypnovel (midazolam 5 mg/ml), added to 1 ml water at a final dose of 2.7 ml/kg. Tracheotomy was performed, and the rats were ventilated with air (Harvard Rodent ventilator), and the chest was opened in the midline. A ligature was positioned around the origin of the pulmonary artery and aorta, and the animal was heparinized (0.1 ml 5,000 units/ml). Once a pulmonary artery cannula was tied in place (tying off the aorta in the process), the left ventricle was opened to allow another cannula to be placed into the left atrium. The time of loss of circulation to the lung was on the order of 10 s. The lung was perfused with a solution of composition 3% BSA, 117 mM NaCl, 2.68 mM KCl, 1.25 mM MgSO4, 1.82 mM CaCl2, 20 mM NaHCO3, 5.55 mM glucose, and 12 mM HEPES. The perfusate volume of 100 ml was circulated by a rotary peristaltic pump from a water-jacketed reservoir maintained at 38°C and gassed with a 95% O2 and 5% CO2 mixture. Perfusate flow was regulated to maintain a perfusion pressure of 7 to 8 mmHg: the reservoir was kept below the level of the rat so that venous return pressure was always negative. Once perfusion was established, ventilation was stopped and the lung filled with a solution of the same composition as the perfusate, with the exclusion of glucose. (Glucose is transported out of the lung lumen in conjunction with sodium ions, and as its concentration falls, it makes liquid absorption rates alter over time.) The volume of liquid instilled into the lung lumen was 15 ml/kg body wt, and it contained a known amount of the impermeant tracer blue dextran (5 mg/ml), as a volume marker. An initial 40-min mixing period of instilling and withdrawing the lung liquid into another heated water-jacketed reservoir was performed to allow for complete degassing of the lung. Subsequently, the lung liquid was sampled (50 μl) every 8 or 10 min after mixing, and the concentration of blue dextran was measured colorimetrically (620 nm), from which total liquid volume at each sampling time could be derived. A regression of total lung liquid volume against time [see reference (83) for details] gave the absorption or secretion rate of the lung liquid. Six samples of lung liquid were taken at 8-min intervals, and this constituted the control slope. At this point, at about 80 min, a compound or compounds were added to the perfusate or to the lung liquid or to both, and a further six samples were taken at 8-min intervals. Terbutaline and amiloride were added separately or together, only to the lung liquid, to achieve a concentration of 10−4 M for terbutaline and 5 × 10−5 M for amiloride. The Na2S solution was freshly made up (always within 30 min of administration) at a concentration of 21 mg/ml (isosmotic with the perfusate and lung liquid) and a sufficient amount was added to both the perfusate and the lung liquid to achieve a concentration of 50 μM in both compartments. Because of the volatility of H2S, aliquots of the Na2S solution were given after every sampling of the lung liquid, i.e., every 8 min. The extra volume added to the lung liquid (3 to 4 μl every 8 min) was corrected for in the subsequent calculations of absorption rates. Perfusion and venous pressures were constantly monitored, and the perfusate flow rates were measured twice during the course of the experiment, although the speed of the perfusion pump was never altered once the experiment commenced. Osmolality of the perfusate was checked during the experiment, as rising osmolality indicates tissue damage, but such an event did not occur in this series of experiments. At the end of the experiments, the lungs were removed and dried to constant weight, and absorption or secretion rates were expressed per gram of dry lung weight. All experiments were performed under license from the United Kingdom Home Office in accordance with the Animals (Scientific Procedures) Act 1986.
Preparation of native Xenopus lung epithelia.
Preparations of amphibian lungs were chosen as a model since these lungs are the only native vertebrate distal (i.e., alveolar) lung tissues that can be prepared for Ussing chamber recordings and have an ion channel/transporter physiology that reflects those of mammals (9, 10, 28, 30, 74). Adult clawed frogs (Xenopus laevis) were purchased from Xenopus Express France (Vernassal, France) and kept in tanks with continuous freshwater supply. Xenopus experiments were performed according to the German law of animal care and were approved by the Regional Board of Giessen (“478_M Xenopus laevis”). For experiments, frogs were anesthetized in 0.2% MS-222 (tricaine methanesulfonate) and subsequently killed by decapitation and sounding of the spinal cord. Lungs were dissected and longitudinally opened to flat sheets by incision from the bronchi along the pulmonary artery. Lung sheets were subsequently mounted between Lucite rings and transferred to perfusion Ussing chambers. The maintenance and treatment of animals adhered to the German law of animal care and was authorized by the Regional Board of Giessen (“478_M Xenopus laevis”).
H441 cell culture.
The human lung adenocarcinoma cell line H441 is a well-established model for the investigation of pulmonary sodium transport processes (5, 46, 68, 90). Cells were purchased from the American Type Culture Collection (distributed by LGC standards, Wesel, Germany) in the 65th passage and cultured exactly, as previously described (6). For transepithelial ion transport studies, cells were seeded in confluent densities onto permeable membrane supports (Snapwell type 3801; Costar, Amsterdam, The Netherlands) under liquid/liquid conditions. After 24 h, the medium on the apical side was aspirated, and the basolateral culture medium was supplemented with 2 × 10−7 M dexamethasone. Cells were cultured for an additional 7–9 days under air-liquid conditions, and the medium was exchanged every 48 h.
Transepithelial ion transport studies.
The excised lung sheets, which were fixed between Lucite rings or H441 monolayers that were grown on permeable supports, were mounted into custom-made perfusion Ussing chambers. The sheets/H441 monolayers separated the chamber halves into an apical and a basolateral compartment, which thereby allowed the specific application of drugs to either side of the epithelium. Each chamber half was perfused separately by a gravity-driven perfusion system. For Xenopus lung experiments, the chambers were perfused with Xenopus Ringer solution, which contained 100 mM NaCl, 3 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, and 10 mM glucose (pH 7.4). Experiments were performed at room temperature. For H441 cell experiments, the Ringer solutions contained 130 mM NaCl, 2.7 mM KCl, 1.5 mM KH2PO4, 1 mM CaCl2, 0.5 mM MgCl2, 10 mM HEPES, and 10 mM glucose (pH 7.35). Experiments were performed at 37°C.
The chambers were connected to a voltage-clamp amplifier (NPI Electronic, Tamm, Germany) by Ag/AgCl electrodes, which were mounted into the chambers via 200-μl pipette tips, filled with 3% of 1 M KCl-agar (Sigma). Only those electrodes were used that had a spontaneous electrical potential of less than 1 mV in perfusion solution. After complete mounting of the tissue/cells and electrodes, the perfusion was started, and the transepithelial potential (VT) was monitored. After equilibration of VT, the tissues were clamped to 0 mV and short-circuit currents (ISC) were recorded on a strip-chart recorder (Kipp&Zonen, Delft, The Netherlands). To determine the transepithelial resistance (RT), 2.5–5-mV voltage pulses were applied to the epithelia, and the resulting current deflections were recorded. RT was then calculated following Ohm's law.
Detection of cellular ATP concentrations.
Cellular ATP concentrations were measured using an ATPlite Luminescence Assay System (Perkin Elmer Life Sciences, Waltham, MA). H441 monolayers were cultured on permeable supports under air-liquid conditions, as described above. Monolayers were incubated for 5 min in Ussing chamber Ringer solution with or without 2.5 × 10−4 M Na2S at 37°C. Positive controls were generated by the addition of 2,4-dinitrophenol (DNP; 10−3 M; Sigma) and 2-deoxy-d-glucose (2DG 3 × 10−2 M; Sigma) to the basolateral culture medium for 60 min at 37°C/5% CO2. After the incubation, monolayers were washed in ice-cold PBS (PAA, Cölbe, Germany) and lysed in 50-μl cell lysis buffer (Perkin Elmer Life Sciences) and 100 μl Ringer solution with shaking. One-hundred-and-fifty microliters of cell lysates were then aliquoted and frozen at −80°C. Cellular ATP concentrations were determined later according to the manufacturer's instructions by incubating the cell lysates with a substrate solution and measuring luminescence with an Infinite 200 microplate reader (Tecan, Mainz, Germany). ATP concentrations were estimated with an ATP standard curve, according to manufacturer's instructions.
Detection of cellular cAMP concentrations.
H441 monolayers were first measured in Ussing chambers, as described above, and exposed to the indicated drugs for 5 min. Immediately afterward, the remaining saline was aspirated, and cells were lysed in 100 μl of 10−1 M HCl. Samples were frozen at −80°C until further procedures were begun. Samples were centrifuged for 15 min to remove cellular debris, and the supernatant containing cAMP was diluted 1:10 with Sample Diluent Buffer supplied by a cyclic AMP enzyme immunoassay kit (Biotrend, Cologne, Germany), according to manufacturer's instructions. cAMP concentrations were determined according to manufacturer's instructions using a cAMP standard curve.
Measurement of apical membrane ENaC currents in H441 cells.
ENaC currents across the apical membrane of H441 monolayers were assessed by a protocol, which was described in detail previously (5). In brief, monolayers were bathed in a potassium-gluconate solution that mimics the cationic composition of the cytosol. This solution contained 122.1 mM K-d-gluconate, 0.2 mM KCl, 10.4 mM NaCl, 1.5 mM KH2PO4, 10.7 mM CaCl2, 0.5 mM MgCl2, 10 mM glucose, and 10 mM HEPES (pH 7.35). Ouabain (2 × 10−3 M; Sigma) was added to the basolateral side. After 10 min of incubation, nystatin (1.5 10−4 M; Sigma) was applied to the basolateral side to permeabilize the basolateral membrane. After 10 min of incubation with nystatin, cells remained untreated or were exposed to forskolin/IBMX (10−5 M/10−4 M), Na2S (5 × 10−5 M) or a combination thereof. After 5 min, a sodium-gluconate solution (where K-d-gluconate was replaced by equimolar concentrations of Na-d-gluconate) was perfused over the apical side to generate an apical-to-basolateral sodium gradient. Subsequently, amiloride (10−5 M) was additionally applied to the apical side to determine apical ENaC-mediated sodium currents.
