

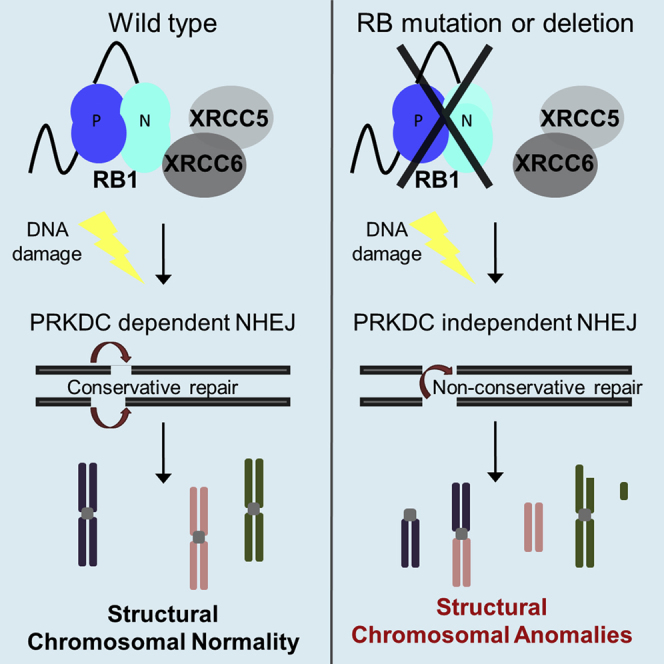
Summary
Deficiencies in DNA double-strand break (DSB) repair lead to genetic instability, a recognized cause of cancer initiation and evolution. We report that the retinoblastoma tumor suppressor protein (RB1) is required for DNA DSB repair by canonical non-homologous end-joining (cNHEJ). Support of cNHEJ involves a mechanism independent of RB1’s cell-cycle function and depends on its amino terminal domain with which it binds to NHEJ components XRCC5 and XRCC6. Cells with engineered loss of RB family function as well as cancer-derived cells with mutational RB1 loss show substantially reduced levels of cNHEJ. RB1 variants disabled for the interaction with XRCC5 and XRCC6, including a cancer-associated variant, are unable to support cNHEJ despite being able to confer cell-cycle control. Our data identify RB1 loss as a candidate driver of structural genomic instability and a causative factor for cancer somatic heterogeneity and evolution.
Graphical Abstract
Highlights
-
•
RB1 associates with XRCC5 and XRCC6, involved in DNA repair by cNHEJ
-
•
RB family loss reduces cNHEJ and boosts repair-associated chromosomal aberrations
-
•
cNHEJ requires RB1’s N-terminal domain but is unrelated to cell-cycle control by RB1
-
•
RB1’s ability to support cNHEJ is targeted by mutation in cancer
Loss of retinoblastoma protein (RB1) is common in various difficult-to-treat cancers. Cook et al. show that RB1 loss significantly impairs repair of DNA via non-homologous end-joining (NHEJ) and, in doing so, promotes genomic instability. These unexpected findings present opportunities for future cancer therapies that exploit this repair defect.
Introduction
DNA double-strand breaks (DSBs), where both strands of DNA are severed, pose an exceptional threat to the stability of a cell’s genome. DSBs are caused by reactive adducts produced during normal metabolic reactions or following exposure to environmental genotoxins, including radiation. Inability to repair DSBs leads to DNA fragmentation and cell death. Unfaithful repair results in genetic instability (GIN) where cells may survive but chromosomes become rearranged, and genetic material mutated, duplicated, or deleted (Negrini et al., 2010).
GIN is a defining characteristic of cancer (Schmitt et al., 2012) promoting initiation and somatic evolution, in turn, linking to disease progression and therapy resistance. To identify molecular events that cause GIN is an important goal in cancer research.
DSBs can be repaired by two mechanistically distinct pathways, homologous recombination (HR) (Heyer et al., 2010) and canonical non-homologous end-joining (cNHEJ) (Lieber, 2010). HR involves single-strand resection of the damaged DNA with re-synthesis that relies on the presence of a homologous sister chromatid. cNHEJ effectively re-joins the severed DNA ends, functioning in the absence of a homologous template. Because cNHEJ has provision to trim DNA ends, it can repair breaks where DNA is chemically modified and complex damage with multiple close-by strand breaks, impeding the use of HR. Although cNHEJ has reduced accuracy compared to HR, most specifically when mending breaks that require end trimming, repair by cNHEJ is conservative as gene positioning and sequence co-linearity within chromosomes are strictly maintained and its simplicity and versatility make it the most prominent route by which DSBs are repaired in higher eukaryotes. cNHEJ also serves other roles, including VDJ recombination in immune cells and telomere maintenance (reviewed in Doksani and de Lange [2014] and Malu et al. [2012]).
Inhibition or mutational loss of cNHEJ or HR leads to use of alternative, inaccurate forms of end-joining (aNHEJ), reliant on resection that exposes serendipitous homology to adjacently positioned yet often noncontiguous chromatin that then is used as a template for repair (reviewed in Aparicio et al. [2014]). Chromosomal alterations are thus generated, including base changes, inversions, translocations, and deletions, with consequence of code alteration, gene gain, loss or deregulation, and the generation of gene fusions. Paired with mitotic checkpoint defects, such misrepair may give rise to progressive GIN.
How canonical HR and NHEJ suppress the use of aNHEJ is not clear. Answers most likely lie in the recruitment of accessory factors by the respective core repair machineries, which protect and manage regulated access to the damaged DNA. These include a diverse collection of chromatin modifying enzymes, comprising effectors of protein acetylation, methylation, and ubiquitination, as well as effectors of chromatin conformation, including the imitation switch (ISWI) chromatin remodeling complexes and the inhibitor of resection 53BP1 (reviewed in Panier and Boulton [2014] and Papamichos-Chronakis and Peterson [2013]).
The retinoblastoma protein (RB1) is an important tumor suppressor. Mutational loss of RB1 is implicated in the development of the childhood eye cancer retinoblastoma but also major cancers including breast and small cell lung cancer, sarcomas, and glioblastoma. Germline mutations in the RB1 encoding gene are associated with a highly penetrant predisposition to retinoblastoma (Lohmann, 2010) and substantially increase the lifetime risk for a spectrum of secondary cancers (Meadows et al., 2009). RB1 belongs to a family of proteins with similar molecular anatomy and related function. RB1 and its paralogs are best recognized for their role in the control of gene transcription by which they affect the cell cycle and a range of other responses, including cell differentiation, epithelial to mesenchymal transition, angiogenesis, cell migration, and metabolism (reviewed in Schaal et al. [2014]). More recently other roles of RB1 have been uncovered, fully or partially independent of its role in controlling gene transcription or the cell cycle, including the activation of apoptosis through association with the cell’s mitochondria (Hilgendorf et al., 2013) and in the control of mitotic fidelity (reviewed in Manning and Dyson [2012]). The latter indicates a key contribution of RB1 in safeguarding numerical chromosomal stability and points to RB1 loss as the cause of aneuploidies observed in cancers with RB1 involvement.
Genomic analysis of retinoblastoma, a cancer that virtually always involves RB1 loss, identified widespread GIN, signified by extensive gain and loss of subchromosomal regions (reviewed in Thériault et al. [2014]), in addition to numerical chromosome abnormalities in a portion of these cancers. These observations suggest RB1 loss, in addition to numerical chromosome instability, may affect chromosome integrity, leading to GIN.
Here, we report that the RB family proteins are required for repair of DNA by cNHEJ. We show that these proteins through use of their amino-terminal domains associate with the core components XRCC5 and 6 of the cNHEJ damage recognition machinery. Our work identifies RB1 and its paralogs as critical factors in cNHEJ with roles of suppressing the use of alternative, inaccurate end-joining in cells. The work identifies RB1 loss as a plausible cause of GIN in cancers with RB1 loss.
Results
Interaction between RB1N and Components Involved in NHEJ
RB1 is built from two major folded domains, the central pocket domain, RB1P, and the amino-terminal domain, RB1N (Hensey et al., 1994), that between them share similarities including fold analogy and conservation of isostructural surfaces (Hassler et al., 2007). RB1P is recognized for its ability to interact with key cellular proteins, including members of the E2 factor (E2F) family of transcription factors and proteins containing an Leu-X-Cys-X-Glu (LXCXE) linear motif (Lee et al., 1998, 2002; Xiao et al., 2003), through which RB1 confers transcriptional regulation, cell-cycle regulation, and tumor suppression (reviewed in Schaal et al. [2014]). The mode of action of RB1N has not been mechanistically defined, although ∼20% of identified frame-preserving tumor mutations reside in this domain, and deletion results in tumor development in mice, predicting importance of this domain in tumor suppression (Goodrich, 2003).
