

Abstract
The genetic disease tuberous sclerosis complex (TSC) is an autosomal dominant disorder caused by loss of function mutations in either TSC1 (hamartin) or TSC2 (tuberin), which serve as negative regulators of mechanistic target of rapamycin complex 1 (mTORC1) activity. TSC patients exhibit developmental brain abnormalities and tuber formations that are associated with neuropsychological and neurocognitive impairments, seizures and premature death. Mechanistically, TSC1 and TSC2 loss of function mutations result in abnormally high mTORC1 activity. Thus, the development of a strategy to inhibit abnormally high mTORC1 activity may have therapeutic value in the treatment of TSC. mTORC1 is a master regulator of growth processes, and its activity can be reduced by withdrawal of growth factors, decreased energy availability, and by the immunosuppressant rapamycin. Recently, glutamine has been shown to alter mTORC1 activity in a TSC1-TSC2 independent manner in cells cultured under amino acid- and serum-deprived conditions. Since starvation culture conditions are not physiologically relevant, we examined if glutamine can regulate mTORC1 in non-deprived cells and in a murine model of TSC. Our results show that glutamine can reduce phosphorylation of S6 and S6 kinase, surrogate indicators of mTORC1 activity, in both deprived and non-deprived cells, although higher concentrations were required for non-deprived cultures. When administered orally to TSC2 knockout mice, glutamine reduced S6 phosphorylation in the brain and significantly prolonged their lifespan. Taken together, these studies suggest that glutamine supplementation can be used as a potential treatment for TSC.
1. Introduction
Tuberous sclerosis complex (TSC) is an autosomal dominant neurodevelopmental disease that causes significant morbidity and mortality. The brains of TSC patients are characterized by focal areas of cortical disorganization called tubers, subependymal nodules, and other developmental lesions. These anatomical defects are often associated with epilepsy, intellectual disability, and autism spectrum disorders. In addition, subependymal nodules can degenerate into low grade subependymal giant cell astrocytomas (SEGAs), which are associated with a wide variety of clinical presentations, in up to 20% of TSC patients [1;5;18]. More than 80% of patients with TSC have loss of function mutations within the genes TSC1 and TSC2, which encode the proteins hamartin and tuberin, respectively [11;23].
Hamartin and tuberin form a heterodimer (TSC1/2) that is a central regulator of the mechanistic Target of Rapamycin Complex 1 (mTORC1). mTORC1 is a multiprotein complex that functions as a cellular kinase, and is composed of a core set of at least 5 key proteins (mTOR, Raptor, PRAS40, Deptor, and mLst8). mTORC1 integrates a diverse array of intra- and extracellular signals, including energy status, growth factors, amino acid and nutrient availability to regulate protein, lipid and nucleotide synthesis, cell growth and autophagy [4;21;29;30;35]. Binding of the small GTPase protein Rheb (Ras-homologue enriched in brain) is required for mTORC1 activity [31]. The TSC1/2 heterodimer controls mTORC1 activity though the GTPase-activating domain of TSC2. TSC1/2 interacts with GTP-bound Rheb, converting it into its inactive GDP-bound form, thus resulting in decreased mTORC1 activity [16;20;22;36]. To a large extent, the molecular pathogenesis of TSC is caused by the cellular consequences of dysregulated, overactive mTORC1 arising from the loss of TSC1/2 inhibitory regulation [14;25;42]. Pharmacological inhibitors of mTORC1, such as rapamycin and other rapalogues, have successfully treated various mouse models of TSC [9;17;24]. These successful preclinical studies have paved the way for many clinical trials for brain, kidney and lung manifestations of TSC [15;41]. Recently, the FDA approved an additional indication for the rapalogue everolimus to treat refractory subependymal giant cell astrocytomas (SEGAs) associated with TSC [10;17;28]. However, in some cases the SEGAs are found to re-emerge after discontinuation of everolimus treatment, and the consequences of long-term treatment with rapalogues remains unclear at this time. Therefore, alternative approaches to the inhibition of mTORC1 may have substantial benefit for the pharmacologic treatment of TSC.
