

Summary
SARM1 function and nicotinamide mononucleotide adenylyltransferase 2 (NMNAT2) loss both promote axon degeneration, but their relative relationship in the process is unknown. Here, we show that NMNAT2 loss and resultant changes to NMNAT metabolites occur in injured SARM1-deficient axons despite their delayed degeneration and that axon degeneration specifically induced by NMNAT2 depletion requires SARM1. Strikingly, SARM1 deficiency also corrects axon outgrowth in mice lacking NMNAT2, independently of NMNAT metabolites, preventing perinatal lethality. Furthermore, NAMPT inhibition partially restores outgrowth of NMNAT2-deficient axons, suggesting that the NMNAT substrate, NMN, contributes to this phenotype. NMNAT2-depletion-dependent degeneration of established axons and restricted extension of developing axons are thus both SARM1 dependent, and SARM1 acts either downstream of NMNAT2 loss and NMN accumulation in a linear pathway or in a parallel branch of a convergent pathway. Understanding the pathway will help establish relationships with other modulators of axon survival and facilitate the development of effective therapies for axonopathies.
Graphical Abstract
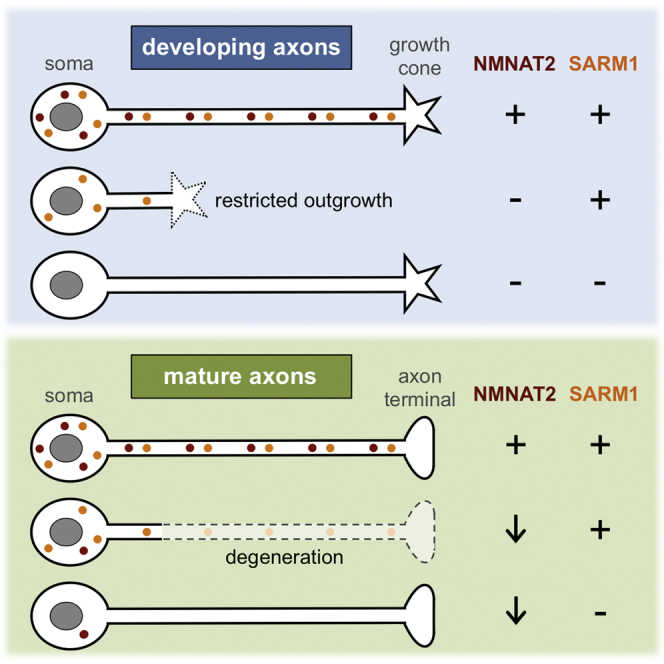
Highlights
-
•
SARM1 is required for WLDS-sensitive axon degeneration caused by NMNAT2 depletion
-
•
SARM1 deficiency rescues developmental axon defects caused by a lack of NMNAT2
-
•
Mice lacking NMNAT2 and SARM1 are viable
-
•
Lowering NMN by inhibiting NAMPT partially rescues growth of NMNAT2-deficient axons
Gilley et al. find that SARM1 promotes axon degeneration after NMNAT2 depletion and limits outgrowth of axons constitutively lacking NMNAT2. The NMNAT substrate NMN also appears to be pro-degenerative in both situations. Restricted outgrowth of NMNAT2-deficient axons is thus mechanistically related to the degeneration of established axons.
Introduction
Axon degeneration is a critical, early event in many neurodegenerative disorders. Studies of WLDS, an aberrant fusion protein with critical nicotinamide mononucleotide adenylyltransferase (NMNAT) activity responsible for the slow Wallerian degeneration phenotype, indicate this often shares molecular features with injury-induced axon degeneration (Conforti et al., 2014). New, endogenous regulators of Wallerian degeneration have emerged, so establishing interactions among them will be important for developing appropriate therapies (Conforti et al., 2014; Neukomm and Freeman, 2014; Pease and Segal, 2014). While a conserved pathway is emerging, the relationship between NMNAT2 and sterile alpha and TIR motif containing 1 (SARM1), key regulators of axon degeneration with opposing influences, is still unknown (Conforti et al., 2014).
NMNAT2 is one of three naturally occurring mammalian NMNATs. Like WLDS, each robustly delays Wallerian degeneration when re-localized or sufficiently overexpressed (Mack et al., 2001; Milde et al., 2013; Sasaki et al., 2009; Yahata et al., 2009), but only depletion of NMNAT2, not NMNAT1 or NMNAT3, triggers WLDS-sensitive neurite degeneration in neuronal cultures (Gilley and Coleman, 2010). This suggests NMNAT2 is the isoform that contributes most to axon survival in a physiological context. Drosophila NMNAT ortholog, dNmnat, appears to have a similar role in flies (Fang et al., 2012). Early loss of short-lived NMNAT2 in injured axons, prior to frank degeneration, has thus been proposed as a trigger for WLDS-sensitive axon degeneration and the point of WLDS influence (Conforti et al., 2014; Gilley and Coleman, 2010). This is supported by studies that show that a critical requirement for PHR1/Highwire ubiquitin ligase during Wallerian degeneration may be linked to its regulation of NMNAT2/dNmnat turnover (Babetto et al., 2013; Xiong et al., 2012) and the proposal that accumulation of the NMNAT substrate NMN, a predicted consequence of NMNAT2 loss, acts as a pro-degenerative signal (Di Stefano et al., 2014). Severe truncation of peripheral nerve axons and CNS axon tracts in NMNAT2-deficient embryos, likely responsible for their perinatal lethality, is also consistent with a critical axon survival function for NMNAT2 in vivo (Gilley et al., 2013; Hicks et al., 2012). Importantly, though, this developmental phenotype appears to be primarily due to an initial outgrowth defect rather than dying-back degeneration of established axons (Gilley et al., 2013).