Owing to the structural similarity of RB1N and RB1P, we reasoned that RB1N, like RB1P, may act by binding partner proteins and that identification of these will reveal the functional significance of RB1N. To capture partner proteins, we utilized as a matrix glutathione S-transferase (GST)-tagged RB1 40-355 (RB1N), previously employed to determine the structure of this domain (Hassler et al., 2007), together with HeLa nuclear extract, and liquid chromatography-tandem mass spectrometry (LC-MS/MS) for partner protein identification. We performed parallel experiments using purified GST tag to distinguish proteins selectively captured by RB1N, which identified 34 proteins that selectively interacted with RB1N (Table S1; Supplemental Experimental Procedures).
Mining for associations of these proteins with cellular processes using the Database for Annotation, Visualization and Integrated Discovery (DAVID) (Dennis et al., 2003) uncovered a number of ontologies enriched in representation by the list of interacting proteins compared to the nuclear proteome (Figure 1A), among them “DNA double-strand repair via NHEJ” and related ontologies, suggesting a connection between RB1 and NHEJ. In particular, XRCC5 (Ku80), XRCC6 (Ku70), and PRKDC (DNA-PK), required for DSB repair by cNHEJ (Lieber et al., 2003), were identified with high confidence (Table S1). Subsequent affinity capture using extract from two other cell lines, HCT116 colon and MCF-7 breast cancer cells, combined with antibody-based detection of XRCC5 and XRCC6 (Figures 1B and 1C) confirmed selective retention of these proteins by GST-RB1N.
Figure 1.

Interaction between RB1N and NHEJ Proteins
(A) Graph depicting enrichment scores calculated using the DAVID gene annotation tool (biological processes). Proteins selectively captured by GST-RB1N (amino acids 40–355) were scored against a self-generated nuclear proteome background. The ontologies shown are represented by three or more proteins with >10-fold enrichment over background.
(B and C) Affinity capture on GST RB1N with analysis by immunoblotting. Nuclear extracts derived from MCF-7 (B) or HCT116 (C).
(D) Schematic representation of full-length RB1 (top) and the folded RB1N (bottom). The position of the twin cyclin-like folds building the scaffold of RB1N and RB1P are indicated. The location of residue alterations yielding RB1NPolyG and RB1N240–242 deletion is indicated.
(E) Ribbon diagram representation of the structure of RB1N (adapted from Hassler et al. [2007]); the unoccupied cyclin wedge region is denoted with a circle, the location of the PolyG alteration is indicated by a dotted line, and the position of RB1N240–242 deletion is denoted in red.
(F) GST-affinity capture using HCT116 cell lysate with GST, RB1N, or RB1NPolyG.
(G) GST-affinity capture using HCT116 cell lysate with GST, RB1N, or RB1N240–242 deletion.
(H) Co-immunoprecipitation of cell endogenous XRCC5 and XRCC6 with RB1 from HCT116. Cells were left untreated or treated with a CDK4/6 selective inhibitor of RB1 phosphorylation PD0332991 (400 nM for 18 hr) followed by irradiation at 5 Gy (IR), with extract preparation 30 min post IR. RAM, irrelevant control antiserum; RB1C, anti-RB1C domain antiserum; In., input lysate.
It has been reported that XRCC5 and XRCC6 can be contaminants associated with affinity-capture experiments (Mellacheruvu et al., 2013). To gain additional evidence for the selectivity of the observed interactions, we made use of a RB1N variant (RB1NPolyG) with a fold-preserving alteration in the B cyclin wedge, predicted by analogy to support protein binding (Figures 1D and 1E) (Hassler et al., 2007). While XRCC6 was readily recovered from HCT116 lysate by wild-type GST-RB1N (Figure 1C), only a minor amount of XRCC6 was recovered using GST-RB1NPolyG (Figure 1F). Identical results were obtained using RB1N240–242 del, a cancer-associated RB1 variant (Shuin et al., 1995) featuring a three amino acid deletion in the same region (Figures 1D and 1G). Together, these experiments provide strong evidence for selectivity of the interactions. They further identify a specific RB1N region, predicted to confer protein binding (Hassler et al., 2007), as responsible for mediating the interaction with XRCC5 and XRCC6.
To document that these interactions arise with cell endogenous components, we immunoprecipitated RB1 from cells (Figure 1H). We used asynchronously growing HCT116 cells or HCT116 cells treated with the CDK4/6 selective inhibitor PD0332991, which enriches for G1 phase cells with underphosphorylated, active RB1 (Fry et al., 2004), followed by ionizing radiation (IR), to induce DSB damage. These experiments confirmed XRCC5 and XRCC6 associate with RB1 and further revealed an increased amount of XRCC5 and XRCC6 is recovered under conditions of RB1 activation and DNA damage, suggesting regulation of the interaction.
Regulation of the Interaction between XRCC6 and RB1
To further confirm RB1/XRCC5/XRCC6 complexes exist within intact cells, we employed proximity ligation (PLA), which visualizes protein interactions in fixed cells based on antibody-mediated detection of the respective interaction partners (Weibrecht et al., 2010). We probed for the interaction between RB1 and XRCC6 in both MCF-7 (Figures 2A and 2B) and HCT116 cells (Figure S1B), which were untreated or treated with PD0332991 or IR alone or in combination. To document assay selectivity, we ablated RB1 using small interfering RNA (siRNA) (Figure 2D; Figures S1A, S1D, and S1E) or omitted one or both primary antibodies (Figures S1F and S1G). These experiments confirmed complexes between XRCC6 and RB1 exist in cells, and that these were located in the cell nucleus.
Figure 2.

Regulation of the Interaction between XRCC6 and RB1 in Cells
(A) PLA probing for XRCC6/RB1 complex in MCF-7 cells. Cells were left untreated or treated with the PD0332991 (400 nM for 18 hr), IR (5 Gy) or PD0332991 (400 nM for 18 hr) followed by IR (5 Gy). Cells were analyzed 30 min post IR. Nuclei visualized with Hoechst 33258. Parallel staining in the absence of primary or secondary antibodies shown in Figure S2A. Identical experiments using HCT116 shown in Figure S1B.
(B) Automated quantification of PLA foci. Data represent counts for a minimum of 100 cells per condition from n = 3 biological repeats. The percentage of cells for each range of interactions is shown. For statistical assessment of results, see Table S2.
(C) Immunoblotting analysis of P-Ser780 RB1, serving as a biomarker for CDK4/6 activity. XRCC5 and XRCC6 and RB1 total protein levels were also assessed. Actin levels are shown as loading controls.
(D) PLA probing for XRCC6/RB1 complex in MCF-7 cells treated with non-targeting (siNT) or RB1 targeting siRNA. Cells were treated with PD0332991 followed by 5 Gy IR as described for (A). For parallel anti-RB1 staining, see Figure S1A. Identical experiments using HCT116 shown in Figures S1D and S1E.
(E) Detection of RB1/XRCC6 complex in cell-cycle-phase-enriched cells. Cells were enriched for the various cell-cycle phases as depicted in Figure S2A. XRCC6/RB1 complexes were detected using PLA. Related data for HCT116 are shown in Figure S2.
(F) Automated quantification of foci number. Cumulative data for n = 3 biological replicates are shown. Data represent evaluation of a minimum of 100 independent cells. Quantification was performed as for (B).
(G) Cell-cycle profiles of the cells analyzed in (E), determined by propidium iodide staining and flow cytometry. Error bars represent ± SD for n = 3 biological replicates.
(H) Immunoblotting analysis depicting protein levels of RB1, XRCC5, and XRCC6 in cells enriched for the various cell-cycle phases. Actin levels are shown as loading controls.