Recent in vitro studies have shown that amino acids are able to regulate mTORC1 signaling through a pathway independent of TSC1/2 [3;32;45]. These studies have suggested that glutamine, the most abundant amino acid found in the circulation, can modulate mTORC1 activity, with both inhibition [12;33] and stimulation [6;34;40] of mTORC1 activity being reported. However, these studies examined the influence of glutamine on mTORC1 activity using cultured cells exposed to serum- and amino acid-free conditions, making it difficult to determine if glutamine can alter mTORC1 activity under normal physiological conditions. In the present study, we examined if glutamine can alter mTORC1 activity in cultured cells maintained in non-deprived conditions. Our in vitro results showed that high concentrations of glutamine inhibited mTORC1 activity, as assessed by decreased phosphorylation of its downstream targets ribosomal protein S6 and S6 kinase. Based on this finding, we then tested if oral glutamine supplementation could be used to decrease mTORC1 activity in the brains of mice with a conditional knockout of Tsc2 in neurons, and prolong their life-span.
2. Materials and Methods
2.1. Reagents
L-glutamine was purchased from Sigma-Aldrich (St. Louis, MO). Antibodies for phosphorylated (Serine 240/244) and total S6, and phosphorylated (Threonine 389) and total S6 kinase were purchased from Cell Signaling Technology (Danvers, MA). Antibodies against the neuronal marker NeuN were obtained from Millipore (Billerica, MA).
2.2. Cell culture and western blotting
Mouse brain endothelial cells (bEnd.3; ATCC, stock #CRL-2299) were grown in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies) supplemented with 10% fetal bovine serum and penicillin/streptomycin (5000 U/5000 μg/ml). Forty-eight hours after reaching confluence, cells were incubated for 2 hr with either serum-free DMEM lacking glutamine or DMEM with serum (Life Technologies, Grand Island, NY). Glutamine at different concentrations was added for 30 min, after which cells were lysed in boiling Laemmli sample buffer, and lysates stored at −20°C until needed for western blot experiments. Lysate protein concentrations were determined by NanoOrange Protein Quantification Assay (Invitrogen, Carlsbad, CA) using bovine serum albumin (BSA) as the standard. Samples were resolved on Tris-glycine SDS-PAGE gels and transferred to Immobilon-P membranes (Millipore, Bedford, MA). Membranes were blocked for 30 min in 5% BSA, and then incubated in primary antibodies overnight at 4°C. Immunoreactivity was detected using species-specific secondary antibodies conjugated to alkaline phosphatase and a chemiluminescence system.
2.3. Tsc2 Conditional Knock-out mice and treatment
All protocols involving the use of animals were in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by The University of Texas Health Science Center Animal Care and Use Committee. The Tsc2flox/flox;CamKIIα-Cre mouse line was generated by crossing Tsc2flox/flox mice [19] to JAX® mice B6.Cg-Tg(CamKIIα-Cre)T29-1Stl/J, stock #005359. The progeny were backcrossed to Tsc2flox/flox mice to get Tsc2flox/flox;CamKIIα-Cre (Tsc2CKO). Control littermates constituted animals lacking CamKIIα-Cre. Glutamine treatment was initiated when Tsc2CKO mice were 1 month of age. A suspension of 3 g/kg glutamine in saline was given twice a day, 5 days a week via gavage. Animals were monitored daily and the date of death noted.
