

Abstract
Isoniazid (INH) is usually administered to treat latent Mycobacterium tuberculosis (Mtb) infections, and is used in combination therapy to treat active tuberculosis disease (TB). Unfortunately, resistance to this drug is hampering its clinical effectiveness. INH is a prodrug that must be activated by Mtb catalase peroxidase (KatG) before it can inhibit InhA (Mtb enoyl-acyl-carrier-protein reductase). Isoniazid-resistant cases of TB found in clinical settings usually involve mutations in or deletion of katG, which abrogate INH activation. Compounds that inhibit InhA without requiring prior activation by KatG would not be affected by this resistance mechanism and hence would display continued potency against these drug-resistant isolates of Mtb. Virtual screening experiments versus InhA in the GO Fight Against Malaria project (GO FAM) were designed to discover new scaffolds that display base stacking interactions with the NAD cofactor. GO FAM experiments included targets from other pathogens, including Mtb, when they had structural similarity to a malaria target. Eight of the sixteen soluble compounds identified by docking against InhA plus visual inspection were modest inhibitors and did not require prior activation by KatG. The best two inhibitors discovered are both fragment-sized compounds and displayed Ki values of 54 and 59 μM, respectively. Importantly, the novel inhibitors discovered have low structural similarity to known InhA inhibitors and, thus, help expand the number of chemotypes on which future medicinal chemistry efforts can be focused. These new fragment hits could eventually help advance the fight against INH-resistant Mtb strains, which pose a significant global health threat.
Keywords: virtual screen, isoniazid-resistant tuberculosis, Mtb InhA, GO Fight Against Malaria project, World Community Grid, AutoDock Vina, TB, fragment-based hit discovery
INTRODUCTION
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb), kills 1.3 million people each year.1 According to the World Health Organization, Mtb infects approximately two billion people.1 Since a third of the global population has a latent Mtb infection, this creates an immense reservoir of disease due to the potential for reactivation.1 There are 8.3 to 9 million new cases of tuberculosis (TB) annually, and half a million children get TB each year. 1 Mtb kills more people in the world than any other bacteria. Of all infectious diseases, only HIV kills more people than Mtb, and TB is the leading cause of death for HIV/AIDS patients.1
Although effective TB drugs have existed for over 60 years, and drug-resistant TB was not a major issue twenty years ago,2 cases of multi drug-resistant TB (MDR-TB) and extensively drug-resistant TB (XDR-TB) continue to increase throughout the world in both frequency and distribution.1–7 MDR-TB cases have nearly doubled in just a few years.4 The global treatment success rate for TB is now less than 50%.1 Each year, one-half million new MDR-TB cases occur (i.e., Mtb infections that are resistant to INH and rifampicin). XDR strains additionally evade fluoroquinolones and at least one of the 2nd-line injectable drugs (amikacin, capreomycin, or kanamycin).1 With the emergence of totally drug-resistant TB (TDR-TB) in several countries, no effective treatment options exist for these patients.3, 5–8
Novel InhA inhibitors effective against isoniazid-resistant mutants would be critical for treating MDR and XDR-TB
InhA, an Mtb enoyl acyl-carrier protein reductase, is the primary target of the front-line drug isoniazid (INH).9, 10 While it is one of the two most important antitubercular drugs and the only drug used for TB prophylaxis, INH suffers from resistance that continues to increase.1, 9, 11, 12 WHO data indicate up to 28% of all TB cases are INH-resistant, and in previously treated TB patients, up to 60% exhibit resistance, making it extremely difficult, time-consuming, and expensive to treat them (if they can be treated at all).1, 2, 13
INH must be activated by Mtb catalase-peroxidase (KatG).14–16 Most clinically relevant INH-resistant Mtb strains involve mutations in or deletions of katG, which abrogate activation of the INH prodrug.17, 18 In some areas, 70% of MDR-TB strains have mutations in katG, as do 100% of sequenced XDR-TB strains.19, 20 Although katG mutations are generally responsible for high-level resistance to INH in clinical isolates, those mutations can be enhanced by additional mutations in the promoter region of inhA, which cause low-level INH-resistance by increasing the amount of InhA produced,21, 22 and are found in up to 28% of INH-resistant clinical isolates (depending on the location of the study).21–31 KatG activates INH to enable formation of a covalent adduct with NAD+ or NADH.14 As has been previously demonstrated, novel InhA inhibitors that do not require prior activation by KatG are not vulnerable to this key mechanism of INH resistance.11, 17
Our virtual screen in this study was motivated by the clear need for a novel InhA-targeting drug that is effective against MDR and XDR-TB strains. A next generation InhA inhibitor, specifically lacking significant cross-resistance with INH by not requiring KatG activation, would be a valuable addition to the antitubercular armamentarium and help stem the tide of TB drug resistance. Guiding our inhibitor discovery and design efforts are numerous X-ray crystal structures of InhA bound to co-factor, substrate mimic, or various inhibitors.10, 32–45 The pursuit of InhA inhibitors lacking the requirement for activation by KatG has transpired through both structure-based and screening approaches.12, 46–49 Notably, the Tonge group 17, 46, 47, 49 and Freundlich et al. 11 have both independently evolved potent triclosan-based inhibitors by leveraging X-ray crystal structures of InhA with bound diaryl ethers. However, a significant limitation of the triclosan scaffold has been the requirement for a phenol moiety, which suffers from rapid Phase II metabolism.50–52 Our virtual screen focused on the NCI library to search for novel scaffolds that inhibit InhA without requiring prior activation by KatG and yet do not contain the problematic phenol group. Although many different factors affect in vivo pharmacokinetics and pharmacodynamics, especially when targeting a pathogen like Mtb that has an unusually thick and waxy cell wall, numerous efflux pumps and detoxification mechanisms, we sought to avoid the known liabilities that some current InhA inhibitors display.
High-throughput docking virtual screening (VS) studies have been used extensively in both academia and the pharmaceutical industry to discover inhibitors of select drug targets (median hit rate of 13% 53) and are complementary to experimental target-based HTS.54 “Docking” flexible models of small molecules computationally probes the energetic landscape governing macromolecular recognition with a target protein, to help guide the discovery and design of novel inhibitors.55–62 Docking flexible models of potential ligands against atomic-scale models of different protein drug targets may reproduce or predict (a) how tightly these compounds bind; (b) where they prefer to bind; and (c) what specific interactions they form at the binding site.
Many VS studies, including some against InhA, have involved computational studies in the absence of experimental validation of their predictions.63–69 In contrast, some pioneering VS against InhA have yielded predictions that were experimentally validated with enzyme inhibition assays70 and/or whole-cell growth assays against Mtb,71, 72 M. vanbaalenii,73 or M. smegmatis.74 These previous, experimentally validated VS against InhA helped establish the feasibility of computer-aided drug discovery against this system and laid the foundation for the research we present.
MATERIALS AND METHODS
The Global Online Fight Against Malaria project and its relevance to TB research
IBM’s World Community Grid is a distributed network of over 2 million internet-connected personal computers in over 80 countries, making it effectively one of the largest supercomputers available. It is entirely devoted to humanitarian research at academic and non-profit institutions. The Global Online Fight Against Malaria (or “GO Fight Against Malaria,” GO FAM) was a project on World Community Grid that A.L.P. designed and executed while at TSRI.75–77 In under two years, the GO FAM project used over 27,385 CPU years to perform virtual screens of 5.6 million commercially available compounds against over 200 structures of targets from 22 classes of validated and potential drug targets for the treatment of malaria and other diseases. Using AutoDock Vina62, GO FAM generated a total of 1.16 billion docking results. These docking studies would have taken over a hundred years to complete on typical academic computing resources. The compound libraries screened were: NCI, Enamine, Asinex, ChemBridge, and Vitas-M Labs, with 3-D models obtained from the ZINC server.78 To the best of our knowledge, GO FAM is the first project to perform over a billion docking jobs, and it has produced the largest VS data sets against malaria and Mtb.
If a malaria target had structural similarity with valid or potential drug targets from other pathogens whose crystal structures were available, the compounds were docked as well against those cognate proteins, including Mtb, E. coli, Staphylococcus aureus, Yersinia pestis, and Brugia malayi. The Mtb subset of GO FAM involved InhA, DHFR (dihydrofolate reductase), OAR (oxo-acyl ACP reductase, or FabG), and cyclophilin A.
On GO FAM we docked a much larger number of compounds against InhA than all previous VS against it combined.65–74 The results presented here encompass only 5.6% of the compounds screened on GO FAM against InhA—we began with the NCI library, because NCI compounds are available to researchers for free, through the NCI’s Developmental Therapeutics Program (DTP).
Screening the NCI library of compounds against InhA on GO Fight Against Malaria
The 316,000 pdbqt files generated for the NCI library (and for the other libraries that represent the 5.6 million compounds docked in the GO FAM experiments) are available at: http://zinc.docking.org/pdbqt. AutoDock Vina62 1.1.2 (or “AD Vina”), which was grid-enabled for World Community Grid by IBM staff, was used to dock each compound in the library against the crystallographic conformation of InhA from 2×23.pdb.39 In positive control re-docking experiments, the co-crystallized inhibitor PT70 docked to the target model of 2×23 with an RMSD = 0.49 Å. Additional (successful) positive control re-docking and cross-docking experiments that utilized AD Vina against other crystal structures of InhA bound to different ligands have been published recently elsewhere.79 This 2×23 structure of InhA was selected for this study, because it is a complex with PT70, a slow, tight-binding inhibitor of InhA with a 7.8 nM Ki and a residence time of 24 minutes. Displaying a long residence time with a pathogenic target imparts favorable properties in vivo.80–82 Using the PT70-induced conformation may enable us to identify and develop novel inhibitors that display long residence times with InhA. The ambitiousness of this goal necessitated the resources of the GO FAM project to screen 5.6 million compounds against this and other crystal structures of InhA. Hydrogen atoms were added to the target model using the MolProbity server.83 Crystallographic waters and counterions were deleted from the pdb file. AutoDockTools 4.284 was then used to generate the pdbqt file. A grid box of 30 × 30 × 30 Å3 centered between the N9 atom of adenine and C1 of the adjacent ribose in the NAD cofactor defined the region that the docked compounds explored. This location and large size were selected to enable the same grid box to be used for all of the different InhA and PfENR targets that were part of these GO FAM experiments. The “exhaustiveness” setting in AD Vina was increased to 20 (from the default of 8), because of the large grid box used.
Target-specific energetic and interaction filtering of VS results
We characterized the predicted binding mode of each compound in the NCI library, by the number and types of energetically favorable interactions with the InhA active site and the estimated free energy of binding, using a software-automated workflow (python and tc-shell scripts) and established protocols.85–87 The results were filtered to harvest compounds that displayed critical interactions (based on analyses of the features displayed by nanomolar inhibitors of InhA in existing crystal structures)11, 35, 39, 41. These filters selected compounds for which the top-scoring binding mode/compound displayed base-stacking interactions between the candidate compound and the NAD+/NADH co-factor and at least two predicted hydrogen bonds to the active site. The selected compounds also had to display an estimated free energy of binding (FEB) ≤ −8.0 kcal/mol, according to the AD Vina scoring function. This set of docking filters harvested 91 compounds, whose binding modes were then visually inspected (see Figure 1).
Figure 1. Workflow Used to Discover Novel InhA Inhibitors in the Virtual Screen with the NCI Library on GO FAM.