A notable and statistically significant increase in both complex abundance and the number of complex-positive cells was observed when cells were pre-treated with PD0332991 followed by IR. Greater than 60% of either MCF-7 or HCT116 cells treated in this way contained six or more foci compared to 20%–25% treated with PD0332991 or IR alone, and <15% of untreated cells (Figures 2B, S1F, and S1G; statistics in Table S2). Immunoblotting confirmed loss of RB1 phosphorylation in PD0332991-treated cells (Figure 2C). Notably, differences in interaction seen by PLA were not paralleled by differences in the levels of XRCC5, XRCC6, or RB1 in the cells, excluding protein abundance changes as the cause for the increase in interactions. We also assessed MCF-7 and HCT116 cells enriched for mid-S or G2/M phase alongside cells enriched for G1 using PD0332991. Flow cytometry confirmed appropriate cell-cycle distributions (Figures 2G and S2E). We observed absence of foci in the vast majority of mid-S and G2/M-enriched cells but presence of foci in many of the G1 enriched cells (Figures 2E–2H and S2; statistics in Table S3). Likewise, these differences were not paralleled by altered abundance of RB1, XRCC5, or XRCC6 (Figures 2H and S2C). Together, these results provide additional evidence for complex formation between the NHEJ components and RB1, which appears to be promoted by RB1 activation and DSBs, in line with our earlier observations using RB1 immunoprecipitation.
RB Family Proteins Promote PRKDC-Dependent NHEJ
Since different methodologies confirmed the interaction of RB1 with components involved in cNHEJ, we assessed whether RB1 affects NHEJ activity in cells. To do so, we used a reporter (Bennardo et al., 2008) (Figure 3A) in which a removable puromycin-resistance gene is inserted between the GFP reading frame and its promoter. I-SceI expression in cells induces removal of the insertion, leaving a DSB that when repaired by NHEJ leads to GFP expression (GFP+), allowing quantification of repair proficiency by flow cytometry. We quantified NHEJ proficiency in HCT116 cells in which siRNA was used to deplete RB1 either alone or together with its paralogs RBL1/p107 and RBL2/p130, known to act redundantly with RB1 in many cell types (Henley and Dick, 2012). To identify cells with reporter uptake, we co-transfected plasmid encoding red fluorescent protein (DsRed). RB1 depletion led to a significant reduction in NHEJ proficiency (Figures 3B and 3C) that was further augmented by co-depletion of RBL1 and RBL2 (Figure 3C), confirming a redundant role of the RB paralogs in NHEJ in HCT116. Cell-cycle analysis performed in parallel for DsRed+ cells of the various treatment groups detected no statistically significant differences, indicating that functional RB loss in these cells, within the time frame and manipulations undertaken, does not cause overt cell-cycle differences (Figure 3D). Hence, cell-cycle differences are not the cause of the difference in reporter repair proficiency that we detect.
Figure 3.

RB Family Proteins Promote PRKDC-Dependent NHEJ
(A and B) Reporter-based quantification of NHEJ repair. Cells were transfected with NHEJ reporter and I-SceI encoding plasmid to introduce DSBs, with repair by NHEJ yielding GFP-positive cells (A). DsRed was used to identify transfected cells. Assays were quantified using flow cytometry. Representative raw images for HCT116 cells treated with siRNA targeting RB1 or RB1, RBL1, and RBL2, or a non-targeting control (siNT) are shown (B) NHEJ repair proficiency is determined by calculating the ratio of GFP-positive (Q2) versus DsRed-positive cell (Q1 + Q2).
(C) Graph depicting repair proficiency in NHEJ reporter-transfected cells. Cells were treated as for (B). NHEJ repair proficiency for cells transfected with siNT was set to 1. Binding of XRCC5 by RBL1 and RBL2 shown in Figure S3.
(D) Cell-cycle profiles for DsRed-positive cells from (C).
(E) Effect of PRKDC inhibition on reporter repair proficiency. HCT116 cells were treated and evaluated as for (C). DMSO or the PRKDC inhibitor NU7441 (5 μM) was added for the duration of the experiment.
(F) Immunodetection documenting RB1 and RB protein family loss in HCT116. HCT116 transfected with siRNA as in (C)–(E). Lysates probed using antibodies as indicated.
(G) NHEJ proficiency in SAOS-2 (RB1-negative) and U2OS (RB1-positive) cells using transient NHEJ reporter transfection as for (C) and (D).
(H) Effect of PRKDC inhibition on NHEJ proficiency. SAOS-2 and U2OS cells with integrated NHEJ reporter were used. Lines were transfected with I-SceI and DsRed plasmids and treated with DMSO or NU7441 as for (E). For all experiments, the average for n = 3 biological replicates is depicted; error bars ±SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 using a paired Student’s t test.
(I) Immunodetection of PRKDC-Ser2056 phosphorylation status. Autophosphorylation of PRKDC-Ser2056 signifies PRKDC activity. Cells were treated with 5 μM NU7441 for 24 hr prior to 5 Gy IR. 2 hr post IR, the cells were harvested into SDS protein loading buffer. Actin and total PRKDC levels are shown as loading controls.
Since our functional analysis indicates RBL1 and/or RBL2 act redundantly to RB1 in NHEJ, we sought to address whether these RB1 paralogs interact with NHEJ components, as observed for RB1. Using GST-fused fragments of RBL1 and RBL2 homologous to RB1N together with HCT116 nuclear extract, we performed affinity-capture experiments as for Figure 1, with control reactions containing GST-RB1N or unfused GST (Figure S3). These experiments clearly document selective capture of XRCC5 by RBL1 and RBL2 arising with comparable efficacy to that seen for RB1 (Figure S3B).
The NHEJ reporter used measures cNHEJ as well as some portion of aNHEJ that is independent of PRKDC (Chiruvella et al., 2013). To distinguish between these different forms of NHEJ repair, we treated reporter-transfected cells with NU7441, a PRKDC inhibitor, which prevents cNHEJ (Zhao et al., 2006). We found that NU7441 reduced NHEJ reporter activity significantly (p < 0.01) in control cells but not in cells with ablation of RB family proteins (Figure 3E). This data provide strong evidence that repair reduction following RB family protein depletion results from loss of cNHEJ with the detected residual reporter activity arising from PRKDC-independent repair.
To probe whether cNHEJ is affected in cancers with mutational RB1 loss, we assessed reporter repair in SAOS-2 osteosarcoma cells, which carry an inactivating mutation in RB1 (Hellwinkel et al., 2005) and U2OS osteosarcoma cells, which have wild-type RB1. Using transient reporter transfections, SAOS-2 cells displayed a significantly lower level of NHEJ capacity (Figure 3G) compared to U2OS cells. We further used SAOS-2 and U2OS lines with stable chromosomal integration of the NHEJ reporter, assessing their respective propensity to undertake PRKDC-dependent, NU7441-sensitive repair. NU7441 did not affect repair proficiency in SAOS-2 cells but significantly reduced repair proficiency in the U2OS cells (Figure 3H). These experiments provide direct evidence for diminished use of cNHEJ in RB1 mutated cancer cells. They in addition confirm repair loss is observable in chromosomally integrated, chromatinized settings. Immunoblotting for PRKDC autophosphorylation (Ser2056) indicated that NU7441 achieved PRKDC inhibition in HCT116, SAOS-2, and U2OS cells (Figure 3I). This analysis further reveals activation of PRKDC occurs in SAOS-2, indicating that PRKDC activation, an early step of cNHEJ repair, arises independently of RB1 function.
The Role of RB1N in NHEJ
To test whether re-expression of RB1 reinstates the capacity of RB1-negative cells to perform cNHEJ, we transfected plasmid encoding human RB1 (Chew et al., 1998) alongside the NHEJ reporter into SAOS-2 cells. We also transfected these cells with plasmid encoding the PolyG alteration shown in our earlier experiments (see Figures 1D and 1E) to disable the interaction with XRCC6. Expression of RB1 significantly increased NHEJ competency compared to vector transfection. In contrast, RB1PolyG was unable to increase NHEJ proficiency (Figure 4A). Furthermore, expression of RB1 but not expression of RB1PolyG restored sensitivity of repair to NU7741, supporting that RB1 but not RB1PolyG reinstates use of cNHEJ in the transfected cells. Parallel immunoblotting revealed equal expression of RB1 and RB1PolyG (Figure 4C). Identical results also were obtained with the XRCC6 binding-defective cancer-associated variant, RB1240–242 del (Figure S4), which like RB1PolyG was unable to support NHEJ despite appropriate expression. RB1 strongly inhibited cell-cycle progression in the transfected SAOS-2 cells, promoting their accumulation and stable arrest in G1 (Figure 4B). This is an expected outcome for RB1-negative cancer cells, where transformation associated hyperactivation of the INK4/ARF locus prevents cell-cycle-dependent RB1 inactivation, and contrasts to cancer and primary cells containing wild-type (WT) RB1 expression that usually are competent to inactivate RB1 (Hinds et al., 1992). Significantly, cell-cycle arrest was seen also where RB1PolyG (Figure 4B) and RB1240–242 del (Figure S4B) were used, with no statistically significant difference detectable compared with RB1. These results provide direct evidence that enablement of cNHEJ by RB1 is not an attribute, or consequence, of G1 cell-cycle arrest, which can be actioned by NHEJ-competent WT RB1 and the NHEJ-incompetent variants alike, but involves a mechanism distinct and genetically separable from that involved in cell-cycle control. This conclusion is supported further by experiments run under conditions that did not involve DNA damage reporter co-expression, revealing identical potency of RB1, RB1PolyG, and RB1240–242 del to support G1 cell-cycle arrest and E2F regulation and to suppress colony outgrowth of SAOS-2 cells (Figures 4D–4H and S4D–S4H).