2.4. Immunohistochemistry
One month old Tsc2CKO mice were given 2 daily doses of either 3 g/kg glutamine or saline, 5 days a week for 2 weeks via gavage. One hour after the last dose, animals were decapitated, their brains quickly removed and dropped-fixed in 4% paraformaldehyde and 15% picric acid. Brains from age-matched control littermates were also collected. Coronal brain sections (40 μm) were incubated with primary antibodies overnight at 4°C which were then detected using species-specific secondary antibodies conjugated to Alexa-fluors. Epifluorescent images of the sections were taken using a Zeiss Axiovert S100 microscope and a MicroFire (Optronics, Goleta, CA) camera. The parameters used for image capture were determined using control littermate samples to minimize background and optimize the signal. These parameters were kept constant across all groups. Three non-overlapping regions in the dentate gyrus of the hippocampus and two sections from each animal (n=5/group) were used for quantification. The fluorescence intensities were measured using Photoshop software (Adobe, San Jose, CA), averaged for each section, then averaged for each animal.
2.5. Statistical analysis
Western blot data was compared using either two-tailed Student’s t-test for unpaired variables or by one-way ANOVA. The analysis of survival was performed using the Kaplan–Meier method. Comparisons between survival curves were carried out using the log-rank test. Data were considered significant at p<0.05.
3. Results
3.1. Glutamine inhibits mTORC1 activity in vitro
Previous studies have reported inhibition of mTORC1 activity by glutamine using cultured cells deprived of serum and/or amino acids, which does not reflect physiological situations. This inhibition was observed at concentrations ranging from a low of 4 mM to a high of 80 mM glutamine [12;33]. We therefore examined if these concentrations of glutamine can inhibit mTORC1 in cells grown under normal culture conditions. Consistent with previous studies, the addition of 5 mM glutamine to serum-starved bEnd.3 cells significantly decreased the phosphorylation of S6 (Figure 1A, p = 0.042) and S6 kinase (Figure 1B, p = 0.018), both downstream targets of mTORC1. The total levels of S6 and S6 kinase were unaffected. In contrast, when 5 mM glutamine was added to cells growing under normal serum conditions, it failed to significantly inhibit mTORC1 activity (Figure 1C–D). Inhibition of mTORC1 under normal culture conditions was observed, however, when a higher concentration of glutamine (40 mM or 5.84 mg/ml) was used (Figure 1C, p = 0.015 and 1D, p = 0.014).
Figure 1. Glutamine decreases the phosphorylation of mTORC1 downstream targets in starved and non-starved brain endothelial bEnd.3 cells.

A and B) bEnd.3 cells exposed to starvation conditions for 2 hr were treated with vehicle (0 mM) or 5 mM glutamine, and lysates collected 30 min later. Glutamine administration significantly decreased the phosphorylation, but not the total levels of the mTORC1 targets A) S6K and B) S6. C and D) bEnd.3 cells under normal culture conditions were treated with vehicle (0 mM), 5 mM or 40 mM glutamine, and lysates collected 30 min later. 40 mM, but not 5 mM, glutamine treatment significantly decreased C) S6K and D) S6 phosphorylation. The total levels of these proteins did not significantly change. Data are presented as mean ± s.e.m after correction against the β-actin loading control, *p<0.05.