RT-PCR detection of H2S-generating enzymes.
Reverse transciptase (RT)-PCR experiments were performed to investigate expression of the H2S generating enzymes CSE, CBS, and 3MST. RNA was isolated from H441 monolayers, as previously described (5). RNA (1 µg) was reverse transcribed with the ImProm-II Reverse Transcription System (Promega, Mannheim, Germany) with oligo(dt) primers, according to manufacturer’s instructions. Reaction conditions were as follows: 5 min at 25°C, 60 min at 42°C, and 5 min at 70°C. For controls, the same procedure was performed without the reverse transcriptase enzyme (-RT). PCR reactions were performed with the GoTaq Polymerase System (Promega), using 2.5 mM dNTP’s, 2.5–3 mM MgCl2, and 10 µM of specific forward and reverse primers. Primer sequences were as follows: CSE forward 5′-ACAGGTACTTCAGGCAAGTGGCATCT-3′ and reverse 5′-CCAGGAACTGGGCAACTGCCAT-3′; CBS forward 5′-GGCGAAGGGGAGCCTGGAGA-3′ and reverse 5′-CGGTGTTCCCGGATGTCGGC-3′; 3MST forward 5′-TTCGACATCGACCAGTGC-3′ and reverse 5′-TGTGAAGGGGATGTTCA-3′). PCR reactions began with initial denaturation for 5 min at 95°C which was followed by 30–33 cycles of denaturation (45 s, at 95°C), annealing (30 s; at 60°C for CSE and CBS; 57°C for 3MST) and elongation (45 s, at 72°C). The reaction was terminated with a final elongation for 10 min at 72°C. Amplicons were separated on a 1.5% agarose gel and visualized with Midori green stain (Biozym; Hessisch Oldendorf, Germany). Afterward, amplicons were isolated from the gel with the WizardSV Gel and PCR clean up system (Promega) and sent for sequencing (Eurofins MWG Operon, Ebersberg, Germany).
Immunoblotting.
For immunoblotting experiments, H441 cells were cultured on 12-well permeable supports (ThinCerts, 0.4 μM, transparent; GreinerBioOne, Monroe, NC) under exactly the same conditions as for transepithelial transport studies. In some experiments, lysates of human embryonic kidney 293 cells, which were a kind gift of Matthias Schönberger (LMU Munich, Germany), were used as a positive control. For Na2S or drug exposure, cells were covered with bicarbonate-buffered saline (with or without drugs), which contained 117 mM NaCl, 25 mM NaHCO3, 4.7 mM KCl, 1.2 mM MgSO4, 2.5 mM CaCl2, and 11 mM d-glucose. The pH of this buffer equilibrated to 7.4 at 5% CO2-atmosphere. After incubation of the cells at 37°C/5% CO2, the permeable supports were immediately transferred to ice-cold PBS. PBS was aspirated, and cells were scraped and lysed in 100 μl of protein lysis buffer. For the detection of phosphorylated proteins, the lysis buffer contained 50 mM Tris·HCl (pH 7.2), 50 mM NaF, 1 mM EGTA, 1 mM EDTA, 1 mM Na-pyrophosphate, 1 mM Na-orthovanadate, 1% Triton-X-100 supplemented with protease inhibitors (Complete, Roche). For the detection of nonphosphorylated proteins, the lysis buffer contained 50 mM Tris·HCl (pH 7.5), 5 mM EDTA, 150 mM NaCl, and 1% Triton X-100 supplemented with protease inhibitors (Complete). Subsequently, cell lysates were snap-frozen. Thawed lysates were centrifuged to remove cellular debris. Protein concentration was determined with Bradford reagent (Applichem, Darmstadt, Germany). Protein (30 μg) was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to either nitrocellulose or PVDF membranes. Membranes were probed with specific antibodies under conditions as listed in Table 1. Bands were visualized with enhanced chemiluminescence solutions (custom-made) and were captured on photoreactive films (Amersham Hyperfilm ECL, GE Healthcare, Little Chalfont, Buckinghamshire, UK). Aliquots of the same sample were separated on two different gels and were probed with the respective antibodies against phosphorylated proteins and total protein (Table 1). All membranes were additionally probed for α-tubulin to control for equal loading. Band intensities were determined using ImageJ (National Institutes of Health, Bethesda, MD).
Table 1.
Working dilutions and immunoblot buffers of the employed antibodies
| Antibody | Supplier | Dilution | Blocking Buffer | Antibody Incubation Buffer |
|---|---|---|---|---|
| Primary antibodies | ||||
| phospho-AMPKα (Thr-172) (40H9); rabbit | Cell Signaling | 1:1000 | 5 % BSA/TBST 50 mM NaF | 5 % BSA/TBST 50 mM NaF |
| AMPKα (23A3); rabbit | Cell Signaling | 1:1000 | 5 % BSA/TBST | 5 % BSA/TBST |
| phospho-CREB (Ser-133); rabbit | Biotrend/Signalway | 1:4000 | 3 % BSA/TBST 50 mM NaF | 3 % BSA/TBST 50 mM NaF |
| CREB (Ab-129); rabbit | Biotrend/Signalway | 1:4000 | 3 % milk/TBST 50 mM NaF | 3 % milk/TBST 50 mM NaF |
| CBS monoclonal (M01), clone 3E1; mouse | Abnova | 1:1000 | 5 % milk/PBST | 5 % milk/PBST |
| CSE monoclonal (M03), clone S51; mouse | Abnova | 1:1000 | 5 % milk/PBST | 5 % milk/PBST |
| α-tubulin; mouse | Sigma | 1:3000–1:10000 | 3–5 % milk/TBST | 3–5% milk/TBST |
| Secondary HRP-linked antibodies | ||||
| anti-rabbit IgG | Cell Signaling | 1:3000 | n/a | primary antibody buffer |
| anti-mouse IgG | Thermo Scientific | 1:3000 | n/a | primary antibody buffer |
TBST, Tris-buffered saline containing 1 % Tween-20; PBST, phosphate-buffered saline containing 1% Tween-20; BSA, bovine serum albumin; CSE, cystathionine-γ-lyase.
Amperometric measurement of H2S production.
Although there are a number of methods that have been used to measure H2S (reviewed in Ref. 59); most—for example, methylene blue, monobromobimane, and HPLC analysis of headspace gas—entail the destruction of the sample and/or are conducted under anaerobic conditions, thereby inflating the H2S concentration. To overcome this shortcoming, we used the amperometric method, which permits measurement of H2S production in real time under more physiological conditions and is arguably best suited for these studies, although it too is limited by the inability to measure intracellular H2S.
H441 cells were cultured on permeable supports exactly as described above. Cells were lysed in lysis buffer, which contained 50 mM Tris·HCl (pH 7.5), 5 mM EDTA, 150 mM NaCl, and 1% Triton X-100 supplemented with protease inhibitors (Complete), and were frozen at −80°C until further procedures. The amperometric (polarographic) H2S sensor was constructed, as described previously (88). The sensor was connected to an Apollo 4000 Free Radical Analyzer (World Precision Instruments, Sarasota FL) with 100-mV polarizing voltage, and data were archived to a PC at 1-s intervals. The resolution was 1.4 × 10−8 M H2S gas (∼10−7 M total sulfide) at pH 7.4. One and one-half ml of lysate was added to a water-jacketed glass metabolism chamber (∼22°C) with ports for H2S and O2 (Clark-type) sensors, and a Teflon microstirring bar. The chamber was then closed, allowing the lysate to consume O2. When the Po2 had decreased to near 0 mmHg, pyridoxal-5-phosphate and cysteine (final concentration 2 × 10−4 M and 10−3 M, respectively) were added, and H2S production and Po2 were continuously monitored for 20–30 min. One and one-half milliliters of air was then injected, which raised Po2 and produced a net H2S consumption. In some experiments, the remaining air was then removed, and as the Po2 fell, H2S production resumed (not shown).
Data analysis and statistics.
Data are presented as means ± SE. The number of experiments is indicated with “n” and is also indicated in bar graphs as numbers in parentheses. Data were analyzed for Gaussian normal distribution with the Kolmogorov-Smirnov test. Student's paired t-test or nonparametric Wilcoxon matched pairs test (both two-tailed) was employed for statistical analysis of dependent experiments (i.e., values before and after drug application). Independent experiments were compared with Student's unpaired t-test or nonparametric Mann-Whitney U-test (two-tailed). Multiple-comparison analysis was performed by one-way ANOVA followed by Tukey's multiple-comparison test. Lung liquid clearance data were analyzed with Student's paired or unpaired t-test (two-tailed). Densitometric data from Western blot experiments were analyzed by Student's unpaired t-test, since this test is robust with respect to a theoretical log-normal distribution of Western blot data (20). P values ≤ 0.05 are considered statistically significant and are indicated by an asterisk. Highly significant P values ≤ 0.01 and ≤ 0.001 are marked with ** and ***, respectively.
RESULTS
H2S inhibits β-adrenergic agonist-stimulated lung liquid clearance in rats.
