

Significance
Alzheimer’s disease (AD) is a growing public health problem, in part because there are no effective therapies. Major roadblocks to the treatment of AD are the lack of adequate diagnostic tools and the absence of viable therapeutic targets. It is now clear that AD is characterized by inflammation. Our results indicate that AD patients and mouse models have increased activation of a set of proteins known as the contact activation system in their circulation. We also demonstrate that the AD-related peptide Aβ can initiate activation of this system in the circulation of animal models. Because the contact activation system contributes to inflammation, our results suggest new pathogenic mechanisms, diagnostic tests, and therapeutic targets for AD.
Keywords: Alzheimer’s disease, factor XII, high molecular-weight kininogen, plasma kallikrein
Abstract
Alzheimer’s disease (AD) is characterized by accumulation of the β-amyloid peptide (Aβ), which likely contributes to disease via multiple mechanisms. Increasing evidence implicates inflammation in AD, the origins of which are not completely understood. We investigated whether circulating Aβ could initiate inflammation in AD via the plasma contact activation system. This proteolytic cascade is triggered by the activation of the plasma protein factor XII (FXII) and leads to kallikrein-mediated cleavage of high molecular-weight kininogen (HK) and release of proinflammatory bradykinin. Aβ has been shown to promote FXII-dependent cleavage of HK in vitro. In addition, increased cleavage of HK has been found in the cerebrospinal fluid of patients with AD. Here, we show increased activation of FXII, kallikrein activity, and HK cleavage in AD patient plasma. Increased contact system activation is also observed in AD mouse model plasma and in plasma from wild-type mice i.v. injected with Aβ42. Our results demonstrate that Aβ42-mediated contact system activation can occur in the AD circulation and suggest new pathogenic mechanisms, diagnostic tests, and therapies for AD.
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder with a complex and still poorly defined etiology. Although multiple factors are likely involved in AD onset and development, a growing body of evidence implicates both neuroinflammation and peripheral inflammation in the disease (1–3). Pathways capable of triggering inflammatory processes are therefore of particular interest to AD etiology and pathogenesis. One such pathway is the contact activation system, which is initiated when the plasma protein factor XII (FXII) is exposed to negatively charged surfaces (contact activation). Contact-activated FXII (FXIIa) triggers plasma kallikrein-mediated cleavage of high molecular-weight kininogen (HK) to release bradykinin, which promotes inflammatory processes including increased blood–brain barrier permeability, edema, and cytokine expression (4) via interaction with receptors B1 and B2 (5). In AD, a possible surface for FXII activation could be the AD-associated peptide beta-amyloid (Aβ), which has been shown to stimulate FXII-dependent plasma kallikrein activity (6, 7) and kallikrein-mediated HK cleavage (6, 8) in vitro.
Although the contact activation system is primarily thought to function in the circulation, there is evidence for its dysregulation in AD brain tissue: FXII is found in Aβ plaques (9), increased plasma kallikrein activity is observed in the AD brain parenchyma (10), and elevated levels of cleaved HK are found in cerebrospinal fluid (CSF) of patients with AD (11). To our knowledge, FXII activation and HK cleavage in the periphery of AD patients have not been demonstrated.
Here, we show increased levels of FXIIa, HK cleavage, and kallikrein activity in the plasma of AD patients compared with nondemented (ND) control plasma. Furthermore, plasma HK cleavage is increased in a mouse model of AD and in wild-type mice i.v. injected with Aβ42, supporting a role for Aβ42 in AD-associated activation of the contact system. Activation of the contact system and associated bradykinin release in the AD circulation could contribute to the inflammatory and vascular dysfunction observed in the disease (3, 12). Plasma HK cleavage may also be a useful, minimally invasive biomarker for identifying AD patients who could benefit from therapeutic strategies directed against FXII.
Results
AD and Control Subject Characteristics.
Two sets of plasma samples from AD patients and ND control individuals were obtained from two AD tissue banks (see Methods). Group 1 consisted of 18 AD and 11 ND samples matched with respect to age and sex (Table S1), and group 2 consisted of 10 AD and 10 ND samples matched with respect to age (Table S2). The extent of dementia in these individuals had been reported using Clinical Dementia Rating (CDR) scores, where 0 = no dementia and 3 = severe dementia (13), as well as Mini Mental State Examination (MMSE) scores, where 30 = no dementia and 0 = severe dementia (14). At the time of blood draw, AD cases in group 1 had an average CDR score of 1.6 ± 1.3 (range, 0–3) and an average MMSE score of 16.5 ± 9.6 (range, 0–30), corresponding to moderate dementia. The presence of CDR 0 and MMSE 30 individuals in this group can be attributed to the fact that several patients (n = 3) were diagnosed with mild cognitive impairment or AD after blood draw. On autopsy, the majority of AD cases in group 1 (77.8%) were Braak stage 5 or 6, corresponding to severe dementia (Table S1) (15). AD cases in group 2 had an average CDR score of 1.0 ± 0.6 at the time of blood draw, with the majority (80%) being CDR 0.5 or 1 (Table S2), corresponding to very mild to mild dementia (13). Group 2 individuals are still living, and therefore Braak stages are not available.
Levels of Cleaved FXII and HK Are Increased in AD Plasma from Group 1.
Plasma was analyzed under reducing conditions, with results reported after normalization to transferrin loading control, plasma levels of which are not altered in AD (16). Cleavage of the FXII zymogen (decreased intensity at 80 kDa) and the corresponding appearance of a FXIIa heavy chain band at 52 kDa were detected in 13 of 18 AD plasmas and 2 of 11 ND plasmas (Fig. 1A). Levels of the 52-kDa heavy chain fragment, the generation of which typically corresponds to FXII activation, were higher in AD than in ND cases (0.47 vs. 0.07; P = 0.029; Fig. 1B). Activation of FXII may occur in conjunction with comorbidities present in AD patients that are absent in controls. However, records of self- or caregiver-reported medical conditions indicate that AD cases did not have higher levels of hypertension, hypercholesterolemia, diabetes, myocardial infarction, or atrial fibrillation than controls (Table S1), arguing against a role for these comorbidities in the increased FXIIa levels observed in AD patient plasma. Interestingly, history of stroke was found in 5 of 18 (almost 30%) of AD cases but was absent in ND cases. Stroke is mediated by thrombosis and/or vessel rupture, both of which generate surfaces for FXII activation such as polyphosphates (17) and RNA (18). However, excluding AD cases with history of stroke did not substantially change the FXIIa levels in AD and ND groups (0.47 vs. 0.07, respectively; P = 0.018; Fig. 1B).