3.2. Oral glutamine administration to Tsc2CKO mice decreases brain mTORC1 activity
Both Tsc1-null and Tsc2-null mice have elevated levels of mTORC1 activity and die prenatally [19;26;27]. Similarly, homozygous Tsc1 and Tsc2 conditional knock-out mice also show elevated levels of mTORC1 signaling and die prematurely [8;37;38;44]. Rapamycin treatment has been shown to reduce mTORC1 activity and enhance survival of these mice [13;39]. Tsc2flox/flox;CamKIIα-Cre mice (Tsc2CKO) express Cre recombinase under control of the neuron-specific CamKIIα kinase promoter. These animals begin to lose Tsc2 starting at postnatal day 18 and start to die prematurely at 5 weeks of age. We examined if oral glutamine could be used to decrease mTORC1 activity in the brain of Tsc2CKO mice and extend their life span. One-month old Tsc2CKO mice were administered 3 g/kg glutamine (n=5) or an equal volume of saline (n=5) via gavage twice a day (morning and afternoon), 5 days per week for 2 weeks. A group of control littermates was used to establish normal levels of pS6. Brain sections from Tsc2CKO mice treated with vehicle showed higher immunoreactivity in the hippocampus, particularly the dentate gyrus, when compared to littermate controls (Figure 2A top and middle panels). Consistent with our in vitro results, the brain sections from glutamine-treated Tsc2CKO mice showed reduced hippocampal phospho-S6 immunoreactivity (Figure 2A bottom panels). Quantification of the phospho-S6 fluorescent intensity revealed a significant increase in immunoreactivity within the dentate gyrus of Tsc2CKO mice as compared to control littermates, and this signal strength was normalized in animals that were fed glutamine (F(2,11) = 10.974, p = 0.002; Figure 2B). No significant difference in NeuN immunoreactivity was detected between any of the groups (F(2,11) = 0.175, n.s.), suggesting no overt neuronal loss between the groups.
Figure 2. Oral glutamine decreases hippocampal mTORC1 activity in Tsc2CKO mice.

A) Representative photomicrographs of the hippocampus from control, Tsc2CKO vehicle- and Tsc2CKO-glutamine-treated mouse brains immunostained with an antibody against phospho-S6 or the neuronal marker NeuN. In control mice (top panels), phospho-S6 immunoreactivity can be seen in the CA1 and CA3 subfields, with limited immunoreactivity in the dentate gyrus. Vehicle-treated Tsc2CKO mice show increased levels of phospho-S6 in the hippocampus compared to control littermates. This increase is most apparent in the CA1 and DG regions. B) Oral administration of glutamine (3 g/kg twice daily) significantly decreases phospho-S6 immunoreactivity in the DG. NeuN immunoreactivity did not change between the groups.
3.3. Daily oral glutamine administration increases survival of Tsc2CKO mice
As Tsc2CKO mice exhibit increased mTORC1 activity in the hippocampus and die prematurely, we questioned if oral glutamine could be used to inhibit mTORC1 activity and, similar to rapamycin treatment, prolong their lifespan. Tsc2CKO mice were treated with 3 g/kg oral glutamine or vehicle as described above (n=11 per group) until death was noted. Our results show that there was a statistical difference between the Kaplan-Meier survival curves of glutamine- and vehicle-treated animals (Figure 3, p = 0.044). It was observed that the time at which 50% of the Tsc2CKO animals die (LT50) was prolonged by approximately 2 weeks in the glutamine-treated group.
Figure 3. Oral glutamine prolongs the lifespan of Tsc2CKO mice.
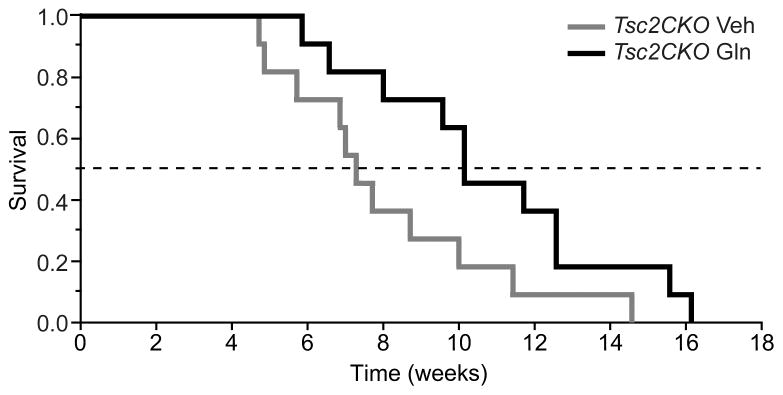
Kaplan-Meier survival curves (n=11/group) showing that oral glutamine administration (3 g/kg, twice daily) significantly increases the lifespan of Tsc2CKO mice. Data are presented as the mean ± s.e.m.