We investigated the effect of exogenous H2S on liquid clearance in rat lungs in situ (Fig. 2). Following a control period, Na2S (5 × 10−5 M, repetitive doses every 8 min) was applied to the alveolar and perfusate compartments. This treatment lowered baseline lung liquid clearance from −0.0255 ± 0.0034 ml·min−1·g−1 to −0.0139 ± 0.0032 ml·min−1·g−1 (n = 4; P = 0.0447; Fig. 2, A and D). By contrast, lung liquid clearance was significantly increased from −0.0238 ± 0.0052 ml·min−1·g−1 to −0.0445 ± 0.0057 ml·min−1·g−1 (n = 4; P = 0.0132) by application of the β-adrenergic agonist terbutaline (10−4 M) to the alveolar compartment (Fig. 2, B and D). Interestingly, this stimulatory effect of terbutaline was completely abrogated in the presence of Na2S (5 × 10−5 M; Fig. 2, C and D). Mean liquid flux of the control period was −0.0297 ± 0.0047 ml·min−1·g−1 and not significantly different following treatment with terbutaline and Na2S (−0.0329 ± 0.0063 ml·min−1·g−1; n = 4; P = 0.5934).
Fig. 2.

Effects of H2S on lung liquid clearance in situ. A–C: representative experiments. Following control periods, either Na2S (5 × 10−5; A), terbutaline (10−4 M; B) or a combination thereof (C) was added. Na2S was applied six times to compensate for loss of H2S from buffer solutions. Numbers below the regression lines indicate the corresponding JV fluxes (ml·min−1·g−1). D: mean values of JV, which indicate that Na2S significantly decreased, whereas terbutaline significantly increased liquid absorption. In the presence of Na2S, terbutaline failed to do so. The “no drug” bars originate from experiments, which evaluated liquid clearance without any drug administration (trace not shown). n = numbers in parentheses. Data were analyzed with Student's paired t-test. *P < 0.05. ns, not significant.
When Na2S was administered in the presence of the ENaC inhibitor amiloride, there was no significant additional effect to that observed when amiloride was given alone (n = 4; P = 0.2995; Fig. 3).
Fig. 3.
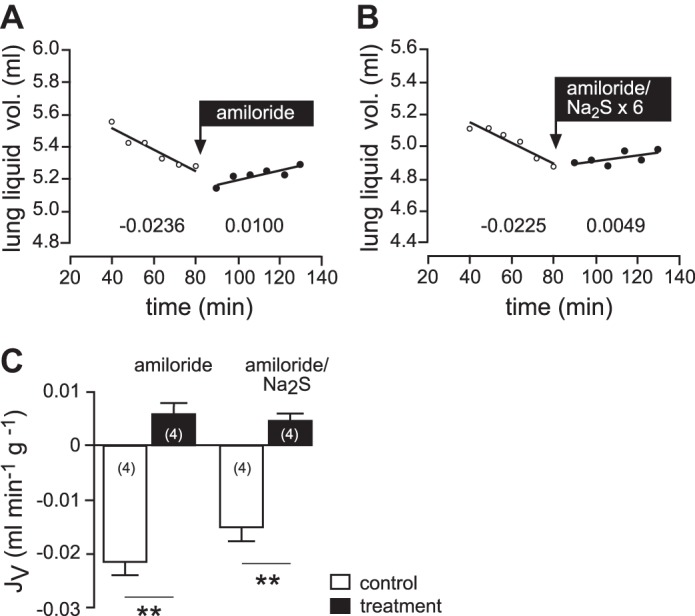
Effects of H2S and amiloride lung liquid clearance in situ. A and B: representative experiments. Following control periods, either amiloride (5 × 10−5; A) or a combination of amiloride and Na2S (5 × 10−5; B) was applied. Numbers below the regression lines indicate the corresponding JV fluxes (ml·min−1·g−1). C: mean values of JV, which indicate that Na2S has no additional effect over that of amiloride alone. n = numbers in parentheses. Data were analyzed with Student's paired and unpaired t-test. **P < 0.01.
H2S inhibits β-adrenergic agonist-stimulated sodium absorption across native lung epithelia.
To investigate whether H2S interferes with terbutaline-stimulated ion transport, transepithelial ion current recordings were performed on freshly excised Xenopus lung preparations. Consistent with previous studies (9), the application of terbutaline (3 × 10−4 M) to the basolateral (blood-faced) side of the lung tissue resulted in an increase in ion current by 1.49 ± 0.12-fold (n = 8; Fig. 4, A and C). The effect of terbutaline was completely inhibited in the presence of 5 × 10−5 M Na2S (Fig. 4, B and C). The ENaC inhibitor amiloride (10−5 M) was applied at the end of the experiments (see Fig. 4, A and B), and amiloride-sensitive fractions were determined as normalized values with respect to the initial current value before the beginning of experiments (to compensate for differences in basal ionic current capacities of the preparations). Normalized amiloride-sensitive current fractions were 0.72 ± 0.03 (n = 7) of control lungs and increased to 1.17 ± 0.09 (n = 8) in terbutaline-treated lungs (Fig. 4D). The amiloride-sensitive current fractions of lungs, which were treated with Na2S (5 × 10−5 M) alone (0.58 ± 0.06; n = 6) or with a combination of Na2S and terbutaline (0.63 ± 0.05; n = 8), were not significantly different from controls (Fig. 4D). There was no change in transepithelial resistance over the time of Na2S (5 × 10−5 M) exposure with respect to control experiments without Na2S (n = 6 and n = 7, respectively; Fig. 4E). These data indicate that H2S inhibits β-adrenergic agonist-induced stimulation of amiloride-sensitive (i.e., ENaC-mediated) sodium absorption.
Fig. 4.

H2S inhibits β-adrenergic agonist-stimulated sodium transport in native epithelial lung preparations. A: representative current trace of an Ussing chamber experiment on freshly dissected Xenopus laevis lungs. After short-circuit current values (ISC) were stable, terbutaline (3 × 10−4 M) was applied to the basolateral compartment. This resulted in a current increase. After the current stabilized again, amiloride (ami.; 10−5 M) was additionally applied to the apical membrane. B: similar experiment as shown in A. Here, terbutaline was applied in the presence of Na2S (5 × 10−5) in the apical compartment. To avoid loss of H2S due to evaporation from buffers, Na2S was applied every 8 min. Note that in the presence of Na2S, terbutaline was ineffective. C: summary of experiments with the data shown in panels A and B, as well as experiments in which lungs were exposed to Na2S (5 × 10−5) alone or to no drugs at all (control). Depicted are values of ISC, which have been normalized to the ISC value before drug application. D: amiloride-sensitive current fractions (Iamiloride) were normalized to the initial ISC value to compensate variations in baseline transport capacity of different lung preparations. Note that Na2S (5 × 10−5) alone had no significant effect on Iamiloride, whereas the stimulatory effect of terbutaline was lost (one-way ANOVA followed by Tukey's multiple-comparison test). ***P < 0.001. E: transepithelial resistances (RT; Ω cm2) of Xenopus lungs, which were exposed to Na2S (5 × 10−5) or no drugs at all (control). n = numbers in parentheses.
H2S inhibits cAMP-stimulated sodium absorption by lung epithelial cells.
The stimulation of sodium absorption by β-adrenergic receptor agonists is due to stimulation of the adenylate cyclase (AC)/cAMP/PKA signaling axis (8, 84). H2S may prevent the stimulation of this signaling pathway by inhibiting β-adrenergic receptor stimulation. To address this question, the human lung epithelial cell line H441 was employed. These cells lack β-adrenergic receptors or corresponding Gs proteins (18). Consistently, H441 cells did not respond to terbutaline (3 × 10−4 M, basolateral application; Fig. 5). Amiloride-sensitive current fractions were 19.08 ± 3.48 μA/cm2 before and 17.35 ± 2.93 μA/cm2 following application of terbutaline (n = 4; P = 0.3750; Fig. 5).
Fig. 5.
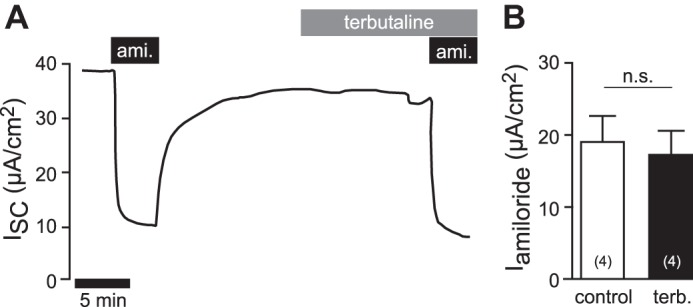
Terbutaline does not increase amiloride-sensitive sodium transport in H441 cells. A: representative current trace of an Ussing chamber experiment on H441 monolayers. First, amiloride (ami; 10 μM) was applied to the apical side to determine amiloride-sensitive ENaC current fractions. After washout of amiloride, terbutaline (3 × 10−4 M) was applied to the basolateral compartment. After 15 min, amiloride (10−5 M) was again applied to the apical side. B: statistical evaluation of experiments as shown in A. Depicted are amiloride-sensitive current fractions (Iamiloride) before (control) and under terbutaline (terb.), which are not found to be significantly different (n = 4; Wilcoxon-matched pairs test). n = numbers in parentheses.