Figure 4.

The Role of RB1N in NHEJ
(A) NHEJ proficiency in SAOS-2 cells expressing RB1 or RB1PolyG variant and response to NU7441 treatment. Data evaluation was as for Figure 3. Related data for RB1240–242 deletion are shown in Figure S4.
(B) Cell-cycle profiles for cells transfected alongside cells from (A). Nocodazole was added 16 hr before harvest to quantify stable G1 arrest.
(C) Immunodetection of RB1 and RB1PolyG in SAOS-2 from (A); loading was normalized for co-transfected DsRed expression plasmid.
(D) Proficiency of RB1PolyG to inhibit cell-cycle progression in SAOS-2. Graph depict percentage of cells with stable G1 arrest, assessed as in (B). SAOS-2 cells transfected with empty vector, RB1, or Poly G expression vector. Related data for RB1240–242 deletion are shown in Figure S4.
(E) Proficiency of RB1PolyG to inhibit colony formation. SAOS-2 transfected with empty RB1 or RB1PolyG encoding vector in combination with vector for puromycin resistance and scored for outgrowth of puromycin resistant colonies. Co-transfected β-galactosidase was used to normalize data to transfection efficacy. Related data for RB1240–242 deletion are shown in Figure S4.
(F) Proficiency of RB1PolyG to regulate E2F activity. E2F reporter activity in SAOS-2 cells. Cells were transfected with RB1, RB1PolyG, or empty expression vector together with E2F promoter luciferase reporter. Co-transfected β-galactosidase was used to normalize data to transfection efficacy. Related data for RB1240–242 deletion are shown in Figure S4.
(G) Immunodetection of RB1 and RB1PolyG in SAOS-2 used in (D) and (E). Loading was normalized using co-transfected EGFP expression plasmid.
(H) Immunodetection of RB1 and RB1PolyG in SAOS-2 used in (F). Loading was normalized to β-galactosidase activity.
For all experiments the average for n = 3 biological replicates is depicted; error bars ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 using a paired Student’s t test.
(I) NHEJ proficiency in TKO MEFs transfected with empty vector or vector expressing RB1 or RB1PolyG variant. Data evaluation was as for (A). Values for MEFs transfected with empty vector were set to 1.
(J) Cell-cycle profiles of reporter transfected TKO MEFs, analyzed as for Figure 4I. Profiles with or without secondary nocodazole block are shown. Data represent transfected cell fractions deduced using DsRed for gating.
(K) Immunodetection of RB1 and RB1PolyG in MEFs from (I); loading was normalized for co-transfected DsRed expression plasmid.
The described observations in SAOS-2 were also corroborated by experiments using mouse embryo fibroblasts (MEFs) with disruption of RB1, RBL1, and RBL2 (TKOs) (Dannenberg et al., 2000). Expression of RB1 significantly increased NHEJ competency, while RB1PolyG, despite adequate expression, did not (Figures 4I and 4L). In these cells, no significant impact on cell-cycle distribution or the ability to progress through the cell cycle to G2 was detected upon expression of either wild-type RB1 or RB1PolyG, in keeping with the ability of these primary cells to undertake cell-cycle-dependent RB1 inactivation, providing further emphasis that the differential support of NHEJ is not explained by differential effects of the two RB1 forms on the cell cycle and is not relying on dominant cell-cycle arrest instilled by RB1 (Figure 4K). In summary, these results provide strong evidence for a role of RB1 in supporting cNHEJ. They further document a mechanism of action distinct from RB1’s function of controlling cell-cycle progression and reveal selective loss of ability to support NHEJ in RB1 variants defective for XRCC5 and XRCC6 binding, including a naturally occurring cancer-associated variant.
RB Family Protein Loss Impairs DNA Damage Clearance
Loss of cNHEJ in cells leads to use of alternative, slower forms of repair, resulting in reduced speed by which DSBs are resolved (Kinashi et al., 2011; Vandersickel et al., 2010). We therefore determined whether repair speed was reduced by RB1 loss using TKO MEFs as well as congenic MEFs with disruption in RB1 (RB1−/−) (Jacks et al., 1992), measuring loss of γH2AX foci as a surrogate for the DSBs resolution (Kuo and Yang, 2008) (Figure 5A).
Figure 5.

RB Family Protein Loss Impairs DNA Damage Clearance
(A) DSB clearance in cells with compromised RB1 function. Wild-type, RB1−/−, or RB1/RBL1/RBL2-null (TKOs) mouse embryonic fibroblasts (MEFs) were exposed to IR of 5 Gy. Prevalence of damage at the indicated times was detected by immunofluorescence staining for γH2AX (green); nuclei were visualized with Hoechst 33258 (blue). Raw images recorded using a high-content imaging platform are shown. Scale bar represents 10 μm.
(B and C) Automated quantitative assessment of damage clearance. γH2AX staining intensity (iSig) was determined for all cells from 30 independent eye fields. (B) Integrated H2AX signal distribution for cells from one representative experiment. The dotted line shows the gating position for cells with above baseline anti-γH2AX fluorescence. DNA content analysis for the same samples shown in Figure S5. (C) Percentage of cells with residual damage determined by gating (dotted line) for cells with above baseline anti-γH2AX fluorescence at each time point. Error bars represent ±SD for n = 3 biological replicates. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 using a paired Student’s t test comparing RB1−/− or TKOs to wild-type cells.
(D) Immunodetection documenting RB protein family loss in MEFs. Lysate from MEFs used for (A)–(C) were probed using antibodies as indicated.
Automated high-content image analysis (Figures 5B–5D) revealed a delay in γH2AX signal loss detectable within 2 hr post IR, indicative of reduced repair speed in both these genetic backgrounds within a time frame comparable to that reported for cells with XRCC5 and XRCC6 loss (Kinashi et al., 2011; Vandersickel et al., 2010). Furthermore, repair delay was exaggerated in TKO compared to RB1−/− MEFs, consistent with our previous observations documenting exacerbated repair loss in HCT116 with combined loss of RB paralogs (Figure 3). Quantification of cells with residual damage confirmed significantly reduced γH2AX clearance in MEFs with RB1 or RB family loss (Figure 5C). Similar levels of γH2AX fluorescence were observed early (15 min) after exposure to IR in all MEF lines, indicating that differences in γH2AX signals at later time points do not reflect a difference in the amount of initial damage (Figure 5C).
The speed by which DSBs are repaired is dependent on cell-cycle position (Chapman et al., 2012). To address whether the differences in repair proficiency observed could be due to differences in the cell-cycle distribution between the different MEF lines, we measured the DNA content by integrating the intensity of the Hoechst DNA stain in the same cells that were quantified for γH2AX intensity. Histograms generated from these measurements revealed typical cell-cycle profiles (Figure S5), but no significant differences in the overall cell-cycle distributions between backgrounds in any of the three experiments as assessed by Wilcoxon rank-sum testing. Similarly, t testing following gating for cells with <2n, 2n, >2–4n, and >4n DNA content (Figure S5) revealed no statistically significant difference for any of these categories at any of the observed time points, ruling out overt cell-cycle variations as an explanation for the differences in DSB clearance observed.
