

Significance
The alarmin IL-33 amplifies immune responses of Th2 and CD8+ cytotoxic T cells against invading pathogens; however, little is known about a potential role of IL-33 in Th1 cell responses. This study demonstrates that activated Th1 effector cells transiently express the IL-33 receptor ST2 upon differentiation in vitro and in vivo during viral infection. ST2 expression on virus-specific Th1 cells depended on the Th1-associated transcription factors T-bet and STAT4. ST2-deficient virus-specific CD4+ T cells showed impaired expansion and Th1 effector function upon viral infection, indicating a direct impact of IL-33 on antiviral Th1 cell responses. These observations redefine the role of ST2 in Th cell activation, with implications for the design of adjuvants and therapies targeting the IL-33–ST2 pathway.
Keywords: IL-33, ST2, CD4 T cell, Th1 cell, virus infection
Abstract
During infection, the release of damage-associated molecular patterns, so-called “alarmins,” orchestrates the immune response. The alarmin IL-33 plays a role in a wide range of pathologies. Upon release, IL-33 signals through its receptor ST2, which reportedly is expressed only on CD4+ T cells of the Th2 and regulatory subsets. Here we show that Th1 effector cells also express ST2 upon differentiation in vitro and in vivo during lymphocytic choriomeningitis virus (LCMV) infection. The expression of ST2 on Th1 cells was transient, in contrast to constitutive ST2 expression on Th2 cells, and marked highly activated effector cells. ST2 expression on virus-specific Th1 cells depended on the Th1-associated transcription factors T-bet and STAT4. ST2 deficiency resulted in a T-cell–intrinsic impairment of LCMV-specific Th1 effector responses in both mixed bone marrow-chimeric mice and adoptive cell transfer experiments. ST2-deficient virus-specific CD4+ T cells showed impaired expansion, Th1 effector differentiation, and antiviral cytokine production. Consequently, these cells mediated little virus-induced immunopathology. Thus, IL-33 acts as a critical and direct cofactor to drive antiviral Th1 effector cell activation, with implications for vaccination strategies and immunotherapeutic approaches.
Intercellular signaling molecules, such as cytokines and damage-associated molecular patterns (DAMPs), are essential for the induction and amplification of immune responses. DAMPs are multifunctional host proteins that indicate tissue damage. They are also referred to as alarmins, because they serve as early warning signals to activate innate and adaptive immune responses (1). The alarmin IL-33, a member of the IL-1 family, is constitutively expressed in the nucleus of endothelial and epithelial cells and is released during tissue damage and necrosis (2). Upon release, IL-33 mediates its activity through a heterodimeric cell surface receptor consisting of the ubiquitous IL-1R accessory protein (IL1RAcP) and the more selectively expressed receptor ST2, also known as T1 and IL-1RL1 (3, 4). The intracellular signaling pathway of this receptor complex includes the recruitment of MyD88 and leads to the activation of NF-κB, ERK, p38, and JNK pathways (2).
IL-33 acts on a wide range of immune cells, but has been implicated predominantly in Th2-associated immune responses (5). Nonetheless, this categorization has recently been challenged by reports describing IL-33 as an enhancer of IFN-γ production by iNKT cells, NK cells, and CD8+ T cells (6–8). In addition, IL-33 signaling is important for the activation and functionality of cytotoxic CD8+ T cells during viral infection (9).
In the present work, we studied a potential impact of IL-33 on Th1 cell responses in vitro and in vivo. In the course of infection with LCMV, which potently induces Th1-differentiated effector CD4+ T cells (10), we found that many of the virus-specific CD4+ T cells expressed the IL-33 receptor ST2. ST2 expression marked activated effector Th1 cells and differed in terms of intensity and stability from the previously described ST2 expression of Th2 cells. At the molecular level, the Th1-associated transcription factors T-bet and STAT4 controlled ST2 expression in vivo and in vitro. ST2 deficiency of LCMV-specific CD4+ T cells resulted in impaired effector Th1 cell differentiation with substantially reduced cell expansion, impaired antiviral cytokine production, and little virus-induced T-cell–mediated immunopathology. Thus, IL-33 acts directly on CD4+ T cells during infection to enhance antiviral effector Th1 cell responses.
Results
Transient ST2 Expression on Virus-Specific CD4+ T Cells Is Associated with Enhanced Th1 Effector Properties.