However, the AC/cAMP/PKA pathway in H441 cells can be stimulated by the AC activator forskolin (10−5 M) and the phosphodiesterase inhibitor IBMX (10−4 M) (41, 79). The administration of forskolin/IBMX (10−5 M/10−4 M) to the apical side of the epithelium stimulated ion currents from 25.70 ± 2.45 μA/cm2 to 34.04 ± 3.12 μA/cm2 (n = 5; P = 0.0006; Fig. 6A). The application of Na2S (5 × 10−5 M) to H441 monolayers resulted in a decrease in ion current from 32.49 ± 2.33 μA/cm2 to 28.21 ± 2.53 μA/cm2 (n = 11; P = 0.0061; and Fig. 6B), which is consistent with previous observations in these cells (6). When forskolin/IBMX was subsequently applied to Na2S-exposed monolayers, there was no stimulation of ion current (Fig. 6B). Similar to the Xenopus lung experiments, normalized amiloride-sensitive currents were determined in H441 monolayers, which were treated with forskolin/IBMX, Na2S (5 × 10−5 M), a combination thereof, or remained untreated (Fig. 6C). Control monolayers had a normalized amiloride-sensitive current of 1.07 ± 0.03 (n = 6). This was significantly increased to 1.62 ± 0.05 (n = 5; P ≤ 0.001) in monolayers, which were treated with forskolin/IBMX. By contrast, monolayers, which were treated with Na2S alone or in combination with forskolin/IBMX, had significantly decreased amiloride-sensitive currents compared with control monolayers [0.76 ± 0.02 (n = 6; P ≤ 0.01) and 0.83 ± 0.09 (n = 5; P ≤ 0.05), respectively]. However, there was no significant difference between Na2S- and Na2S/forskolin/IBMX-treated monolayers, indicating that forskolin/IBMX was ineffective in the presence of Na2S. Na2S did not impair transepithelial resistance of H441 monolayers (before Na2S: 402.00 ± 37.25 Ω cm2; after Na2S: 391.30 ± 35.14 Ω cm2; n = 6; P = 1.0000; Fig. 6D). Furthermore, the inhibition of the forskolin/IBMX effect by Na2S was dose-dependent within a concentration range of 0.5–5 × 10−5 M Na2S (Fig. 6E).
Fig. 6.

H2S inhibits adenylate cyclase/cAMP-stimulated sodium transport in H441 cells. A: representative current trace of an Ussing chamber experiment on H441 monolayers. First, amiloride (ami.; 10−5 M) was applied to the apical membrane (duration indicated by the box). After removal of the drug, forskolin (fsk.; 10−5 M) and IBMX (10−4 M) were apically applied for 5 min. This resulted in an increase in short-circuit current (ISC), which was again sensitive to amiloride. B: similar experiment as shown in A. Here, Na2S (5 × 10−5) was applied after washout of amiloride. Subsequently, monolayers were stimulated with fsk./IBMX in the presence of Na2S. There was no stimulation of ISC by fsk./IBMX. C: statistical evaluation of experiments shown in A, as well as experiments in which Na2S alone was applied or no drugs at all (control). Depicted are amiloride-sensitive current fractions (Iamiloride), which were normalized to the first Iamiloride before drug exposure. Statistical analysis was performed by one-way ANOVA followed by Tukey's multiple comparison test. D: transepithelial resistances (RT; Ω cm2) of H441 monolayers before (control) and after 10 min of exposure to Na2S (50 μM). Nonparametric Wilcoxon-matched pairs test was performed for statistical analysis. E: effect of Na2S is dose-dependent. Depicted are percent changes in ISC due to the application of forskolin/IBMX (fsk./IBMX) in the presence of different concentrations of Na2S. Asterisks indicate levels of significance with respect to 0 μM Na2S (nonparametric Kruskal-Wallis test followed by Dunn's multiple-comparison test). *P < 0.05, **P < 0.01, ***P < 0.001. n = number within parentheses.
H2S acts independently of adenylate cyclase and PKA.
Because H2S inhibited forskolin/IBMX-stimulated transepithelial sodium absorption, it was investigated whether H2S interferes with the activity of AC or PKA. The substrate for cAMP production by AC is ATP. An impaired cellular energy metabolism due to H2S (19) may account for the observed inhibition of AC-induced effects in H441 cells. Two experimental strategies were pursued to address this hypothesis (Fig. 7, A and B). Intracellular ATP concentrations of H441 cells were measured with a luminometric assay (Fig. 7A). H441 monolayers, which were exposed to a high concentration of Na2S (2.5 × 10−4 M, apical and basolateral side) for 5 min, had intracellular ATP concentrations of 200.85 ± 20.62 × 10−6 M (n = 4). These were, although not statistically different, even higher than those of saline-exposed control monolayers, which were 129.93 ± 23.40 × 10−6 M (n = 4; P = 0.2000). By contrast, a combination of 2,4-dinitrophenol (10−3 M) and 2-deoxy-d-glucose (3 × 10−2 M), which served as a positive control, decreased ATP levels to 5.32 ± 1.31 × 10−6 M within 60 min (n = 4).
Fig. 7.

H2S does not deplete ATP levels in H441 cells. A: intracellular ATP concentrations of H441 cells, which were exposed to dinitrophenol (DNP, 1 × 10−3 M)/2-deoxy-d-glucose (2DG; 3 × 10−2 M) to the basolateral culture medium for 60 min or Ringer solution without (control) or with Na2S (2.5 × 10−4 M) for 5 min. Nonparametric Mann-Whitney U-test was employed for statistical analysis. B, top: representative Western blots against total AMPK, phosphorylated (pAMPK), and tubulin (loading control) of H441 monolayers, which were exposed to DNP/2DG, Ringer solution (control, ctrl) or Ringer solution with Na2S (2.5 × 10−4 M). B, bottom: densitometric analysis of blots as shown. Band intensities are given as a ratio of tubulin-normalized pAMPK/AMPK. n = numbers within parentheses. Student's unpaired t-test was employed for statistical analysis (P = 0.8840).
In addition, the activation status of the cellular energy sensor AMPK was investigated by using specific antibodies against total and active (Thr-172 phosphorylated) AMPK (Fig. 7B). Whereas 2,4-dinitrophenol and 2-deoxy-d-glucose led to a strong activation of AMPK in H441 monolayers, there was no such effect in monolayers that were exposed to 2.5 × 10−4 M Na2S within 10 min. These data indicate that there is no ATP depletion of H441 monolayers in response to Na2S.
H2S has been demonstrated to either stimulate (43, 54) or inhibit (55, 91) AC activity. A possible impact of H2S on AC was addressed by measuring cellular cAMP concentrations of H441 monolayers after electrophysiological recordings in Ussing chambers (Fig. 8A). H441 monolayers, which have been stimulated with forskolin/IBMX (10−5 M/10−4 M), had increased cAMP levels (559.62 ± 34.90 pmol/ml; n = 4) compared with unstimulated control monolayers (61.82 ± 0.84 pmol/ml; n = 3; Fig. 8B, bottom). However, although the additional administration of Na2S completely blocked the forskolin/IBMX-induced current activation (Fig. 8A, left; P = 0.0159), cAMP levels of Na2S-treated monolayers (515.26 ± 15.31 pmol/ml; n = 5) were not significantly different from monolayers that have been treated with forksolin/IBMX alone (Fig. 8A, right; P = 0.2857). These data indicate that decreased cAMP concentrations are not the reason for the loss of ENaC activation by forskolin/IBMX in Na2S-exposed H441.
Fig. 8.

H2S does not prevent cAMP formation and PKA activity in H441 cells. A, left: ISC values of H44s monolayers in Ussing chambers. Cells were either untreated (control), stimulated with forskolin (fsk.)/IBMX, or fsk./IBMX together with Na2S (5 × 10−5). Shown are values of ISC, which are normalized to the ISC before drug application. A, right: intracellular cAMP concentrations of the same H441 monolayers, which have been used for the Ussing chamber measurements (left). Statistical analysis was performed by nonparametric Mann-Whitney U-test. B: similar experiment as shown in Fig. 7B. PKA activity was indirectly assessed by immunoblotting against phosphorylated and total CREB. Student's unpaired t-test was employed for statistical analysis of the densitometry data (P = 0.8502). *P < 0.05. n = numbers in parentheses.
Therefore, it was investigated whether H2S affects PKA. PKA activity was indirectly measured by immunoblot experiments against the phosphorylated form of cAMP response element binding protein (CREB; Ser-133 phosphorylated), which is a target for active PKA (Fig. 8B). Since H441 cells had high levels of phosphorylated CREB under the employed culture conditions (data not shown), dexamethasone was removed for 24 h prior to experiments. Under these conditions H441 cells maintain the sodium-absorbing phenotype, albeit to a smaller extent (data not shown) and a functional cAMP/PKA signaling system (41). Under these conditions, treatment of cells with forskolin/IBMX (10−5 M/10−4 M; 10 min; apical side) increased CREB phosphorylation. This effect was not altered in the presence of Na2S (2.5 × 10−4 M, apical side). These data indicate that H2S is not inhibiting PKA and acts downstream of this kinase.
H2S inhibits the cAMP-mediated increase in apical ENaC currents.