Effect of RB Protein Loss in the Maintenance of Chromosomal Integrity
To obtain direct evidence that RB protein loss impairs DNA repair and assess whether RB protein loss adversely affects chromosomal integrity as expected for cells with cNHEJ defect (Iliakis et al., 2004), we determined chromatin repair and chromosomal radiation sensitivity using comet analysis and metaphase spreads (Figure 6). Comet analysis detected a significant increase in cells with excess unligated chromatin, detectable as late as 8 hr after irradiation in cells with RB family loss. Similar results were obtained for TKO MEFs compared to WT MEF and HCT116 in which RB family proteins were depleted using siRNA compared to either mock-treated HCT116 or HCT116 treated with a nontargeting oligonucelotide (Figure 6; for raw data, see Figure S6). No significant difference was observed in the amount or distribution of DNA damage immediately after irradiation, indicating defective DNA repair as opposed to increased damage susceptibility in cells with RB family loss. We also assessed radiation-induced excess first division chromosomal aberrations, initially using HCT116 cells with siRNA-mediated RB family loss. We observed a significant increase in the frequency of chromosomal aberrations compared to mock-transfected cells, comprised of increased chromosome-type aberrations and surplus chromatid breaks (Table S4). Identical results were obtained when we treated these same cells with siRNA targeting DNA ligase 4 (LIG4), involved in DNA strand ligation in cNHEJ. We also probed for loss of chromosomal integrity in irradiated TKO MEFs, revealing a consistent and significant increase of chromosome-type aberrations compared to congenic wild-type MEFs (Figure S6; Table S4). Together these results strongly support the notion that loss of RB function significantly impairs cellular DSB repair and significantly increases the incidence of structural chromosomal aberrations and chromosomal instability.
Figure 6.

RB Family Loss Impairs Break Repair and Increases Radiation Induced Chromosomal Aberration
(A) Analysis of DNA strand break induction and repair by Comet assay. Data for wild-type and RB family defective (TKO) MEFs and data for HCT116 colorectal cancer cells transfected with siRNA targeting RB family proteins RB1, RBL1, and RBL2, a non-targeting control (NT), or in the absence of oligonucleotide are shown. Cells were treated with either 10 or 15 Gy and harvested for analysis at the indicated time. Data shown are derived from two independently processed technical replicates. 25 cells per replicate were analyzed.
(B) Immunodetection documenting RB protein family loss in MEFs and HCT116. Lysates were prepared from MEFs and HCT116 used for (A), (C), (D), and Figure S6 and probed using antibodies as indicated.
(C) Chromosome aberration analysis. HCT116 treated with siRNA as for (A) were irradiated with the dose indicated. Cells were processed for analysis 16 hr post IR. Colcemid was added for the final 10 hr. Table S4 provides full numerical details of all analyses. Errors represent SEM. Related analysis of wild-type and TKO MEFs shown in Figure S6.
(D) Examples of Giemsa-stained metaphases, documenting aberrations detected in HCT116. Examples of Giemsa-stained metaphases from HCT116 cells documenting exemplary aberration scored. Aberrations denoted by arrows; dc, dicentric; ac, excess acentric fragment; ctb, chromatid break.
Error shown represents SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 using an unpaired heteroscedacstic Student’s t test.
Discussion
We here document a thus-far-unrecognized role of the RB family in supporting DNA repair by cNHEJ. We show that RB family protein loss is associated with canonical features observed in cells with defective cNHEJ, including loss of PRKDC-dependent damage repair, delayed damage clearance, and excess genotoxin-induced structural chromosomal aberrations.
Loss of cNHEJ promotes chromosomal rearrangements presumed to arise from increased use of error-prone aNHEJ (Rothkamm et al., 2003). Our experiments documenting delayed repair kinetics and excess same cycle chromosomal aberrations in cells with RB family protein loss is consistent with this notion. We note that the excess of chromosomal aberrations is more pronounced and diverse in transformed HCT116 cells compared to primary MEFs. Differences in type and level of chromosomal aberrations between primary and transformed cells with cNHEJ loss have been noted previously (Burma et al., 2006) and may be explained by loss of DNA integrity surveillance in cancers cells, permitting survival and cell-cycle progression despite reduced repair.
A substantial body of recent work has documented loss of mitotic fidelity in cellular models with RB1 loss (Manning and Dyson, 2012) and loss of mitotic fidelity is well suited to explain the development of chromosomal aneuploidy seen in retinoblastoma tumors. However, loss of mitotic fidelity cannot explain the spectrum of GIN seen in retinoblastoma. Genomic profiling of malignant retinoblastoma compared to premalignant retinoma lesions suggests complex GIN is associated with malignant progression in this disease (Dimaras et al., 2008; Sampieri et al., 2008) and GIN, comprising extensive copy-number changes and rearrangements, is prominent in late onset, sporadic disease, with presence in early onset, familial cases, although to a lesser degree (Thériault et al., 2014). These observations identify GIN as a relevant, albeit facultative event in retinoblastoma progression consistent with a concept whereby defective repair following from RB1 loss combines with stochastic factors, including environmental genotoxin exposure and checkpoint loss, that cooperate to unleash GIN in this disease. Significantly, recent work documented excess micronuclei formation, indicative of excess acentric chromosome fragments, in primary murine osteoblasts with RB1 loss, exacerbated by IR (Gonzalez-Vasconcellos et al., 2013), which is in agreement with our observations of excess genetic aberration in cells with loss of RB function. Importantly, evidence of excess chromosomal aberrations was apparent in cells with heterozygous loss of RB1, inferring systemic repair defects may manifest in individuals with constitutional RB1 mutation where heterozygous defects are present in somatic tissue. Defective DSB repair with excess genotoxin-driven genetic aberration documented here and in Gonzalez-Vasconcellos et al. (2013) may explain the recognized risk for secondary cancers in such patients following external beam radiotherapy and genotoxic chemotherapy, for which a mechanistic justification has thus far been lacking (Kleinerman et al., 2012; Wong et al., 2014).
Our data allow us to draw several mechanistic conclusions pertaining to the contribution of RB1 to cNHEJ. We identify two different fold-preserving variants affecting RB1N that are defective for the interaction with XRCC6. Significantly, we document that these RB1 variants are unable to support NHEJ in primary MEFs with RB family loss and in cancer-derived SAOS-2 osteosarcoma cells with spontaneous inactivation of RB1. Together these results provide strong support that the physical interaction with cNHEJ proteins underlies the mechanism through which RB1 affects this form of repair. Importantly, RB1 variants defective in supporting NHEJ were capable of blocking cell-cycle progression and E2F transcriptional activation as effectively as WT RB1, providing strong support that neither loss of cell-cycle inhibition nor loss of E2F regulation provide a mechanistic explanation for the contribution of RB proteins in NHEJ that we describe. A recent report demonstrates stimulation of HR following enforced expression of RB1 in a RB1-mutated, retinoblastoma-derived cell line, which could suggest a potentially broader role of RB1 in DSB repair (Yang et al., 2013). Whether RB1 has a direct critical role in HR or whether cell-cycle changes seen to arise in the cells following RB1 expression indirectly affected repair pathway choice is not known. HR components were not identified in the mass spectrometry approach we used suggesting the mechanistic contribution of RB1 to NHEJ and HR is distinct. As noted DSB repair involves extensive chromatin modification. RB1 and its paralogs can facilitate chromatin modification through recruitment of chromatin modifiers that mostly interact with RB1P and the RB1 carboxyterminal region (reviewed in Manning and Dyson [2012] and Talluri and Dick [2012]). Intriguingly, several of these modifiers are known to be involved in cNHEJ, including the ISWI remodeling machine (Aydin et al., 2014), the HDAC1 and 2 histone deacetylases (Miller et al., 2010), the SUV4 histone methyl transferase (Tuzon et al., 2014), and the inhibitor of resection 53BP1 (Carr et al., 2014). A proposition for a mechanism of action how RB family proteins support cNHEJ could be that they, by way of interaction with the damage recognition complex, recruit these chromatin modifiers to the site of DNA repair.
A considerable body of evidence supports the view that cNHEJ is active and has major roles during G1 and early S (Branzei and Foiani, 2008), the cell-cycle phases where RB family proteins are naturally active or are known to be activated by DNA damage-associated inhibition of the CDK4, 6, and 2 cyclin-dependent kinases. Recent work has shown that cNHEJ is also used in G2 (Kakarougkas and Jeggo, 2014), where RB proteins are not known to be active. Our quantitative data based on monitoring reporter repair argue the contribution of RB family proteins is considerable, with little PRKDC-dependent repair activity observed in cells with RB loss. However, G2 phase cells are naturally underrepresented in actively proliferating cell populations. Hence, the quantitative contribution of G2 cells to the total repair activity within the population is expected to be small and may remain underreported by the reporter-based assessment used here.