We set out to investigate whether CD4+ effector T cells can express ST2 during a Th1-promoting infection. To do so, we analyzed endogenous CD4+ T cells in LCMV-infected WT mice, as well as adoptively transferred LCMV-TCR transgenic (tg) CD4+ T cells with a congenic marker (Thy1.1+). Given that regulatory T cells (Tregs) are known to express ST2 (11), we excluded CD4+ FoxP3+ T cells from our flow cytometry analysis. Endogenous CD4+ effectors were defined by their down-regulation of CD62L and showed transient expression of ST2 that peaked at day 8 postinfection (Fig. 1 A and B). The analysis of CD4+ T cells at the peak of ST2 expression confirmed that ST2 was expressed exclusively on activated CD62Llo CD4+ T cells, and not on resting CD62Lhi CD4+ T cells (Fig. 1C). Transient ST2 expression during LCMV infection was found on transferred LCMV-TCRtg CD4+ T cells as well (Fig. 1D). Here the peak of ST2 expression was observed at day 6 postinfection, followed by a gradual decrease until day 14.
Fig. 1.
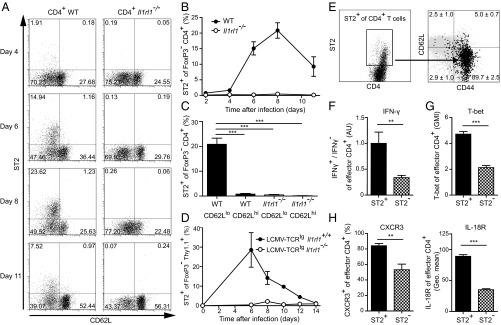
Transient ST2 expression on virus-specific CD4+ T cells is associated with enhanced Th1 effector properties. (A–C) Kinetic flow cytometry analysis of ST2 expression of FoxP3− CD4+ T cells in peripheral blood during LCMV infection (2 × 106 PFU). (A) Analysis of one representative mouse per time point (n = 3). (B) Symbols represent mean ± SD values (n = 3). (C) Frequencies of ST2-expressing FoxP3− CD4+ T cells at day 8 postinfection. Bars represent mean + SD (n = 3). (D) Il1rl1+/+ and Il1rl1−/− naive LCMV-TCRtg CD4+ Thy1.1+ T cells were transferred into WT recipients, followed by LCMV infection (200 PFU). Shown are the results of kinetic analysis of ST2 expression of FoxP3− CD4+ Thy1.1+ cells in peripheral blood. Symbols represent mean ± SD values (n = 3–5). (E) ST2 expression of splenic total CD4+ T cells at day 8 postinfection. ST2+ CD4+ T cells are shown in black with average ± SD frequencies; the expression pattern of total CD4+ T cells is shown in gray. Data are from one representative mouse (n = 3). (F–H) Comparison of splenic ST2+ and ST2− effector CD4+ T cells analyzed by flow cytometry at day 8 postinfection. Effector CD4+ T cells were defined by CD62Llo or CD44+ expression. (F) Expression levels of IFN-γ after GP64 peptide restimulation. The ratio of IFN-γ+ to IFN-γ− of ST2+ or ST2− CD44+ CD4+ T cells was calculated, and the average value of the ST2+ fraction was set to an arbitrary unit of 1. (G) Geometric mean index (GMI) of T-bet protein. (H) Expression of CXCR3 and IL-18R. Bars represent mean + SD (n = 3). All data are representative of two or three independent experiments.
To characterize ST2+ CD4+ T cells during virus-induced Th1 differentiation, we analyzed endogenous CD4+ T cells in WT mice at the peak of ST2 expression on day 8 postinfection (cf. Fig. 1B). At this time point, the majority of ST2+ CD4+ T cells were CD62Llo CD44+ effector cells (Fig. 1E). In addition, we found several functional differences between ST2+ and ST2− effector CD4+ T cells. The ratio between IFN-γ producers and nonproducers was higher within the ST2+ CD44+ CD4+ T cells (Fig. 1F). The lineage-specifying transcription factor of Th1 cells, T-bet (12), was preferentially expressed within the ST2+ fraction of effector CD4+ T cells, just like the Th1-associated receptors CXCR3 (13) and IL-18R (14) (Fig. 1 G and H). In contrast, the expression of GATA-3, FoxP3, and Eomesodermin did not differ substantially between ST2+ and ST2− effector CD4+ T cells (Fig. S1). Taken together, these data indicate that Th1 cells transiently express the IL-33 receptor ST2 at the peak of LCMV infection, and that the ST2+ fraction of the CD4+ T cells is highly activated and exhibits enhanced Th1 effector properties.