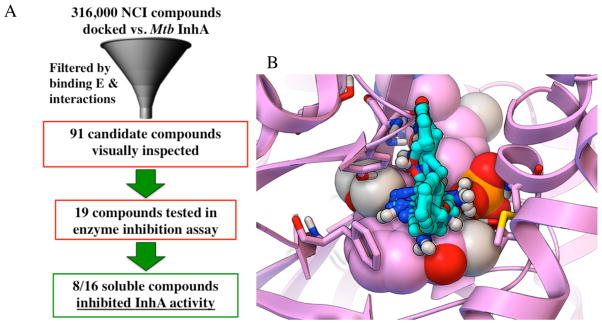
(A) The workflow used energetic and interaction-based filters (e.g., requiring compounds to display an estimated free energy of binding ≤ −8.0 kcal/mol, base stack with the NAD cofactor, and form at least two hydrogen bonds with the active site) to filter the VS results and harvest NCI compounds for visual inspection. Candidates that passed visual inspection were then ordered and tested in InhA inhibition assays. Eight of the sixteen soluble compounds inhibited InhA activity by 27 to 71% at 100 μM. (B) The predicted binding modes for all eight novel InhA inhibitors are displayed as sticks-and-balls with cyan carbons, while the InhA target is shown in magenta. The NAD cofactor is rendered as CPK, and the key residues Gly96, Ser123, Phe149, Tyr158, Thr196, and Met199 are shown as thin sticks.
Visual inspection is a subjective process, but experience in macromolecular recognition and knowledge of the strengths and weaknesses of different modeling approaches used to view, measure, and judge/prioritize the docked results can be helpful. The use of human knowledge (“in cerebro” quality control) to prioritize computer-aided (“in silico”) predictions has been a successful strategy in previous blind docking challenges, such as SAMPL2 and SAMPL4.87–89 Our visual inspection process incorporated multiple criteria, in an attempt to decrease the number of false positives that often result from virtual screens. Unfavorable aspects of a docking result included compounds that (a) have docked modes displaying distorted geometries (i.e., peptide bonds in the ligand models were allowed to freely rotate during docking, since the target was rigid, but docked modes displaying peptide bonds that were 30 to 90° from cis or trans were rejected); (b) have one or more large hydrophobic groups (e.g., phenyl or t-butyl) exposed to solvent; or (c) display more than three unfavorable electrostatic repulsion interactions with polar or charged groups in the target. Favorable aspects of a docked pose included compounds where (d) a majority of their heteroatoms are involved in favorable electrostatic interactions or hydrogen bonds (as discussed in our recent manuscript)79, since heteroatoms will likely need to be added during the optimization process, without violating Lipinski’s rules90; (e) the hydrophobic groups displayed van der Waals interactions with non-polar regions of the target (as measured by the AD Vina scoring function62, characterized using distance-dependent and atom pair-specific criteria implemented in the Fox software85); and (f) aromatic rings displayed pi-pi (base-stacking or T-stacking)91–93 or pi-cation interactions with the target92–94, as characterized by the Fox software85. The AD Vina scoring function uses ambiguous atom types (i.e., hydrogen bond donors are treated as donor and/or acceptor) and a spherical hydrogen bond potential (instead of an angle-dependent potential).62 Consequently, to verify the number of hydrogen bonds that the docking filters detected, the donor-acceptor distance and donor-hydrogen-acceptor angle (which should be between 120–180°) for predicted hydrogen bonds were all measured manually in PMV.84 Because rotatable polar hydrogen atoms in the ligands are placed in arbitrary torsion angles by AD Vina,62 and since the model of the target was rigid, structural intuition was also employed when deciding which hydrogen bonds the docked mode displayed. Special emphasis was placed on candidates that (g) displayed hydrogen bonds with invariant residues and backbone atoms, since previous studies with MDR HIV protease and MDR P. falciparum DHFR have shown that displaying these features renders the evolution of drug resistance less likely.95–111
InhA inhibition and kinetics experiments
An InhA inhibition assay was performed on the selected candidate compounds. Briefly, the candidate inhibitor was assayed at 100 μM in a reaction buffer (30 mM PIPES, 150 mM NaCl, 1 mM EDTA, at pH 6.8) containing 30 μM trans-2-dodecenoyl coenzyme A (DD CoA), 250 μM NADH, and 100 nM InhA. The enzymatic activity at 100 μM inhibitor concentration was quantified, and the three compounds that displayed the largest inhibition of enzyme activity were selected for further IC50 measurements. In general, IC50 values were determined by varying the concentration of inhibitors in the aforementioned reaction mixture. The data were analyzed using equation 1, where I is the inhibitor concentration and y is percent activity.
| Equation 1 |
To provide mechanistic insight, the top two compounds were chosen for subsequent measurements of their inhibition constant (Ki). The third best inhibitor displayed solubility problems at higher concentrations. The Ki value was calculated by determining the kcat and Km (DD CoA) values at several fixed inhibitor concentrations using the same assay condition described above. The data were analyzed using the standard equations for competitive and non-competitive inhibition (equations 2 and 3).
| Equation 2 |
| Equation 3 |
Comparing the new InhA inhibitors to known InhA inhibitors
To assess the chemical property space of the eight InhA inhibitors found, a Principal Component Analysis (PCA) was performed on the combination of the 157 known InhA inhibitors in TB Mobile 2 and the eight inhibitors discovered.112, 113 The sdf of the TB Mobile data set was used for the Principal Component Analysis(PCA), which was calculated using Discovery Studio 3.5114 and used eight interpretable descriptors (AlogP, molecular weight, number of rotatable bonds, number of rings, number of aromatic rings, number of hydrogen bond acceptors, number of hydrogen bond donors, and molecular fractional polar surface area).
The pairwise Tanimoto similarity115 was calculated for each of the new InhA inhibitors discovered in the NCI library versus each of the 154 known InhA inhibitors from TB Mobile 112. This again used the TB Mobile dataset sdf for which MDL descriptors were calculated, followed by Tanimoto similarity using the “find similar molecules by fingerprints” protocol in Discovery Studio 3.5114. This then enabled us to calculate the minimum, maximum and average similarity as measures of proximity to known InhA inhibitors.
The pairwise Tanimoto similarity115 was also calculated for each of the 8 new InhA inhibitors versus each of the 7 other compounds in order to determine the number of distinct scaffolds, according to a cut off > 0.7116 (see Supporting Information Table S1).
Minimum inhibitory concentration (MIC90) assay vs. Mtb
For these whole-cell in vitro studies, new batches of NCI 99389 and 111591 were obtained from the NCI. LC-MS data confirmed that each sample had the expected molecular weight and was >97% pure by HPLC @ 250 nm (see the Supporting Information, Figures S1–S2 and S4–S5). 1H NMR (600 MHz), using d6-DMSO for 99389 and d6-acetone for 111591 as NMR solvents, confirmed that each sample had the expected structure (see the Supporting Information, Figures S3 and S6). MIC90 values of the compounds were determined following the microplate-based Alamar Blue assay (MABA) method as previously described.117 50 mM compound stock in DMSO was dissolved in sterile Middlebrook 7H9-OADC broth, making a 1 mM pre-test solution. 100 μL pre-test solution was added into wells in column 1 of a sterile polystyrene 96-well round-bottom plate (CLS3795, Corning, NY). Wells in column 2 to 12 received 50 μL of sterile 7H9-OADC broth. Serial two-fold dilutions of compounds were performed, and column 12 was set as drug-free (inoculum-only) control. Final concentrations of compounds were as follows: for INH, 0.012 to 25 μg/mL; for NCI 99389 and 111591, 0.50 to 500 μM. M. tuberculosis wild type strain H37Rv and inhA over-expression strain mc2491410 were 1:1000 diluted in 7H9+OADC medium at mid-logarithmic stage of growth (OD595nm=0.4). 50 μL of diluted bacteria suspensions were inoculated into each well. Plates were sealed with Parafilm and incubated at 37 °C for seven days. 20 μL alamarBlue® reagent (Invitrogen, Frederick, MD) freshly mixed with 12.5 μL 20% Tween 80 was added into each well, followed by 24-hour incubation in 37 °C. Absorbance was read at 570 nm, with reference wavelength 600 nm, using a microplate reader (ELX808, Biotek Instruments). The MIC90 endpoint was defined as the lowest concentration of the test agent that produced at least 90% reduction in absorbance compared with that of the drug-free control.
Cellular toxicity assay
The cytotoxicity of 99389 and 111591 were determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliuim bromide (MTT) cytotoxicity assay with the Vybrant MTT Cell Proliferation Assay Kit (Molecular Probes).13 Vero cells (African green monkey kidney epithelial cells; ATCC) were plated in a sterilized 96-well plate (Costar 3595, Corning, NY) at 2 × 105 cells/well with volume 50 μL and incubated for 3 hours in a 37 °C incubator. 99389 and 111591 were dissolved in DMSO at a final concentration of 10 mg/mL. In a separate 96-well plate, two-fold serial dilutions were performed with Dulbecco’s modified eagle medium (Gibco) supplemented with 10% fetal bovine serum. 50 μL of diluted compounds were added to appropriate test plate wells to generate final concentrations ranging from 0.78 to 100 μg/mL. Column 12 was utilized for a drug-free control. After 48 hours treatment, 10 μL of 12 mM MTT stock solution was added into each well and incubated at 37 °C for 4 hours. 100 μL of 0.1 g/mL SDS•HCl solution was added into each well subsequently to halt the reaction, followed by 4-hour incubation at 37 °C. The absorbance at 570 nm was read with a VersaMax ELISA microplate reader (Molecular Devices). The CC50 was extrapolated by plotting absorbance at 570 nm versus concentration of untreated Vero cells control.
RESULTS AND DISCUSSION
This pilot study of a small subset (i.e., 5.6%) of the GO FAM results vs. InhA has led to the discovery of several novel inhibitors (see Figures 2–4). 19 compounds were ordered, but three of them were insoluble at 100 μM in DMSO and could not be assayed (see Table 1). Eight of the 16 soluble candidates were modest inhibitors of InhA (i.e., showing 27% to 71% inhibition at 100 μM). Using the same assay conditions and protocol (i.e., without pre-incubating the compound and InhA), the very potent positive control compound PT70 displayed 75% inhibition at 100 nM (i.e., the well-optimized lead compound PT70 was >1000 times more potent than our best new inhibitor). If scaffolds are defined as small molecule chemotypes that possess less than 70% similarity according to Tanimoto values116, these eight inhibitors represented five novel scaffolds against InhA (see Figure 4 and Supporting Information Table S1). As required by the docking filters, all eight compounds are predicted to base stack with the NAD cofactor (see Figures 2–3), instead of forming a covalent adduct with it. As demonstrated by the InhA activity assays, they do not require prior activation by Mtb KatG. “Hits” were classified in a way that involved the potency for a particular size of compound. “Fragment hits” (novel inhibitors with a MW < 300 Daltons)118 tend to display a potency in the 100’s of μM to low mM range (according to Ki or Kd).88, 118–124 Consequently, we defined fragment hits as novel inhibitors with a MW < 300 g/mol that displayed a Ki value < 100 μM. Two of our inhibitors are “fragment hits”; they are structurally distinct from known InhA inhibitors (see Figures 2, 5 and Table 2); and they displayed Ki values of 54.1 ± 5.4 and 59.2 ± 8.7 μM, respectively (see Figure 6). Thus, using a stringent metric, 2 out of 16 compounds were novel fragment hits against InhA (i.e., a 12.5% hit rate), representing two promising new scaffolds.
Figure 2. Predicted binding modes of the two most potent new InhA inhibitors discovered in GO FAM experiment 5.