SARM1, in contrast, has a pro-degenerative function. It is required for the normal, rapid progression of degeneration after injury and other WLDS-sensitive insults (Gerdts et al., 2013; Osterloh et al., 2012). Multimerization, mediated by its SAM (sterile α motif) domain, appears to be required for a pro-degenerative function dependent on its TIR (Toll-interleukin-1 receptor) domain (Gerdts et al., 2013). Crucially, SARM1 depletion is currently the only known manipulation that delays WLDS-sensitive axon degeneration as robustly as expression of WLDS or other NMNATs.
In this study, we used axon injury in nerves and neurons from Sarm1−/− mice to establish the relationship between NMNAT2 loss and a SARM1-dependent function during WLDS-sensitive degeneration. Remarkably, we also find that mice lacking both NMNAT2 and SARM1 are viable, enabling us to examine the mechanistic origins of the developmental axon outgrowth defect associated with constitutive NMNAT2 deficiency.
Results
SARM1 Promotes Axon Degeneration Downstream of NMNAT2 Loss
NMNAT2 levels have previously been found to decline rapidly in the distal stumps of transected wild-type SCG and DRG neurites prior to their degeneration at ∼8 hr (Gilley and Coleman, 2010; Yang et al., 2013). Here, we find equivalent loss of NMNAT2 in wild-type and Sarm1−/− SCG neurites by 4 hr after cut (Figure 1A), even though degeneration of transected Sarm1−/− neurites is delayed for at least 3 days (Osterloh et al., 2012). Levels of the NMNAT substrate, NMN, and product, NAD, have also been shown to respectively rise and fall in the distal stumps of transected wild-type sciatic nerves (Di Stefano et al., 2014) prior to axon degeneration, which occurs at around 36 hr (Beirowski et al., 2005). This is consistent with a predicted loss of NMNAT2 in injured axons in vivo. Here, we find that these changes are not prevented in Sarm1−/− nerves at 30 hr after lesion (Figure 1B), despite Sarm1−/− axons being preserved for at least 14 days (Osterloh et al., 2012). In fact, by 30 hr, NMN has risen significantly more than in wild-type nerves, and by 120 hr, NAD levels have dropped below those in wild-type nerves at 30 hr (a short time before they degenerate). Together, these data suggest SARM1 promotes Wallerian degeneration downstream or independently of NMNAT2 loss and consequent changes in NMN and NAD levels.
Figure 1.

SARM1 Acts Downstream of NMNAT2 Loss during Axon Degeneration
(A) Representative immunoblot of uncut (0 hr) and 4-hr-cut wild-type and Sarm1−/− SCG neurite extracts probed for NMNAT2 and β-actin (sample control). NMNAT2 migrates at ∼32 kDa. Quantification of normalized NMNAT2 levels (to β-actin), is shown below with 4-hr data presented relative to uncut (set at 1). Means ± SEM are plotted (n = 3; p = 0.86 wild-type versus Sarm1−/− at 4 hr).
(B) NMN and NAD levels in uncut, and 30-hr- or 120-hr-lesioned wild-type and Sarm1−/− sciatic nerves. Means ± SEM are plotted (n = 3–5). Levels in 120-hr-cut wild-type nerves were not determined (ND) as their axons are degenerated.
(C) Representative images of distal neurites of wild-type and Sarm1−/− SCG neurons 24 and 72 hr after injection with 100 ng/μl Nmnat2 siRNA (siNmnat2) and 10 ng/μl pEGFP-C1 (expressed EGFP allows visualization of the neurites of injected neurons).
(D) Quantification of survival of wild-type (WT) and Sarm1−/− SCG neurites (left) and cell bodies (right) for 3 days after injection with pEGFP-C1 and siNmnat2 or non-targeting siRNA (siNT) as in (C). Means ± SEM are plotted (n = 3–4).