Fig. 1.

Activation of the FXIIa-driven contact system in AD patient plasma from group 1. (A) Western blot analysis of FXIIa and transferrin loading control in plasma of 18 AD patients and 11 ND controls from group 1 (representative samples shown), showing FXII zymogen (80 kDa) and the FXIIa heavy chain (52 kDa). Lane loaded with FXII-deficient human plasma (FXII-def) shows that the bands just below and above the FXIIa band are nonspecific. (B) FXIIa levels normalized to transferrin were significantly higher in AD (P = 0.029) than ND plasma. When AD cases with a history of stroke (n = 5) were excluded from the analysis (AD SE), FXIIa levels remained significantly higher than in ND plasma (P = 0.018). (C) Western blot analysis of HK in representative samples showing intact HK (120 kDa), HK light chain (56 kDa), and light chain fragment (45 kDa). Lane labeled “HK” is loaded with purified HK for positive control. (D) Intact HK levels normalized to transferrin were significantly lower in AD (P < 0.0001) than ND plasma. When AD cases with a history of stroke (n = 5) were excluded from the analysis, intact HK levels in AD SE remained significantly higher than in ND plasma (P = 0.0002). Red points in AD and AD SE groups represent individuals who developed cognitive decline at least 1 y after blood draw. (E) Kallikrein-like activity was higher in AD plasma compared with ND (P = 0.0006). (F) Kallikrein-like activity was inversely correlated to intact HK levels (r = −0.81; P < 0.0001). (G) HK levels normalized to transferrin were higher in both individuals with CERAD score B (P = 0.003) and those with CERAD score C (P < 0.0001) than in individuals with CERAD score 0. Samples were analyzed three separate times, with similar results. Results are presented as vertical scatter plots with medians, with statistical significance determined using the Mann–Whitney test for two-group comparisons and the Kruskal-Wallis test with Dunn’s Multiple Comparison posttest for comparisons between multiple groups.
The presence of FXIIa in plasma (Fig. 1A) was accompanied by HK cleavage, seen as diminished signal intensity of intact HK bands at 120 kDa (Fig. 1C). AD plasma as a group had much lower levels of intact HK than ND plasma (0.01 vs. 1.19; P < 0.0001; Fig. 1D), even when AD cases with history of stroke were excluded (0.01 vs. 1.19; P = 0.0002; Fig. 1D). Decreases in intact (single-chain) HK levels were accompanied by the appearance of cleaved HK fragments: the cleaved HK light chain band migrating at 56 kDa and an additional 45-kDa band representing a degradation product of 56-kDa cleaved HK light chain. Because HK cleavage products are rapidly degraded (19), samples with high levels of HK cleavage did not necessarily have proportionally higher levels of HK light chain, making quantification of the HK breakdown products uninformative. Interestingly, some samples (e.g., AD1 and AD2) that did not have detectable FXIIa showed evidence of HK cleavage, demonstrating that HK cleavage is a more sensitive indicator of contact activation than FXII activation.
To further examine whether the reduction in intact HK levels observed by Western blot is a result of contact system activation, we measured the activity of plasma kallikrein, the enzyme responsible for HK cleavage. Kallikrein-like activity, measured by chromogenic substrate assay, was higher in AD plasma compared with ND plasma [40.9% vs. 4.5% of dextran sulfate 500 kDa (DXS)-activated plasma; P = 0.0006; Fig. 1E] and correlated with HK cleavage (r = −0.81; P < 0.0001; Fig. 1F). Because kallikrein generation is triggered by FXIIa, this result also indicates that FXII cleavage detected by Western blot represents FXII activation.
When levels of intact HK were plotted as a function of Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) score [a measure of Aβ plaque pathology in the brain on autopsy; 0 = normal brain; B = probable AD; C = definite AD (20)], plasma from individuals with a CERAD score of 0 had higher levels of intact HK compared with plasma from individuals with a CERAD score of B (1.19 vs. 0.01; P < 0.05) or C (1.19 vs. 0.01; P < 0.001) (Fig. 1G). The fact that HK cleavage is apparent in the plasma of individuals with CERAD B and does not increase further in those with CERAD C suggests that FXIIa-driven inflammation begins early in disease progression and is mostly developed by the time individuals reach CERAD B status. The idea that HK cleavage is an early event in AD is also suggested by its presence in plasma from three individuals who were cognitively normal at time of blood draw but went on to develop cognitive impairment (red points in Fig. 1D). Indeed, the presence of preclinical AD may explain the FXII activation and HK cleavage observed in two ND samples (Fig. 1 B and D).
Levels of Cleaved HK Are Increased in AD Plasma from Group 2.
We next tested plasma samples from a different tissue bank and from patients with AD at earlier clinical stages of disease (on average). Although FXIIa was not detected by Western blot in AD or ND plasma from group 2 (Fig. S1), intact HK levels were lower in AD than ND (0.59 vs. 0.85; P < 0.0001; Fig. 2 A and B), as seen in group 1. Kallikrein-like activity was also increased in AD plasma (1.2% vs. 0.96% of DXS-activated plasma; P = 0.03; Fig. 2C), corroborating the decreased levels of intact HK seen by Western blot.
Fig. 2.