We hypothesized that H2S prevents the PKA-induced increase in ENaC activity in the apical membrane. H441 monolayers were permeabilized at the basolateral membrane with 1.5 × 10−4 M nystatin (in the presence of the Na+-K+-ATPase inhibitor ouabain; 2 × 10−3 M) and exposed to Na2S (5 × 10−5), forskolin/IBMX, a combination thereof, or remained unstimulated (control). After drug exposure, an apical to basolateral sodium gradient was established to measure sodium channel activity across the apical membrane. Amiloride (10−5 M) was finally applied to the apical side to determine amiloride-sensitive ENaC currents (Fig. 9). Amiloride-sensitive current fractions were normalized to the initial current value before permeabilization to compensate for differences in sodium transport capacity of the monolayers (Fig. 9B). Control monolayers had normalized apical ENaC currents of 0.64 ± 0.04 (n = 5). Monolayers that had been exposed to forskolin/IBMX had significantly increased current fractions of 1.11 ± 0.07 (n = 5; P < 0.001). By contrast, neither monolayers that had been treated with Na2S (0.49 ± 0.06; n = 5) nor a combination of Na2S and forskolin/IBMX (0.66 ± 0.05; n = 5), had significantly different ENaC current fractions with respect to control experiments. These data indicate that Na2S alone did not affect apical membrane ENaC activity, whereas it prevented the stimulation of apical membrane ENaC by forskolin/IBMX.
Fig. 9.

H2S prevents increase in apical membrane ENaC activity by forskolin/IBMX. A: representative current traces of Ussing chamber experiments on basolaterally permeabilized H441 monolayers. The basolateral solution contained nystatin (1.5 × 10−4 M), ouabain (2 × 10−3 M), and high K+. After permeabilization, monolayers were incubated with forskolin/IBMX (fsk./IBMX), Na2S (5 × 10−5 M), a combination thereof (Na2S/fsk./IBMX), or no drugs at all (control), and the apical membrane was subsequently changed to a high Na+ solution (gray bar) to establish an apical to basolateral sodium gradient. Amiloride (ami.; 10−5 M) was applied afterward to determine amiloride-sensitive fractions (Iamiloride) of the apical membrane. B: statistical analysis of data shown in A. Depicted are apical membrane Iamiloride, which were normalized to the initial ISC of intact monolayers before permeabilization (to compensate for alterations in baseline sodium transport capacity). Statistical analysis was performed by one-way ANOVA followed by Tukey's multiple-comparison test. ***P < 0.001. n = number within parentheses.
Endogenous H2S production in lung epithelial cells.
Finally, it was investigated whether the employed Na2S/H2S concentrations are in a range that could be biologically/physiologically relevant. To clarify whether H2S production could, in principle, take place in sodium-transporting epithelial cells, the expression of H2S-generating enzymes was investigated in H441 cells (Fig. 10). Expression of mRNA for all H2S-generating enzymes was detected in H441 cells by RT-PCR, as well as protein production of CSE and CBS by Western blot analysis (Fig. 10, A–C). However, we were not able to detect protein expression of 3-MST by immunoblotting. In addition, H2S production was directly measured with an amperometric sensor in H441 lysates (Fig. 10D). The detected amounts of H2S reflect the concentration range in which inhibition of β-adrenergic agonist-stimulated sodium absorption was observed (Fig. 6E).
Fig. 10.

Expression of H2S-generating enzymes and H2S production in H441 cells. A, left: representative result of a RT-PCR demonstrating expression of two transcript variants of cystathionine-γ-lyase (CSE1, CSE2). A, right: representative immunoblot demonstrating specific bands at the expected size for CSE in three H441 monolayer lysates. Human embryonic kidney (HEK)-293 cell lysates served as a positive control. B, left: representative result of a RT-PCR demonstrating expression of cystathionine-β-synthase (CBS). B, right: representative immunoblot demonstrating specific bands at the expected size for CBS in H441 and HEK cells. C: representative result of a RT-PCR demonstrating expression of 3-mercaptopyruvate sulfurtransferase (3MST). D: representative result (n = 2) of an amperometric measurement of H2S production by H441 lysates. After a control period, l-cysteine (1 × 10−3 M) and pyridoxalphosphate (PLP; 2 × 10−4 M) was applied. In the presence of oxygen (air), H2S concentrations decline.
DISCUSSION
We found that Na2S/H2S significantly decreased baseline lung liquid clearance in rats. This is consistent with the previously described inhibition of pulmonary transepithelial sodium absorption (which is the osmotic driving force for liquid absorption) by H2S (2, 6, 24). Furthermore, H2S seems to completely abolish the stimulation of lung liquid clearance by the β-adrenergic receptor agonist terbutaline. β-adrenergic agents have long been known to stimulate absorption of luminal lung liquid in both the fetus (13, 61, 83) and the postnatal animal (11, 69), and more recent reports show the same effect of terbutaline, i.e., stimulating amiloride-sensitive sodium absorption (9, 89). It is well established that β-adrenergic receptor agonists increase the abundance of ENaCs in the apical membrane of pulmonary epithelial cells by a cAMP-dependent pathway (8, 90). This is consistent with the observed increase in amiloride-sensitive sodium absorption and, consequently, with the clearance of lung liquid in response to terbutaline. Na2S decreased baseline liquid clearance but—at the same concentration—appears to completely inhibit β-adrenergic agonist stimulated liquid clearance. However, the rat preparation measures net liquid movement across the lung epithelium, so we cannot be absolutely certain that the effect of H2S on terbutaline in this particular preparation may not be the sum of effects of these two agents on separate mechanisms of liquid transport. The fact that Na2S has no additional effect to that of amiloride alone indicates, nevertheless, that it is affecting transport involving ENaC. The evidence from the experiments on Xenopus lung preparations, in which Na2S alone did not significantly inhibit amiloride-sensitive baseline ENaC currents, whereas the stimulatory effect of terbutaline was lost, suggests that H2S is able to inhibit the action of terbutaline independently of baseline sodium transport or ENaC activity. By contrast, Na2S treatment did decrease amiloride-sensitive ENaC currents in H441 monolayers. We have previously shown that this is due to inhibition of Na+-K+-ATPase activity and basolateral potassium channels (6, 24). The discrepancy between Xenopus lungs and H441 monolayers is likely due to different sensitivities of the preparation to sulfur salts. The IC50 for Na2S in H441 cells is 1.46 × 10−4 M (6), whereas in Xenopus lungs, the IC50 for comparable sulfur salt (NaHS) is 1.18 × 10−3 M (24). Indirect effects on cAMP-stimulated sodium transport due to basolateral transport inhibition by H2S could be excluded in H441 monolayers, which were basolaterally permeabilized in the presence of the Na+-K+-ATPase inhibitor ouabain and a high-potassium solution in the basolateral Ussing chamber compartment. Under these conditions, H2S did not affect baseline amiloride-sensitive currents (i.e., apical membrane ENaC). This is consistent with previous observations that H2S does not affect the baseline activity of membrane-located ENaC in H441 cells or heterologously expressed ENaC in Xenopus oocytes (6). The stimulation of apical membrane ENaC by cAMP-elevating drugs, however, was again prevented by Na2S, indicating that H2S specifically prevents the activation of ENaC in response to β-adrenergic agonists or cAMP-elevating drugs.
The signaling cascade underlying β-adrenergic receptor activation and stimulation of ENaC trafficking includes the formation of cAMP by AC and activation of PKA (8, 84). This is consistent with the observed increase in amiloride-sensitive ENaC currents in H441 cells, which have been treated with the cAMP-elevating drugs forskolin and IBMX. Again, this stimulation of ENaC trafficking in H441 cells was inhibited by Na2S. The lack of an effect of terbutaline in these cells further indicates that H2S rather indirectly affects β-adrenergic receptor activation or its coupled G proteins in rat lungs and native lung epithelia. Interestingly, despite the blocked ENaC trafficking in the presence of H2S, we could not detect any impairment of cellular energy metabolism or AC-mediated cAMP formation and PKA activation. This suggests that H2S is likely acting downstream of PKA activity. Consistent with the generally observed secretory epithelial response to H2S, the uncoupling of ENaC stimulation by PKA may allow H2S to stimulate e.g., CFTR-mediated chloride secretion by cAMP/PKA-mediated signaling while simultaneously preventing enhanced sodium absorption. However, in the perfused lung preparation the observation that Na2S has no additional effect on lung liquid movement than that of amiloride alone suggests that the action of H2S is solely on the functioning of ENaC; in this preparation, at least there is no evidence of an increased stimulation of chloride secretion or an effect of H2S on other intracellular mechanisms. Furthermore, a potential contribution of CFTR or other chloride channels to the observed effects of H2S on sodium absorption can be excluded since there is no electrochemical gradient for chloride fluxes in basolaterally permeabilized H441 monolayers.
An important point that needs to be addressed is the potential toxicity of H2S. Submicromolar concentrations of H2S inhibit cytochrome c oxidase and, hence, cellular respiration, but 10−5 M is needed when it is located in intact mitochondria, and in many instances, 10−4 M H2S or considerably higher concentrations are needed to inhibit oxidative phosphorylation in intact cells (22). We did not detect any changes in intracellular ATP concentrations or activation of the cellular stress sensor AMPK [which would respond to changes in cellular respiration or production of reactive oxygen species, such as superoxide (36)]. Furthermore, there was no impairment of transepithelial resistances in epithelial preparations or paracellular leakage in rat lung experiments (which would lead to an exponential increase in lung liquid volume; data not shown). Therefore, it is unlikely that the observed effects of H2S are attributed to its toxicity.