The docked modes produced by AutoDock Vina are displayed as ball-and-sticks with cyan carbon atoms, and the InhA target (2X23.pdb) is displayed as magenta ribbons. The NAD cofactor is displayed in CPK, and the key residues Gly96, Ser123, Phe149, Tyr158, Thr196, and Met199 are shown as thin sticks. A close-up view of the predicted binding mode of the top fragment hit, NCI 99389 (Kiapp = 54.1 ± 5.4 μM), is shown in (A), while the full view is displayed in (B). In (C) the docked mode of NCI 99389 is compared to the experimentally-determined binding mode of PT70, the inhibitor that crystallized with InhA in 2×23.pdb, which is displayed as ball-and-sticks with magenta carbons. In (D) the predicted binding mode of the 2nd most potent fragment hit, NCI 111591 (Kiapp = 59.2 ± 8.7 μM), is displayed.
Figure 4. Summary of the 2D structures, docking scores, and InhA inhibitory activities of the eight new inhibitors discovered.

The most potent new InhA inhibitor discovered is shown in the top-left corner, while the least potent new inhibitor is displayed in the bottom-right corner. These eight new inhibitors correspond to five novel scaffolds versus InhA (i.e., NCI 111588 – 111591 represent analogs of one scaffold, according to a Tanimoto cut-off of 0.7; see Supporting Information Table S1). FEB signifies the estimated free energy of binding value from AutoDock Vina’s scoring function, in kcal/mol. The Lig. Eff. is the calculated ligand efficiency from AutoDock Vina, in kcal/mol/heavy atom. The % inhibition of InhA activity was produced when each compound was present at 100 μM. The region of each compound that was predicted to base stack with the NAD cofactor is highlighted with a red circle.
Table 1.
Compound ID’s, 2D structures, and InhA inhibition data.
| NCI ID | Structure | Kiapp (μM) & mechanism | InhA inhibition at 100 μM inhibitor |
|---|---|---|---|
| 660846 |
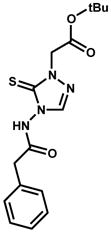
|
2.7% | |
| 683622 |
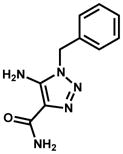
|
9.2% | |
| 609097 |
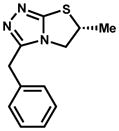
|
10.3% | |
| 314884 |
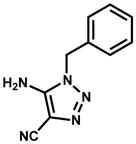
|
10.3% | |
| 371850 |
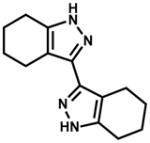
|
Not soluble | |
| 11142 |
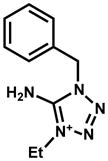
|
4.5% | |
| 75300 |
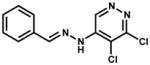
|
Inhibitor precipitated in buffer | |
| 75301 |
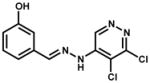
|
Inhibitor precipitated in buffer | |
| 99389 (ZINC01654204) |
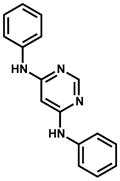
|
54.1 ± 5.4 competitive | 71.0% (estimated ~ 40 μM) IC50 |
| 111588 (ZINC0135077) |
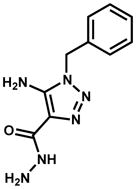
|
29.1% | |
| 111589 (ZINC04994329) |
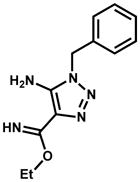
|
33.7% | |
| 111590 (ZINC00129134) |
|
30.9% | |
| 111591 (ZINC01703321) |
|
59.2 ± 8.7 noncompetitive | 42.8% |
| 112144 (ZINC04878446) |
|
205.6 ± 46 (precipitation observed at high concentrations) | 34.6% |
| 130836 |
|
9.1% | |
| 135809 (ZINC01722139) |
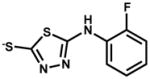
|
29.1% | |
| 196166 (ZINC01734860) |
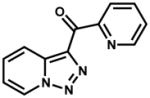
|
27.3% | |
| 213837 |
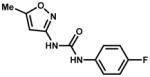
|
10.9% | |
| 293934 |
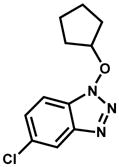
|
9.1% |
Note: Each compound was initially tested at a single concentration. Only the top inhibitors (highlighted with bold font) were subjected to additional experimental characterization. For the top eight new inhibitors, both the NCI ID and the ZINC ID are displayed.
Figure 3. Predicted binding modes of the least potent new InhA inhibitors discovered in GO FAM experiment 5.

The docked modes produced by AutoDock Vina are displayed as ball-and-sticks with cyan carbon atoms. The InhA target (2×23.pdb) is displayed as magenta ribbons, with the NAD cofactor as CPK. The key residues Gly96, Ser123, Phe149, Tyr158, Thr196, and Met199 are shown as thin sticks. The predicted binding modes of the following new InhA inhibitors are depicted: (A) NSC 112144 (Kiapp = 205.6 ± 46 μM), (B) NSC 111589, (C) NSC 111590, (D) NSC 111588, (E) NSC 135809, and (F) NSC 196166.
Figure 5. Comparing the chemical space of the new InhA inhibitors to known InhA inhibitors.

A Principal Component Analysis (PCA) was performed on the combination of the 157 known InhA inhibitors in the TB Mobile 2 data set and the 8 novel InhA inhibitors discovered. Three PCs explain 84.8% of the variance observed. The 157 InhA inhibitors in the TB Mobile 2 data set are displayed in magenta. The two most potent new InhA inhibitors discovered are depicted in green, and the other six novel InhA inhibitors identified are in blue. A red circle highlights the location of NCI 111591. The PCA indicates that the 8 new InhA inhibitors have similar chemical properties to known InhA inhibitors, but they are generally not within the main clusters of these previously characterized InhA inhibitors.
Table 2.
Tanimoto Similarity to 154 Known InhA Inhibitors in the TB Mobile data set
| Compound IDs | Average Similarity | Maximum Similarity | Minimum Similarity |
|---|---|---|---|
| 1) NCI 99389 ZINC01654204 |
0.3087 | 0.4130 | 0.1458 |
| 2) NCI 111591 ZINC01703321 |
0.3681 | 0.4634 | 0.1311 |
| 3) NCI 112144 ZINC04878446 |
0.4503 | 0.5738 | 0.15 |
| 4) NCI 111589 ZINC04994329 |
0.4307 | 0.5732 | 0.1642 |
| 5) NCI 111590 ZINC00129134 |
0.4296 | 0.5522 | 0.1935 |
| 6) NCI 111588 ZINC0135077 |
0.4542 | 0.5897 | 0.1406 |
| 7) NCI 135809 ZINC01722139 |
0.2731 | 0.4366 | 0.1061 |
| 8) NCI 196166 ZINC01734860 |
0.3551 | 0.4833 | 0.1724 |
Figure 6. Kinetic data on the two most potent InhA inhibitors discovered indicate NCI 99389 is a competitive inhibitor, while 111591 is noncompetitive.

IC50 values were measured for the top 2 fragment hits, followed by a detailed mechanistic study to measure the Ki values. NCI 99389 showed a Kiapp of 54.1 ± 5.4 μM and a competitive binding mechanism, indicating that the inhibitor competed with the CoA substrate and bound directly to the enzyme. Conversely, NCI111591 had a Kiapp of 59.2 ± 8.7 μM and a non-competitive binding mechanism, suggesting a more complex scenario where the inhibitor could bind to both the holo enzyme and to the substrate-enzyme complex.
The most promising new scaffold, NCI 99389, is a 4,6-diaminopyrimidine and was predicted to form the following quaternary interactions in the docking studies: base stacking with the nicotinamide ring of the NAD cofactor (similar to the crystallographic binding mode of PT70)39; T-stacking with the side-chain of Phe149; hydrogen-bonding with the hydroxyl of Tyr158 (similar to PT70)39; hydrogen-bonding with the 2′-hydroxyl of the ribose adjacent to the nicotinamide ring of NAD (similar to PT70)39; hydrogen-bonding with the sulfur in the side-chain of Met199; favorable electrostatic interactions with the hydroxyl of Tyr158; and favorable electrostatic interactions with the phosphate group proximal to the ribose of NAD (see Figure 2.A). As a reference, in addition to the aforementioned hydrogen bonding and base stacking interactions that the crystallographic binding mode of PT70 displays, PT70 also contains a six carbon alkyl tail that packs very well into a hydrophobic pocket of InhA. Despite using a large grid box in these docking calculations (i.e., allowing the ligands to sample a large volume of the protein), docking calculations suggest a clear preference for 99389 to bind in a similar fashion to the crystallographic binding mode of PT70 (see Figure 2.C). Experimental structure verification will be necessary to firmly establish the actual binding mode. Since this compound is an un-optimized fragment hit and lacks the long alkyl tail that PT70 contains, it might not produce the same “closed” conformation that PT70 induces39, 125 (which might give 99389 a different binding mode than what these docking calculations predicted), and/or it might have to pay a larger enthalpic penalty to induce the ligand-bound conformation that InhA forms (which, when combined with the numerous hydrophobic contacts that it lacks, might also explain part of the >1,000-fold difference in potency versus PT70). NCI 99389 displayed an IC50 of approximately 40 μM against InhA (see Table 1). In subsequent kinetic experiments (see Figure 6 and Table 1), it displayed an apparent Ki of 54.1 ± 5.4 μM, and it demonstrated a competitive mechanism of inhibition with respect to substrate.
The second most promising new scaffold discovered, NCI 111591, is a 5-amino-1H-1,2,3-triazole and had a docked mode that displayed the following quaternary interactions with InhA: base stacking with the nicotinamide ring of the NAD cofactor; hydrogen-bonding with the hydroxyl of Tyr158; hydrogen-bonding with the 2′-hydroxyl of the ribose adjacent to the nicotinamide ring of NAD; hydrogen-bonding with the carbonyl oxygen of the nicotinamide ring; favorable electrostatic interactions with the sulfur in the side-chain of Met199; favorable electrostatic interactions with the phosphate group proximal to the ribose of NAD; and favorable electrostatic interactions with the 2′-hydroxyl of the ribose that is adjacent to the nicotinamide ring of NAD (see Figure 2.D). When present at 100 μM, NCI 11591 inhibited InhA activity 42.8%. It displayed an apparent Ki of 59.2 ± 8.7 μM. As shown in Figure 6, 111591 followed a non-competitive mechanism of inhibition (i.e., it can bind when the substrate is “not on or on,” meaning it could potentially bind to the holo enzyme, to the InhA:substrate complex, and/or to the InhA:NAD+:product complex). To further confirm the inhibition mode, data were fit to both Equation 2 and Equation 3 for comparison. The Ki values generated from the non-competitive fit and competitive fit were 59.2 ± 8.7 μM and 54.0 ± 51.0 μM, respectively. The much greater error for the competitive fit unambiguously confirmed the inhibition mechanism as non-competitive.