Although NMNAT2 loss has been strongly implicated as a trigger for Wallerian degeneration, this is difficult to prove definitively, as injury induces many simultaneous changes. We therefore assessed whether SARM1 is required during related, WLDS-sensitive axon degeneration induced specifically by small interfering RNA (siRNA)-mediated depletion of NMNAT2 (Gilley and Coleman, 2010). We found distal neurites of Sarm1−/− neurons are completely protected from Nmnat2 siRNA-induced degeneration for at least 72 hr, and also from the later, slower loss of cell viability (Figures 1C and 1D). NMNAT2 depletion can thus trigger SARM1-dependent axon degeneration in the absence of injury or other initiating stimuli supporting a situation in which either SARM1 itself, or a SARM1-dependent activity of a convergent pathway, promotes WLDS-sensitive axon degeneration downstream of NMNAT2 loss.
SARM1 Promotes Axon Defects in Mice Lacking NMNAT2
We next bred mice doubly homozygous for the Nmnat2gtE gene trap allele (effectively a null allele; Gilley et al., 2013) and a Sarm1 knockout allele (Kim et al., 2007) to investigate whether SARM1 might also contribute to axon defects associated with constitutive NMNAT2 deficiency during development.
Intriguingly, whole-mount labeling of distal phrenic nerve branches in the diaphragm and of intercostal nerves in Nmnat2gtE/gtE;Sarm1−/− embryos/newborn pups revealed clear and reproducible rescue of the peripheral nerve axon truncation defect associated with NMNAT2 deficiency in Nmnat2gtE/gtE animals (Figures 2A and 2B). The underlying cause of this defect in Nmnat2gtE/gtE embryos is restricted axon extension (Gilley et al., 2013); therefore, to obtain a quantitative measure of rescue by SARM1 deficiency, we compared neurite outgrowth in SCG explant cultures from Nmnat2gtE/gtE and Nmnat2gtE/gtE;Sarm1−/− embryos with that of relevant controls (Figures 2C and 2D). Strikingly, Nmnat2gtE/gtE;Sarm1−/− neurite outgrowth matched that in Sarm1−/− and Nmnat2+/gtE;Sarm1−/− littermate cultures, as well as in wild-type and Nmnat2+/gtE cultures assessed in parallel. Similar rescue was also seen in DRG cultures (not shown). The axon extension defect associated with a lack of NMNAT2 thus appears fully corrected in late-stage embryos and newborn mice deficient for SARM1. Consistent with this, while Nmnat2gtE/gtE (and Nmnat2gtE/gtE;Sarm1+/−) pups die at birth, Nmnat2gtE/gtE;Sarm1−/− mice in contrast are viable and reach weaning at the expected Mendelian ratios (Figure 2E). Adult Nmnat2gtE/gtE;Sarm1−/− mice are also fertile and, despite subtle gender- and Sarm1-genotype-specific differences, their weights at 10 weeks are within a healthy range and similar to control groups (Figure 2F). Crucially, RT-PCR and immunoblotting confirmed that the Nmnat2gtE allele remains effectively silenced on a Sarm1−/− background (Figures 2G and 2H). This rules out reduced efficacy of the gene trap as being responsible for rescue of the phenotype.
Figure 2.

Normal Development and Survival of Nmnat2gtE/gtE;Sarm1−/− Mice
(A) Neurofilament-L immunostaining of diaphragms from E18.5 Nmnat2gtE/gtE and Nmnat2gtE/gtE;Sarm1−/− embryos (montages representative of n = 3 for each genotype). Boxed areas are magnified (right). Distal branches of phrenic nerves were present in wild-type, Nmnat2+/gtE, Sarm1−/−, and Nmnat2+/gtE;Sarm1−/− embryos as expected (not shown).
(B) DiI-labeled intercostal nerves in ribcages from P0 Nmnat2gtE/gtE and Nmnat2gtE/gtE;Sarm1−/− pups (montages, representative of n = 3 for each genotype). Nerve truncation was seen in all Nmnat2gtE/gtE pups but was rescued in all Nmnat2gtE/gtE;Sarm1−/− pups.
(C) Quantification of radial neurite outgrowth over 7 days for E18.5 SCG explant cultures of the genotypes listed. Means ± SEM are plotted (n = 3–6 embryos of each genotype, average of both ganglia). Extension of the majority of Nmnat2gtE/gtE neurites (mass) and a more robust sub-population (max) are both shown.
(D) Representative images of Nmnat2gtE/gtE and Nmnat2gtE/gtE;Sarm1−/− neurite outgrowth at 7 days in vitro in E18.5 SCG explant cultures. Mass and max extension of Nmnat2gtE/gtE neurites are marked (continuous and dashed lines).
(E) Viability of offspring from Nmnat2+/gtE or Nmnat2+/gtE;Sarm1−/− crosses.
(F) Weights of 10-week-old male or female Nmnat2gtE/gtE;Sarm1−/− mice relative to indicated control groups. Both individual weights and means ± SEM are plotted. The weights of different genotypes in control groups do not vary significantly.