Activation of the FXIIa-driven contact system in AD patient plasma from group 2. (A) Western blot analysis of HK and transferrin loading control in plasma of 10 AD patients and 10 ND controls from group 2. (B) Intact HK levels normalized to transferrin were significantly lower in AD (P < 0.0001) compared with ND plasma. (C) Kallikrein-like activity was higher in AD plasma compared with ND (P = 0.03). (D) Levels of CSF Aβ42, which decrease with AD, were positively correlated with intact HK levels (r = 0.63; P = 0.003). (E) Levels of CSF tau, which increase with AD, were not correlated with intact HK levels (r = −0.11; P = 0.64). (F) Levels of CSF p-tau, which increase with AD, were not correlated with intact HK levels (r = −0.25; P = 0.28). Samples were analyzed three separate times with similar results. Results are presented as vertical scatter plots with medians, with statistical significance determined using the Mann–Whitney test.
Levels of CSF Aβ42, total tau (tau), and phosphorylated tau (p-tau) in group 2 were available from the Washington University Alzheimer’s Disease Research Center. Decreased CSF Aβ42 levels are thought to be the earliest CSF marker of incipient AD, appearing as early as ∼15 y before onset of symptoms and remaining low as disease progresses, whereas CSF tau and p-tau levels (related to neurofibrillary tangle formation and neurodegeneration) begin to rise closer to the appearance of cognitive decline (21). We therefore performed correlation analyses for these CSF biomarkers and intact HK levels. Levels of intact plasma HK were positively correlated with CSF Aβ42 (r = 0.63; P = 0.003; Fig. 2D), consistent with increased HK cleavage in the plasma of individuals with lower CSF Aβ42. Intact plasma HK did not correlate with CSF tau (r = −0.11; P = 0.64; Fig. 2E) or p-tau (r = −0.25; P = 0.28; Fig. 2F), suggesting that increased HK cleavage may be an early event in AD progression that precedes substantial changes to neuronal injury markers.
Blood Draw Variables and Contact System Activation.
Undetectable FXIIa and less dramatic HK cleavage in group 2 than group 1 may stem from the earlier disease stage of group 2 cases (CDR score: group 2, 1.0 ± 0.6 vs. group 1, 1.6 ± 1.3) and from differences in blood collection. Although we show that the degree of contact system activation may increase with disease progression (Fig. 2D), the method of blood collection appears to also play a role, since ND plasma in group 2 had lower kallikrein activity than ND plasma in group 1 (Figs. 1E and 2C).
In group 1, blood was drawn into heparinized Vacutainer tubes via vacuum, whereas group 2 blood was drawn into EDTA-coated syringes via aspiration. Heparin can promote contact system activation in a purified protein system (22) and in plasma diluted to 30% (23), but not in plasma diluted to 90% (24), arguing against heparin-mediated activation of undiluted blood during collection. In contrast, EDTA is a Zn++ chelator and may therefore prevent ongoing contact activation after blood collection, resulting in the detection of less FXIIa and intact HK in group 2. To test these possibilities, we evaluated the activation potential of blood from wild-type C57BL/6 mice collected into heparin or EDTA. Both EDTA- and heparin-anticoagulated plasma treated with the FXII activator DXS had comparably decreased levels of intact HK (Fig. 3), indicating that heparin-mediated promotion or EDTA-mediated inhibition of ex vivo FXII activation cannot explain the differences between groups 1 and 2. However, the possibility remains that long-term storage of frozen plasma with EDTA vs. heparin could lead to differences in ex vivo contact activation.
Fig. 3.
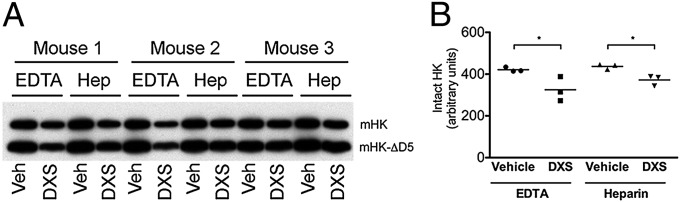
Blood draw into EDTA or heparin (hep) tubes does not affect ex vivo contact activation. (A) Western blot analysis of WT mouse plasma with an antibody against murine HK (mHK) light chain. Mouse plasma contains mHK (110 kDa) and mHK-∆D5 (mHK lacking domain 5; 82 kDa) (52). (B) Intact mHK (sum of mHK and mHK-ΔD5 bands) is decreased in samples activated with DXS compared with vehicle in both EDTA and heparin conditions (P < 0.05).
Another possible explanation is that plasma collected into Vacutainer tubes (with vacuum) has increased FXII-driven thrombin generation (25) and earlier clot formation (26) compared with blood drawn into S-Monovette tubes, which use a syringe-like mechanism. Thus, blood draw methods used for group 1 may have resulted in ex vivo amplification of smaller differences in contact factor activation present in vivo, which did not occur in group 2.
Levels of Cleaved HK Are Increased in Plasma from a Mouse Model of AD and in Plasma from Wild-Type Mice Injected with Aβ42.
AD patients are a heterogeneous population with various disease etiologies and comorbidities. To analyze FXII activation and HK cleavage in a more homogeneous model, we examined plasma from the Tg6799 mouse model of AD, in which AD pathology is driven by the overexpression of human Aβ (27) (hereafter referred to as AD mice). Because detection of FXIIa by Western blot in mouse plasma is technically challenging given the poor ability of antibodies to recognize murine FXIIa fragments (Fig. S2), we focused on HK cleavage as a marker of contact system activation. Compared with nontransgenic wild-type littermate controls (WT), AD mice had decreased levels of intact HK (2.0 vs. 1.6; P = 0.0012; Fig. 4 A and B). These data corroborate HK cleavage in human AD patient plasma and also support the idea that the increased HK cleavage in plasma from patients with AD is related to Aβ-driven AD pathology, and not to comorbidities present in AD patients.
Fig. 4.