The predicted binding modes for the six less potent (and thus less promising) new InhA inhibitors discovered are presented in Figure 3. They were all predicted to base-stack with the nicotinamide ring of the NAD cofactor and to form a hydrogen bond with the hydroxyl of Tyr158. Five of the six inhibitors (i.e., all except NCI 111590) docked to form both a hydrogen bond and an additional favorable electrostatic interaction with the 2′-hydroxyl of the ribose adjacent to the nicotinamide ring of NAD, but the predicted pose of 111590 only formed a favorable electrostatic interaction with that hydroxyl. 112144 was also predicted to make a hydrogen bond with the carbonyl of Gly96, but it was predicted to display unfavorable electrostatic repulsion with both phosphate groups of the NAD cofactor. The amino and ether oxygen atoms of 111589 were predicted to form both favorable and unfavorable electrostatic interactions, respectively, with the sulfur in the side-chain of Met199, and it displayed an internal hydrogen bond between that amino and ether oxygen. The conserved regions of the scaffold in 111590 and 111588 had docked modes that superimposed, and they formed favorable electrostatic interactions with the sulfur in the side-chain of Met199 and with the hydroxyl of Ser123. If a slight conformational change occurs in the side-chain of Phe149, they could both T-stack with it. Docking of 135809 suggested favorable electrostatic interactions could be formed with the phosphate group adjacent to the ribose of the NAD cofactor, but it formed unfavorable electrostatic repulsion with both the sulfur in the side-chain of Met199 and the carbonyl of the nicotinamide ring of the NAD cofactor. The weaker InhA activity of 135809 compared to the other compounds with the most similar structures to it (see Figure 4 and Supporting Information Table S1) might be due to these unfavorable electrostatic repulsions and perhaps to the less stable/less likely protonation state of the central thiadiazole ring (i.e., of the different independent models for the different protonation states of this compound that were screened, the model that passed the docking filters had a protonated thiadiazole, but ChemDraw calculations predict that this compound should not be protonated at neutral pH). 196166 was the least potent InhA inhibitor, and its docked mode displayed three unfavorable electrostatic repulsions with the phosphate adjacent to the ribose of the NAD cofactor. The terminal pyridine also displayed unfavorable electrostatic repulsion with the sulfur in the side-chain of Met161, but the pyridine that is part of the two fused rings might T-stack with Phe149.
The 8 inhibitors discovered were compared to a set of 157 previously-characterized InhA inhibitors available in TB Mobile 2.112, 113 Principal Component Analysis (PCA) showed that these 8 InhA GO FAM inhibitors were generally not part of the main clusters of known InhA inhibitors, but they do have similar chemical properties to them (see Figure 5). Each inhibitor was also individually compared to all known InhA inhibitors in the TB Mobile data set to obtain sets of pairwise Tanimoto coefficients, which were then averaged. Their average Tanimoto similarities ranged from 0.27 to 0.45 (with 1.0 indicating identical 2D structures). The two most promising new scaffolds discovered, NCI 99389 and 111591, had a maximum Tanimoto similarity to a known InhA inhibitor of 0.413 and 0.463, with a minimum similarity of 0.146 and 0.131, and average similarity values of 0.309 and 0.368, respectively. These distinct cheminformatic analyses support these 8 GO FAM compounds as novel InhA inhibitors based on current literature data.
The top two fragment hits were then studied with (a) whole-cell in vitro Mtb growth experiments, using both the wild type strain H37Rv and the inhA-over-expressing strain mc24914,10 and with (b) Vero cell cytotoxicity experiments (see Table 3). Unfortunately, the top InhA inhibitor discovered, 99389, displayed an MIC90 of 500 μM against wild type Mtb. The MIC > 500 μM it exhibited against the inhA-overexpressor strain of Mtb suggests that InhA might be the primary target against whole-cell Mtb. However, this compound lacked sufficient efficacy against Mtb and displayed considerable cytotoxicity with Vero cells, with a CC50 < 3.0 μM. As expected, the positive control INH displayed almost a 10-fold decrease in potency against the inhA overexpressor: MIC90 of 0.4 μM (0.05 μg/mL) against wild type Mtb and 3 μM (0.4 μg/mL) against strain mc24914. The second most promising InhA inhibitor discovered, NCI 111591, displayed an MIC90 of 125 μM against both wild type Mtb and the inhA-overexpressing strain of Mtb. Although 111591 was 4-fold more potent against whole-cell Mtb, the lack of a shift in its potency against the inhA overexpressor indicates that InhA is likely not its primary target in Mtb. In the PCA that compared the new inhibitors to the 157 known InhA inhibitors in TB Mobile 2112, 113, 111591 was further from the main clusters of known InhA inhibitors, while 99389 was at the edge of the central cluster (see Figure 5). NCI 111591 was less toxic against Vero cells, with a CC50 of 26 μM; however, it still displayed an insufficient selectivity index of 0.21.
Table 3.
Summary of Anti-Mtb Efficacy and Mammalian Cell Cytotoxicity
| NCI ID | Structure | Vero cell cytotoxicity: CC50 (μM) | MIC90 vs Mtb(μM) | |
|---|---|---|---|---|
| H37Rv wild type | mc24914 inhA overexpressor | |||
|
| ||||
| 99389 |
|
< 3.0 | 500 | > 500 |
|
| ||||
| 111591 |
|
26 | 125 | 125 |
Although several InhA inhibitors with nM potency have been reported previously,11, 15, 41, 126 novel chemotypes that inhibit InhA are needed for the following reasons: (a) the presence of the phenol group in triclosan derivatives 11, 41, 126 poses a metabolic liability for in vivo applications, 50–52 (b) other advanced InhA leads have fared poorly when administered in mouse models of TB,15 and (c) drug-resistant strains of Mtb (i.e., MDR-TB, XDR-TB, and TDR-TB) continue to evolve and spread throughout the world.1–7 In addition, since 28% to 60% of TB cases are INH-resistant, 1, 2, 13 and InhA is one of the most validated targets for treating TB, new chemotypes that inhibit InhA without displaying cross-resistance with INH could eventually seed the development of urgently needed new drug combinations for the treatment of active and latent TB infections.
The novel fragment hits we discovered against InhA displayed low μM potency, with Ki values of 54.1 and 59.2 μM for the two most promising new scaffolds. Using stringent criteria to define a hit (a Ki value < 100 μM for a fragment-sized compound), this VS had a 12.5% hit rate. Thus, from a computational chemistry perspective, this VS was a success, since the median hit rate from hundreds of published VS is ~ 13%.53 More importantly, these novel InhA inhibitors are all predicted to base stack with the NAD cofactor (instead of forming a covalent adduct with it). Our InhA inhibition assays demonstrated that these novel inhibitors do not require prior activation by Mtb KatG, which means that they should not be susceptible to the main mechanism of INH resistance found in clinical settings. In addition, the novel inhibitors discovered all lack the presence of the phenol group that poses a metabolic liability for triclosan derivatives. Consequently, the two most promising new chemotypes discovered might eventually enable the development of new InhA inhibitors that are effective against MDR-TB, XDR-TB, and TDR-TB. Since it is slightly weaker than NCI 99389, we consider NCI 11591 to be the second most promising new scaffold against InhA discovered in the present study. However, given its superior MIC against Mtb and its lower toxicity against Vero cells, NCI 111591 seems to be a more promising scaffold versus whole-cell Mtb.
Although these new chemotypes discovered against InhA are somewhat weak inhibitors (i.e., they are “fragment hits” and not drug-sized “leads”), they were identified from a diverse, commercial library using freely available compounds. In addition, they are both fragment-sized compounds (i.e., MW < 300 g/mol)118. Fragment-based hit discovery was not our initial goal, but it was the ultimate result of this pilot study. Unlike traditional high-throughput screens, fragment-based drug discovery is founded on screening a smaller number of smaller-sized compounds, to advance the goal of discovering novel fragment hits with Ki or Kd values in the high μM to low mM range.119 Those initial novel fragments can then be optimized using structure-based and medicinal chemistry strategies to develop potent leads, some of which have advanced to become a clinical candidate118 or an FDA approved drug.127 In the pioneering “SAR by NMR” study, the novel fragment hits discovered displayed Kd values of 100 μM to 9.5 mM.124 Although a few studies have discovered fragment hits with potencies as great as 24 μM,128 49 μM,128 60 μM,128 or 80 μM,129 most fragment hits have potencies (i.e., Ki or Kd values) in the 100 to 300 μM range, and many are in the low mM.88, 118–120, 122, 123, 128–130 Thus, although our two fragment hits have a very weak potency when compared to optimized lead compounds (such as PT70), our hits have not yet been optimized against the InhA target or against whole-cell Mtb, and they are actually more potent than most novel fragment hits.
Our most promising new fragment hit against InhA has a fairly simple structure and should be amenable to structure-based, medicinal chemistry-guided optimization. Due to their lack of whole-cell efficacy against Mtb and their inadequate selectivity indexes for Vero cell cytotoxicity, both of our top fragment hits will need considerable optimization before advancing to the lead compound stage. Several previous studies performed structure-activity-relationship (SAR) experiments to guide the development of more potent InhA inhibitors.41, 43, 47, 48, 126 Those studies suggest that our most promising new fragment hit could perhaps be developed into a more potent InhA inhibitor by appending appropriate functionality off one or both of the phenyl rings to make additional energetically favorable interactions with residues such as Gly96, Phe97, Phe149, Met155, Pro156, Ala157, Pro193, Ala198, Met199, Ile202, Val203, Leu207, Gln214, Ile215, Leu218, or the NAD cofactor.
The similarity in the substructures that are predicted to base stack with the NAD cofactor (see Figure 4) suggests that click chemistry, especially target-guided click chemistry,131–134 might be a useful approach to aid the discovery and development of novel InhA inhibitors. Target-guided click chemistry is based on the principle that the azide and alkyne-containing fragments will interact with each other and click together (to form the triazole ring) only if those fragments are correctly positioned and have high affinity and long residence times in the target enzyme.131–134 Since inhibitors that display long residence times with pathogenic targets can have more favorable properties in vivo,39, 80–82, 125 and since our computational results indicate that the triazole ring is predicted to form key base stacking and hydrogen bonding interactions with InhA (instead of just serving as a linker), we suggest that in situ click chemistry should be investigated in future studies against InhA.
CONCLUSIONS
Our Virtual Screen of the NCI library with a published crystal structure (PDB ID: 2×2339) led to the discovery of two promising and novel fragment hits that inhibit InhA activity. This pilot study demonstrated the utility of the (public domain/open access) GO FAM docking data against InhA and of our approach to its analysis. Novel inhibitors of the key TB drug target InhA were discovered in an efficient manner, requiring the experimental assessment of fewer than 20 candidate compounds. These open access GO FAM data against InhA and other targets for treating TB and malaria represent a valuable resource for the drug discovery community.
Supplementary Material
Acknowledgments
We are very grateful for the seed funding provided by the IBM International Foundation (from part of the prize money that Watson won on Jeopardy!), which was used to create the GO Fight Against Malaria project (GO FAM). This research was also funded in part by the “AutoDock Development and Maintenance” grant to A.J.O. (R01 GM069832). We thank Sargis Dallakyan for maintaining the server that was used to submit these virtual screens to the GO FAM project and to receive the results from it. We thank the IBM World Community Grid team (including Juan A. Hindo, Viktors Berstis, Al Seippel, Keith J. Uplinger, Kevin Reed, Tedi Hahn, Jonathan D. Armstrong, and Erika Tuttle) for devoting their time to this humanitarian effort. We are also grateful for the World Community Grid volunteers, who donated their CPU time to support the GO FAM calculations. All GO FAM results are legally public domain and, thus, accessible to any scientist by contacting A.J.O or A.L.P.
We thank the Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute, National Institutes of Health (NIH), an agency of the U.S Public Health Service (PHS), for providing free access to these NCI compounds. We thank Thomas Mayo and Katalin Nadassy of Biovia., for providing free access to and assistance with Discovery Studio and Pipeline Pilot. J.S.F. is grateful to Professor Bill Jacobs (Albert Einstein College of Medicine) for his lab’s kind gift of M. tuberculosis strain mc24914.
TB Mobile was partially funded by Award Number 2R42AI088893-02 “Identification of novel therapeutics for tuberculosis combining cheminformatics, diverse databases and logic based pathway analysis” to S.E. from the National Institutes of Allergy and Infectious Diseases. Funding was also provided in part by NIH 3DP2OD008459-01S1 to J.S.F. and by GM102864 to P.J.T.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare that they have no competing financial interest.