ST2 Expression Is Weaker and Less Stable on Th1 Cells than on Th2 Cells.
Having detected ST2 on effector Th1 cells in vivo, we next examined whether in vitro-generated Th1 cells also express ST2. After two rounds of in vitro differentiation, we found ST2 protein on both Th1 and Th2 cells (Fig. 2A). The frequency of ST2+ cells was comparable in the two subsets (Fig. 2B), whereas the amounts of ST2 protein (Fig. 2C) and Il1rl1 mRNA (Fig. 2D) were clearly higher in Th2 cells than in Th1 cells. Both subsets expressed homogenously and exclusively their lineage-specifying transcription factors T-bet (Th1) and GATA-3 (Th2), and did not express the Treg transcription factor FoxP3 (Fig. 2E). ST2 receptor signaling was functional in Th1 cells, as demonstrated by the finding that exposure to IL-33 increased phospho-p38 levels (Fig. S2).
Fig. 2.

ST2 expression is weaker and less stable on Th1 cells than on Th2 cells. (A–E) Naive LCMV-TCRtg CD4+ T cells were differentiated to Th1 and Th2 cells for 8 d. (A) Representative dot plots of ST2 expression on Th1 and Th2 cells. Il1rl1−/− Th1 cells served as staining controls. (B–D) Mean + SD of two or three independent cultures for flow cytometry analysis of ST2 frequency (B), geometric mean of ST2 protein within ST2+ cells (C), and quantitation of Il1rl1 mRNA (D). (E) Flow cytometry analysis of ST2 and T-bet, GATA-3, and FoxP3. Th1 cells are shown in blue; Th2 cells, in red. The GMI of the respective protein is displayed in the upper right corner of the plot. (F–I) Naive LCMV-TCRtg CD4+ T cells were differentiated to Th1 and Th2 cells for 8 d and placed into resting condition in fresh medium with IL-7 for another 5 d. Analysis gates were set on living cells. The frequency of ST2-expressing cells and the GMI of T-bet and GATA-3 protein are shown for Th1 cells (F) and Th2 cells (G) normalized as percentage of d0. (H) Frequencies of Th1 cells expressing IFN-γ, IL-4, and IL-17 after restimulation with PMA and ionomycin. (I) ST2+ and ST2− Th1 cells were sorted on day 8 and plated separately under resting conditions with IL-7. Total numbers of living cells were assessed over time. Symbols represent the mean ± SD of two or three pooled, independent experiments.
To determine the stability of ST2 expression, we further cultured Th1 and Th2 cells under resting conditions with IL-7 but in the absence of polarizing cytokines. ST2 expression on Th1 cells decreased within the first 24 h and continuously thereafter (Fig. 2F). In contrast, Th2 cells maintained and even slightly increased their ST2 expression (Fig. 2G). Th1 cells maintained their differentiated phenotype while losing ST2 expression (Fig. 2H and Fig. S3). Importantly, when Th1 cells were sorted into ST2+ and ST2− subpopulations and plated separately, the survival of the two subsets in culture was indistinguishable (Fig. 2I), making it unlikely that preferential cell death of ST2+ Th1 cells had confounded the analysis of ST2 down-regulation. Thus, in vitro-generated Th1 cells can express ST2, which is down-regulated by these cells during resting conditions.
T-bet and STAT4 Regulate ST2 Expression on Th1 cells in Vivo and in Vitro.
Ectopic GATA-3 expression experiments and studies on inducible GATA-3–deficient cells have shown that ST2 expression in Th2 and Treg cells is dependent on GATA-3 (15, 16). In contrast, ST2+ Th1 cells did not express elevated GATA-3 levels, and their expression pattern of ST2 differed from that of Th2 cells (Fig. 2). Thus, we hypothesized that other transcriptional regulators are essential to Th1-associated ST2 expression. STAT4 and T-bet are both crucial in Th1 differentiation (12, 17). When adoptively transferred into WT recipients, T-bet–deficient (Tbx21−/−) and STAT4-deficient (Stat4−/−) LCMV-TCRtg effector CD4+ T cells exhibited impaired ST2 expression on days 6–10 after LCMV infection (Fig. 3A). In contrast, IFN-γR signals were dispensable for ST2 expression, as demonstrated by the finding that IFN-γR–deficient (Ifngr1−/−) LCMV-TCRtg CD4+ Thy1.1+ cells exhibited normal ST2 levels when tested analogously (Fig. S4).