4. Discussion
Tuberous Sclerosis Complex (TSC) is a genetic disorder that results in over-activation of mTORC1 leading to significant neurologic morbidity and mortality. While rapalogues such as everolimus have shown very promising results in treating several of the manifestations of TSC, these drugs can have significant side effects. The recent finding that amino acids can modulate mTORC1 activity raises the intriguing question of whether they could also be used to treat some aspects of TSC. The results presented in this manuscript revealed three key findings on glutamine modulation of mTORK1 activity: 1) glutamine can inhibit mTORC1 signaling in cells grown in either serum-deprived or normal culture conditions, although relatively high concentrations are required to alter mTORC1 activity in non-deprived conditions, 2) oral administration of glutamine can be used to decrease mTORC1 activity in the brain, and 3) oral administration of glutamine to Tsc2CKO mutant mice prolongs the lifespan of these animals. The results presented in this study are the first to show that glutamine supplementation can decrease brain mTROC1 activity in Tsc2CKO mutant mice and suggest that this amino acid may be beneficial in the treatment of TSC.
The pathogenesis of TSC is thought to be caused by mutations of the normal allele leading to loss of heterozygosity (LOH). Although Tsc2-knockout mouse strains have been developed to mimic this phenomenon, homozygous deletion of Tsc2 is associated with embryonic lethality. To circumvent this problem, we generated CamKIIα-Cre-driven conditional knock-out mice [19]. These mice develop normally as the Tsc2 alleles are present during development, while the subsequent deletion of the Tsc2 gene during postnatal week 3 (when CamKIIα-Cre is starting to be expressed) results in high levels of mTORC1 activity and premature death. Glutamine was found to reduce mTORC1 activity and prolong the lifespan of these mutant mice. These findings suggest that glutamine supplementation may have therapeutic value in the treatment of not only TSC, but also diseases in which dysregulation of the mTORC1 pathway has been implicated, including cancer, Alzheimer’s disease, and Parkinson’s disease [7].
One limitation of the present study is that we are uncertain as to the precise mechanism through which glutamine inhibits mTORC1 activity. Using amino acid-deprived cell culture models, a few mechanisms have been proposed for amino acid regulation of mTORC1. For example, it has been proposed that high amino acid levels in the lysosome recruits mTORC1 to the lysosome where it can then be activated by Rheb [3]. In contrast, when amino acid levels are limiting, GATOR1 (a Rag-GAP) switches active Rag heterodimers into an inactive state. This results in mTORC1 being released from the lysosomal surface and away from Rheb. However, a study by Averous et al., reports that mTORC1 activity does not correlate with its lysosomal localization in serum fed-cells [2]. Other mechanisms for glutamine regulation of mTORC1 have been proposed, including increasing the influx of leucine via the heterodimeric co-transporter SLC7A5-SLC3A2 [34], increasing the levels of α-ketoglutarate [43], and decreasing AKT activity [12]. Further research will be required to identify the signaling pathway(s) responsible for the decrease in brain mTORC1 activity that we observed following oral glutamine administration.
Supplementary Material
Highlights.
Glutamine inhibits mTORC1 activity in serum-deprived and normal culture conditions
Oral administration of glutamine decreases mTORC1 activity in Tsc2CKO mice brains
Oral administration of glutamine to Tsc2CKO mice prolongs their life span
Glutamine may have therapeutic benefit in the treatment of tuberous sclerosis
Acknowledgments
This work was supported by grants (NS053588 and NS087149) from the NIH. We thank Dr. Julia Hill for her helpful comments.