The following material is available free of charge via the Internet at http://pubs.acs.org:
Figures S1–S6 (which present LC-MS and NMR data on the two fragment hits); Table S1 (a Tanimoto comparison of each of the eight new inhibitors compared to the other 7 inhibitors); as well as video S1 (99389_vs_InhA_AlexLPerryman_JSF_PT_AJO_v2.qt), an animation of the predicted binding mode of the top InhA inhibitor discovered, NCI 99389.
References
- 1.WHO. Global Tuberculosis Report. World Health Organization; Geneva: 2013. [Google Scholar]
- 2.Abubakar I, Zignol M, Falzon D, Raviglione M, Ditiu L, Masham S, Adetifa I, Ford N, Cox H, Lawn SD, Marais BJ, McHugh TD, Mwaba P, Bates M, Lipman M, Zijenah L, Logan S, McNerney R, Zumla A, Sarda K, Nahid P, Hoelscher M, Pletschette M, Memish ZA, Kim P, Hafner R, Cole S, Migliori GB, Maeurer M, Schito M, Zumla A. Drug-Resistant Tuberculosis: Time for Visionary Political Leadership. The Lancet Infectious Diseases. 2013;13:529–539. doi: 10.1016/S1473-3099(13)70030-6. [DOI] [PubMed] [Google Scholar]
- 3.Velayati AA, Farnia P, Masjedi MR. The Totally Drug Resistant Tuberculosis (Tdr-Tb) Int J Clin Exp Med. 2013;6:307–309. [PMC free article] [PubMed] [Google Scholar]
- 4.Dheda K, Migliori GB. The Global Rise of Extensively Drug-Resistant Tuberculosis: Is the Time to Bring Back Sanatoria Now Overdue? The Lancet. 2012;379:773–775. doi: 10.1016/S0140-6736(11)61062-3. [DOI] [PubMed] [Google Scholar]
- 5.Gothi D, Joshi JM. Resistant Tb: Newer Drugs and Community Approach. Recent Pat Antiinfect Drug Discovery. 2011;6:27–37. doi: 10.2174/157489111794407859. [DOI] [PubMed] [Google Scholar]
- 6.Udwadia ZF, Amale RA, Ajbani KK, Rodrigues C. Totally Drug-Resistant Tuberculosis in India. Clinical Infectious Diseases. 2012;54:579–581. doi: 10.1093/cid/cir889. [DOI] [PubMed] [Google Scholar]
- 7.Velayati AA, Masjedi MR, Farnia P, Tabarsi P, Ghanavi J, ZiaZarifi AH, Hoffner SE. Emergence of New Forms of Totally Drug-Resistant Tuberculosis Bacilli: Super Extensively Drug-Resistant Tuberculosis or Totally Drug-Resistant Strains in Iran. CHEST Journal. 2009;136:420–425. doi: 10.1378/chest.08-2427. [DOI] [PubMed] [Google Scholar]
- 8.Shah NS, Richardson J, Moodley P, Babaria P, Ramtahal M, Heysell SK, Li X, Moll AP, Friedland G, Sturm AW, Gandhi NR. Increasing Drug Resistance in Extensively Drug-Resistant Tuberculosis, South Africa. Emerg Infect Dis. 2011;17:510–513. doi: 10.3201/eid1703.101363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vilchèze C, Jacobs WR., Jr The Mechanism of Isoniazid Killing: Clarity through the Scope of Genetics. Ann Rev Microbiol. 2007;61:35–50. doi: 10.1146/annurev.micro.61.111606.122346. [DOI] [PubMed] [Google Scholar]
- 10.Vilcheze C, Wang F, Arai M, Hazbon MH, Colangeli R, Kremer L, Weisbrod TR, Alland D, Sacchettini JC, Jacobs WR. Transfer of a Point Mutation in Mycobacterium Tuberculosis Inha Resolves the Target of Isoniazid. Nat Med. 2006;12:1027–1029. doi: 10.1038/nm1466. [DOI] [PubMed] [Google Scholar]
- 11.Freundlich JS, Wang F, Vilchèze C, Gulten G, Langley R, Schiehser GA, Jacobus DP, Jacobs WR, Sacchettini JC. Triclosan Derivatives: Towards Potent Inhibitors of Drug-Sensitive and Drug-Resistant Mycobacterium Tuberculosis. ChemMedChem. 2009;4:241–248. doi: 10.1002/cmdc.200800261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.North EJ, Jackson M, Lee RE. New Approaches to Target the Mycolic Acid Biosynthesis Pathway for the Development of Tuberculosis Therapeutics. Curr Pharm Des. 2014;20:4357–78. doi: 10.2174/1381612819666131118203641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vilcheze C, Baughn AD, Tufariello J, Leung LW, Kuo M, Basler CF, Alland D, Sacchettini JC, Freundlich JS, Jacobs WR., Jr Novel Inhibitors of Inha Efficiently Kill Mycobacterium Tuberculosis under Aerobic and Anaerobic Conditions. Antimicrob Agents Chemother. 2011;55:3889–98. doi: 10.1128/AAC.00266-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heym B, Zhang Y, Poulet S, Young D, Cole ST. Characterization of the Katg Gene Encoding a Catalase-Peroxidase Required for the Isoniazid Susceptibility of Mycobacterium Tuberculosis. J Bacteriology. 1993;175:4255–4259. doi: 10.1128/jb.175.13.4255-4259.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Encinas L, O’Keefe H, Neu M, Remuinan MJ, Patel AM, Guardia A, Davie CP, Perez-Macias N, Yang H, Convery MA, Messer JA, Perez-Herran E, Centrella PA, Alvarez-Gomez D, Clark MA, Huss S, O’Donovan GK, Ortega-Muro F, McDowell W, Castaneda P, Arico-Muendel CC, Pajk S, Rullas J, Angulo-Barturen I, Alvarez-Ruiz E, Mendoza-Losana A, Pages LB, Castro-Pichel J, Evindar G. Encoded Library Technology as a Source of Hits for the Discovery and Lead Optimization of a Potent and Selective Class of Bactericidal Direct Inhibitors of Mycobacterium Tuberculosis Inha. J Med Chem. 2014;57:1276–1288. doi: 10.1021/jm401326j. [DOI] [PubMed] [Google Scholar]
- 16.Zhang L, Ye Y, Duo L, Wang T, Song X, Lu X, Ying B, Wang L. Application of Genotype Mtbdrplus in Rapid Detection of the Mycobacterium Tuberculosis Complex as Well as Its Resistance to Isoniazid and Rifampin in a High Volume Laboratory in Southern China. Mol Biol Rep. 2011;38:2185–92. doi: 10.1007/s11033-010-0347-0. [DOI] [PubMed] [Google Scholar]
- 17.Tonge PJ, Kisker C, Slayden RA. Development of Modern Inha Inhibitors to Combat Drug Resistant Strains of Mycobacterium Tuberculosis. Curr Top Med Chem. 2007;7:489–98. doi: 10.2174/156802607780059781. [DOI] [PubMed] [Google Scholar]
- 18.Hazbon MH, Brimacombe M, Bobadilla del Valle M, Cavatore M, Guerrero MI, Varma-Basil M, Billman-Jacobe H, Lavender C, Fyfe J, Garcia-Garcia L, Leon CI, Bose M, Chaves F, Murray M, Eisenach KD, Sifuentes-Osornio J, Cave MD, Ponce de Leon A, Alland D. Population Genetics Study of Isoniazid Resistance Mutations and Evolution of Multidrug-Resistant Mycobacterium Tuberculosis. Antimicrob Agents Chemother. 2006;50:2640–9. doi: 10.1128/AAC.00112-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ajbani K, Rodrigues C, Shenai S, Mehta A. Mutation Detection and Accurate Diagnosis of Extensively Drug-Resistant Tuberculosis: Report from a Tertiary Care Center in India. J Clinical Microbiology. 2011;49:1588–1590. doi: 10.1128/JCM.00113-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chia BS, Lanzas F, Rifat D, Herrera A, Kim EY, Sailer C, Torres-Chavolla E, Narayanaswamy P, Einarsson V, Bravo J, Pascale JM, Ioerger TR, Sacchettini JC, Karakousis PC. Use of Multiplex Allele-Specific Polymerase Chain Reaction (Mas-Pcr) to Detect Multidrug-Resistant Tuberculosis in Panama. PLoS ONE. 2012;7:e40456. doi: 10.1371/journal.pone.0040456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fenner L, Egger M, Bodmer T, Altpeter E, Zwahlen M, Jaton K, Pfyffer GE, Borrell S, Dubuis O, Bruderer T, Siegrist HH, Furrer H, Calmy A, Fehr J, Stalder JM, Ninet B, Bottger EC, Gagneux S. Effect of Mutation and Genetic Background on Drug Resistance in Mycobacterium Tuberculosis. Antimicrob Agents Chemother. 2012;56:3047–53. doi: 10.1128/AAC.06460-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tessema B, Beer J, Emmrich F, Sack U, Rodloff AC. Analysis of Gene Mutations Associated with Isoniazid, Rifampicin and Ethambutol Resistance among Mycobacterium Tuberculosis Isolates from Ethiopia. BMC Infect Dis. 2012;12:37–43. doi: 10.1186/1471-2334-12-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shubladze N, Tadumadze N, Bablishvili N. Molecular Patterns of Multidrug Resistance of in Georgia. Int J Mycobacteriol. 2013;2:73–78. doi: 10.1016/j.ijmyco.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hung NV, Ando H, Thuy TT, Kuwahara T, Hang NT, Sakurada S, Thuong PH, Lien LT, Keicho N. Clonal Expansion of Mycobacterium Tuberculosis Isolates and Coexisting Drug Resistance in Patients Newly Diagnosed with Pulmonary Tuberculosis in Hanoi, Vietnam. BMC Res Notes. 2013;6:444–51. doi: 10.1186/1756-0500-6-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tseng ST, Tai CH, Li CR, Lin CF, Shi ZY. The Mutations of Katg and Inha Genes of Isoniazid-Resistant Mycobacterium Tuberculosis Isolates in Taiwan. J Microbiol Immunol Infect. 2013:163–71. doi: 10.1016/j.jmii.2013.08.018. [DOI] [PubMed] [Google Scholar]
- 26.Huyen MN, Cobelens FG, Buu TN, Lan NT, Dung NH, Kremer K, Tiemersma EW, van Soolingen D. Epidemiology of Isoniazid Resistance Mutations and Their Effect on Tuberculosis Treatment Outcomes. Antimicrob Agents Chemother. 2013;57:3620–7. doi: 10.1128/AAC.00077-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yadav R, Sethi S, Dhatwalia SK, Gupta D, Mewara A, Sharma M. Molecular Characterisation of Drug Resistance in Mycobacterium Tuberculosis Isolates from North India. Int J Tuberc Lung Dis. 2013;17:251–7. doi: 10.5588/ijtld.12.0319. [DOI] [PubMed] [Google Scholar]
- 28.Varghese B, Hillemann A, Wijayanti DR, Shoukri M, Al-rabiah F, Al-Omari R, Al-Hajoj S. New Insight into the Molecular Characterization of Isoniazid and Rifampicin Resistant Mycobacterium Tuberculosis Strains from Saudi Arabia. Infect Genet Evol. 2012;12:549–56. doi: 10.1016/j.meegid.2012.01.024. [DOI] [PubMed] [Google Scholar]
- 29.Ali A, Hasan R, Jabeen K, Jabeen N, Qadeer E, Hasan Z. Characterization of Mutations Conferring Extensive Drug Resistance to Mycobacterium Tuberculosis Isolates in Pakistan. Antimicrob Agents Chemother. 2011;55:5654–9. doi: 10.1128/AAC.05101-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Balabanova Y, Nikolayevskyy V, Ignatyeva O, Kontsevaya I, Rutterford CM, Shakhmistova A, Malomanova N, Chinkova Y, Mironova S, Fedorin I, Drobniewski FA. Survival of Civilian and Prisoner Drug-Sensitive, Multi- and Extensive Drug- Resistant Tuberculosis Cohorts Prospectively Followed in Russia. PLoS ONE. 2011;6:e20531. doi: 10.1371/journal.pone.0020531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kozhamkulov U, Akhmetova A, Rakhimova S, Belova E, Alenova A, Bismilda V, Chingissova L, Ismailov S, Ramanculov E, Momynaliev K. Molecular Characterization of Rifampicin- and Isoniazid-Resistant Mycobacterium Tuberculosis Strains Isolated in Kazakhstan. Jpn J Infect Dis. 2011;64:253–5. [PubMed] [Google Scholar]