(G) Representative RT-PCR analysis of Nmnat2, Sarm1, and Actb (sample control) mRNA levels in E18.5 embryo brains of the indicated genotypes. Quantification of normalized Nmnat2 levels (to Actb), relative to wild-type levels, is shown below. Means ± SEM are plotted (n = 3).
(H) Representative immunoblots (of n = 3) showing NMNAT2, SARM1, and ßIII-tubulin (sample control) expression in brains from E18.5 embryo of the indicated genotypes.
As in injured nerves, SARM1 deficiency does not prevent expected changes to NMNAT metabolites in the brains of embryos lacking NMNAT2 (Figure 3A). In fact, the relative increase in NMN in Nmnat2gtE/gtE;Sarm1−/− brains is greater than in Nmnat2gtE/gtE brains, presumably due to rescued outgrowth of CNS axons, where changes, away from the influence of nuclear NMNAT1, should be greatest. A small, but significant, reduction in NAD is also evident in Nmnat2gtE/gtE;Sarm1−/− brains. In contrast, heterozygosity for the Nmnat2gtE allele does not significantly alter brain NMN or NAD levels on either Sarm1 background. Interestingly, there is a trend toward higher NMN and NAD levels in Sarm1−/− brains, independent of NMNAT2-related changes, and the total adenylate pool (ATP + ADP + AMP) is increased (1,764 ± 113 nmol/g tissue in Sarm1−/− brains compared to 1,314 ± 97 in Sarm1+/+ brains; p = 0.009). Thus, SARM1 itself may have some influence over NAD and/or energy metabolism.
Figure 3.

NMN and NAD Changes Resulting from a Lack of NMNAT2 during Development Occur in the Absence of SARM1
(A) NMN and NAD levels (nmol/g tissue) and NMN/NAD ratios in brains from E18.5 embryos/P0 pups of the indicated genotypes. Means ± SEM are plotted (n = 5–7 each genotype). Data for samples wild-type for Sarm1 (red tones) have been presented previously in a different format (Di Stefano et al., 2014). Re-use of the same dataset here allows direct comparison with the Sarm1−/− background data (blue tones), which was obtained in parallel.
(B) NMN and NAD levels (nmol/mg extracted protein) and NMN/NAD ratios in separate neurite or ganglia fractions from 7-days-in-vitro explant cultures of DRGs from E13.5/14.5 embryos of the indicated genotypes. Ganglia fractions contain short proximal neurite stumps as well as cell bodies. Means ± SEM are plotted (n = 3–5 each genotype). See also Figure S1, where data in (B) are presented as relative changes.
In case the complexity of whole brain concealed more marked changes specifically in axons, we also assessed nucleotide levels separately in DRG neurites and ganglia (Figures 3B and S1). Indeed, consistent with NMNAT2 being the major axonal NMNAT, we detected a greater relative increase in NMN and decrease in NAD in Nmnat2gtE/gtE;Sarm1−/− neurites than in brains. In contrast, changes in ganglia fractions were far more modest, indicating NMNAT2 has less influence in the cell body (and associated proximal neurite stumps). Surprisingly, accumulation of NMN was not evident in the limited neurite outgrowth in Nmnat2gtE/gtE cultures. However, the NMN/NAD ratio is increased to the same extent as in Nmnat2gtE/gtE;Sarm1−/− neurites, due to a much greater reduction in NAD, consistent with comparable loss of NMNAT activity. We speculate that, even though stunted Nmnat2gtE/gtE neurites do not show signs of overt degeneration, their integrity could still be compromised, resulting in NMN and NAD efflux and the lower than expected levels of both nucleotides. Low NAD in Nmnat2gtE/gtE ganglia fractions suggests this may also extend to proximal neurite stumps and possibly even cell bodies. Finally, as in brains, heterozygosity for the Nmnat2gtE allele does not significantly alter NMN and NAD levels in either DRG fraction. This is consistent with overt normality of Nmnat2+/gtE axons in both contexts.
Overall, these results indicate that SARM1 is critically involved in the manifestation of the phenotype of Nmnat2gtE/gtE mice, with it, or a dependent activity, acting downstream of predominantly axonal alterations to NMN and NAD resulting from the lack of NMNAT2. Crucially, this suggests significant mechanistic overlap between WLDS-sensitive axon degeneration and the axon outgrowth defect in these mice.
NMN Contributes to the Limited Outgrowth of NMNAT2-Deficient Neurites
We have previously implicated rising NMN, rather than declining NAD, as a critical pro-degenerative event in injured axons, in part by showing that blocking NMN synthesis with NAMPT inhibitor FK866 and scavenging NMN via ectopic expression of bacterial NMN deamidase (converting it to NaMN) both delay Wallerian degeneration (Di Stefano et al., 2014). We therefore used these reagents to investigate whether NMN also limits outgrowth and survival of axons lacking NMNAT2.