Abbreviations
- mTORC
mechanistic Target of Rapamycin Complex
- Rheb
Ras-homologue enriched in brain
- S6
ribosomal protein S6
- S6K
ribosomal protein S6 kinase
- TS
Tuberous Sclerosis
- TSC
Tuberous Sclerosis Complex
Footnotes
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Adriaensen ME, Schaefer-Prokop CM, Stijnen T, Duyndam DA, Zonnenberg BA, Prokop M. Prevalence of subependymal giant cell tumors in patients with tuberous sclerosis and a review of the literature. Eur J Neurol. 2009;16:691–696. doi: 10.1111/j.1468-1331.2009.02567.x. [DOI] [PubMed] [Google Scholar]
- 2.Averous J, Lambert-Langlais S, Carraro V, Gourbeyre O, Parry L, B’Chir W, Muranishi Y, Jousse C, Bruhat A, Maurin AC, Proud CG, Fafournoux P. Requirement for lysosomal localization of mTOR for its activation differs between leucine and other amino acids. Cell Signal. 2014;26:1918–1927. doi: 10.1016/j.cellsig.2014.04.019. [DOI] [PubMed] [Google Scholar]
- 3.Bar-Peled L, Sabatini DM. Regulation of mTORC1 by amino acids. Trends Cell Biol. 2014;24:400–406. doi: 10.1016/j.tcb.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Betz C, Hall MN. Where is mTOR and what is it doing there? J Cell Biol. 2013;203:563–574. doi: 10.1083/jcb.201306041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan JA, Zhang H, Roberts PS, Jozwiak S, Wieslawa G, Lewin-Kowalik J, Kotulska K, Kwiatkowski DJ. Pathogenesis of tuberous sclerosis subependymal giant cell astrocytomas: biallelic inactivation of TSC1 or TSC2 leads to mTOR activation. J Neuropathol Exp Neurol. 2004;63:1236–1242. doi: 10.1093/jnen/63.12.1236. [DOI] [PubMed] [Google Scholar]
- 6.Chiu M, Tardito S, Barilli A, Bianchi MG, Dall’Asta V, Bussolati O. Glutamine stimulates mTORC1 independent of the cell content of essential amino acids. Amino Acids. 2012;43:2561–2567. doi: 10.1007/s00726-012-1312-0. [DOI] [PubMed] [Google Scholar]
- 7.Chong ZZ, Shang YC, Zhang L, Wang S, Maiese K. Mammalian target of rapamycin: hitting the bull’s-eye for neurological disorders. Oxid Med Cell Longev. 2010;3:374–391. doi: 10.4161/oxim.3.6.14787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crino PB. Evolving neurobiology of tuberous sclerosis complex. Acta Neuropathol. 2013;125:317–332. doi: 10.1007/s00401-013-1085-x. [DOI] [PubMed] [Google Scholar]
- 9.Curatolo P, Moavero R. mTOR Inhibitors in Tuberous Sclerosis Complex. Curr Neuropharmacol. 2012;10:404–415. doi: 10.2174/157015912804143595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Curran MP. Everolimus: in patients with subependymal giant cell astrocytoma associated with tuberous sclerosis complex. Paediatr Drugs. 2012;14:51–60. doi: 10.2165/11207730-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 11.Dabora SL, Jozwiak S, Franz DN, Roberts PS, Nieto A, Chung J, Choy YS, Reeve MP, Thiele E, Egelhoff JC, Kasprzyk-Obara J, Domanska-Pakiela D, Kwiatkowski DJ. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet. 2001;68:64–80. doi: 10.1086/316951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deldicque L, Sanchez CC, Horman S, De P, Bertrand IL, Hue L, Francaux M. Antagonistic effects of leucine and glutamine on the mTOR pathway in myogenic C2C12 cells. Amino Acids. 2008;35:147–155. doi: 10.1007/s00726-007-0607-z. [DOI] [PubMed] [Google Scholar]