Nevertheless, the precise cellular target for H2S remains unclear. It has been suggested that there is a pool of subapically located ENaC-containing vesicles, which rapidly translocate to the apical epithelial plasma membrane in response to cAMP/PKA stimulation (23, 90). In basolaterally permeabilized H441 monolayers, H2S was able to prevent the stimulation of apical membrane ENaC by cAMP-elevating drugs. Given that under these conditions Na2S did not significantly affect baseline ENaC activity, H2S may interact with the delivery of ENaC channels to the apical epithelial membrane. However, the events that link cAMP-dependent PKA activation to stimulated ENaC trafficking to the apical membrane have yet to be elucidated (for excellent review, see Ref. 8). Potential targets for H2S may be proteins that regulate the trafficking or membrane fusion of channel-containing vesicles. Sitdikova and colleagues (53) suggested that H2S may regulate vesicular trafficking by thiol-dependent modifications of SNARE (soluble N-ethylmaleimide-sensitive-factor attachment receptor) complexes or associated proteins (53). Consistent with this idea, a recent study demonstrated reduced granule exocytosis in H2S-exposed platelets, which may be due to interference of H2S with SNARE-mediated vesicle exocytosis (34). Aside from the trafficking machinery, the translocation of ENaC-containing vesicles in lung epithelial cells may involve additional, yet unidentified, pathways involving protein tyrosine kinase (8, 51, 90), and H2S has recently been implicated in the modulation of tyrosine kinase-dependent signaling pathways (32, 78).
In conclusion, the present study demonstrates that H2S inhibits the proabsorptive effect of β-adrenergic agonists on lung liquid clearance by preventing the increase in ENaC-mediated sodium absorption. However, it has to be questioned whether or not H2S plays a significant role under physiological conditions. The expression of H2S-generating enzymes has been described in rat lungs (48). As indicated in the present study, sodium-transporting lung epithelial cells also express H2S-generating enzymes. These sodium-absorbing cells have the capacity to produce H2S in concentrations that reflect the range of the observed inhibition of ENaC trafficking. Although these data confirm that the H2S concentrations that were employed in this study are in a biologically relevant range, the contribution of endogenously generated H2S to ion transport regulation in the lung needs to be demonstrated. Under normal physiological conditions, H2S concentrations are low (56, 58), and the future challenge will be the identification of physiological stimulators of endogenous H2S production in the lung.
Perspectives and Significance
The findings from this study have several implications for lung physiology and pathophysiology: 1) H2S might play an important role in the hypoxic lung. For example, modulation of endogenous H2S generation affects acute hypoxic vasoconstriction in the perfused rat lung (48). As also indicated in this study, significant amounts of H2S can be generated under hypoxic conditions (57, 60). We have shown that sodium-absorbing epithelia have the capacity to produce H2S in concentrations that reflect the range of the observed inhibition of ENaC. H2S might, thus, contribute to hypoxic inhibition of sodium transport in pulmonary epithelial cells (35, 47, 65, 77, 82). 2) In low concentrations, H2S exerts anti-inflammatory, anti-oxidant, and vasoactive properties and is currently being evaluated as a potential therapeutic option in animal models of various lung diseases (15, 49, 81), including acute lung injury (ALI) (25–27, 29, 64). Most of those studies investigated effects upon systemic administration of H2S-liberating molecules or sulfur salts, such as Na2S/NaHS. A study by Toombs et al. (80) demonstrated significant H2S concentrations in the exhaled air of humans upon systemic administration of Na2S. The authors detected H2S levels of up to 100 ppb (80), which correspond to low micromolar H2S concentrations. This likely reflects an alveolar H2S content that is in the observed effective concentration range for inhibition of cAMP-stimulated ENaC activity. Impaired lung liquid clearance and the development of pulmonary edema are characteristics of patients with ALI (87). An impaired β-adrenergic agonist-induced stimulation of lung liquid clearance in the presence of H2S is an important aspect to be considered when the therapeutic potential of H2S or H2S-liberating molecules is evaluated. 3) H2S can be generated by the lung pathogens Pseudomonas aeruginosa, Bacillus anthracis, or Staphylococcus aureus (72). Interestingly, chronic Pseudomonas aeruginosa lung infections abrogate the proabsorptive effect of β-adrenergic stimulation on lung liquid clearance (12). Bacteria-derived H2S may, thus, impair the proabsorptive capacity of lung epithelia in lung infections.
GRANTS
This work is supported by a grant from the German Research Foundation (DFG; AL1453/1-2) to M. Althaus, as well as grants to D. V. Walters (the Dewey Bequest) and to K. R. Olson (NSF IOS 1051627).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: A.M.A., J.-P.B., A.R.B., M.C.O.-W., L.W., K.R.O., D.V.W., and M.A. performed experiments; A.M.A., J.-P.B., A.R.B., M.C.O.-W., L.W., K.R.O., D.V.W., and M.A. analyzed data; A.M.A., J.-P.B., A.R.B., M.C.O.-W., L.W., K.R.O., D.V.W., and M.A. approved final version of manuscript; A.R.B., M.C.O.-W., K.R.O., D.V.W., and M.A. interpreted results of experiments; A.R.B., M.C.O.-W., K.R.O., D.V.W., and M.A. prepared figures; K.R.O., D.V.W., and M.A. conception and design of research; K.R.O., D.V.W., and M.A. drafted manuscript; M.A. edited and revised manuscript.
ACKNOWLEDGMENTS
The authors would like to thank Mirjam Buss and Anja Schnecko for technical assistance, Franziska Rinker and Franziska Sennert for help with some experiments, and Pawel Piotr Szczesniak for helpful comments on the manuscript. David Lovell is acknowledged for professional advice on statistical analysis.
REFERENCES
- 1.Alexandrou D, Walters DV. The role of Cl− in the regulation of ion and liquid transport in the intact alveolus during β-adrenergic stimulation. Exp Physiol 98: 576–584, 2013. [DOI] [PubMed] [Google Scholar]
- 2.Althaus M. Gasotransmitters: novel regulators of epithelial Na+ transport? Front Physiol 3: 83, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Althaus M. ENaC inhibitors and airway re-hydration in cystic fibrosis: state of the art. Curr Mol Pharmacol 6: 3–12, 2013. [DOI] [PubMed] [Google Scholar]
- 4.Althaus M, Clauss WG, Fronius M. Amiloride-sensitive sodium channels and pulmonary edema. Pulm Med 2011: 830320, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Althaus M, Pichl A, Clauss WG, Seeger W, Fronius M, Morty RE. Nitric oxide inhibits highly selective sodium channels and the Na+/K+-ATPase in H441 cells. Am J Respir Cell Mol Biol 44: 53–65, 2011. [DOI] [PubMed] [Google Scholar]
- 6.Althaus M, Urness K, Clauss W, Baines D, Fronius M. The gasotransmitter hydrogen sulphide decreases Na+ transport across pulmonary epithelial cells. Br J Pharmacol 166: 1946–1963, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Azad AK, Rauh R, Vermeulen F, Jaspers M, Korbmacher J, Boissier B, Bassinet L, Fichou Y, Des Georges M, Stanke F, Boeck de K, Dupont L, Balascáková M, Hjelte L, Lebecque P, Radojkovic D, Castellani C, Schwartz M, Stuhrmann M, Schwarz M, Skalicka V, Monestrol de I, Girodon E, Férec C, Claustres M, Tümmler B, Cassiman J, Korbmacher C, Cuppens H. Mutations in the amiloride-sensitive epithelial sodium channel in patients with cystic fibrosis-like disease. Hum Mutat 30: 1093–1103, 2009. [DOI] [PubMed] [Google Scholar]
- 8.Baines D. Kinases as targets for ENaC regulation. Curr Mol Pharmacol 6: 50–64, 2013. [DOI] [PubMed] [Google Scholar]
- 9.Baxendale-Cox LM. Terbutaline increases open channel density of epithelial sodium channel (ENaC) in distal lung. Respir Physiol 116: 1–8, 1999. [DOI] [PubMed] [Google Scholar]
- 10.Berger J, Hardt M, Clauss WG, Fronius M. Basolateral Cl− uptake mechanisms in Xenopus laevis lung epithelium. Am J Physiol Regul Integr Comp Physiol 299: R92–R100, 2010. [DOI] [PubMed] [Google Scholar]
- 11.Berthiaume Y, Staub NC, Matthay MA. Beta-adrenergic agonists increase lung liquid clearance in anesthetized sheep. J Clin Invest 79: 335–343, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boyer S, Faure K, Ader F, Husson MO, Kipnis E, Prangere T, Leroy X, Guery BP. Chronic pneumonia with Pseudomonas aeruginosa and impaired alveolar fluid clearance. Respir Res 6: 17, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown MJ, Olver RE, Ramsden CA, Strang LB, Walters DV. Effects of adrenaline and of spontaneous labour on the secretion and absorption of lung liquid in the fetal lamb. J Physiol 344: 137–152, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, Rossier BC. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 367: 463–467, 1994. [DOI] [PubMed] [Google Scholar]