Activation of the FXIIa-driven contact system in plasma from an AD mouse model and from wild-type mice injected with Aβ42. (A) Plasma from AD mice (n = 7) and littermate controls (WT; n = 7) was analyzed by Western blot with an antibody against mHK light chain. Representative blot showing HK from one AD and one WT mouse (full blot in Fig. S4). (B) The levels of intact mHK (sum of mHK and mHK-∆D5 bands) normalized to transferrin were lower in AD than WT mice (P = 0.0012). (C) Representative transmission electron microscopy images of Aβ42, scrambled (scr) Aβ42, and amylin used for injections. (Scale bar, 100 nm.) (D) Representative blot probed with an antibody against mHK light chain showing C57BL/6 mice injected with vehicle, Aβ42, Aβ42 scr, or amylin (full blot in Fig. S5). (E) The levels of intact HK (sum of mHK and mHK-∆D5 bands) normalized to transferrin were significantly lower (P < 0.001) in mice injected with Aβ42 than in vehicle-, Aβ42 scr-, or amylin-injected mice. (F) Kallikrein activity as measured by chromogenic substrate was increased in plasma from mice injected with Aβ42, but not Aβ42 scr or amylin, compared with plasma from mice injected with vehicle (P < 0.001). (G) Plasma Aβ42 levels in C57BL/6 mice injected with Aβ42 were significantly higher than in mice injected with vehicle (P = 0.001). In mice injected with vehicle, levels of Aβ42 were below the detection limit of the ELISA (0.0156 ng/mL). (H) The dose of Aβ42 (0, 3, 6, or 9 mg/kg Aβ42) injected into WT mice (n = 4 per dose) correlated with kallikrein activity levels in plasma (r = 0.92, P < 0.0001). (I) Kallikrein activity was increased in WT (n = 4 per group; P < 0.01), but not in FXII−/− mice injected with 6 mg/kg Aβ42 (n = 6 per group). Results are presented as vertical scatter plots with medians for B, G, and I and with means for E and F, with statistical significance determined using the Mann–Whitney test for B and G, ANOVA with Dunnett’s posttest for E and F, and Kruskal-Wallis with Dunn’s posttest for I.
We next investigated whether the increased HK cleavage found in plasma from AD patients and mouse models could be mediated by Aβ42, an aggregation-prone, negatively charged peptide. To control for nonspecific effects, scrambled Aβ42 as well as amylin, another aggregating peptide, were used. Peptide preparations were shown by transmission electron microscopy to be composed of similar-sized structures (Fig. 4C). Incubation of Aβ42 but not scrambled Aβ42, amylin, or vehicle with WT (C57BL/6) mouse plasma resulted in HK cleavage ex vivo (Fig. S3), confirming results obtained with human plasma (6, 8). C57BL/6 mice were then i.v. injected with the same peptides or vehicle. Plasma from mice injected with Aβ42 but not scrambled Aβ42 or amylin had decreased levels of intact HK (2.8 ± 0.8 vs. 4.5 ± 1.0; P < 0.01; Fig. 4 D and E) and increased kallikrein-like activity compared to plasma from mice injected with vehicle (73.0 ± 38.2% vs. 4.9 ±1.9% of DXS-activated plasma; P < 0.001; Fig. 4F). The presence of Aβ42 in the plasma of injected mice was confirmed by ELISA (17.1 ± 9.0 ng/mL in Aβ42-injected mice compared with undetectable levels in vehicle-injected mice; lower limit of detection = 0.0156 ng/mL; Fig. 4G). Aβ42-mediated activation of the contact system in vivo, as determined by kallikrein activity in plasma, was both dose-dependent (r = 0.92; P < 0.0001; Fig. 4H) and FXII-dependent, since injection of Aβ42 into FXII−/− mice did not result in increased kallikrein activity (Fig. 4I). Our combined results indicate that circulating Aβ42 functions as a FXII contact activator capable of triggering kallikrein activity and HK cleavage in vivo and support the hypothesis that increased HK cleavage in AD patient and mouse model plasma is a result of Aβ42-mediated FXII activation.
Discussion
It is clear that AD is a complex, multifactorial disease, and it is likely that multiple disease mechanisms contribute to pathology. Here, we demonstrate increased activation of the contact system (as determined by FXII cleavage, kallikrein activity, and HK cleavage) in AD patient plasma, which represents a new potential mechanism of inflammatory pathology in AD. Although our study examines a relatively small number of samples, we demonstrated increased contact system activation in samples from two separate tissue banks, which provides the basis for further investigation of the role of the FXII-driven contact activation system in AD.
We also provide evidence that this phenomenon is related to AD-specific mechanisms, and not to comorbidities found in patients with AD, since mouse models of AD (in which pathology is driven by overexpression of Aβ) also had increased plasma contact system activation. Although it is possible that AD-related pathological changes downstream of Aβ42, and not Aβ42 itself, contribute to FXII activation in plasma, our results with i.v. injection of Aβ42 support the interpretation that contact system activation in AD is mediated by circulating Aβ42 in a FXII-dependent manner. In the Tg6799 mouse model of AD used in this study, levels of plasma Aβ42 are ∼2 ng/mL (28) compared with an absence of Aβ42 in littermate controls. These levels are only eightfold lower than the concentration of Aβ42 measured in WT mice i.v. injected with Aβ42 that exhibited plasma contact activation. The fact that scrambled Aβ42 had no effect on contact system activation suggests that a combination of total negative charge and density of negative charge play a role in Aβ42-mediated FXII activation.
If increased contact system activation in AD patient plasma is a result of the interaction between FXII and Aβ42, levels of plasma Aβ42 would be expected to be elevated before AD onset and during AD development. Although studies of high-risk populations for AD (familial AD patients, family members of AD patients, and Down syndrome individuals) demonstrate increased plasma Aβ42 concentrations compared with controls (29), the relationship between circulating Aβ42 levels and sporadic AD progression is less clear: cross-sectional studies of plasma Aβ42 have produced inconsistent results, whereas longitudinal studies have generally found higher Aβ42 levels before and just at the onset of AD symptoms with a subsequent decrease (29, 30). This suggests that increases in plasma Aβ42 could be an early event in AD development and supports Aβ42-mediated FXII activation as an early pathological process preceding the onset of symptoms. This is reflected in our results showing a positive correlation between the early disease biomarker CSF Aβ42 and HK cleavage. Although elevated levels of Aβ42 before AD onset would directly facilitate contact system activation, its levels in more advanced disease stages do not necessarily need to be elevated, since changes to the vasculature that precede and accompany AD (31) may create an environment that allows Aβ42 levels that cannot activate FXII in normal individuals to produce a pathological effect.