- 13.Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, Ramesh V, Silva AJ. Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat Med. 2008;14:843–848. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feliciano DM, Lin TV, Hartman NW, Bartley CM, Kubera C, Hsieh L, Lafourcade C, O’Keefe RA, Bordey A. A circuitry and biochemical basis for tuberous sclerosis symptoms: from epilepsy to neurocognitive deficits. Int J Dev Neurosci. 2013;31:667–678. doi: 10.1016/j.ijdevneu.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gipson TT, Johnston MV. Plasticity and mTOR: towards restoration of impaired synaptic plasticity in mTOR-related neurogenetic disorders. Neural Plast. 2012:486402. doi: 10.1155/2012/486402. Epub;%2012 Apr 30 2012, 486402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han JM, Sahin M. TSC1/TSC2 signaling in the CNS. FEBS Lett. 2011;585:973–980. doi: 10.1016/j.febslet.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hasskarl J. Everolimus. Recent Results Cancer Res. 201:373–92. doi: 10.1007/978-3-642-54490-3_23.(2014)373-392. [DOI] [PubMed] [Google Scholar]
- 18.Henske EP, Wessner LL, Golden J, Scheithauer BW, Vortmeyer AO, Zhuang Z, Klein-Szanto AJ, Kwiatkowski DJ, Yeung RS. Loss of tuberin in both subependymal giant cell astrocytomas and angiomyolipomas supports a two-hit model for the pathogenesis of tuberous sclerosis tumors. Am J Pathol. 1997;151:1639–1647. [PMC free article] [PubMed] [Google Scholar]
- 19.Hernandez O, Way S, McKenna J, III, Gambello MJ. Generation of a conditional disruption of the Tsc2 gene. Genesis. 2007;45:101–106. doi: 10.1002/dvg.20271. [DOI] [PubMed] [Google Scholar]
- 20.Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412:179–190. doi: 10.1042/BJ20080281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang K, Fingar DC. Growing knowledge of the mTOR signaling network. Semin Cell Dev Biol. doi: 10.1016/j.semcdb.2014.09.011. %19 pii S1084–9521 2014 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones AC, Shyamsundar MM, Thomas MW, Maynard J, Idziaszczyk S, Tomkins S, Sampson JR, Cheadle JP. Comprehensive mutation analysis of TSC1 and TSC2-and phenotypic correlations in 150 families with tuberous sclerosis. Am J Hum Genet. 1999;64:1305–1315. doi: 10.1086/302381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Julich K, Sahin M. Mechanism-based treatment in tuberous sclerosis complex. Pediatr Neurol. 2014;50:290–296. doi: 10.1016/j.pediatrneurol.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kassai H, Sugaya Y, Noda S, Nakao K, Maeda T, Kano M, Aiba A. Selective activation of mTORC1 signaling recapitulates microcephaly, tuberous sclerosis, and neurodegenerative diseases. Cell Rep. 2014;7:1626–1639. doi: 10.1016/j.celrep.2014.04.048. [DOI] [PubMed] [Google Scholar]
- 26.Kobayashi T, Minowa O, Kuno J, Mitani H, Hino O, Noda T. Renal carcinogenesis, hepatic hemangiomatosis, and embryonic lethality caused by a germ-line Tsc2 mutation in mice. Cancer Res. 1999;59:1206–1211. [PubMed] [Google Scholar]
- 27.Kobayashi T, Minowa O, Sugitani Y, Takai S, Mitani H, Kobayashi E, Noda T, Hino O. A germ-line Tsc1 mutation causes tumor development and embryonic lethality that are similar, but not identical to, those caused by Tsc2 mutation in mice. Proc Natl Acad Sci USA. 2001;98:8762–8767. doi: 10.1073/pnas.151033798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krueger DA, Care MM, Agricola K, Tudor C, Mays M, Franz DN. Everolimus long-term safety and efficacy in subependymal giant cell astrocytoma. Neurology. 2013;80:574–580. doi: 10.1212/WNL.0b013e3182815428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122:3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. Rheb binds and regulates the mTOR kinase. Curr Biol. 2005;15:702–713. doi: 10.1016/j.cub.2005.02.053. [DOI] [PubMed] [Google Scholar]