Fig. 3.

T-bet and STAT4 regulate ST2 expression on Th1 cells in vivo and in vitro. (A) Plain control, Tbx21−/−, and Stat4−/− naive LCMV-TCRtg CD4+ Thy1.1+ cells were transferred into WT recipients and infected with LCMV (200 PFU). Shown is a time course analysis of ST2 expression on circulating effector FoxP3− CD4+ Thy1.1+ cells, effector T cells as defined by CD62Llo or CD44+ expression. Symbols represent mean ± SEM values of two pooled, independent experiments (n = 3–8). (B–D) Naive control (B), Tbx21−/− (C), and Stat4−/− (D) LCMV-TCRtg CD4+ T cells were differentiated to Th1 cells for 4 d and then kinetically analyzed by flow cytometry for pSTAT4 (triangle), T-bet (square), and ST2 (diamond) expression until day 8 of culture. Representative data from one of three experiments are shown.
Based on impaired ST2 expression in vivo, we also analyzed in vitro-differentiated Tbx21−/− and Stat4−/− Th1 cells. Despite their respective genetic defects, differentiation of both genotypes resulted in activated cells with clear Th1 characteristics (Fig. S5). Control Th1 cells expressed T-bet as expected and showed increased ST2 expression throughout the second round of differentiation (Fig. 3B). Tbx21−/− Th1 cells phosphorylated STAT4 but displayed impaired ST2 expression (Fig. 3C). Similarly, Stat4−/− Th1 cells failed to express ST2 despite enhanced T-bet expression early in the second round of differentiation (Fig. 3D). IL-2 can contribute to ST2 expression in Th2 cells in a STAT5-dependent fashion (15), but it failed to rescue ST2 in Tbx21−/− and Stat4−/− Th1 cells (Fig. S6). These findings suggest that both T-bet and STAT4 are required for optimal ST2 expression in Th1 cells.
ST2-deficient CD4+ T Cells Are Impaired in Expansion and Cytokine Production After Viral Infection.
IL-33 is released to the extracellular space during infection, thereby acting as an alarmin that should be accessible to differentiating ST2+ Th1 cells. Indeed, Il1rl1−/− mice generated fewer cytokine-producing CD4+ T cells than WT controls (Fig. 4A), which was evident in both the percentage and absolute number of CD4+ T cells expressing IFN-γ, TNF-α, IL-2, and combinations thereof (Fig. S7 A and B).
Fig. 4.

ST2 cell-intrinsically enhances the expansion of polyfunctional antiviral CD4+ T cells. (A) WT and Il1rl1−/− mice were infected with LCMV. On day 9, we enumerated GP64-specific splenic CD4+ T cells expressing IFN-γ, TNF-α, IL-2, and combinations thereof. (B–F) Irradiated WT recipients were reconstituted with WT (CD45.1+) and Il1rl1−/− (CD45.2+) bone marrow and subjected to flow cytometry analysis either before LCMV infection (B, Left) or 9 d after LCMV infection (B, Right and C–F). (B) Ratio of WT and Il1rl1−/− CD4+ T cells, either CD44lo or CD44hi. Symbols represent individual mice. (C) Frequencies of I-Ab GP66-77 tetramer-binding CD44lo or CD44hi CD4+ T cells in the WT and Il1rl1−/− compartments. (D) Representative FACS plots of specific I-Ab GP66-77 tetramer binding compared with irrelevant hCLIP-loaded I-Ab tetramer. Values indicate the percentage (mean ± SD) of tetramer binding within the WT and Il1rl1−/− CD4+CD44hi compartments. (E) Frequencies of WT and Il1rl1−/− CD4+ T cells expressing IFN-γ, TNF-α, IL-2, or combinations thereof after GP64 peptide restimulation. (F) IFN-γ geometric mean of IFN-γ–producing WT and Il1rl1−/− CD4+ T cells. Bars represent mean + SEM values of four mice (A) or six mice (B–F). A, E, and F show representative results from two independent experiments. The paired Student t test was used in F.