- 15.Cao H, Zhou X, Zhang J, Huang X, Zhai Y, Zhang X, Chu L. Hydrogen sulfide protects against bleomycin-induced pulmonary fibrosis in rats by inhibiting NF-κB expression and regulating Th1/Th2 balance. Toxicol Lett 224: 387–394, 2014. [DOI] [PubMed] [Google Scholar]
- 16.Carlton DP, Cummings JJ, Chapman DL, Poulain FR, Bland RD. Ion transport regulation of lung liquid secretion in foetal lambs. J Dev Physiol 17: 99–107, 1992. [PubMed] [Google Scholar]
- 17.Chapman DL, Carlton DP, Cummings JJ, Poulain FR, Bland RD. Intrapulmonary terbutaline and aminophylline decrease lung liquid in fetal lambs. Pediatr Res 29: 357–361, 1991. [DOI] [PubMed] [Google Scholar]
- 18.Chen L, Song W, Davis IC, Shrestha K, Schwiebert E, Sullender WM, Matalon S. Inhibition of Na+ transport in lung epithelial cells by respiratory syncytial virus infection. Am J Respir Cell Mol Biol 40: 588–600, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Collman JP, Ghosh S, Dey A, Decréau RA. Using a functional enzyme model to understand the chemistry behind hydrogen sulfide induced hibernation. Proc Natl Acad Sci USA 106: 22,090–22,095, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Degasperi A, Birtwistle MR, Volinsky N, Rauch J, Kolch W, Kholodenko BN. Evaluating strategies to normalise biological replicates of Western blot data. PLos One 9: e87293, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeLeon ER, Stoy GF, Olson KR. Passive loss of hydrogen sulfide in biological experiments. Anal Biochem 421: 203–207, 2012. [DOI] [PubMed] [Google Scholar]
- 22.Dombkowski RA, Russell MJ, Schulman AA, Doellman MM, Olson KR. Vertebrate phylogeny of hydrogen sulfide vasoactivity. Am J Physiol Regul Integr Comp Physiol 288: R243–R252, 2005. [DOI] [PubMed] [Google Scholar]
- 23.Edinger RS, Bertrand CA, Rondandino C, Apodaca GA, Johnson JP, Butterworth MB. The epithelial sodium channel (ENaC) establishes a trafficking vesicle pool responsible for its regulation. PLos One 7: e46593, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Erb A, Althaus M. Actions of hydrogen sulfide on sodium transport processes across native distal lung epithelia (Xenopus laevis). PLos One 9: e100971, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Esechie A, Enkhbaatar P, Traber DL, Jonkam C, Lange M, Hamahata A, Djukom C, Whorton EB, Hawkins HK, Traber LD, Szabo C. Beneficial effect of a hydrogen sulphide donor (sodium sulfide) in an ovine model of burn- and smoke-induced acute lung injury. Br J Pharmacol 158: 1442–1453, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Faller S, Ryter SW, Choi AMK, Loop T, Schmidt R, Hoetzel A. Inhaled hydrogen sulfide protects against ventilator-induced lung injury. Anesthesiology 113: 104–115, 2010. [DOI] [PubMed] [Google Scholar]
- 27.Faller S, Zimmermann KK, Strosing KM, Engelstaedter H, Buerkle H, Schmidt R, Spassov SG, Hoetzel A. Inhaled hydrogen sulfide protects against lipopolysaccharide-induced acute lung injury in mice. Med Gas Res 2: 26, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fischer H, Clauss W. Regulation of Na+ channels in frog lung epithelium: a target tissue for aldosterone action. Pflügers Arch 416: 62–67, 1990. [DOI] [PubMed] [Google Scholar]
- 29.Francis RC, Vaporidi K, Bloch KD, Ichinose F, Zapol WM. Protective and detrimental effects of sodium sulfide and hydrogen sulfide in murine ventilator-induced lung injury. Anesthesiology 115: 1012–1021, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fronius M, Berk A, Clauss W, Schnizler M. Ion transport across Xenopus alveolar epithelium is regulated by extracellular ATP, UTP and adenosine. Respir Physiol Neurobiol 139: 133–144, 2004. [DOI] [PubMed] [Google Scholar]
- 31.Garty H, Palmer LG. Epithelial sodium channels: function, structure, and regulation. Physiol Rev 77: 359–396, 1997. [DOI] [PubMed] [Google Scholar]
- 32.Ge S, Zhao M, Wu D, Chen Y, Wang Y, Zhu J, Cai W, Zhu Y, Zhu Y. Hydrogen sulfide targets EGFR Cys797/Cys798 residues to induce Na+/K+-ATPase endocytosis and inhibition in renal tubular epithelial cells and increases sodium excretion in chronic salt-loaded rats. Antioxid Redox Signal 21: 2061–2082, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gibson G, Cummings J, Macfarlane G. Growth and activities of sulphate-reducing bacteria in gut contents of healthy subjects and patients with ulcerative colitis. FEMS Microbiol Lett 86: 103–112, 1991. [Google Scholar]
- 34.Grambow E, Mueller-Graf F, Delyagina E, Frank M, Kuhla A, Vollmar B. Effect of the hydrogen sulfide donor GYY4137 on platelet activation and microvascular thrombus formation in mice. Platelets 25: 166–174, 2014. [DOI] [PubMed] [Google Scholar]
- 35.Güney S, Schuler A, Ott A, Höschele S, Zügel S, Baloglu E, Bärtsch P, Mairbäurl H. Dexamethasone prevents transport inhibition by hypoxia in rat lung and alveolar epithelial cells by stimulating activity and expression of Na+-K+-ATPase and epithelial Na+ channels. Am J Physiol Lung Cell Mol Physiol 293: L1332–L1338, 2007. [DOI] [PubMed] [Google Scholar]
- 36.Han Y, Wang Q, Song P, Zhu Y, Zou M. Redox regulation of the AMP-activated protein kinase. PLos One 5: e15420, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hennig B, Diener M. Actions of hydrogen sulphide on ion transport across rat distal colon. Br J Pharmacol 158: 1263–1275, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hollenhorst MI, Richter K, Fronius M. Ion transport by pulmonary epithelia. J Biomed Biotechnol 2011: 174306, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hooper SB, Kitchen MJ, Wallace MJ, Yagi N, Uesugi K, Morgan MJ, Hall C, Siu KKW, Williams IM, Siew M, Irvine SC, Pavlov K, Lewis RA. Imaging lung aeration and lung liquid clearance at birth. FASEB J 21: 3329–3337, 2007. [DOI] [PubMed] [Google Scholar]
- 40.Hummler E, Barker P, Gatzy J, Beermann F, Verdumo C, Schmidt A, Boucher R, Rossier BC. Early death due to defective neonatal lung liquid clearance in α-ENaC-deficient mice. Nat Genet 12: 325–328, 1996. [DOI] [PubMed] [Google Scholar]
- 41.Ismail NAS, Baines DL, Wilson SM. The phosphorylation of endogenous Nedd4-2 In Na+-absorbing human airway epithelial cells. Eur J Pharmacol 732: 32–42, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kerem E, Bistritzer T, Hanukoglu A, Hofmann T, Zhou Z, Bennett W, MacLaughlin E, Barker P, Nash M, Quittell L, Boucher R, Knowles MR. Pulmonary epithelial sodium-channel dysfunction and excess airway liquid in pseudohypoaldosteronism. N Engl J Med 341: 156–162, 1999. [DOI] [PubMed] [Google Scholar]
- 43.Kimura H. Hydrogen sulfide induces cyclic AMP and modulates the NMDA receptor. Biochem Biophys Res Commun 267: 129–133, 2000. [DOI] [PubMed] [Google Scholar]
- 44.Kimura H. Metabolic turnover of hydrogen sulfide. Front Physiol 3: 101, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koefoed-Johnsen V, Ussing HH. The nature of the frog skin potential. Acta Physiol Scand 42: 298–308, 1958. [DOI] [PubMed] [Google Scholar]
- 46.Korbmacher JP, Michel C, Neubauer D, Thompson K, Mizaikoff B, Frick M, Dietl P, Wittekindt OH. Amiloride-sensitive fluid resorption in NCI-H441 lung epithelia depends on an apical Cl− conductance. Physiol Rep 2: e00201, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Loeh B, Baloglu E, Ke A, Bärtsch P, Mairbäurl H. β2-adrenergic stimulation blunts inhibition of epithelial ion transport by hypoxia of rat alveolar epithelial cells. Cell Physiol Biochem 25: 123–134, 2010. [DOI] [PubMed] [Google Scholar]
- 48.Madden JA, Ahlf SB, Dantuma MW, Olson KR, Roerig DL. Precursors and inhibitors of hydrogen sulfide synthesis affect acute hypoxic pulmonary vasoconstriction in the intact lung. J Appl Physiol 112: 411–418, 2012. [DOI] [PubMed] [Google Scholar]
- 49.Madurga A, Mizikova I, Ruiz-Camp J, Vadász I, Herold S, Mayer K, Fehrenbach H, Seeger W, Morty RE. Systemic hydrogen sulfide administration partially restores normal alveolarization in an experimental animal model of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 306: L684–L697, 2014. [DOI] [PubMed] [Google Scholar]
- 50.Mall M, Grubb BR, Harkema JR, O'Neal WK, Boucher RC. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat Med 10: 487–493, 2004. [DOI] [PubMed] [Google Scholar]
- 51.Marunaka Y, Niisato N, Taruno A, Ohta M, Miyazaki H, Hosogi S, Nakajima K, Kusuzaki K, Ashihara E, Nishio K, Iwasaki Y, Nakahari T, Kubota T. Regulation of epithelial sodium transport via epithelial Na+ channel. J Biomed Biotechnol 2011: 978196, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mimoun S, Andriamihaja M, Chaumontet C, Atanasiu C, Benamouzig R, Blouin JM, Tomé D, Bouillaud F, Blachier F. Detoxification of H2S by differentiated colonic epithelial cells: implication of the sulfide oxidizing unit and of the cell respiratory capacity. Antioxid Redox Signal 17: 1–10, 2012. [DOI] [PubMed] [Google Scholar]