First, we tested the prediction that survival of Nmnat2gtE/gtE;Sarm1−/− SCG neurites should be impaired by ectopic re-expression of SARM1 due to re-activation of a SARM1-dependent degeneration pathway downstream of NMNAT2 deficiency, and assessed whether these events are NMN-dependent using co-expression of NMN deamidase (Figures 4A and 4B). While ectopic re-expression of SARM1 (C-terminal GFP tagged) in Sarm1−/− neurons (with normal NMNAT2) unexpectedly induced some neurite degeneration by 24 hr, it induced much more extensive degeneration of Nmnat2gtE/gtE;Sarm1−/− neurites (lacking NMNAT2) and, crucially, this was significantly reduced by co-expression of NMN deamidase (N-terminal GFP tagged). This suggests that, as well as promoting Wallerian degeneration, NMN also drives SARM1-dependent degeneration in this context. Importantly, these experiments additionally confirm that rescue of Nmnat2gtE/gtE;Sarm1−/− axons is due to SARM1 deficiency and not other, undefined changes resulting from mouse breeding.
Figure 4.

Pharmacological and Genetic Inhibition of NMN Accumulation Promotes Growth and Survival of NMNAT2-Deficient Neurites
(A) Representative images of neurites of Sarm1−/− or Nmnat2gtE/gtE;Sarm1−/− SCG neurons 24 hr after injection with 6.25 μg/μl Texas red dextran (for rapidly labeling neurites of injected neurons) together with 40 ng/μl GFP-NMN deamidase expression construct and 10 ng/μl pEGFP-C1 (GFP-NMNd (& GFP)), 10 ng/μl SARM1-GFP expression construct and 40 ng/μl pEGFP-C1 (SARM1-GFP (& GFP)), or 10 ng/μl SARM1-GFP and 40 ng/μl NMN deamidase-GFP expression constructs (SARM1-GFP & GFP-NMNd). SARM1-GFP induces degeneration of the proximal ∼1–2 mm of neurites, presumably shortly after exiting the soma.
(B) Quantification of neurite survival in experiments described in (A). Means ± SEM are plotted (n = 3–7 each treatment). Neurite survival after GFP-NMNd (& GFP) expression is not significantly different from GFP alone (not shown).
(C) Quantification of neurite outgrowth over 7 days for explant cultures of DRGs taken from E18.5 Nmnat2gtE/gtE embryos (left) or wild-type embryos (right) treated with FK866 and/or NaAD after 2 days in vitro. Means ± SEM are plotted (n = 3–6 each treatment, statistical significance shown relative to untreated).
(D) Representative phase contrast images showing the physical appearance of treated Nmnat2gtE/gtE DRG neurites (if present) at 5 days in vitro (3 days after compound addition). See also Figures S2 and S3.
Next, we used FK866 to assess whether NMN accumulation also limits Nmnat2gtE/gtE neurite outgrowth (Figures 4C and 4D). Interestingly, FK866 stimulated additional outgrowth of Nmnat2gtE/gtE DRG neurites, but only for about 24 hr, after which neurites underwent complete degeneration. Because FK866 causes simultaneous reductions to both NMN and NAD in neuronal cultures (Di Stefano et al., 2014), we considered that this later degeneration could be due to an extreme reduction in NAD in neurites now lacking both NAMPT and NMNAT2 activities and/or secondary to neuronal death caused by reduced somal NAD. To overcome this, we co-treated cultures with potential sources of NAD. NAD itself was ineffective (Figure S2), likely due to inefficient entry into neurons and/or failed conversion of more readily internalized NAD precursors (generated by extracellular degradation) back into NAD when NMNAT2 is absent (Di Stefano et al., 2014; Nikiforov et al., 2011), possibly even leading to further NMN accumulation in Nmnat2gtE/gtE neurites. However, NaAD, an alternative source of NAD via endogenous NAD synthetase (Di Stefano et al., 2014), did promote additional outgrowth and survival of Nmnat2gtE/gtE neurites when added with FK866 (but, importantly, not when added alone). Together, these data suggest increased NMN does contribute to the restricted outgrowth of Nmnat2gtE/gtE neurites, although rescue of Nmnat2gtE/gtE neurite outgrowth by FK866 and NaAD still falls some way short of the outgrowth of wild-type neurites treated in the same way (despite this also being slightly inhibitory) (Figure 4C). Therefore, either inhibition of NAMPT by FK866 or clearance of pre-accumulated NMN is relatively ineffective and/or outgrowth is not exclusively limited by NMN-dependent events.
Discussion
Despite their shared ability to profoundly influence axon survival after injury, no link between NAD metabolism and SARM1 function had previously been firmly established. Our studies show not only that either SARM1 itself, or a SARM1-dependent convergent activity, acts downstream of NMNAT2 loss and resulting changes to key NMNAT metabolites to promote WLDS-sensitive axon degeneration but also that this also occurs downstream of NMNAT2 deficiency and dependent changes during development to impair axon outgrowth and limit mouse survival. Intriguingly, an NMNAT2 depletion-dependent accumulation of NMNAT substrate, NMN, also appears to be a critical pro-degenerative signal in both situations (this study; Di Stefano et al., 2014), strongly suggesting they are each mediated by activation of the same NMN- and SARM1-dependent degeneration pathway. Importantly, our data appear to rule out any lasting stabilization of NAD or reduced accumulation of NMN as part of the mechanism underlying protection of SARM1-deficient axons, although intriguing increases in total adenylate pool and steady-state levels of NMN and NAD in Sarm1−/− brains may yet be found to be involved.