NHEJ plays a key role in VDJ recombination, and mutations in components of cNHEJ, including PRKDC and Artemis, result in severe combined immunodeficiency (SCID) affecting B and T cell maturation in humans and mice (Woodbine et al., 2014). While there is evidence that mechanistic differences between DSB repair and VDJ exist (Malu et al., 2012), it is possible that RB1 loss alone or together with its paralogs affects VDJ recombination in addition to DSB repair. Homozygous loss of RB1 is embryonic lethal in mice (Jacks et al., 1992) and presumably in human. RB1 loss arises in individuals with somatic or germline heterozygous mutations upon subsequent loss of the wild-type allele through secondary mutation or mitotic nondisjunction. Given the rarity of these events, it would not be expected that VDJ deficiency and associated pathologies are observable, or clinically relevant, in this context. However, several studies have assessed the effect of engineered RB1 and RB family deletion on the hematopoietic system in the mouse (Daria et al., 2008; Viatour et al., 2008; Walkley and Orkin, 2006). Although these studies did not explicitly address whether VDJ recombination is lost, phenotypes consistent with such loss were observed. Two independent studies examining RB1 deletion note a considerable decrease in B cells with IGG/IGM rearrangement, although the number of B cells precursors with un-rearrange IGG/IGM was not unaltered (Daria et al., 2008; Walkley and Orkin, 2006). No effect on T cell maturation was observed, which might be explained by redundant functioning of RB family proteins in this lineage. Radical loss of common lymphoid precursors was observed following combined deletion of the RB family (Viatour et al., 2008), indicating additional and profound early lineage reliance on RB family proteins at a stage predating T cell differentiation and VDJ recombination, precluding conclusions as to the role of the RB family in VDJ recombination in this lineage.
Our experiments document that the role of RB1 in cNHEJ involves the RB1N domain. Although a role of RB1N in tumor suppression had been anticipated (Blanquet et al., 1995; Lee et al., 1992; Lohmann et al., 1997), a mechanistic basis of how defects in this region contribute to cancer development has remained elusive. This work now demonstrates a defined functional role of this domain. The involvement of RB1N in supporting cNHEJ provides a plausible mechanistic explanation for tumorigenicity associated with mutations in this region.
Our results have important conceptual implications in that they identify RB family proteins as cofactors supporting cNHEJ and predict impairment of this repair pathway in RB1-negative cancers with consequence of structural chromosomal instability that promotes tumor evolution in response to genotoxic stress. They also present potential opportunities for therapy that exploit the greater reliance of such cancers on alternative routes of repair.
Experimental Procedures
Cells, DNA plasmids, siRNA, and antibodies and general procedures involving these are described in the Supplemental Experimental Procedures.
GST Affinity Capture and Mass Spectrometry
GST fusion proteins were expressed in E. coli and produced as described (Hassler et al., 2007). Purified GST tagged proteins were bound to Glutathione Sepharose 4 Fast Flow resin and used as an affinity matrix for interacting protein capture from nuclear cell extract. Affinity-captured proteins were either assessed using tryptic proteolysis followed by MS analysis (QSTAR Elite, Applied Biosystems) or immunoblotting. Raw MS data were analyzed using Mascot version 2.1 (Matrix Science) followed by Scaffold 3 (Proteome Software).
Co-immunoprecipitation
Nuclear extracts were prepared from purified cell nuclei using micrococcal nuclease to release chromatin-bound protein complexes. Cell nuclei were made as for GST affinity capture. RB1 was immunoprecipitated using rabbit serum against a human RB1 C-terminal fragment (aa 763–928) (Zarkowska and Mittnacht, 1997) crosslinked to Protein A/G Plus Agarose (Thermo Fisher Scientific).
Proximity Ligation Analysis
PLA was performed using Duolink technology (Cambridge Biosciences) in accordance with the manufacturer’s recommendations.
γH2AX Staining and Quantitation
Cells were cultured in black-walled 96-well plates (PerkinElmer) and fixed and processed as described in Richardson et al. (2012). Cells were probed with anti-γH2AX for 3 hr at room temperature followed by Alexa Fluor 488 fluorescent secondary antibody (Invitrogen) containing 2 μM Hoechst 33342 (Sigma). Plates were imaged using an automated high-content platform (GE Healthcare). The average intensity (iSig) of the γH2AX signal was recorded for all individual cells present in six eye fields in each of five wells. The integrated intensity for Hoechst 33342 was extracted using CellProfiler Image analysis freeware.
NHEJ Reporter Assay
NHEJ reporter assays were performed as described (Bennardo et al., 2008). For parallel cell-cycle analysis NHEJ reporter/DsRed-transfected cells were subjected to fluorescence-activated cell sorting for DsRed-positive cells. DsRed-positive cells were subjected to propidium iodide (PI) staining of DNA followed by analysis using flow cytometry. Alternatively, transfected cells were subjected to life-cell staining of DNA using Hoechst 33342 dye. Prior to analysis cells were incubated for 45 min in media containing 10 μg/ml Hoechst 33342 followed by dual color flow cytometry analysis with data collection gated for DsRed-positive cells. Data for 5,000–10,000 individual cells were collected in each case.
Cell-based assays assessing the effect of RB1 on E2F regulation and cell proliferation are described in the Supplemental Experimental Procedures.
Chromosomal Radiation Sensitivity Analysis
HCT116 cells were transfected with siRNA 48 hr prior to irradiation. Colcemid (Sigma-Aldrich Karyomax) was added at 20 ng/ml 6 hr (HCT116) or 24 hr (MEFs) post IR. Cells were harvested for analysis 16 hr (HCT116) or 28 hr (MEFs) post IR using trypsin. Giemsa-stained metaphase spreads were prepared and analyzed as previously described (Nuta et al., 2014).
Comet Analysis
Cells irradiated on ice were either harvested immediately (0 hr) or following further incubation at 37°C. Cells were embedded in low melting agarose on glass slides followed by lysis in 100 mM EDTA, 2.5 M NaCl, 10 mM Tris-HCl (pH 10.5), 1% Triton X-100 followed by electrophoresis in 50 mM NaOH, 1 mM disodium EDTA (pH 12.5). Slides were stained with propidium iodide, visualized at 20× magnification, and analyzed using Komet Analysis software 6.0 (Andor Technology).
Additional details for all experimental procedures are provided in the Supplemental Experimental Procedures.
Author Contributions
S.M. and P.H.H. planned and supervised the project. R.C., P.H.H., and S.M. wrote the manuscript. R.C., J.A.H, K.R., P.H.H., and S.M. designed experiments. V.J.S. and J.A.H. supervised Comet analysis, K.R. supervised analysis of chromosomal radiation sensitivity performed by J.M. R.C. performed the majority of experiments. G.Z. performed Comet and GST-fusion protein interaction analyses and supported chromosomal radiation sensitivity analysis. M.T.L. contributed to RB1 mutant analysis. S.R performed cell synchronization experiments. R.C., G.Z., K.R., P.H.H., and S.M. analyzed data.
Acknowledgments
The authors thank Professor J. M. Stark for providing the NHEJ reporter plasmids, Professor H. teRiele for TKO MEFs, and Drs. S.R. Stockwell and D. Wetterskog for support with high-content image analysis. The work was supported through Cancer Research UK (C107/10433 and CRUK/A15043 to S.M. and C2259/A16569 to J.H.), Worldwide Cancer Research (12-1280 to S.M.) and the Wellcome Trust (WT089028 to P.H.H.). R.C. was supported by an ICR fellowship. J.M. and K.R. were supported by the National Institute for Health Research. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The views expressed in this publication are those of the authors and not necessarily those of Cancer Research UK, Worldwide Cancer Research, the Wellcome Trust, the NHS, the National Institute for Health Research or the Department of Health.
Published: March 26, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Supplemental Information includes Supplemental Experimental Procedures, six figures, and four tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2015.02.059.
Contributor Information
Paul H. Huang, Email: paul.huang@icr.ac.uk.
Sibylle Mittnacht, Email: s.mittnacht@ucl.ac.uk.