To directly address the CD4+ T-cell–intrinsic role of ST2 in the antiviral response, we reconstituted lethally irradiated WT mice with WT and Il1rl1−/− bone marrow. In the resulting chimeras, Il1rl1−/− CD4+ T cells were slightly more abundant than WT CD4+ T cells (differentiated by the CD45.1 congenic marker) in both the naive CD44lo compartment and the effector/memory (CD44hi) pool (Fig. 4B and Fig. S7C). This finding indicates that IL-33 signals are not required for homeostatic maintenance of either CD4+ T-cell compartment. At day 9 after the LCMV challenge, however, WT CD44hi CD4+ T cells outnumbered CD44hi Il1rl1−/− CD4+ T cells by approximately fourfold (Fig. 4B). Conversely, both CD44lo compartments contracted proportionally (Fig. 4B and Fig. S7C). In concert with these observations, LCMV GP66-77 MHC class II tetramer-binding cells were significantly more frequent within the WT CD4+ T-cell compartment than within the Il1rl1−/− CD4+ T-cell compartment, and were virtually uniformly CD44hi cells (Fig. 4 C and D).
Restimulation with an immunodominant LCMV-derived peptide epitope revealed that cytokine-expressing cells were significantly less frequent within the Il1rl1−/− CD4+ T-cell compartment than within the WT CD4+ T-cell compartment (Fig. 4E). These differences were evident at the level of absolute effector CD4+ T-cell counts as well (Fig. S7D). Furthermore, we noted that IFN-γ expression levels in Il1rl1−/− CD4+ T cells were modestly yet consistently diminished (Fig. 4F). Thus, IL-33 signals serve critical cell-intrinsic functions in polyfunctional CD4+ T-cell responses to viral infection.
Adoptively Transferred ST2-deficient CD4+ T Cells Cause Less Virus-Induced Immunopathology.
Impaired antiviral CD4+ T-cell responses, as observed in Il1rl1−/− mice and mixed bone marrow-chimeric mice, are suggestive of an intrinsically defective Th1 differentiation of Il1rl1−/− CD4+ T cells. Independent support for this concept stemmed from Th1 polarization experiments, which were conducted with limiting IL-12 concentrations. In this setting, Il1rl1−/− CD4+ T cells exhibited lower T-bet, IFN-γ, CD44, and IL-18R expression compared with Il1rl1+/+ control cells (Fig. S8).
To address the functional impact of this finding, we exploited the LCMV wasting disease model, in which effector CD4+ T cells can mediate weight loss (18, 19). We adoptively transferred Il1rl1+/+ or Il1rl1−/− LCMV-TCRtg CD4+ Thy1.1+ cells into WT mice and monitored their body weight after LCMV infection. Recipients of Il1rl1+/+ LCMV-TCRtg CD4+ Thy1.1+ cells lost significantly more weight than recipients of Il1rl1−/− LCMV-TCRtg CD4+ Thy1.1+ cells (Fig. 5A). Analysis of the transferred cells at day 6 postinfection demonstrated lower cell numbers and reduced T-bet expression by the Il1rl1−/− CD4+ Thy1.1+ cells (Fig. 5 B and C). Furthermore, cytokine expression by the transferred CD4+ Thy1.1+ cells was diminished in the Il1rl1−/− fraction (Fig. 5D and Fig. S9). The Il1rl1−/− CD4+ T cells also displayed reduced expression of CXCR3 and CD44 (Fig. 5 E and F), indicating diminished potential to migrate to inflamed sites of infection and impaired activation, respectively. The endogenous CD8+ T-cell response did not differ between the recipients of Il1rl1+/+ and Il1rl1−/− CD4+ T cells, as demonstrated by a similar frequency of LCMV-specific GP33 tetramer-positive CD8+ T cells and similar CD44 expression by these cells (Fig. 5 G and H). These findings indicate that the impaired expansion, activation, and effector differentiation of Il1rl1−/− CD4+ T cells result in a decreased immunopathological potential after LCMV infection.
Fig. 5.

Adoptively transferred ST2-deficient CD4+ T cells cause less virus-induced immunopathology. Naive Il1rl1+/+ and Il1rl1−/− LCMV-TCRtg CD4+ Thy1.1+ cells were transferred into WT recipients, which were subsequently infected with LCMV (200 PFU). (A) Body weight was monitored daily; symbols represent mean ± SD (n = 4). Differences in body weight between Il1rl1+/+ and Il1rl1−/− CD4+ Thy1.1+ cell recipients were analyzed by two-way ANOVA for the entire time course. (B–F) Analysis of splenic transferred Il1rl1+/+ and Il1rl1−/− CD4+ Thy1.1+ cells at day 6 postinfection. Shown are total CD4+ Thy1.1+ cell numbers (B), T-bet GMI (C), IFN-γ+ TNF-α+ IL-2+ frequencies after GP64 peptide restimulation (D), and geometric means of CXCR3 (E) and CD44 (F). (G and H) Frequencies of splenic, endogenous GP33-tretramer+ CD8+ T cells (G) and CD44+ GP33-tetramer+ CD8+ T cells (H). Bars represent mean + SD (n = 4). All data are representative of two independent experiments.