- 32.Dias MV, Vasconcelos IB, Prado AM, Fadel V, Basso LA, de Azevedo WF, Jr, Santos DS. Crystallographic Studies on the Binding of Isonicotinyl-Nad Adduct to Wild-Type Isoniazid Resistant 2-Trans-Enoyl-Acp (Coa) Reductase from Mycobacterium Tuberculosis. J Struct Biol. 2007;159:369–80. doi: 10.1016/j.jsb.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 33.He X, Alian A, Stroud R, Ortiz de Montellano PR. Pyrrolidine Carboxamides as a Novel Class of Inhibitors of Enoyl Acyl Carrier Protein Reductase from Mycobacterium Tuberculosis. J Med Chem. 2006;49:6308–23. doi: 10.1021/jm060715y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oliveira JS, Pereira JH, Canduri F, Rodrigues NC, de Souza ON, de Azevedo WF, Jr, Basso LA, Santos DS. Crystallographic and Pre-Steady-State Kinetics Studies on Binding of Nadh to Wild-Type and Isoniazid-Resistant Enoyl-Acp(Coa) Reductase Enzymes from Mycobacterium Tuberculosis. J Mol Biol. 2006;359:646–66. doi: 10.1016/j.jmb.2006.03.055. [DOI] [PubMed] [Google Scholar]
- 35.Shirude PS, Madhavapeddi P, Naik M, Murugan K, Shinde V, Nandishaiah R, Bhat J, Kumar A, Hameed S, Holdgate G, Davies G, McMiken H, Hegde N, Ambady A, Venkatraman J, Panda M, Bandodkar B, Sambandamurthy VK, Read JA. Methyl-Thiazoles: A Novel Mode of Inhibition with the Potential to Develop Novel Inhibitors Targeting Inha in Mycobacterium Tuberculosis. J Med Chem. 2013;56:8533–42. doi: 10.1021/jm4012033. [DOI] [PubMed] [Google Scholar]
- 36.Wang F, Langley R, Gulten G, Dover LG, Besra GS, Jacobs WR, Jr, Sacchettini JC. Mechanism of Thioamide Drug Action against Tuberculosis and Leprosy. J Exp Med. 2007;204:73–8. doi: 10.1084/jem.20062100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rozwarski DA, Vilcheze C, Sugantino M, Bittman R, Sacchettini JC. Crystal Structure of the Mycobacterium Tuberculosis Enoyl-Acp Reductase, Inha, in Complex with Nad+ and a C16 Fatty Acyl Substrate. J Biol Chem. 1999;274:15582–9. doi: 10.1074/jbc.274.22.15582. [DOI] [PubMed] [Google Scholar]
- 38.Rozwarski DA, Grant GA, Barton DH, Jacobs WR, Jr, Sacchettini JC. Modification of the Nadh of the Isoniazid Target (Inha) from Mycobacterium Tuberculosis. Science. 1998;279:98–102. doi: 10.1126/science.279.5347.98. [DOI] [PubMed] [Google Scholar]
- 39.Luckner SR, Liu N, am Ende CW, Tonge PJ, Kisker C. A Slow, Tight Binding Inhibitor of Inha, the Enoyl-Acyl Carrier Protein Reductase from Mycobacterium Tuberculosis. J Biol Chem. 2010;285:14330–7. doi: 10.1074/jbc.M109.090373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He X, Alian A, Ortiz de Montellano PR. Inhibition of the Mycobacterium Tuberculosis Enoyl Acyl Carrier Protein Reductase Inha by Arylamides. Bioorg Med Chem. 2007;15:6649–58. doi: 10.1016/j.bmc.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sullivan TJ, Truglio JJ, Boyne ME, Novichenok P, Zhang X, Stratton CF, Li HJ, Kaur T, Amin A, Johnson F, Slayden RA, Kisker C, Tonge PJ. High Affinity Inha Inhibitors with Activity against Drug-Resistant Strains of Mycobacterium Tuberculosis. ACS Chem Biol. 2006;1:43–53. doi: 10.1021/cb0500042. [DOI] [PubMed] [Google Scholar]
- 42.Hartkoorn RC, Sala C, Neres J, Pojer F, Magnet S, Mukherjee R, Uplekar S, Boy-Rottger S, Altmann KH, Cole ST. Towards a New Tuberculosis Drug: Pyridomycin - Nature’s Isoniazid. EMBO Mol Med. 2012;4:1032–42. doi: 10.1002/emmm.201201689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Freundlich JS, Wang F, Vilcheze C, Gulten G, Langley R, Schiehser GA, Jacobus DP, Jacobs WR, Jr, Sacchettini JC. Triclosan Derivatives: Towards Potent Inhibitors of Drug-Sensitive and Drug-Resistant. Mycobacterium Tuberculosis ChemMedChem. 2009;4:241–8. doi: 10.1002/cmdc.200800261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hartkoorn RC, Pojer F, Read JA, Gingell H, Neres J, Horlacher OP, Altmann KH, Cole ST. Pyridomycin Bridges the Nadh- and Substrate-Binding Pockets of the Enoyl Reductase Inha. Nat Chem Biol. 2014;10:96–8. doi: 10.1038/nchembio.1405. [DOI] [PubMed] [Google Scholar]
- 45.Argyrou A, Vetting MW, Blanchard JS. New Insight into the Mechanism of Action of and Resistance to Isoniazid: Interaction of Mycobacterium Tuberculosis Enoyl-Acp Reductase with Inh-Nadp. J Am Chem Soc. 2007;129:9582–3. doi: 10.1021/ja073160k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pan P, Tonge PJ. Targeting Inha, the Fasii Enoyl-Acp Reductase: Sar Studies on Novel Inhibitor Scaffolds. Current Topics in Med Chem. 2012;12:672–693. doi: 10.2174/156802612799984535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boyne ME, Sullivan TJ, amEnde CW, Lu H, Gruppo V, Heaslip D, Amin AG, Chatterjee D, Lenaerts A, Tonge PJ, Slayden RA. Targeting Fatty Acid Biosynthesis for the Development of Novel Chemotherapeutics against Mycobacterium Tuberculosis: Evaluation of a-Ring-Modified Diphenyl Ethers as High-Affinity Inha Inhibitors. Antimicrob Agents Chemother. 2007;51:3562–7. doi: 10.1128/AAC.00383-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuo MR, Morbidoni HR, Alland D, Sneddon SF, Gourlie BB, Staveski MM, Leonard M, Gregory JS, Janjigian AD, Yee C, Musser JM, Kreiswirth B, Iwamoto H, Perozzo R, Jacobs WR, Jr, Sacchettini JC, Fidock DA. Targeting Tuberculosis and Malaria through Inhibition of Enoyl Reductase: Compound Activity and Structural Data. J Biol Chem. 2003;278:20851–9. doi: 10.1074/jbc.M211968200. [DOI] [PubMed] [Google Scholar]
- 49.Lu H, Tonge PJ. Inhibitors of Fabi, an Enzyme Drug Target in the Bacterial Fatty Acid Biosynthesis Pathway. Accounts of Chem Res. 2008;41:11–20. doi: 10.1021/ar700156e. [DOI] [PubMed] [Google Scholar]
- 50.England K, am Ende C, Lu H, Sullivan TJ, Marlenee NL, Bowen RA, Knudson SE, Knudson DL, Tonge PJ, Slayden RA. Substituted Diphenyl Ethers as a Broad-Spectrum Platform for the Development of Chemotherapeutics for the Treatment of Tularaemia. J Antimicrob Chemother. 2009;64:1052–61. doi: 10.1093/jac/dkp307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu JL, Liu J, Cai Z. Determination of Triclosan Metabolites by Using in-Source Fragmentation from High-Performance Liquid Chromatography/Negative Atmospheric Pressure Chemical Ionization Ion Trap Mass Spectrometry. Rapid Commun Mass Spectrom. 2010;24:1828–34. doi: 10.1002/rcm.4558. [DOI] [PubMed] [Google Scholar]
- 52.Wang LQ, Falany CN, James MO. Triclosan as a Substrate and Inhibitor of 3′-Phosphoadenosine 5′-Phosphosulfate-Sulfotransferase and Udp-Glucuronosyl Transferase in Human Liver Fractions. Drug metabolism and disposition: the biological fate of chemicals. 2004;32:1162–9. doi: 10.1124/dmd.104.000273. [DOI] [PubMed] [Google Scholar]
- 53.Zhu T, Cao S, Su PC, Patel R, Shah D, Chokshi HB, Szukala R, Johnson ME, Hevener KE. Hit Identification and Optimization in Virtual Screening: Practical Recommendations Based on a Critical Literature Analysis. J Med Chem. 2013;56:6560–6572. doi: 10.1021/jm301916b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tanrikulu Y, Kruger B, Proschak E. The Holistic Integration of Virtual Screening in Drug Discovery. Drug Discovery Today. 2013;18:358–64. doi: 10.1016/j.drudis.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 55.Goodsell DS, Olson AJ. Automated Docking of Substrates to Proteins by Simulated Annealing. Proteins. 1990;8:195–202. doi: 10.1002/prot.340080302. [DOI] [PubMed] [Google Scholar]
- 56.Goodsell DS, Lauble H, Stout CD, Olson AJ. Automated Docking in Crystallography: Analysis of the Substrates of Aconitase. Proteins. 1993;17:1–10. doi: 10.1002/prot.340170104. [DOI] [PubMed] [Google Scholar]
- 57.Goodsell DS, Morris GM, Olson AJ. Automated Docking of Flexible Ligands: Applications of Autodock. J Mol Recognit. 1996;9:1–5. doi: 10.1002/(sici)1099-1352(199601)9:1<1::aid-jmr241>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 58.Morris GM, Goodsell DS, Huey R, Olson AJ. Distributed Automated Docking of Flexible Ligands to Proteins: Parallel Applications of Autodock 2.4. J Comput Aided Mol Des. 1996;10:293–304. doi: 10.1007/BF00124499. [DOI] [PubMed] [Google Scholar]
- 59.Olson AJ, Goodsell DS. Automated Docking and the Search for Hiv Protease Inhibitors. SAR QSAR Environ Res. 1998;8:273–85. doi: 10.1080/10629369808039144. [DOI] [PubMed] [Google Scholar]
- 60.Soares TA, Goodsell DS, Briggs JM, Ferreira R, Olson AJ. Docking of 4-Oxalocrotonate Tautomerase Substrates: Implications for the Catalytic Mechanism. Biopolymers. 1999;50:319–28. doi: 10.1002/(SICI)1097-0282(199909)50:3<319::AID-BIP7>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 61.Osterberg F, Morris GM, Sanner MF, Olson AJ, Goodsell DS. Automated Docking to Multiple Target Structures: Incorporation of Protein Mobility and Structural Water Heterogeneity in Autodock. Proteins. 2002;46:34–40. doi: 10.1002/prot.10028. [DOI] [PubMed] [Google Scholar]
- 62.Trott O, Olson AJ. Autodock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J Comput Chem. 2010;31:455–61. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kumar M, Vijayakrishnan R, Subba Rao G. In Silico Structure-Based Design of a Novel Class of Potent and Selective Small Peptide Inhibitor of Mycobacterium Tuberculosis Dihydrofolate Reductase, a Potential Target for Anti-Tb Drug Discovery. Mol Divers. 2010;14:595–604. doi: 10.1007/s11030-009-9172-6. [DOI] [PubMed] [Google Scholar]