Supplemental Information
References
- Aparicio T., Baer R., Gautier J. DNA double-strand break repair pathway choice and cancer. DNA Repair (Amst.) 2014;19:169–175. doi: 10.1016/j.dnarep.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aydin O.Z., Vermeulen W., Lans H. ISWI chromatin remodeling complexes in the DNA damage response. Cell Cycle. 2014;13:3016–3025. doi: 10.4161/15384101.2014.956551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennardo N., Cheng A., Huang N., Stark J.M. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008;4:e1000110. doi: 10.1371/journal.pgen.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanquet V., Turleau C., Gross-Morand M.S., Sénamaud-Beaufort C., Doz F., Besmond C. Spectrum of germline mutations in the RB1 gene: a study of 232 patients with hereditary and non hereditary retinoblastoma. Hum. Mol. Genet. 1995;4:383–388. doi: 10.1093/hmg/4.3.383. [DOI] [PubMed] [Google Scholar]
- Branzei D., Foiani M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008;9:297–308. doi: 10.1038/nrm2351. [DOI] [PubMed] [Google Scholar]
- Burma S., Chen B.P., Chen D.J. Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair (Amst.) 2006;5:1042–1048. doi: 10.1016/j.dnarep.2006.05.026. [DOI] [PubMed] [Google Scholar]
- Carr S.M., Munro S., Zalmas L.P., Fedorov O., Johansson C., Krojer T., Sagum C.A., Bedford M.T., Oppermann U., La Thangue N.B. Lysine methylation-dependent binding of 53BP1 to the pRb tumor suppressor. Proc. Natl. Acad. Sci. USA. 2014;111:11341–11346. doi: 10.1073/pnas.1403737111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman J.R., Taylor M.R., Boulton S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell. 2012;47:497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- Chew Y.P., Ellis M., Wilkie S., Mittnacht S. pRB phosphorylation mutants reveal role of pRB in regulating S phase completion by a mechanism independent of E2F. Oncogene. 1998;17:2177–2186. doi: 10.1038/sj.onc.1202443. [DOI] [PubMed] [Google Scholar]
- Chiruvella K.K., Liang Z., Wilson T.E. Repair of double-strand breaks by end joining. Cold Spring Harb. Perspect. Biol. 2013;5:a012757. doi: 10.1101/cshperspect.a012757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannenberg J.H., van Rossum A., Schuijff L., te Riele H. Ablation of the retinoblastoma gene family deregulates G(1) control causing immortalization and increased cell turnover under growth-restricting conditions. Genes Dev. 2000;14:3051–3064. doi: 10.1101/gad.847700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daria D., Filippi M.D., Knudsen E.S., Faccio R., Li Z., Kalfa T., Geiger H. The retinoblastoma tumor suppressor is a critical intrinsic regulator for hematopoietic stem and progenitor cells under stress. Blood. 2008;111:1894–1902. doi: 10.1182/blood-2007-02-071746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G., Jr., Sherman B.T., Hosack D.A., Yang J., Gao W., Lane H.C., Lempicki R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:3. [PubMed] [Google Scholar]
- Dimaras H., Khetan V., Halliday W., Orlic M., Prigoda N.L., Piovesan B., Marrano P., Corson T.W., Eagle R.C., Jr., Squire J.A., Gallie B.L. Loss of RB1 induces non-proliferative retinoma: increasing genomic instability correlates with progression to retinoblastoma. Hum. Mol. Genet. 2008;17:1363–1372. doi: 10.1093/hmg/ddn024. [DOI] [PubMed] [Google Scholar]
- Doksani Y., de Lange T. The role of double-strand break repair pathways at functional and dysfunctional telomeres. Cold Spring Harb. Perspect. Biol. 2014;6:a016576. doi: 10.1101/cshperspect.a016576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry D.W., Harvey P.J., Keller P.R., Elliott W.L., Meade M., Trachet E., Albassam M., Zheng X., Leopold W.R., Pryer N.K., Toogood P.L. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol. Cancer Ther. 2004;3:1427–1438. [PubMed] [Google Scholar]
- Gonzalez-Vasconcellos I., Anastasov N., Sanli-Bonazzi B., Klymenko O., Atkinson M.J., Rosemann M. Rb1 haploinsufficiency promotes telomere attrition and radiation-induced genomic instability. Cancer Res. 2013;73:4247–4255. doi: 10.1158/0008-5472.CAN-12-3117. [DOI] [PubMed] [Google Scholar]
- Goodrich D.W. How the other half lives, the amino-terminal domain of the retinoblastoma tumor suppressor protein. J. Cell. Physiol. 2003;197:169–180. doi: 10.1002/jcp.10358. [DOI] [PubMed] [Google Scholar]
- Hassler M., Singh S., Yue W.W., Luczynski M., Lakbir R., Sanchez-Sanchez F., Bader T., Pearl L.H., Mittnacht S. Crystal structure of the retinoblastoma protein N domain provides insight into tumor suppression, ligand interaction, and holoprotein architecture. Mol. Cell. 2007;28:371–385. doi: 10.1016/j.molcel.2007.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellwinkel O.J., Müller J., Pollmann A., Kabisch H. Osteosarcoma cell lines display variable individual reactions on wildtype p53 and Rb tumour-suppressor transgenes. J. Gene Med. 2005;7:407–419. doi: 10.1002/jgm.684. [DOI] [PubMed] [Google Scholar]
- Henley S.A., Dick F.A. The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle. Cell Div. 2012;7:10. doi: 10.1186/1747-1028-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensey C.E., Hong F., Durfee T., Qian Y.W., Lee E.Y., Lee W.H. Identification of discrete structural domains in the retinoblastoma protein. Amino-terminal domain is required for its oligomerization. J. Biol. Chem. 1994;269:1380–1387. [PubMed] [Google Scholar]
- Heyer W.D., Ehmsen K.T., Liu J. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010;44:113–139. doi: 10.1146/annurev-genet-051710-150955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgendorf K.I., Leshchiner E.S., Nedelcu S., Maynard M.A., Calo E., Ianari A., Walensky L.D., Lees J.A. The retinoblastoma protein induces apoptosis directly at the mitochondria. Genes Dev. 2013;27:1003–1015. doi: 10.1101/gad.211326.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinds P.W., Mittnacht S., Dulic V., Arnold A., Reed S.I., Weinberg R.A. Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell. 1992;70:993–1006. doi: 10.1016/0092-8674(92)90249-c. [DOI] [PubMed] [Google Scholar]
- Iliakis G., Wang H., Perrault A.R., Boecker W., Rosidi B., Windhofer F., Wu W., Guan J., Terzoudi G., Pantelias G. Mechanisms of DNA double strand break repair and chromosome aberration formation. Cytogenet. Genome Res. 2004;104:14–20. doi: 10.1159/000077461. [DOI] [PubMed] [Google Scholar]
- Jacks T., Fazeli A., Schmitt E.M., Bronson R.T., Goodell M.A., Weinberg R.A. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- Kakarougkas A., Jeggo P.A. DNA DSB repair pathway choice: an orchestrated handover mechanism. Br. J. Radiol. 2014;87:20130685. doi: 10.1259/bjr.20130685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinashi Y., Takahashi S., Kashino G., Okayasu R., Masunaga S., Suzuki M., Ono K. DNA double-strand break induction in Ku80-deficient CHO cells following boron neutron capture reaction. Radiat. Oncol. 2011;6:106. doi: 10.1186/1748-717X-6-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinerman R.A., Yu C.L., Little M.P., Li Y., Abramson D., Seddon J., Tucker M.A. Variation of second cancer risk by family history of retinoblastoma among long-term survivors. J. Clin. Oncol. 2012;30:950–957. doi: 10.1200/JCO.2011.37.0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo L.J., Yang L.X. Gamma-H2AX - a novel biomarker for DNA double-strand breaks. In Vivo. 2008;22:305–309. [PubMed] [Google Scholar]
- Lee E.Y., Chang C.Y., Hu N., Wang Y.C., Lai C.C., Herrup K., Lee W.H., Bradley A. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–294. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- Lee J.O., Russo A.A., Pavletich N.P. Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from HPV E7. Nature. 1998;391:859–865. doi: 10.1038/36038. [DOI] [PubMed] [Google Scholar]