- 53.Mitrukhina OB, Yakovlev AV, Sitdikova GF. The effects of hydrogen sulfide on the processes of exo- and endocytosis of synaptic vesicles in the mouse motor nerve endings. Biochem Moscow Suppl Ser A7: 170–173, 2013. [Google Scholar]
- 54.Muzaffar S, Jeremy JY, Sparatore A, Del Soldato P, Angelini GD, Shukla N. H2S-donating sildenafil (ACS6) inhibits superoxide formation and gp91phox expression in arterial endothelial cells: role of protein kinases A and G. Br J Pharmacol 155: 984–994, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nagpure BV, Bian J. Hydrogen sulfide inhibits A2A adenosine receptor agonist induced β-amyloid production in SH-SY5Y neuroblastoma cells via a cAMP-dependent pathway. PLos One 9: e88508, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Olson KR. The therapeutic potential of hydrogen sulfide: separating hype from hope. Am J Physiol Regul Integr Comp Physiol 301: R297–R312, 2011. [DOI] [PubMed] [Google Scholar]
- 57.Olson KR. Hydrogen sulfide as an oxygen sensor. Clin Chem Lab Med 51: 623–632, 2013. [DOI] [PubMed] [Google Scholar]
- 58.Olson KR. Hydrogen sulfide: both feet on the gas and none on the brake? Front Physiol 4: 2, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Olson KR, DeLeon ER, Liu F. Controversies and conundrums in hydrogen sulfide biology. Nitric Oxide 41: 11–26, 2014. [DOI] [PubMed] [Google Scholar]
- 60.Olson KR, Whitfield NL, Bearden SE, St Leger J, Nilson E, Gao Y, Madden JA. Hypoxic pulmonary vasodilation: a paradigm shift with a hydrogen sulfide mechanism. Am J Physiol Regul Integr Comp Physiol 298: R51–R60, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Olver RE, Ramsden CA, Strang LB, Walters DV. The role of amiloride-blockable sodium transport in adrenaline-induced lung liquid reabsorption in the fetal lamb. J Physiol 376: 321–340, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Olver RE, Strang LB. Ion fluxes across the pulmonary epithelium and the secretion of lung liquid in the foetal lamb. J Physiol 241: 327–357, 1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Olver RE, Walters DV, Wilson SM. Developmental regulation of lung liquid transport. Annu Rev Physiol 66: 77–101, 2004. [DOI] [PubMed] [Google Scholar]
- 64.Otulakowski G, Kavanagh BP. Hydrogen sulfide in lung injury: therapeutic hope from a toxic gas? Anesthesiology 113: 4–6, 2010. [DOI] [PubMed] [Google Scholar]
- 65.Planès C, Escoubet B, Blot-Chabaud M, Friedlander G, Farman N, Clerici C. Hypoxia downregulates expression and activity of epithelial sodium channels in rat alveolar epithelial cells. Am J Respir Cell Mol Biol 17: 508–518, 1997. [DOI] [PubMed] [Google Scholar]
- 66.Pouokam E, Diener M. Mechanisms of actions of hydrogen sulphide on rat distal colonic epithelium. Br J Pharmacol 162: 392–404, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pouokam E, Steidle J, Diener M. Regulation of colonic ion transport by gasotransmitters. Biol Pharm Bull 34: 789–793, 2011. [DOI] [PubMed] [Google Scholar]
- 68.Ramminger SJ, Richard K, Inglis SK, Land SC, Olver RE, Wilson SM. A regulated apical Na+ conductance in dexamethasone-treated H441 airway epithelial cells. Am J Physiol Lung Cell Mol Physiol 287: L411–L419, 2004. [DOI] [PubMed] [Google Scholar]
- 69.Ramsden CA, Markiewicz M, Walters DV, Gabella G, Parker KA, Barker PM, Neil HL. Liquid flow across the epithelium of the artificially perfused lung of fetal and postnatal sheep. J Physiol 448: 579–597, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rashid S, Heer JK, Garle MJ, Alexander SPH, Roberts RE. Hydrogen sulphide-induced relaxation of porcine peripheral bronchioles. Br J Pharmacol 168: 1902–1910, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reiffenstein RJ, Hulbert WC, Roth SH. Toxicology of hydrogen sulfide. Annu Rev Pharmacol Toxicol 32: 109–134, 1992. [DOI] [PubMed] [Google Scholar]
- 72.Shatalin K, Shatalina E, Mironov A, Nudler E. H2S: a universal defense against antibiotics in bacteria. Science 334: 986–990, 2011. [DOI] [PubMed] [Google Scholar]
- 73.Siew ML, Wallace MJ, Allison BJ, Kitchen MJ, te Pas AB, Islam MS, Lewis RA, Fouras A, Yagi N, Uesugi K, Hooper SB. The role of lung inflation and sodium transport in airway liquid clearance during lung aeration in newborn rabbits. Pediatr Res 73: 443–449, 2013. [DOI] [PubMed] [Google Scholar]
- 74.Sommer D, Bogdan R, Berger J, Peters DM, Morty RE, Clauss WG, Fronius M. CFTR-dependent Cl− secretion in Xenopus laevis lung epithelium. Respir Physiol Neurobiol 158: 97–106, 2007. [DOI] [PubMed] [Google Scholar]
- 75.Soukup B, Benjamin A, Orogo-Wenn M, Walters D. Physiological effect of protein kinase C on ENaC-mediated lung liquid regulation in the adult rat lung. Am J Physiol Lung Cell Mol Physiol 302: L133–L139, 2012. [DOI] [PubMed] [Google Scholar]
- 76.Stipanuk MH, Beck PW. Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem J 206: 267–277, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tan CD, Smolenski RT, Harhun MI, Patel HK, Ahmed SG, Wanisch K, Yáñez-Muñoz RJ, Baines DL. AMP-activated protein kinase (AMPK)-dependent and -independent pathways regulate hypoxic inhibition of transepithelial Na+ transport across human airway epithelial cells. Br J Pharmacol 167: 368–382, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tao B, Liu S, Zhang C, Fu W, Cai W, Wang Y, Shen Q, Wang M, Chen Y, Zhang L, Zhu Y, Zhu Y. VEGFR2 functions as an H2S-targeting receptor protein kinase with its novel Cys1045-Cys1024 disulfide bond serving as a specific molecular switch for hydrogen sulfide actions in vascular endothelial cells. Antioxid Redox Signal 19: 448–464, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thomas CP, Campbell JR, Wright PJ, Husted RF. cAMP-stimulated Na+ transport in H441 distal lung epithelial cells: role of PKA, phosphatidylinositol 3-kinase, and sgk1. Am J Physiol Lung Cell Mol Physiol 287: L843–L851, 2004. [DOI] [PubMed] [Google Scholar]
- 80.Toombs CF, Insko MA, Wintner EA, Deckwerth TL, Usansky H, Jamil K, Goldstein B, Cooreman M, Szabo C. Detection of exhaled hydrogen sulphide gas in healthy human volunteers during intravenous administration of sodium sulphide. Br J Clin Pharmacol 69: 626–636, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vadivel A, Alphonse RS, Ionescu L, Machado DS, O'Reilly M, Eaton F, Haromy A, Michelakis ED, Thébaud B. Exogenous hydrogen sulfide (H2S) protects alveolar growth in experimental O2-induced neonatal lung injury. PLoS One 9: e90965, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vivona ML, Matthay M, Chabaud MB, Friedlander G, Clerici C. Hypoxia reduces alveolar epithelial sodium and fluid transport in rats: reversal by β-adrenergic agonist treatment. Am J Respir Cell Mol Biol 25: 554–561, 2001. [DOI] [PubMed] [Google Scholar]
- 83.Walters DV, Olver RE. The role of catecholamines in lung liquid absorption at birth. Pediatr Res 12: 239–242, 1978. [DOI] [PubMed] [Google Scholar]
- 84.Walters DV, Ramsden CA, Olver RE. Dibutyryl cAMP induces a gestation-dependent absorption of fetal lung liquid. J Appl Physiol 68: 2054–2059, 1990. [DOI] [PubMed] [Google Scholar]
- 85.Wang R. Two's company, three's a crowd: can H2S be the third endogenous gaseous transmitter? FASEB J 16: 1792–1798, 2002. [DOI] [PubMed] [Google Scholar]
- 86.Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev 92: 791–896, 2012. [DOI] [PubMed] [Google Scholar]
- 87.Ware LB, Matthay MA. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med 163: 1376–1383, 2001. [DOI] [PubMed] [Google Scholar]
- 88.Whitfield NL, Kreimier EL, Verdial FC, Skovgaard N, Olson KR. Reappraisal of H2S/sulfide concentration in vertebrate blood and its potential significance in ischemic preconditioning and vascular signaling. Am J Physiol Regul Integr Comp Physiol 294: R1930–R1937, 2008. [DOI] [PubMed] [Google Scholar]
- 89.Wilkinson WJ, Benjamin AR, Proost de I, Orogo-Wenn MC, Yamazaki Y, Staub O, Morita T, Adriaensen D, Riccardi D, Walters DV, Kemp PJ. Alveolar epithelial CNGA1 channels mediate cGMP-stimulated, amiloride-insensitive, lung liquid absorption. Pflügers Arch 462: 267–279, 2011. [DOI] [PubMed] [Google Scholar]
- 90.Woollhead AM, Baines DL. Forskolin-induced cell shrinkage and apical translocation of functional enhanced green fluorescent protein-human alphaENaC in H441 lung epithelial cell monolayers. J Biol Chem 281: 5158–5168, 2006. [DOI] [PubMed] [Google Scholar]
- 91.Yang H, Wu Z, Wood M, Whiteman M, Bian J. Hydrogen sulfide attenuates opioid dependence by suppression of adenylate cyclase/cAMP pathway. Antioxid Redox Signal 20: 31–41, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]