- 64.Kumar A, Siddiqi MI. Virtual Screening against Mycobacterium Tuberculosis Dihydrofolate Reductase: Suggested Workflow for Compound Prioritization Using Structure Interaction Fingerprints. J Mol Graph Model. 2008;27:476–88. doi: 10.1016/j.jmgm.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 65.Lu XY, Chen YD, Jiang YJ, You QD. Discovery of Potential New Inha Direct Inhibitors Based on Pharmacophore and 3d-Qsar Analysis Followed by in Silico Screening. Eur J Med Chem. 2009;44:3718–30. doi: 10.1016/j.ejmech.2009.03.043. [DOI] [PubMed] [Google Scholar]
- 66.Lu XY, Chen YD, You QD. 3d-Qsar Studies of Arylcarboxamides with Inhibitory Activity on Inha Using Pharmacophore-Based Alignment. Chem Biol Drug Des. 2010;75:195–203. doi: 10.1111/j.1747-0285.2009.00926.x. [DOI] [PubMed] [Google Scholar]
- 67.Subba Rao G, Vijayakrishnan R, Kumar M. Structure-Based Design of a Novel Class of Potent Inhibitors of Inha, the Enoyl Acyl Carrier Protein Reductase from Mycobacterium Tuberculosis: A Computer Modelling Approach. Chem Biol Drug Des. 2008;72:444–9. doi: 10.1111/j.1747-0285.2008.00722.x. [DOI] [PubMed] [Google Scholar]
- 68.Punkvang A, Saparpakorn P, Hannongbua S, Wolschann P, Pungpo P. Elucidating Drug-Enzyme Interactions and Their Structural Basis for Improving the Affinity and Potency of Isoniazid and Its Derivatives Based on Computer Modeling Approaches. Molecules. 2010;15:2791–813. doi: 10.3390/molecules15042791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kinnings SL, Liu N, Tonge PJ, Jackson RM, Xie L, Bourne PE. A Machine Learning-Based Method to Improve Docking Scoring Functions and Its Application to Drug Repurposing. J Chem Inf Model. 2011;51:408–19. doi: 10.1021/ci100369f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pauli I, dos Santos RN, Rostirolla DC, Martinelli LK, Ducati RG, Timmers LF, Basso LA, Santos DS, Guido RV, Andricopulo AD, Norberto de Souza O. Discovery of New Inhibitors of Mycobacterium Tuberculosis Inha Enzyme Using Virtual Screening and a 3d-Pharmacophore-Based Approach. J Chem Inf Model. 2013;53:2390–401. doi: 10.1021/ci400202t. [DOI] [PubMed] [Google Scholar]
- 71.Mohan SB, Ravi Kumar BV, Dinda SC, Naik D, Prabu Seenivasan S, Kumar V, Rana DN, Brahmkshatriya PS. Microwave-Assisted Synthesis, Molecular Docking and Antitubercular Activity of 1,2,3,4-Tetrahydropyrimidine-5-Carbonitrile Derivatives. Bioorg Med Chem Lett. 2012;22:7539–42. doi: 10.1016/j.bmcl.2012.10.032. [DOI] [PubMed] [Google Scholar]
- 72.Muddassar M, Jang JW, Hong SK, Cho YS, Kim EE, Keum KC, Oh T, Cho SN, Pae AN. Identification of Novel Antitubercular Compounds through Hybrid Virtual Screening Approach. Bioorg Med Chem. 2010;18:6914–21. doi: 10.1016/j.bmc.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 73.Izumizono Y, Arevalo S, Koseki Y, Kuroki M, Aoki S. Identification of Novel Potential Antibiotics for Tuberculosis by in Silico Structure-Based Drug Screening. Eur J Med Chem. 2011;46:1849–56. doi: 10.1016/j.ejmech.2011.02.047. [DOI] [PubMed] [Google Scholar]
- 74.Kinjo T, Koseki Y, Kobayashi M, Yamada A, Morita K, Yamaguchi K, Tsurusawa R, Gulten G, Komatsu H, Sakamoto H, Sacchettini JC, Kitamura M, Aoki S. Identification of Compounds with Potential Antibacterial Activity against Mycobacterium through Structure-Based Drug Screening. J Chem Inf Model. 2013;53:1200–12. doi: 10.1021/ci300571n. [DOI] [PubMed] [Google Scholar]
- 75.Perryman AL. I’ll Take “Curing Malaria” for $1,000, Alex. In: Fishkind A, Gordon R, editors. Citizen IBM. IBM; 2011. [Google Scholar]
- 76.Perryman AL. Go Fight against Malaria. http://www.worldcommunitygrid.org/research/gfam/overview.do.
- 77.Perryman AL, Olson AJ. Global Online Fight against Malaria Project. http://GOFightAgainstMalaria.scripps.edu.
- 78.Irwin JJ, Shoichet BK. Zinc--a Free Database of Commercially Available Compounds for Virtual Screening. J Chem Inf Model. 2005;45:177–82. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stec J, Vilcheze C, Lun S, Perryman AL, Wang X, Freundlich JS, Bishai W, Jacobs WR, Jr, Kozikowski AP. Biological Evaluation of Potent Triclosan-Derived Inhibitors of the Enoyl-Acyl Carrier Protein Reductase Inha in Drug-Sensitive and Drug-Resistant Strains of Mycobacterium Tuberculosis. ChemMedChem. 2014;9:2528–37. doi: 10.1002/cmdc.201402255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chang A, Schiebel J, Yu W, Bommineni GR, Pan P, Baxter MV, Khanna A, Sotriffer CA, Kisker C, Tonge PJ. Rational Optimization of Drug-Target Residence Time: Insights from Inhibitor Binding to the Staphylococcus Aureus Fabi Enzyme-Product Complex. Biochemistry. 2013;52:4217–28. doi: 10.1021/bi400413c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lu H, Tonge PJ. Drug-Target Residence Time: Critical Information for Lead Optimization. Curr Opin Chem Biol. 2010;14:467–74. doi: 10.1016/j.cbpa.2010.06.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lu H, England K, am Ende C, Truglio JJ, Luckner S, Reddy BG, Marlenee NL, Knudson SE, Knudson DL, Bowen RA, Kisker C, Slayden RA, Tonge PJ. Slow-Onset Inhibition of the Fabi Enoyl Reductase from Francisella Tularensis: Residence Time and in Vivo Activity. ACS Chem Biol. 2009;4:221–31. doi: 10.1021/cb800306y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. Molprobity: All-Atom Structure Validation for Macromolecular Crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. Autodock4 and Autodocktools4: Automated Docking with Selective Receptor Flexibility. J Comput Chem. 2009;30:2785–91. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Forli S, Olson AJ. Raccoon. Molecular Graphics Laboratory; 2010. [Google Scholar]
- 86.Cosconati S, Forli S, Perryman AL, Harris R, Goodsell DS, Olson AJ. Virtual Screening with Autodock: Theory and Practice. Expert Opin Drug Discovery. 2010;5:597–607. doi: 10.1517/17460441.2010.484460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Perryman AL, Santiago DN, Forli S, Santos-Martins D, Olson AJ. Virtual Screening with Autodock Vina and the Common Pharmacophore Engine of a Low Diversity Library of Fragments and Hits against the Three Allosteric Sites of Hiv Integrase: Participation in the Sampl4 Protein-Ligand Binding Challenge. J Comput Aided Mol Des. 2014;28:429–41. doi: 10.1007/s10822-014-9709-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mobley DL, Liu S, Lim NM, Wymer KL, Perryman AL, Forli S, Deng N, Su J, Branson K, Olson AJ. Blind Prediction of Hiv Integrase Binding from the Sampl4 Challenge. J Comput Aided Mol Des. 2014;28:327–45. doi: 10.1007/s10822-014-9723-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Voet AR, Kumar A, Berenger F, Zhang KY. Combining in Silico and in Cerebro Approaches for Virtual Screening and Pose Prediction in Sampl4. J Comput Aided Mol Des. 2014;28:363–73. doi: 10.1007/s10822-013-9702-2. [DOI] [PubMed] [Google Scholar]
- 90.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Advanced Drug Delivery Reviews. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 91.Hunter CA. Sequence-Dependent DNA Structure. The Role of Base Stacking Interactions. J Mol Biol. 1993;230:1025–54. doi: 10.1006/jmbi.1993.1217. [DOI] [PubMed] [Google Scholar]
- 92.Jiang X, Loo DD, Hirayama BA, Wright EM. The Importance of Being Aromatic: Pi Interactions in Sodium Symporters. Biochemistry. 2012;51:9480–7. doi: 10.1021/bi301329w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Durrant JD, McCammon JA. Binana: A Novel Algorithm for Ligand-Binding Characterization. J Mol Graph Model. 2011;29:888–93. doi: 10.1016/j.jmgm.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sponer J, Leszczynski J, Hobza P. Electronic Properties, Hydrogen Bonding, Stacking, and Cation Binding of DNA and Rna Bases. Biopolymers. 2001;61:3–31. doi: 10.1002/1097-0282(2001)61:1<3::AID-BIP10048>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 95.Lefebvre E, Schiffer CA. Resilience to Resistance of Hiv-1 Protease Inhibitors: Profile of Darunavir. AIDS Rev. 2008;10:131–42. [PMC free article] [PubMed] [Google Scholar]
- 96.Nalam MN, Ali A, Altman MD, Reddy GS, Chellappan S, Kairys V, Ozen A, Cao H, Gilson MK, Tidor B, Rana TM, Schiffer CA. Evaluating the Substrate-Envelope Hypothesis: Structural Analysis of Novel Hiv-1 Protease Inhibitors Designed to Be Robust against Drug Resistance. J Virol. 2010;84:5368–78. doi: 10.1128/JVI.02531-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kairys V, Gilson MK, Lather V, Schiffer CA, Fernandes MX. Toward the Design of Mutation-Resistant Enzyme Inhibitors: Further Evaluation of the Substrate Envelope Hypothesis. Chem Biol Drug Des. 2009;74:234–45. doi: 10.1111/j.1747-0285.2009.00851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Altman MD, Ali A, Reddy GS, Nalam MN, Anjum SG, Cao H, Chellappan S, Kairys V, Fernandes MX, Gilson MK, Schiffer CA, Rana TM, Tidor B. Hiv-1 Protease Inhibitors from Inverse Design in the Substrate Envelope Exhibit Subnanomolar Binding to Drug-Resistant Variants. J Am Chem Soc. 2008;130:6099–113. doi: 10.1021/ja076558p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chellappan S, Kiran Kumar Reddy GS, Ali A, Nalam MN, Anjum SG, Cao H, Kairys V, Fernandes MX, Altman MD, Tidor B, Rana TM, Schiffer CA, Gilson MK. Design of Mutation-Resistant Hiv Protease Inhibitors with the Substrate Envelope Hypothesis. Chem Biol Drug Des. 2007;69:298–313. doi: 10.1111/j.1747-0285.2007.00514.x. [DOI] [PubMed] [Google Scholar]