Discussion
In this study, we have shown that the IL-33 receptor ST2 is induced on Th1 effector cells upon differentiation in vitro and in vivo during LCMV infection. ST2 expression on Th1 cells is transient and is associated with the cellular activation status. In Th1 cells, STAT4 and T-bet cooperate to drive ST2 expression. The absence of ST2 on CD4+ T cells impairs Th1 cell activation during viral infection and results in decreased expansion, impaired effector function, and reduced T-cell–mediated immunopathology.
ST2 protein has been reported to be expressed on in vitro-differentiated Th2 but not Th1 cells (20). In addition, ST2+ CD4+ T cells isolated from uninfected mice display mostly Th2 characteristics, such as expression of IL-4 and IL-5 (21). Using improved detection methods, we now show that ST2 is also expressed on activated Th1 cells, albeit at a lower expression level. Moreover, Th1 cells intrinsically down-regulate ST2 expression when transferred to resting conditions. Comparably higher stability and intensity of ST2 expression on Th2 cells may explain why in earlier ex vivo isolations from uninfected mice, ST2+ CD4+ T cells exhibited mainly Th2 cell characteristics (21). Very likely, Th1 cells originating from previous infections of those mice had lost ST2 expression while transitioning to a resting state, precluding detection of the ST2 receptor on these cells.
The lineage decision of Th cell subsets is controlled by “master regulator” transcription factors, e.g., T-bet in Th1 (12) and GATA-3 in Th2 cells (22). Nonetheless, T-bet and GATA-3 also share a considerable set of target genes (23). GATA-3 has been identified as an inducer of ST2 expression in Th2 and Treg cells (15, 16). In contrast to Th2 cells, Th1 cells express only very low levels of GATA-3 and show a transient ST2 expression pattern; therefore, transcription factors other than GATA-3 likely play a role in Th1-related ST2 expression. Indeed, we identified both T-bet and STAT4 as key drivers of ST2 expression in Th1 cells in vitro and in vivo during LCMV infection. This finding is in line with transcription factor-binding data from ChIP-Seq experiments with in vitro-differentiated Th1 cells indicating the binding of T-bet (24) and STAT4 (25) to the ST2 gene locus. Thus, the induction of ST2 expression may be accomplished by direct activation of Il1rl1 enhancers and promoters and/or permissive epigenetic changes by T-bet and STAT4.
ST2 expression on CD4+ T cells coincides with increases in splenic IL-33 mRNA during viral infection (9), suggesting an impact of IL-33 on effector CD4+ T-cell differentiation. This notion is supported by several results. First, the ST2+ fraction of the Th1 effector cells exhibited more pronounced Th1 cell characteristics and increased cell activation compared with their ST2− counterparts at the peak of LCMV infection. Second, in WT and Il1rl1−/− mixed bone marrow chimeras, IL-33 signals were critical for the expansion and antiviral cytokine production of CD4+ T cells in LCMV infection. Finally, adoptively transferred LCMV-specific Il1rl1−/− CD4+ T cells showed decreased expansion, activation, and Th1 cell differentiation at the peak of LCMV infection, which translated into a reduced immunopathological potential of these cells.
Il1rl1−/− CD4+ T cells exhibited decreased activation and Th1 effector characteristics during LCMV infection, but Th1 cell differentiation per se was not completely abrogated. This finding is in line with the function of IL-33 as a general alarmin and not as a Th1-specific factor. This is reflected by the plethora of different target cells of IL-33 (5) and the downstream signaling of its receptor, leading to the activation of rather broad transcriptional regulators like JNK, NF-κB, and p38 MAPK (2). Nevertheless, an impact of two of those factors on optimal Th1 effector differentiation has already been documented. NF-κB plays a T-cell–intrinsic role in optimizing Th1 cell differentiation (26), and the p38 MAP kinase pathway is required for optimal expression of IFN-γ in effector Th1 cells (27).