HK cleavage in AD patient plasma was not detected previously (11). The inclusion of polybrene in blood collection tubes and the younger age of AD patients in that study may explain the discrepancy between our results. The elevated HK cleavage and kallikrein activity observed in AD patient plasma in our study need to be validated in a larger cohort that includes individuals with familial and sporadic AD, as well as individuals with other neurodegenerative diseases. Furthermore, blood collection protocols will need to be optimized to take advantage of the potential amplification of in vivo contact system activation observed in plasma from group 1, and standardized to minimize differences in plasma activation during collection and storage. Most likely, HK cleavage would need to be used in conjunction with other disease markers to serve as a diagnostic tool for AD, since FXII activation and HK cleavage may also be features of unrelated disease states. Increased levels of FXIIa have been found in chronic conditions such as systemic amyloidosis (7) and hyperlipidemia (32), as well as after acute conditions such as ischemic stroke (33) and myocardial infarction (34). Increased cleavage of HK has been observed in chronic conditions such as systemic lupus erythematosus (35) and certain cancers (36), and during acute attacks in hereditary angioedema (37). Evaluation of larger populations would help determine appropriate cutoff values for FXII activation and HK cleavage that may differentiate between AD and other disease states.
We propose that chronic activation of the FXIIa-driven contact system by Aβ42 in the AD circulation can result in constant low levels of bradykinin-mediated inflammatory processes. These include increased blood–brain barrier permeability, increased extravasation of plasma proteins in the brain parenchyma, and cytokine up-regulation, all processes that are known to occur in AD (38, 39). More importantly (and given that plasma Aβ42 levels are elevated before the appearance of clinical symptoms), these events could precipitate AD onset and lead to the pathological changes associated with AD. For example, extravasation of plasma proteins into the brain parenchyma causes glial activation (40, 41), and the accompanying edema can cause narrowing of vessels, resulting in cerebral blood flow (CBF) disturbances (42). Glial activation (43, 44) and CBF disruption (45, 46) are early events in AD development that can directly contribute to neuronal dysfunction (12, 38, 47).
Monitoring the levels of plasma FXII/HK cleavage could be used as a diagnostic tool to help identify AD patients with this biochemical signature. If FXII activation is found to contribute to disease progression, identification of these AD patients may pave the way for new therapies aimed at neutralizing FXIIa, such as anti-FXIIa antibodies that specifically block FXIIa activity and interfere with FXIIa-driven bradykinin formation in plasma (48).
Methods
Human Plasma Samples.
Experiments with human plasma were approved by the Rockefeller Institutional Review Board. Plasma from AD patients and ND controls was obtained from the University of Kentucky Sanders-Brown Center on Aging (group 1) and Washington University Knight Alzheimer’s Disease Research Center (ADRC) (group 2). For group 1, blood from participants giving written, informed consent was drawn into heparinized plastic Vacutainer tubes with a 23- or 21-gauge needle. AD cases were defined by a clinical diagnosis of AD, as well as a postmortem CERAD neuritic plaque score (20) of B or C, corresponding to probable or definite AD, respectively. ND cases had CERAD scores of 0 and no clinical diagnosis of AD. AD and ND cases were sex- and age-matched (Table S1). For group 2, blood was drawn, using EDTA-coated syringes, into polypropylene tubes containing a final concentration of 5 mM EDTA. Plasma was prepared by centrifuging blood at 2,000 × g for 15 min and flash frozen on dry ice before storage at −80 °C. Group 2 AD cases were defined by a CDR score (13) ≥0.5 and CSF Aβ42 levels <500 pg/mL, and ND cases were defined by a CDR score of 0 and CSF Aβ42 levels >500 pg/mL (Table S2). CSF Aβ42 cutoff values for AD vs. ND were based on correlations between CSF Aβ42 levels and cortical amyloid load, as assessed by positron emission tomography with Pittsburgh Compound B (49).
Analysis of Contact System Activation in Human Plasma by Western Blot.
Total plasma protein concentration was measured by bicinchoninic acid assay (BCA), and equal amounts of total protein from each sample were analyzed by reducing Western blot with monoclonal antibodies against FXII heavy chain (Haemotologic Technologies), HK light chain (Abcam), and transferrin (Abcam), in that order. Blots were stripped between antibody incubations and developed using Enhanced Chemoluminescent substrate (Perkin-Elmer). Protein levels were quantified using densitometric analysis with ImageJ (NIH).
Measurement of Kallikrein-like Activity in Human and Mouse Plasma.
Plasma kallikrein-like activity was measured using the chromogenic substrate S-2302 (Chromogenix), based on the method described in ref. 50, with some modifications. Plasma (diluted 1:30) in 20 mM Hepes with 140 mM NaCl was mixed with S-2302 (0.67 mM final concentration) in duplicate, and absorbance at 405 nm was read for 30 min at room temperature in a Molecular Devices Spectramax 384 Plus spectrophotometer. Rate of substrate conversion over time was calculated for each plasma sample by the data acquisition software (Softmax 6.1) and expressed as a percentage of the rate found for normal human or wild-type mouse plasma activated with DXS (Sigma). S-2302 can also be cleaved by FXIIa, FXIa, and plasmin. To determine whether S-2302 cleavage was mediated by members of the contact activation pathway (kallikrein, FXIIa, or FXIa) or plasmin, plasmin activity in all samples was determined, using a different substrate (Pefachrome-5329; Pentapharm), and found not to differ between AD and ND (Fig. S6).
Mouse Lines.