Overall, our studies suggest either that changes resulting from NMNAT2 loss directly activate a pro-degenerative SARM1 function in axons in a linear pathway or that parallel SARM1-dependent and NMNAT2 loss-dependent signals merge at a common downstream step in a convergent pathway (Figure S3). Importantly, the critical SARM1-dependent event in either model likely acts upstream of calpain activation (Yang et al., 2013). Given that NMNAT2 depletion alone can activate SARM1-dependent neurite degeneration, the convergent pathway model would appear to require that SARM1 has constitutive pro-degenerative activity, with activation or disinhibition of a critical downstream step only occurring after NMNAT2 loss. While a proposed regulatory role for the N-terminal domain of SARM1 (Gerdts et al., 2013) appears more consistent with activation of a pro-degenerative function in a linear pathway, our finding that exogenous expression of SARM1(-GFP) induces some degeneration even when NMNAT2 is present (in Sarm1−/− neurons) suggests it could have some constitutive pro-degenerative activity (although conditions in these assays could also influence this outcome). Understanding precisely how endogenous SARM1 functions in the context of NMNAT2 loss and NMN accumulation during axon degeneration, and how this relates to other cellular roles of SARM1 (Conforti et al., 2014), will therefore be an important goal for future studies.
We had previously proposed that the early, limited extension of axons lacking NMNAT2 reflects restricted supply of an NMNAT-activity-dependent molecule required for axon growth, with NAD being a prime candidate (Gilley et al., 2013). However, initially improved outgrowth of Nmnat2gtE/gtE neurites further deprived of NAD by FK866 challenges this idea, and the involvement of SARM1 instead suggests these neurites stop extending at the point where SARM1-dependent degeneration becomes active. In light of our findings, we speculate that NMN concentration may determine this activity. We predict that, due to a diminishing influence of nuclear NMNAT1, NMN in NMNAT2-deficient axons will rise as a function of distance from the soma, with some axon outgrowth possible up to the point at which a critical threshold is exceeded and SARM1-dependent degeneration is triggered.
NAD plays a key role in many diverse cellular functions (Di Stefano and Conforti, 2013), so the fact that significant reductions in NAD do not prevent the delayed degeneration of cut SARM1-deficient axons or limit the normal growth and survival of intact axons lacking both NMNAT2 and SARM1 (in culture and in mice) was somewhat surprising. This, together with the ability of FK866 to delay degeneration of cut axons (Di Stefano et al., 2014) and temporarily rescue Nmnat2gtE/gtE neurite outgrowth, despite further depletion of NAD, clearly indicates that fairly substantial reductions in NAD are not necessarily detrimental. However, it would be surprising if greater NAD depletion did not limit axon survival in some situations.
On its own, survival of Nmnat2gtE/gtE;Sarm1−/− mice for several months with no overt problems suggests very strong correction of the defects associated with constitutive NMNAT2 deficiency during early development. However, it will still be critical to establish the precise extent and duration of this rescue in an aging study. Nevertheless, these mice already represent a unique resource for the identification or confirmation of other early pro-degenerative events triggered by NMNAT2 loss and NMN accumulation, given that our data suggest an activated degeneration pathway is paused at a SARM1-dependent step in Nmnat2gtE/gtE;Sarm1−/− axons. For example, SARM1 appears to act downstream of reactive oxygen species accumulation, ATP depletion, and Ca2+ influx when cell death and axon degeneration are induced by the mitochondrial toxin carbonyl cyanide m-chlorophenyl hydrazone (CCCP) (Summers et al., 2014), but it will be important to establish whether these changes also occur downstream of NMNAT2 loss during WLDS-sensitive degeneration. They should also be particularly useful for studying non-axonal phenotypes associated with NMNAT2 deficiency that may be SARM1 independent, such as its role in cardiac hypertrophy (Cai et al., 2012).
To conclude, these studies have now firmly established the order of two core events in an emerging WLDS-sensitive and SARM1-dependent axon degeneration pathway. As additional steps are added, it will become increasingly apparent how this relates to other cell-death programs and how it might be most effectively targeted in axonal disorders. Crucially, the lack of any overt defects in adult Nmnat2gtE/gtE;Sarm1−/− mice suggest that if effective targeting of this pathway can be achieved, it could potentially provide long-term relief from symptoms without major side effects.
Experimental Procedures
Animals, Genotyping, and Primary Neuronal Cultures
Animal work was carried out in accordance with the Animals (Scientific Procedures) Act, 1986, under Project License PPL 70/7620. Primers used for PCR genotyping of mice are listed in the Supplemental Experimental Procedures.