- Lee C., Chang J.H., Lee H.S., Cho Y. Structural basis for the recognition of the E2F transactivation domain by the retinoblastoma tumor suppressor. Genes Dev. 2002;16:3199–3212. doi: 10.1101/gad.1046102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end joining pathway. Annu. Rev. Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber M.R., Ma Y., Pannicke U., Schwarz K. Mechanism and regulation of human non-homologous DNA end joining. Nat. Rev. Mol. Cell Biol. 2003;4:712–720. doi: 10.1038/nrm1202. [DOI] [PubMed] [Google Scholar]
- Lohmann D. Retinoblastoma. Adv. Exp. Med. Biol. 2010;685:220–227. doi: 10.1007/978-1-4419-6448-9_21. [DOI] [PubMed] [Google Scholar]
- Lohmann D.R., Gerick M., Brandt B., Oelschläger U., Lorenz B., Passarge E., Horsthemke B. Constitutional RB1-gene mutations in patients with isolated unilateral retinoblastoma. Am. J. Hum. Genet. 1997;61:282–294. doi: 10.1086/514845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malu S., Malshetty V., Francis D., Cortes P. Role of non-homologous end joining in V(D)J recombination. Immunol. Res. 2012;54:233–246. doi: 10.1007/s12026-012-8329-z. [DOI] [PubMed] [Google Scholar]
- Manning A.L., Dyson N.J. RB: mitotic implications of a tumour suppressor. Nat. Rev. Cancer. 2012;12:220–226. doi: 10.1038/nrc3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meadows A.T., Friedman D.L., Neglia J.P., Mertens A.C., Donaldson S.S., Stovall M., Hammond S., Yasui Y., Inskip P.D. Second neoplasms in survivors of childhood cancer: findings from the Childhood Cancer Survivor Study cohort. J. Clin. Oncol. 2009;27:2356–2362. doi: 10.1200/JCO.2008.21.1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellacheruvu D., Wright Z., Couzens A.L., Lambert J.P., St-Denis N.A., Li T., Miteva Y.V., Hauri S., Sardiu M.E., Low T.Y. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods. 2013;10:730–736. doi: 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller K.M., Tjeertes J.V., Coates J., Legube G., Polo S.E., Britton S., Jackson S.P. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end joining. Nat. Struct. Mol. Biol. 2010;17:1144–1151. doi: 10.1038/nsmb.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrini S., Gorgoulis V.G., Halazonetis T.D. Genomic instability—an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010;11:220–228. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- Nuta O., Moquet J., Bouffler S., Lloyd D., Sepai O., Rothkamm K. Impact of long-term exposure to sodium arsenite on cytogenetic radiation damage. Mutagenesis. 2014;29:123–129. doi: 10.1093/mutage/get070. [DOI] [PubMed] [Google Scholar]
- Panier S., Boulton S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014;15:7–18. doi: 10.1038/nrm3719. [DOI] [PubMed] [Google Scholar]
- Papamichos-Chronakis M., Peterson C.L. Chromatin and the genome integrity network. Nat. Rev. Genet. 2013;14:62–75. doi: 10.1038/nrg3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson E., Stockwell S.R., Li H., Aherne W., Cuomo M.E., Mittnacht S. Mechanism-based screen establishes signalling framework for DNA damage-associated G1 checkpoint response. PLoS ONE. 2012;7:e31627. doi: 10.1371/journal.pone.0031627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothkamm K., Krüger I., Thompson L.H., Löbrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell. Biol. 2003;23:5706–5715. doi: 10.1128/MCB.23.16.5706-5715.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampieri K., Mencarelli M.A., Epistolato M.C., Toti P., Lazzi S., Bruttini M., De Francesco S., Longo I., Meloni I., Mari F. Genomic differences between retinoma and retinoblastoma. Acta Oncol. 2008;47:1483–1492. doi: 10.1080/02841860802342382. [DOI] [PubMed] [Google Scholar]
- Schaal C., Pillai S., Chellappan S.P. The Rb-E2F transcriptional regulatory pathway in tumor angiogenesis and metastasis. Adv. Cancer Res. 2014;121:147–182. doi: 10.1016/B978-0-12-800249-0.00004-4. [DOI] [PubMed] [Google Scholar]
- Schmitt M.W., Prindle M.J., Loeb L.A. Implications of genetic heterogeneity in cancer. Ann. N Y Acad. Sci. 2012;1267:110–116. doi: 10.1111/j.1749-6632.2012.06590.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuin T., Torigoe S., Kubota Y., Kishida T., Hosaka M., Horikoshi T., Yao M., Kondo K., Sakai N., Danenberg K. Retinoblastoma gene mutation in primary human renal cell carcinoma. Oncol. Res. 1995;7:63–66. [PubMed] [Google Scholar]
- Talluri S., Dick F.A. Regulation of transcription and chromatin structure by pRB: here, there and everywhere. Cell Cycle. 2012;11:3189–3198. doi: 10.4161/cc.21263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thériault B.L., Dimaras H., Gallie B.L., Corson T.W. The genomic landscape of retinoblastoma: a review. Clin. Experiment. Ophthalmol. 2014;42:33–52. doi: 10.1111/ceo.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuzon C.T., Spektor T., Kong X., Congdon L.M., Wu S., Schotta G., Yokomori K., Rice J.C. Concerted activities of distinct H4K20 methyltransferases at DNA double-strand breaks regulate 53BP1 nucleation and NHEJ-directed repair. Cell Rep. 2014;8:430–438. doi: 10.1016/j.celrep.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandersickel V., Depuydt J., Van Bockstaele B., Perletti G., Philippe J., Thierens H., Vral A. Early increase of radiation-induced γH2AX foci in a human Ku70/80 knockdown cell line characterized by an enhanced radiosensitivity. J. Radiat. Res. (Tokyo) 2010;51:633–641. doi: 10.1269/jrr.10033. [DOI] [PubMed] [Google Scholar]
- Viatour P., Somervaille T.C., Venkatasubrahmanyam S., Kogan S., McLaughlin M.E., Weissman I.L., Butte A.J., Passegué E., Sage J. Hematopoietic stem cell quiescence is maintained by compound contributions of the retinoblastoma gene family. Cell Stem Cell. 2008;3:416–428. doi: 10.1016/j.stem.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkley C.R., Orkin S.H. Rb is dispensable for self-renewal and multilineage differentiation of adult hematopoietic stem cells. Proc. Natl. Acad. Sci. USA. 2006;103:9057–9062. doi: 10.1073/pnas.0603389103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weibrecht I., Leuchowius K.J., Clausson C.M., Conze T., Jarvius M., Howell W.M., Kamali-Moghaddam M., Söderberg O. Proximity ligation assays: a recent addition to the proteomics toolbox. Expert Rev. Proteomics. 2010;7:401–409. doi: 10.1586/epr.10.10. [DOI] [PubMed] [Google Scholar]
- Wong J.R., Morton L.M., Tucker M.A., Abramson D.H., Seddon J.M., Sampson J.N., Kleinerman R.A. Risk of subsequent malignant neoplasms in long-term hereditary retinoblastoma survivors after chemotherapy and radiotherapy. J. Clin. Oncol. 2014;32:3284–3290. doi: 10.1200/JCO.2013.54.7844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodbine L., Gennery A.R., Jeggo P.A. The clinical impact of deficiency in DNA non-homologous end joining. DNA Repair (Amst.) 2014;16:84–96. doi: 10.1016/j.dnarep.2014.02.011. [DOI] [PubMed] [Google Scholar]
- Xiao B., Spencer J., Clements A., Ali-Khan N., Mittnacht S., Broceño C., Burghammer M., Perrakis A., Marmorstein R., Gamblin S.J. Crystal structure of the retinoblastoma tumor suppressor protein bound to E2F and the molecular basis of its regulation. Proc. Natl. Acad. Sci. USA. 2003;100:2363–2368. doi: 10.1073/pnas.0436813100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Tian S., Brown B., Chen P., Hu H., Xia L., Zhang J., Cai X., Chen Z., Pan X. The Rb1 gene inhibits the viability of retinoblastoma cells by regulating homologous recombination. Int. J. Mol. Med. 2013;32:137–143. doi: 10.3892/ijmm.2013.1374. [DOI] [PubMed] [Google Scholar]
- Zarkowska T., Mittnacht S. Differential phosphorylation of the retinoblastoma protein by G1/S cyclin-dependent kinases. J. Biol. Chem. 1997;272:12738–12746. doi: 10.1074/jbc.272.19.12738. [DOI] [PubMed] [Google Scholar]
- Zhao Y., Thomas H.D., Batey M.A., Cowell I.G., Richardson C.J., Griffin R.J., Calvert A.H., Newell D.R., Smith G.C., Curtin N.J. Preclinical evaluation of a potent novel DNA-dependent protein kinase inhibitor NU7441. Cancer Res. 2006;66:5354–5362. doi: 10.1158/0008-5472.CAN-05-4275. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.