- 100.Prabu-Jeyabalan M, King NM, Nalivaika EA, Heilek-Snyder G, Cammack N, Schiffer CA. Substrate Envelope and Drug Resistance: Crystal Structure of Ro1 in Complex with Wild-Type Human Immunodeficiency Virus Type 1 Protease. Antimicrob Agents Chemother. 2006;50:1518–21. doi: 10.1128/AAC.50.4.1518-1521.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.King NM, Prabu-Jeyabalan M, Nalivaika EA, Wigerinck P, de Bethune MP, Schiffer CA. Structural and Thermodynamic Basis for the Binding of Tmc114, a Next-Generation Human Immunodeficiency Virus Type 1 Protease Inhibitor. J Virol. 2004;78:12012–21. doi: 10.1128/JVI.78.21.12012-12021.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.King NM, Prabu-Jeyabalan M, Nalivaika EA, Schiffer CA. Combating Susceptibility to Drug Resistance: Lessons from Hiv-1 Protease. Chem Biol. 2004;11:1333–8. doi: 10.1016/j.chembiol.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 103.Nalam MN, Ali A, Reddy GS, Cao H, Anjum SG, Altman MD, Yilmaz NK, Tidor B, Rana TM, Schiffer CA. Substrate Envelope-Designed Potent Hiv-1 Protease Inhibitors to Avoid Drug Resistance. Chem Biol. 2013;20:1116–24. doi: 10.1016/j.chembiol.2013.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shen Y, Altman MD, Ali A, Nalam MN, Cao H, Rana TM, Schiffer CA, Tidor B. Testing the Substrate-Envelope Hypothesis with Designed Pairs of Compounds. ACS Chem Biol. 2013;8:2433–41. doi: 10.1021/cb400468c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nalam MN, Schiffer CA. New Approaches to Hiv Protease Inhibitor Drug Design Ii: Testing the Substrate Envelope Hypothesis to Avoid Drug Resistance and Discover Robust Inhibitors. Curr Opin HIV AIDS. 2008;3:642–6. doi: 10.1097/COH.0b013e3283136cee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chusacultanachai S, Thiensathit P, Tarnchompoo B, Sirawaraporn W, Yuthavong Y. Novel Antifolate Resistant Mutations of Plasmodium Falciparum Dihydrofolate Reductase Selected in Escherichia Coli. Mol Biochem Parasitol. 2002;120:61–72. doi: 10.1016/s0166-6851(01)00440-6. [DOI] [PubMed] [Google Scholar]
- 107.Japrung D, Leartsakulpanich U, Chusacultanachai S, Yuthavong Y. Conflicting Requirements of Plasmodium Falciparum Dihydrofolate Reductase Mutations Conferring Resistance to Pyrimethamine-Wr99210 Combination. Antimicrob Agents Chemother. 2007;51:4356–60. doi: 10.1128/AAC.00577-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kamchonwongpaisan S, Vanichtanankul J, Taweechai S, Chitnumsub P, Yuthavong Y. The Role of Tryptophan-48 in Catalysis and Binding of Inhibitors of Plasmodium Falciparum Dihydrofolate Reductase. Int J Parasitol. 2007;37:787–93. doi: 10.1016/j.ijpara.2006.12.017. [DOI] [PubMed] [Google Scholar]
- 109.Maitarad P, Kamchonwongpaisan S, Vanichtanankul J, Vilaivan T, Yuthavong Y, Hannongbua S. Interactions between Cycloguanil Derivatives and Wild Type and Resistance-Associated Mutant Plasmodium Falciparum Dihydrofolate Reductases. J Comput Aided Mol Des. 2009;23:241–52. doi: 10.1007/s10822-008-9254-z. [DOI] [PubMed] [Google Scholar]
- 110.Yuthavong Y, Yuvaniyama J, Chitnumsub P, Vanichtanankul J, Chusacultanachai S, Tarnchompoo B, Vilaivan T, Kamchonwongpaisan S. Malarial (Plasmodium Falciparum) Dihydrofolate Reductase-Thymidylate Synthase: Structural Basis for Antifolate Resistance and Development of Effective Inhibitors. Parasitology. 2005;130:249–59. doi: 10.1017/s003118200400664x. [DOI] [PubMed] [Google Scholar]
- 111.Lin YC, Perryman AL, Olson AJ, Torbett BE, Elder JH, Stout CD. Structural Basis for Drug and Substrate Specificity Exhibited by Fiv Encoding a Chimeric Fiv/Hiv Protease. Acta Crystallogr D Biol Crystallogr. 2011;67:540–8. doi: 10.1107/S0907444911011681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ekins S, Clark AM, Sarker M. Tb Mobile: A Mobile App for Anti-Tuberculosis Molecules with Known Targets. J Cheminform. 2013;5:13. doi: 10.1186/1758-2946-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Clark AM, Sarker M, Ekins S. New Target Prediction and Visualization Tools Incorporating Open Source Molecular Fingerprints for Tb Mobile 2.0. J Cheminform. 2014;6:38–54. doi: 10.1186/s13321-014-0038-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Inc., A. S. Discovery Studio Modeling Environment, Release 4.0. Accelrys Software Inc; San Diego, CA: 2013. [Google Scholar]
- 115.Willett P. Similarity-Based Virtual Screening Using 2d Fingerprints. Drug Discovery Today. 2006;11:1046–53. doi: 10.1016/j.drudis.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 116.Ballell L, Bates RH, Young RJ, Alvarez-Gomez D, Alvarez-Ruiz E, Barroso V, Blanco D, Crespo B, Escribano J, Gonzalez R, Lozano S, Huss S, Santos-Villarejo A, Martin-Plaza JJ, Mendoza A, Rebollo-Lopez MJ, Remuinan-Blanco M, Lavandera JL, Perez-Herran E, Gamo-Benito FJ, Garcia-Bustos JF, Barros D, Castro JP, Cammack N. Fueling Open-Source Drug Discovery: 177 Small-Molecule Leads against Tuberculosis. ChemMedChem. 2013;8:313–21. doi: 10.1002/cmdc.201200428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Franzblau SG, Witzig RS, McLaughlin JC, Torres P, Madico G, Hernandez A, Degnan MT, Cook MB, Quenzer VK, Ferguson RM, Gilman RH. Rapid, Low-Technology Mic Determination with Clinical Mycobacterium Tuberculosis Isolates by Using the Microplate Alamar Blue Assay. J Clin Microbiol. 1998;36:362–6. doi: 10.1128/jcm.36.2.362-366.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chessari G, Woodhead AJ. From Fragment to Clinical Candidate--a Historical Perspective. Drug Discovery Today. 2009;14:668–75. doi: 10.1016/j.drudis.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 119.Hajduk PJ, Greer J. A Decade of Fragment-Based Drug Design: Strategic Advances and Lessons Learned. Nat Rev Drug Discovery. 2007;6:211–9. doi: 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]
- 120.Austin C, Pettit SN, Magnolo SK, Sanvoisin J, Chen W, Wood SP, Freeman LD, Pengelly RJ, Hughes DE. Fragment Screening Using Capillary Electrophoresis (Cefrag) for Hit Identification of Heat Shock Protein 90 Atpase Inhibitors. J Biomolecular Screening. 2012;17:868–76. doi: 10.1177/1087057112445785. [DOI] [PubMed] [Google Scholar]
- 121.Teotico DG, Babaoglu K, Rocklin GJ, Ferreira RS, Giannetti AM, Shoichet BK. Docking for Fragment Inhibitors of Ampc β-Lactamase. Proc Natl Acad Sci USA. 2009;106:7455–7460. doi: 10.1073/pnas.0813029106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Murray CW, Callaghan O, Chessari G, Cleasby A, Congreve M, Frederickson M, Hartshorn MJ, McMenamin R, Patel S, Wallis N. Application of Fragment Screening by X-Ray Crystallography to Beta-Secretase. J Med Chem. 2007;50:1116–23. doi: 10.1021/jm0611962. [DOI] [PubMed] [Google Scholar]
- 123.Chen Y, Shoichet BK. Molecular Docking and Ligand Specificity in Fragment-Based Inhibitor Discovery. Nat Chem Biol. 2009;5:358–64. doi: 10.1038/nchembio.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Discovering High-Affinity Ligands for Proteins: Sar by Nmr. Science. 1996;274:1531–4. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 125.Li HJ, Lai CT, Pan P, Yu W, Liu N, Bommineni GR, Garcia-Diaz M, Simmerling C, Tonge PJ. A Structural and Energetic Model for the Slow-Onset Inhibition of the Mycobacterium Tuberculosis Enoyl-Acp Reductase Inha. ACS Chem Biol. 2014;9:986–93. doi: 10.1021/cb400896g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.am Ende CW, Knudson SE, Liu N, Childs J, Sullivan TJ, Boyne M, Xu H, Gegina Y, Knudson DL, Johnson F, Peloquin CA, Slayden RA, Tonge PJ. Synthesis and in Vitro Antimycobacterial Activity of B-Ring Modified Diaryl Ether Inha Inhibitors. Bioorg Med Chem Lett. 2008;18:3029–33. doi: 10.1016/j.bmcl.2008.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, Spevak W, Zhang C, Zhang Y, Habets G, Burton EA, Wong B, Tsang G, West BL, Powell B, Shellooe R, Marimuthu A, Nguyen H, Zhang KY, Artis DR, Schlessinger J, Su F, Higgins B, Iyer R, D’Andrea K, Koehler A, Stumm M, Lin PS, Lee RJ, Grippo J, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, Chapman PB, Flaherty KT, Xu X, Nathanson KL, Nolop K. Clinical Efficacy of a Raf Inhibitor Needs Broad Target Blockade in Braf-Mutant Melanoma. Nature. 2010;467:596–9. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.de Kloe GE, Bailey D, Leurs R, de Esch IJ. Transforming Fragments into Candidates: Small Becomes Big in Medicinal Chemistry. Drug Discovery Today. 2009;14:630–46. doi: 10.1016/j.drudis.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 129.Congreve M, Chessari G, Tisi D, Woodhead AJ. Recent Developments in Fragment-Based Drug Discovery. J Med Chem. 2008;51:3661–80. doi: 10.1021/jm8000373. [DOI] [PubMed] [Google Scholar]
- 130.Teotico DG, Babaoglu K, Rocklin GJ, Ferreira RS, Giannetti AM, Shoichet BK. Docking for Fragment Inhibitors of Ampc Beta-Lactamase. Proc Natl Acad Sci USA. 2009;106:7455–60. doi: 10.1073/pnas.0813029106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Krasinski A, Radic Z, Manetsch R, Raushel J, Taylor P, Sharpless KB, Kolb HC. In Situ Selection of Lead Compounds by Click Chemistry: Target-Guided Optimization of Acetylcholinesterase Inhibitors. J Am Chem Soc. 2005;127:6686–92. doi: 10.1021/ja043031t. [DOI] [PubMed] [Google Scholar]
- 132.Mamidyala SK, Finn MG. In Situ Click Chemistry: Probing the Binding Landscapes of Biological Molecules. Chem Soc Rev. 2010;39:1252–61. doi: 10.1039/b901969n. [DOI] [PubMed] [Google Scholar]
- 133.Manetsch R, Krasinski A, Radic Z, Raushel J, Taylor P, Sharpless KB, Kolb HC. In Situ Click Chemistry: Enzyme Inhibitors Made to Their Own Specifications. J Am Chem Soc. 2004;126:12809–18. doi: 10.1021/ja046382g. [DOI] [PubMed] [Google Scholar]
- 134.Sharpless KB, Manetsch R. In Situ Click Chemistry: A Powerful Means for Lead Discovery. Expert Opin Drug Discovery. 2006;1:525–38. doi: 10.1517/17460441.1.6.525. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.