Taken together, our data indicate a direct and enhancing activity of the alarmin IL-33 on virus-induced effector Th1 cells. These findings have considerable implications for the design of therapies that include the application of IL-33 or the blockade of its receptor ST2.
Materials and Methods
Mice and Infection.
Il1rl1−/− (28), Tbx21−/− (12), Stat4−/− (12), Ifngr1−/− (29), and SMARTA1 TCR-tg mice (30) were bred on a C57BL/6 background. The mice were infected i.v. with 200 or 2 × 106 plaque-forming units (PFU) of LCMV-WE. Animal experiments were performed at the Charité–Universitätsmedizin Berlin and at the Universities of Geneva and Basel in accordance with the German and Swiss laws for animal protection, respectively. They were approved by the Landesamt für Gesundheit und Soziales of Berlin, the Direction Générale de la Santé of the Canton of Geneva, and the Cantonal Veterinary Office of the Canton of Basel.
Primary T-Cell Cultures.
Naive CD4+ CD62Lhi CD44− cells were sorted from pooled spleen and lymph node cells by FACS. T cells were cultured as described in SI Materials and Methods.
Flow Cytometry Analysis.
Flow cytometry detection of cell surface ST2 was done after a two-step amplification procedure, increasing fluorescence intensity, as described previously (9). Further stainings are described in SI Materials and Methods.
Statistical Analysis.
Unless stated otherwise, two-way ANOVA was used for multiple comparisons, and the unpaired two-tailed Student t test was used for comparisons of two groups (GraphPad Prism 5.02). *P = 0.01–0.05, statistically significant; **P = 0.001–0.01, very significant; ***P < 0.001, extremely significant.
Supplementary Material
Acknowledgments
We thank S. Ebel and V. Holecska for their expert technical assistance. Cell sorting was carried out at the Flow Cytometry Core Facility at the German Rheumatism Research Center Berlin. This work was supported by the German Research Foundation (SFB650, Grant TP28). C.B. is a fellow of the International Max Planck Research School for Infectious Diseases and Immunology. A.N.H. was supported by a European Molecular Biology Organization (EMBO) long-term fellowship and a Marie Curie fellowship. D.D.P. was supported by the Klaus Tschira Foundation and the Swiss National Science Foundation. M.L. is a Lichtenberg Fellow funded by the Volkswagen Foundation.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1418549112/-/DCSupplemental.
References
- 1.Oppenheim JJ, Yang D. Alarmins: Chemotactic activators of immune responses. Curr Opin Immunol. 2005;17(4):359–365. doi: 10.1016/j.coi.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 2.Palmer G, Gabay C. Interleukin-33 biology with potential insights into human diseases. Nat Rev Rheumatol. 2011;7(6):321–329. doi: 10.1038/nrrheum.2011.53. [DOI] [PubMed] [Google Scholar]
- 3.Schmitz J, et al. IL-33, an interleukin-1–like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23(5):479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 4.Ali S, et al. IL-1 receptor accessory protein is essential for IL-33–induced activation of T lymphocytes and mast cells. Proc Natl Acad Sci USA. 2007;104(47):18660–18665. doi: 10.1073/pnas.0705939104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: Back to the future. Immunity. 2013;39(6):1003–1018. doi: 10.1016/j.immuni.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang Q, et al. IL-33 synergizes with TCR and IL-12 signaling to promote the effector function of CD8+ T cells. Eur J Immunol. 2011;41(11):3351–3360. doi: 10.1002/eji.201141629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bourgeois E, et al. The pro-Th2 cytokine IL-33 directly interacts with invariant NKT and NK cells to induce IFN-gamma production. Eur J Immunol. 2009;39(4):1046–1055. doi: 10.1002/eji.200838575. [DOI] [PubMed] [Google Scholar]
- 8.Smithgall MD, et al. IL-33 amplifies both Th1- and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK cells. Int Immunol. 2008;20(8):1019–1030. doi: 10.1093/intimm/dxn060. [DOI] [PubMed] [Google Scholar]
- 9.Bonilla WV, et al. The alarmin interleukin-33 drives protective antiviral CD8+ T cell responses. Science. 2012;335(6071):984–989. doi: 10.1126/science.1215418. [DOI] [PubMed] [Google Scholar]