The Tg6799 mouse model of AD (Jackson) was used, which is double transgenic for APP/Presenilin 1 and expresses five familial AD mutations: three in APP (K670N/M671L, Swedish; I716V, Florida; V717I, London) and two in Presenilin 1 (M146L, L286V) under the mouse thy1 promoter. Tg6799 mice develop amyloid plaques at 2 mo of age and cognitive impairment by 4–5 mo of age (27). Nontransgenic littermates were used as controls. For Aβ42 injection experiments, 2-mo-old C57BL/6 mice (Jackson) and FXII−/− mice backcrossed to C57BL/6 mice for >10 generations (51) were used.
Analysis of Contact System Activation in AD and Wild-Type Mouse Plasma.
All animal experiments were conducted in accordance with the guidelines of the US National Institutes of Health Guide for the Care and Use of Laboratory Animals and with approval from the Animal Care and Use Committee of The Rockefeller University. Tg6799 mice (n = 7) or littermate control mice (WT; n = 7) at 6 mo of age were anesthetized with atropine (500 mg/kg body weight) and avertin (0.04 mg/kg body weight), intraperitoneally. Blood (100 µL) was collected via retroorbital bleeding through gel-repel-coated (Sigma) and polybrene-coated (Santa Cruz) capillaries into EDTA-coated tubes (BD) containing 5 mM EDTA. Plasma was prepared by centrifugation (1,500 × g for 15 min, twice) and stored in polypropylene tubes containing 5 mM EDTA. Total protein concentration was determined by BCA, and plasma from each mouse containing 20 μg total protein was analyzed by Western blot, using monoclonal antibodies against HK light chain (R&D) and transferrin (Abcam), in that order. Blots were stripped between antibody incubations. Protein levels were quantified by densitometric analysis.
For experiments investigating the effect of different anticoagulants on ex vivo contact system activation, blood from C57BL/6 mice (n = 3) was collected into EDTA-coated tubes (as described above) or heparin-coated tubes (Sarstedt). Plasma was prepared as described above and stored in polypropylene tubes containing an additional 5 mM EDTA (final concentration) or 10 U/mL heparin (final concentration), respectively. Plasma was activated with DXS, 0.1 μg/mL final concentration, or vehicle for 20 min at 37 °C; reactions were stopped with reducing sample buffer and analyzed by Western blot.
For in vitro and i.v. injection experiments, Aβ42, scrambled Aβ42, and amylin (Anaspec) were prepared as follows: peptides were resuspended in a minimum amount of 1% NH4OH and then diluted to 1 mg/mL with PBS. Peptide concentration was determined by BCA, and the state of aggregation was determined by transmission electron microscopy (see following). Plasma from C57BL/6 mice collected into EDTA, as described above, was incubated with 20 μM Aβ42, scrambled Aβ42, amylin, or vehicle for 1 h at 37 °C and analyzed by Western blot. Aβ42 (3, 6, or 9 mg/kg body weight), scrambled Aβ42 (6 mg/kg), amylin (6 mg/kg), or vehicle were administered via retroorbital injection into 2-mo-old C57BL/6 or FXII−/− mice under anesthesia, using avertin and atropine as described above. After 6 h, blood was collected and processed as described above. Levels of plasma Aβ42 were determined at 6 h postinjection, using an Aβ42-specific ELISA kit (Life Technologies) according to the manufacturer’s instructions.
Transmission Electron Microscopy.
Samples were diluted to 0.1 mg/mL, applied to glow-discharged CF200-Cu grids (Electron Microscopy Sciences), washed three times with ultrapure water, and negatively stained with 2% uranyl acetate. Images were acquired using a JEOL JEM 100CX Transmission Microscope at The Rockefeller University Electron Microscopy Resource Center.
Statistical Analysis.
Comparisons between groups were performed using the two-tailed Mann–Whitney test for nonparametric data. Comparisons between multiple groups were performed using Kruskal-Wallis test, followed by Dunn’s Multiple Comparison Test. Comparisons of kallikrein-like activity between groups were performed using the one-tailed Mann–Whitney test. For i.v. injection experiments, comparisons between groups were performed using ANOVA, followed by Dunnett’s Multiple Comparison Test for multiple groups with a single control. Data are presented as vertical scatter plots with medians and reported as medians or means ± SD in the text. Correlations between CSF biomarker measures, kallikrein-like activity, and Western blot results were examined using Pearson’s correlation coefficient (r). P values ≤ 0.05 were considered significant (*), with values ≤ 0.01 designated **, values ≤ 0.001 designated ***, and values ≤ 0.0001 designated ****. Analyses were performed using GraphPad Prism 5 software.
Supplementary Material
Acknowledgments
We are grateful to P. Nelson and E. Abner from the University of Kentucky Sanders-Brown Center on Aging (NIH P30 AG028383) and A. Fagan from the Washington University ADRC for help with plasma sample selection and discussion. We acknowledge the following Washington University studies led by J. C. Morris for providing plasma samples: The Adult Children Study (NIH P01 AG02627606), Healthy Aging and Senile Dementia (NIH P01 AG003991), and ADRC (NIH P50 AG005681). We thank A. Richards for expert and dedicated experimental assistance and E. Norris, M. Cortes-Canteli, A. Kruyer, H.J. Ahn, M. Ponda, and P. Greengard for helpful discussion. This work was supported by NIH Grant NS50537, the Alzheimer’s Drug Discovery, Thome Memorial Medical, Litwin, Rudin Family, Blanchette Hooker Rockefeller, and Mellam Family foundations; the Cure Alzheimer’s Fund; and John A. Herrmann (S.S.); the Nicholson Exchange program (D.Z. and S.S.); and Vetenskapsrådet (K2013-65X-21462-04-5), Deutsche Forschungsgemeinschaft (SFB 877 and SFB 841), and European Research Council Grant (ERC-StG-2012-311575 F-12) (T.R.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1423764112/-/DCSupplemental.