Explant cultures of superior cervical ganglia (SCGs) from embryonic day 18.5 (E18.5) mouse embryos and postnatal day 0 (P0) to P2 pups and dorsal root ganglia (DRGs) from E13.5/14.5 and E17.5/E18.5 embryos and P0 pups, and dissociated SCG neuron cultures (P0–P2), were set up as described previously (Gilley et al., 2013; Gilley and Coleman, 2010).
Neurite Outgrowth Assays
Neurite outgrowth assays were performed and quantified as described previously (Gilley et al., 2013). NAMPT inhibitor FK866 (100 nM, generously supplied by the National Institute of Mental Health), NaAD (250 μM, Sigma-Aldrich), NAD (1 mM, Sigma-Aldrich), or NMN (1 mM, Sigma-Aldrich) were added to E17.5/E18.5 Nmnat2gtE/gtE DRG explant cultures at 2 days in vitro as described.
RT-PCR Analysis and Immunoblotting
Whole brains from E18.5 mouse embryos and neurite fractions from SCG explant cultures were processed as described previously for the generation of RNA and/or protein extracts (Gilley et al., 2013; Gilley and Coleman, 2010). Primers for RT-PCR, and primary antibodies and concentrations for immunoblotting, are listed in the Supplemental Experimental Procedures.
Determination of NMN and NAD Levels
NMN and NAD measurements in whole brains from E18.5 embryos or newborn (P0) pups, uncut or lesioned sciatic nerves from adult mice, and neurite and ganglia fractions from DRG cultures (prepared as for SCG cultures in Gilley and Coleman, 2010) were performed using UV-C18 high-performance liquid chromatography (HPLC) and/or spectrofluorometric HPLC analysis essentially as described previously (Di Stefano et al., 2014).
Microinjections, Labeling/Immunostaining, and Microscopy
Injections and quantification of neurite/cell-body survival were performed essentially as described previously (Gilley and Coleman, 2010). Plasmids and siRNAs injected are listed in the figure legends and in the Supplemental Experimental Procedures.
Anterograde labeling of intercostal nerve axons with the lipophilic dye DiI (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate) in fixed P0 mouse pups and whole-mount neurofilament light-chain immunostaining of fixed diaphragms from E17.5/E18.5 embryos were performed as described previously (Gilley et al., 2013).
Phase-contrast and fluorescence images of primary cultures and stained/labeled tissues were acquired as described previously (Gilley et al., 2013). Image processing is described in the Supplemental Experimental Procedures.
Statistical Analysis
Appropriate statistical tests were performed using Prism software (GraphPad Software). The Student’s t test, ANOVA with Tukey’s or Dunnett’s post hoc correction (as applicable), and Pearson’s chi-square test were used in this study. A p value > 0.05 was considered not significant, and ∗ indicates p < 0.05, ∗∗ indicates p < 0.01, and ∗∗∗ indicates p < 0.001.
Author Contributions
J.G. and M.P.C. conceived the study. J.G. designed and performed all experiments with the exception of nucleotide measurements, obtained by G.O., and in vitro and in vivo neurite and nerve injury experiments, designed and performed by I.N.-F. J.G. and M.P.C. co-wrote the manuscript with input from G.O.
Acknowledgments
We thank Dr. Yi-Ping Hsueh for the SARM1 antibody; Dr. Laura Conforti, Dr. Michele Di Stefano, and Prof. Nadia Raffaelli for helpful discussions and reagents; and Dr. Anne Segonds-Pichon for help with statistical analysis. This work was funded by an Institute Strategic Programme Grant from the Biotechnology and Biological Sciences Research Council.
Published: March 26, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures and three figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2015.02.060.