- 10.Varga SM, Welsh RM. High frequency of virus-specific interleukin-2–producing CD4(+) T cells and Th1 dominance during lymphocytic choriomeningitis virus infection. J Virol. 2000;74(9):4429–4432. doi: 10.1128/jvi.74.9.4429-4432.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turnquist HR, et al. IL-33 expands suppressive CD11b+ Gr-1(int) and regulatory T cells, including ST2L+ Foxp3+ cells, and mediates regulatory T cell-dependent promotion of cardiac allograft survival. J Immunol. 2011;187(9):4598–4610. doi: 10.4049/jimmunol.1100519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szabo SJ, et al. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100(6):655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 13.Loetscher P, et al. CCR5 is characteristic of Th1 lymphocytes. Nature. 1998;391(6665):344–345. doi: 10.1038/34814. [DOI] [PubMed] [Google Scholar]
- 14.Xu D, et al. Selective expression and functions of interleukin 18 receptor on T helper (Th) type 1 but not Th2 cells. J Exp Med. 1998;188(8):1485–1492. doi: 10.1084/jem.188.8.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo L, et al. IL-1 family members and STAT activators induce cytokine production by Th2, Th17, and Th1 cells. Proc Natl Acad Sci USA. 2009;106(32):13463–13468. doi: 10.1073/pnas.0906988106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schiering C, et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature. 2014;513(7519):564–568. doi: 10.1038/nature13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. 1996;382(6587):174–177. doi: 10.1038/382174a0. [DOI] [PubMed] [Google Scholar]
- 18.Kamperschroer C, Quinn DG. The role of proinflammatory cytokines in wasting disease during lymphocytic choriomeningitis virus infection. J Immunol. 2002;169(1):340–349. doi: 10.4049/jimmunol.169.1.340. [DOI] [PubMed] [Google Scholar]
- 19.Quinn DG, Zajac AJ, Frelinger JA, Muller D. Transfer of lymphocytic choriomeningitis disease in beta 2 microglobulin-deficient mice by CD4+ T cells. Int Immunol. 1993;5(10):1193–1198. doi: 10.1093/intimm/5.10.1193. [DOI] [PubMed] [Google Scholar]
- 20.Xu D, et al. Selective expression of a stable cell surface molecule on type 2 but not type 1 helper T cells. J Exp Med. 1998;187(5):787–794. doi: 10.1084/jem.187.5.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Löhning M, et al. T1/ST2 is preferentially expressed on murine Th2 cells, independent of interleukin 4, interleukin 5, and interleukin 10, and important for Th2 effector function. Proc Natl Acad Sci USA. 1998;95(12):6930–6935. doi: 10.1073/pnas.95.12.6930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89(4):587–596. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 23.Jenner RG, et al. The transcription factors T-bet and GATA-3 control alternative pathways of T-cell differentiation through a shared set of target genes. Proc Natl Acad Sci USA. 2009;106(42):17876–17881. doi: 10.1073/pnas.0909357106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakayamada S, et al. Early Th1 cell differentiation is marked by a Tfh cell-like transition. Immunity. 2011;35(6):919–931. doi: 10.1016/j.immuni.2011.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wei L, et al. Discrete roles of STAT4 and STAT6 transcription factors in tuning epigenetic modifications and transcription during T helper cell differentiation. Immunity. 2010;32(6):840–851. doi: 10.1016/j.immuni.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Corn RA, et al. T cell-intrinsic requirement for NF-kappa B induction in postdifferentiation IFN-gamma production and clonal expansion in a Th1 response. J Immunol. 2003;171(4):1816–1824. doi: 10.4049/jimmunol.171.4.1816. [DOI] [PubMed] [Google Scholar]
- 27.Rincón M, et al. Interferon-gamma expression by Th1 effector T cells mediated by the p38 MAP kinase signaling pathway. EMBO J. 1998;17(10):2817–2829. doi: 10.1093/emboj/17.10.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Townsend MJ, Fallon PG, Matthews DJ, Jolin HE, McKenzie AN. T1/ST2-deficient mice demonstrate the importance of T1/ST2 in developing primary T helper cell type 2 responses. J Exp Med. 2000;191(6):1069–1076. doi: 10.1084/jem.191.6.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Müller U, et al. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264(5167):1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 30.Oxenius A, Bachmann MF, Zinkernagel RM, Hengartner H. Virus-specific MHC-class II-restricted TCR-transgenic mice: Effects on humoral and cellular immune responses after viral infection. Eur J Immunol. 1998;28(1):390–400. doi: 10.1002/(SICI)1521-4141(199801)28:01<390::AID-IMMU390>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.