References
- 1.von Bernhardi R. Glial cell dysregulation: A new perspective on Alzheimer disease. Neurotox Res. 2007;12(4):215–232. doi: 10.1007/BF03033906. [DOI] [PubMed] [Google Scholar]
- 2.Wyss-Coray T. Inflammation in Alzheimer disease: Driving force, bystander or beneficial response? Nat Med. 2006;12(9):1005–1015. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- 3.Träger U, Tabrizi SJ. Peripheral inflammation in neurodegeneration. J Mol Med (Berl) 2013;91(6):673–681. doi: 10.1007/s00109-013-1026-0. [DOI] [PubMed] [Google Scholar]
- 4.Leeb-Lundberg LM, Marceau F, Müller-Esterl W, Pettibone DJ, Zuraw BL. International union of pharmacology. XLV. Classification of the kinin receptor family: From molecular mechanisms to pathophysiological consequences. Pharmacol Rev. 2005;57(1):27–77. doi: 10.1124/pr.57.1.2. [DOI] [PubMed] [Google Scholar]
- 5.Renné T, Schmaier AH, Nickel KF, Blombäck M, Maas C. In vivo roles of factor XII. Blood. 2012;120(22):4296–4303. doi: 10.1182/blood-2012-07-292094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shibayama Y, et al. Zinc-dependent activation of the plasma kinin-forming cascade by aggregated beta amyloid protein. Clin Immunol. 1999;90(1):89–99. doi: 10.1006/clim.1998.4621. [DOI] [PubMed] [Google Scholar]
- 7.Maas C, et al. Misfolded proteins activate factor XII in humans, leading to kallikrein formation without initiating coagulation. J Clin Invest. 2008;118(9):3208–3218. doi: 10.1172/JCI35424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bergamaschini L, et al. The region 1-11 of Alzheimer amyloid-beta is critical for activation of contact-kinin system. Neurobiol Aging. 2001;22(1):63–69. doi: 10.1016/s0197-4580(00)00174-3. [DOI] [PubMed] [Google Scholar]
- 9.Yasuhara O, Walker DG, McGeer PL. Hageman factor and its binding sites are present in senile plaques of Alzheimer’s disease. Brain Res. 1994;654(2):234–240. doi: 10.1016/0006-8993(94)90484-7. [DOI] [PubMed] [Google Scholar]
- 10.Ashby EL, Love S, Kehoe PG. Assessment of activation of the plasma kallikrein-kinin system in frontal and temporal cortex in Alzheimer’s disease and vascular dementia. Neurobiol Aging. 2012;33(7):1345–1355. doi: 10.1016/j.neurobiolaging.2010.09.024. [DOI] [PubMed] [Google Scholar]
- 11.Bergamaschini L, et al. Activation of the contact system in cerebrospinal fluid of patients with Alzheimer disease. Alzheimer Dis Assoc Disord. 1998;12(2):102–108. doi: 10.1097/00002093-199806000-00008. [DOI] [PubMed] [Google Scholar]
- 12.Pimentel-Coelho PM, Rivest S. The early contribution of cerebrovascular factors to the pathogenesis of Alzheimer’s disease. Eur J Neurosci. 2012;35(12):1917–1937. doi: 10.1111/j.1460-9568.2012.08126.x. [DOI] [PubMed] [Google Scholar]
- 13.Morris JC. The Clinical Dementia Rating (CDR): Current version and scoring rules. Neurology. 1993;43(11):2412–2414. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- 14.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 15.Hyman BT. New neuropathological criteria for Alzheimer disease. Arch Neurol. 1998;55(9):1174–1176. doi: 10.1001/archneur.55.9.1174. [DOI] [PubMed] [Google Scholar]
- 16.Squitti R, et al. Ceruloplasmin/Transferrin ratio changes in Alzheimer’s disease. Int J Alzheimers Dis. 2010;2011:231595. doi: 10.4061/2011/231595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Müller F, et al. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell. 2009;139(6):1143–1156. doi: 10.1016/j.cell.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kannemeier C, et al. Extracellular RNA constitutes a natural procoagulant cofactor in blood coagulation. Proc Natl Acad Sci USA. 2007;104(15):6388–6393. doi: 10.1073/pnas.0608647104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Renné T. 2013. The FXII-driven Plasma Contact System. Hemostasis and Thrombosis. Basic Princeples and Clinical Practice, eds Marder V, Aird WC, Bennett JS, Schulman S, White, II GC (Wolters Kluwer, Philadelphia, PA), Vol 6, pp 242–253.