Supplemental Information
References
- Babetto E., Beirowski B., Russler E.V., Milbrandt J., DiAntonio A. The Phr1 ubiquitin ligase promotes injury-induced axon self-destruction. Cell Rep. 2013;3:1422–1429. doi: 10.1016/j.celrep.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beirowski B., Adalbert R., Wagner D., Grumme D.S., Addicks K., Ribchester R.R., Coleman M.P. The progressive nature of Wallerian degeneration in wild-type and slow Wallerian degeneration (WldS) nerves. BMC Neurosci. 2005;6:6. doi: 10.1186/1471-2202-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y., Yu S.S., Chen S.R., Pi R.B., Gao S., Li H., Ye J.T., Liu P.Q. Nmnat2 protects cardiomyocytes from hypertrophy via activation of SIRT6. FEBS Lett. 2012;586:866–874. doi: 10.1016/j.febslet.2012.02.014. [DOI] [PubMed] [Google Scholar]
- Conforti L., Gilley J., Coleman M.P. Wallerian degeneration: an emerging axon death pathway linking injury and disease. Nat. Rev. Neurosci. 2014;15:394–409. doi: 10.1038/nrn3680. [DOI] [PubMed] [Google Scholar]
- Di Stefano M., Conforti L. Diversification of NAD biological role: the importance of location. FEBS J. 2013;280:4711–4728. doi: 10.1111/febs.12433. [DOI] [PubMed] [Google Scholar]
- Di Stefano M., Nascimento-Ferreira I., Orsomando G., Mori V., Gilley J., Brown R., Janeckova L., Vargas M.E., Worrell L.A., Loreto A. A rise in NAD precursor nicotinamide mononucleotide (NMN) after injury promotes axon degeneration. Cell Death Differ. 2014 doi: 10.1038/cdd.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y., Soares L., Teng X., Geary M., Bonini N.M. A novel Drosophila model of nerve injury reveals an essential role of Nmnat in maintaining axonal integrity. Curr. Biol. 2012;22:590–595. doi: 10.1016/j.cub.2012.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J., Summers D.W., Sasaki Y., DiAntonio A., Milbrandt J. Sarm1-mediated axon degeneration requires both SAM and TIR interactions. J. Neurosci. 2013;33:13569–13580. doi: 10.1523/JNEUROSCI.1197-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J., Coleman M.P. Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 2010;8:e1000300. doi: 10.1371/journal.pbio.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J., Adalbert R., Yu G., Coleman M.P. Rescue of peripheral and CNS axon defects in mice lacking NMNAT2. J. Neurosci. 2013;33:13410–13424. doi: 10.1523/JNEUROSCI.1534-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks A.N., Lorenzetti D., Gilley J., Lu B., Andersson K.E., Miligan C., Overbeek P.A., Oppenheim R., Bishop C.E. Nicotinamide mononucleotide adenylyltransferase 2 (Nmnat2) regulates axon integrity in the mouse embryo. PLoS ONE. 2012;7:e47869. doi: 10.1371/journal.pone.0047869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y., Zhou P., Qian L., Chuang J.Z., Lee J., Li C., Iadecola C., Nathan C., Ding A. MyD88-5 links mitochondria, microtubules, and JNK3 in neurons and regulates neuronal survival. J. Exp. Med. 2007;204:2063–2074. doi: 10.1084/jem.20070868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack T.G., Reiner M., Beirowski B., Mi W., Emanuelli M., Wagner D., Thomson D., Gillingwater T., Court F., Conforti L. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat. Neurosci. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- Milde S., Fox A.N., Freeman M.R., Coleman M.P. Deletions within its subcellular targeting domain enhance the axon protective capacity of Nmnat2 in vivo. Sci Rep. 2013;3:2567. doi: 10.1038/srep02567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neukomm L.J., Freeman M.R. Diverse cellular and molecular modes of axon degeneration. Trends Cell Biol. 2014;24:515–523. doi: 10.1016/j.tcb.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikiforov A., Dölle C., Niere M., Ziegler M. Pathways and subcellular compartmentation of NAD biosynthesis in human cells: from entry of extracellular precursors to mitochondrial NAD generation. J. Biol. Chem. 2011;286:21767–21778. doi: 10.1074/jbc.M110.213298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterloh J.M., Yang J., Rooney T.M., Fox A.N., Adalbert R., Powell E.H., Sheehan A.E., Avery M.A., Hackett R., Logan M.A. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science. 2012;337:481–484. doi: 10.1126/science.1223899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pease S.E., Segal R.A. Preserve and protect: maintaining axons within functional circuits. Trends Neurosci. 2014;37:572–582. doi: 10.1016/j.tins.2014.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y., Vohra B.P., Baloh R.H., Milbrandt J. Transgenic mice expressing the Nmnat1 protein manifest robust delay in axonal degeneration in vivo. J. Neurosci. 2009;29:6526–6534. doi: 10.1523/JNEUROSCI.1429-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers D.W., DiAntonio A., Milbrandt J. Mitochondrial dysfunction induces Sarm1-dependent cell death in sensory neurons. J. Neurosci. 2014;34:9338–9350. doi: 10.1523/JNEUROSCI.0877-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X., Hao Y., Sun K., Li J., Li X., Mishra B., Soppina P., Wu C., Hume R.I., Collins C.A. The Highwire ubiquitin ligase promotes axonal degeneration by tuning levels of Nmnat protein. PLoS Biol. 2012;10:e1001440. doi: 10.1371/journal.pbio.1001440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahata N., Yuasa S., Araki T. Nicotinamide mononucleotide adenylyltransferase expression in mitochondrial matrix delays Wallerian degeneration. J. Neurosci. 2009;29:6276–6284. doi: 10.1523/JNEUROSCI.4304-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Weimer R.M., Kallop D., Olsen O., Wu Z., Renier N., Uryu K., Tessier-Lavigne M. Regulation of axon degeneration after injury and in development by the endogenous calpain inhibitor calpastatin. Neuron. 2013;80:1175–1189. doi: 10.1016/j.neuron.2013.08.034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.