- 32.Menon S, Dibble CC, Talbott G, Hoxhaj G, Valvezan AJ, Takahashi H, Cantley LC, Manning BD. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell. 2014;156:771–785. doi: 10.1016/j.cell.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakajo T, Yamatsuji T, Ban H, Shigemitsu K, Haisa M, Motoki T, Noma K, Nobuhisa T, Matsuoka J, Gunduz M, Yonezawa K, Tanaka N, Naomoto Y. Glutamine is a key regulator for amino acid-controlled cell growth through the mTOR signaling pathway in rat intestinal epithelial cells. Biochem Biophys Res Commun. 2005;326:174–180. doi: 10.1016/j.bbrc.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 34.Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, Myer VE, MacKeigan JP, Porter JA, Wang YK, Cantley LC, Finan PM, Murphy LO. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell. 2009;136:521–534. doi: 10.1016/j.cell.2008.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takei N, Nawa H. mTOR signaling and its roles in normal and abnormal brain development. Front Mol Neurosci. 7:28. doi: 10.3389/fnmol.2014.00028. eCollection;%2014. (2014) 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol. 2003;13:1259–1268. doi: 10.1016/s0960-9822(03)00506-2. [DOI] [PubMed] [Google Scholar]
- 37.Uhlmann EJ, Wong M, Baldwin RL, Bajenaru ML, Onda H, Kwiatkowski DJ, Yamada K, Gutmann DH. Astrocyte-specific TSC1 conditional knockout mice exhibit abnormal neuronal organization and seizures. Ann Neurol. 2002;52:285–296. doi: 10.1002/ana.10283. [DOI] [PubMed] [Google Scholar]
- 38.Way SW, McKenna J, III, Mietzsch U, Reith RM, Wu HC, Gambello MJ. Loss of Tsc2 in radial glia models the brain pathology of tuberous sclerosis complex in the mouse. Hum Mol Genet. 2009;18:1252–1265. doi: 10.1093/hmg/ddp025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Way SW, Rozas NS, Wu HC, McKenna J, III, Reith RM, Hashmi SS, Dash PK, Gambello MJ. The differential effects of prenatal and/or postnatal rapamycin on neurodevelopmental defects and cognition in a neuroglial mouse model of tuberous sclerosis complex. Hum Mol Genet. 2012;21:3226–3236. doi: 10.1093/hmg/dds156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Willems L, Jacque N, Jacquel A, Neveux N, Maciel TT, Lambert M, Schmitt A, Poulain L, Green AS, Uzunov M, Kosmider O, Radford-Weiss I, Moura IC, Auberger P, Ifrah N, Bardet V, Chapuis N, Lacombe C, Mayeux P, Tamburini J, Bouscary D. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood. 2013;122:3521–3532. doi: 10.1182/blood-2013-03-493163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wong M. A critical review of mTOR inhibitors and epilepsy: from basic science to clinical trials. Expert Rev Neurother. 2013;13:657–669. doi: 10.1586/ern.13.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wong M. Mammalian target of rapamycin (mTOR) pathways in neurological diseases. Biomed J. 2013;36:40–50. doi: 10.4103/2319-4170.110365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yao K, Yin Y, Li X, Xi P, Wang J, Lei J, Hou Y, Wu G. Alpha-ketoglutarate inhibits glutamine degradation and enhances protein synthesis in intestinal porcine epithelial cells. Amino Acids. 2012;42:2491–2500. doi: 10.1007/s00726-011-1060-6. [DOI] [PubMed] [Google Scholar]
- 44.Zeng LH, Rensing NR, Zhang B, Gutmann DH, Gambello MJ, Wong M. Tsc2 gene inactivation causes a more severe epilepsy phenotype than Tsc1 inactivation in a mouse model of tuberous sclerosis complex. Hum Mol Genet. 2011;20:445–454. doi: 10.1093/hmg/ddq491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011;334:678–683. doi: 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.