- 20.Mirra SS, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41(4):479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 21.Musiek ES, Holtzman DM. Origins of Alzheimer’s disease: Reconciling cerebrospinal fluid biomarker and neuropathology data regarding the temporal sequence of amyloid-beta and tau involvement. Curr Opin Neurol. 2012;25(6):715–720. doi: 10.1097/WCO.0b013e32835a30f4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Silverberg M, Diehl SV. The autoactivation of factor XII (Hageman factor) induced by low-Mr heparin and dextran sulphate. The effect of the Mr of the activating polyanion. Biochem J. 1987;248(3):715–720. doi: 10.1042/bj2480715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hojima Y, Cochrane CG, Wiggins RC, Austen KF, Stevens RL. In vitro activation of the contact (Hageman factor) system of plasma by heparin and chondroitin sulfate E. Blood. 1984;63(6):1453–1459. [PubMed] [Google Scholar]
- 24.Pixley RA, Cassello A, De La Cadena RA, Kaufman N, Colman RW. Effect of heparin on the activation of factor XII and the contact system in plasma. Thromb Haemost. 1991;66(5):540–547. [PubMed] [Google Scholar]
- 25.Loeffen R, et al. Preanalytic variables of thrombin generation: Towards a standard procedure and validation of the method. J Thromb Haemost. 2012;10(12):2544–2554. doi: 10.1111/jth.12012. [DOI] [PubMed] [Google Scholar]
- 26.Ramström S. Clotting time analysis of citrated blood samples is strongly affected by the tube used for blood sampling. Blood Coagul Fibrinolysis. 2005;16(6):447–452. doi: 10.1097/01.mbc.0000178827.52242.89. [DOI] [PubMed] [Google Scholar]
- 27.Oakley H, et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J Neurosci. 2006;26(40):10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ou-Yang MH, Van Nostrand WE. The absence of myelin basic protein promotes neuroinflammation and reduces amyloid β-protein accumulation in Tg-5xFAD mice. J Neuroinflammation. 2013;10:134. doi: 10.1186/1742-2094-10-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mayeux R, Schupf N. Blood-based biomarkers for Alzheimer’s disease: Plasma Aβ40 and Aβ42, and genetic variants. Neurobiol Aging. 2011;32(Suppl 1):S10–S19. doi: 10.1016/j.neurobiolaging.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gabelle A, et al. Plasma amyloid-β levels and prognosis in incident dementia cases of the 3-City Study. J Alzheimers Dis. 2013;33(2):381–391. doi: 10.3233/JAD-2012-121147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Humpel C. Chronic mild cerebrovascular dysfunction as a cause for Alzheimer’s disease? Exp Gerontol. 2011;46(4):225–232. doi: 10.1016/j.exger.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cardigan RA, Crook M, Mackie IJ, Machin SJ. Plasma levels of factor XIIa and factor VIIa are increased but not related in primary hyperlipidaemia. Blood Coagul Fibrinolysis. 2001;12(3):187–192. doi: 10.1097/00001721-200104000-00004. [DOI] [PubMed] [Google Scholar]
- 33.Siegerink B, Govers-Riemslag JW, Rosendaal FR, Ten Cate H, Algra A. Intrinsic coagulation activation and the risk of arterial thrombosis in young women: Results from the Risk of Arterial Thrombosis in relation to Oral contraceptives (RATIO) case-control study. Circulation. 2010;122(18):1854–1861. doi: 10.1161/CIRCULATIONAHA.110.943738. [DOI] [PubMed] [Google Scholar]
- 34.Kohler HP, Carter AM, Stickland MH, Grant PJ. Levels of activated FXII in survivors of myocardial infarction—association with circulating risk factors and extent of coronary artery disease. Thromb Haemost. 1998;79(1):14–18. [PubMed] [Google Scholar]
- 35.Weiser P, et al. Activated contact system and abnormal glycosaminoglycans in lupus and other auto- and non-autoimmune diseases. Prog Mol Biol Transl Sci. 2010;93:443–472. doi: 10.1016/S1877-1173(10)93019-6. [DOI] [PubMed] [Google Scholar]
- 36.Pan J, et al. Glycosaminoglycans and activated contact system in cancer patient plasmas. Prog Mol Biol Transl Sci. 2010;93:473–495. doi: 10.1016/S1877-1173(10)93020-2. [DOI] [PubMed] [Google Scholar]
- 37.Cugno M, et al. Activation of the coagulation cascade in C1-inhibitor deficiencies. Blood. 1997;89(9):3213–3218. [PubMed] [Google Scholar]
- 38.Wyss-Coray T, Rogers J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med. 2012;2(1):a006346. doi: 10.1101/cshperspect.a006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farrall AJ, Wardlaw JM. Blood-brain barrier: Ageing and microvascular disease—systematic review and meta-analysis. Neurobiol Aging. 2009;30(3):337–352. doi: 10.1016/j.neurobiolaging.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 40.Ryu JK, McLarnon JG. A leaky blood-brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer’s disease brain. J Cell Mol Med. 2009;13(9A):2911–2925. doi: 10.1111/j.1582-4934.2008.00434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burda JE, Sofroniew MV. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron. 2014;81(2):229–248. doi: 10.1016/j.neuron.2013.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci. 2011;12(12):723–738. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Olsson B, et al. Microglial markers are elevated in the prodromal phase of Alzheimer’s disease and vascular dementia. J Alzheimers Dis. 2013;33(1):45–53. doi: 10.3233/JAD-2012-120787. [DOI] [PubMed] [Google Scholar]
- 44.Carter SF, et al. 2012. Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-L-deprenyl: A multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J Nucl Med. 2012;53(1):37-46.
- 45.Johnson KA, Albert MS. Perfusion abnormalities in prodromal AD. Neurobiol Aging. 2000;21(2):289–292. doi: 10.1016/s0197-4580(00)00137-8. [DOI] [PubMed] [Google Scholar]
- 46.Wierenga CE, Hays CC, Zlatar ZZ. Cerebral blood flow measured by arterial spin labeling MRI as a preclinical marker of Alzheimer’s disease. J Alzheimers Dis. 2014;42(Suppl 4):S411–S419. doi: 10.3233/JAD-141467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de la Torre JC. Cerebral hemodynamics and vascular risk factors: Setting the stage for Alzheimer’s disease. J Alzheimers Dis. 2012;32(3):553–567. doi: 10.3233/JAD-2012-120793. [DOI] [PubMed] [Google Scholar]
- 48.Larsson M, et al. 2014. A factor XIIa inhibitory antibody provides thromboprotection in extracorporeal circulation without increasing bleeding risk. Sci Transl Med. 6(222):222ra17.
- 49.Fagan AM, et al. Decreased cerebrospinal fluid Abeta(42) correlates with brain atrophy in cognitively normal elderly. Ann Neurol. 2009;65(2):176–183. doi: 10.1002/ana.21559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gallimore MJ, Friberger P. Simple chromogenic peptide substrate assays for determining prekallikrein, kallikrein inhibition and kallikrein “like” activity in human plasma. Thromb Res. 1982;25(3):293–298. [PubMed] [Google Scholar]
- 51.Pauer HU, et al. Targeted deletion of murine coagulation factor XII gene-a model for contact phase activation in vivo. Thromb Haemost. 2004;92(3):503–508. doi: 10.1160/TH04-04-0250. [DOI] [PubMed] [Google Scholar]
- 52.Merkulov S, et al. Deletion of murine kininogen gene 1 (mKng1) causes loss of plasma kininogen and delays thrombosis. Blood. 2008;111(3):1274–1281. doi: 10.1182/blood-2007-06-092338. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.