

Significance
Glucocorticoid receptor (GR) belongs to the nuclear receptor superfamily and functions as a transcription factor. GR regulates various physiological processes, including cell proliferation, energy homeostasis, and inflammation. In this study, we provide molecular evidence for the role of GR in the regulation of mRNA stability, which we term GR-mediated mRNA decay (GMD). Efficient GMD requires a ligand, a GR loaded onto target mRNA, upstream frameshift 1 (UPF1), and proline-rich nuclear receptor coregulatory protein 2. GMD functions in the chemotaxis of human monocytes by targeting chemokine (C-C motif) ligand 2 mRNA. Thus, we unravel a previously unappreciated role of GR, which is traditionally considered a transcription factor, in posttranscriptional regulation.
Keywords: glucocorticoid receptor, PNRC2, UPF1, glucocorticoid receptor-mediated mRNA decay, Nonsense-mediated mRNA decay
Abstract
Glucocorticoid receptor (GR), which was originally known to function as a nuclear receptor, plays a role in rapid mRNA degradation by acting as an RNA-binding protein. The mechanism by which this process occurs remains unknown. Here, we demonstrate that GR, preloaded onto the 5′UTR of a target mRNA, recruits UPF1 through proline-rich nuclear receptor coregulatory protein 2 (PNRC2) in a ligand-dependent manner, so as to elicit rapid mRNA degradation. We call this process GR-mediated mRNA decay (GMD). Although GMD, nonsense-mediated mRNA decay (NMD), and staufen-mediated mRNA decay (SMD) share upstream frameshift 1 (UPF1) and PNRC2, we find that GMD is mechanistically distinct from NMD and SMD. We also identify de novo cellular GMD substrates using microarray analysis. Intriguingly, GMD functions in the chemotaxis of human monocytes by targeting chemokine (C-C motif) ligand 2 (CCL2) mRNA. Thus, our data provide molecular evidence of a posttranscriptional role of the well-studied nuclear hormone receptor, GR, which is traditionally considered a transcription factor.
At the cellular level, glucocorticoid receptor (GR), which belongs to the nuclear receptor superfamily, functions as a transcription factor regulating various physiological processes (1–3). In the presence of a glucocorticoid, which diffuses through the plasma membrane into the cytoplasm, cytosolic GR binds to the glucocorticoid. The resulting glucocorticoid–GR complex is activated and then enters the nucleus. Once in the nucleus, GR dimerizes, binds to specific cis-acting elements, and recruits coregulatory proteins for transcriptional activation or repression (4, 5).
The majority of the coregulatory proteins commonly contain a nuclear receptor box (NR box, also called an LXXLL motif), which is important for interactions between coregulatory proteins and nuclear receptors (4–6). The proline-rich nuclear receptor coregulatory protein (PNRC), however, is an exception because it interacts with nuclear receptors through an SH3-binding motif [SD(E)PPSPS] rather than an NR box (7, 8). Two PNRC paralogs, PNRC1 and PNRC2, have been identified in mammalian cells (7, 8). PNRC1 and PNRC2 are believed to play similar roles in nuclear receptor-mediated signaling because they interact with similar groups of nuclear receptors.
Although PNRC2 is known to function as a coregulatory protein for nuclear receptors, it has a distinct function in mRNA decay pathways including nonsense-mediated mRNA decay (NMD), staufen (STAU)-mediated mRNA decay (SMD), and replication-dependent histone mRNA degradation (HMD) (9–13). NMD serves as a mechanism of both mRNA quality control and posttranscriptional regulation by selectively recognizing and degrading cellular transcripts that are abnormal or that contain a premature translation termination codon (PTC), as reviewed elsewhere (14–16). A key NMD factor, UPF1, is recruited to a terminating ribosome at a PTC. UPF1 then recruits general RNA-degrading enzymes via adaptor/effector proteins such as PNRC2, suppressor with morphogenetic effect on genitalia 5 (SMG5), SMG6, and SMG7, resulting in rapid mRNA degradation. In SMD, UPF1 is recruited to mRNA via its interaction with STAU, which recognizes a stem-loop structure in the 3′UTR or an RNA duplex structure formed by intermolecular mRNA–mRNA or mRNA–long noncoding RNA interactions. The recruited UPF1 interacts with PNRC2 and triggers rapid mRNA degradation (17–19). NMD and SMD are closely related pathways because both require UPF1 and both occur in a translation-dependent manner. HMD, another UPF1-dependent mRNA degradation pathway, also requires a translation event, as reviewed elsewhere (20).
Here, we demonstrate a novel mRNA decay pathway induced by ligand-bound GR loaded onto the 5′UTR of target mRNAs. We call this process GR-mediated mRNA decay (GMD). We show that GMD is a mechanistically unique pathway of UPF1-dependent mRNA decay because it occurs in a translation-independent manner. In addition, efficient joining of UPF1 to GMD machinery requires a glucocorticoid-induced GR–PNRC2 interaction. We also provide evidence that the regulation of GMD efficiency by a glucocorticoid is necessary for chemotaxis of human monocytes; these data are suggestive of the physiological importance of GMD.
Results
PNRC2 Interacts with GR in a Ligand-Dependent Manner, Recruiting UPF1 and Decapping Enzyme 1a to the GR Complex.
It has been reported that GR binds directly to a subset of mRNAs (21, 22). It has also been reported that GR binds to PNRC2 in a ligand-dependent manner (in a yeast two-hybrid system), albeit under nonphysiological conditions (8, 23). We have shown that PNRC2 physically interacts with UPF1 and decapping enzyme 1a (DCP1A; a component of the decapping complex), thereby triggering NMD (11). According to these previous reports, we hypothesized that GR is complexed with UPF1 and DCP1A via its direct interaction with PNRC2 in a ligand-dependent manner.
To test this possibility, we first looked for differences in the composition of the GR-containing complex either in the absence or in the presence of dexamethasone (Dex), which is a potent synthetic glucocorticoid (24). Immunoprecipitation (IP) experiments were performed using α-GR antibody and RNase A-treated total extracts of HEK293T cells transiently expressing MYC-PNRC2 and FLAG-UPF1. The cells were either pretreated or not treated with Dex before IP. The levels of transiently expressed MYC-PNRC2 and FLAG-UPF1 were comparable to, and lower than, those of endogenous PNRC2 and UPF1, respectively (Fig. S1). RT-PCR of endogenous GAPDH mRNA using α-[32P]-dATP and specific oligonucleotides demonstrated efficient removal of cellular RNAs by RNase A treatment before IPs (Fig. 1A, Lower). The IP results revealed that MYC-PNRC2, FLAG-UPF1, and endogenous DCP1A coimmunopurified with endogenous GR in nine-, seven-, and sixfold greater amounts, respectively, after Dex treatment (Fig. 1A, Upper). Other NMD factors, such as UPF2, UPF3X, and eIF4AIII, and an SMD factor, STAU1, were not detectably enriched in GR IP, regardless of Dex treatment. These results suggest that Dex treatment triggers efficient formation of a GR complex containing PNRC2, UPF1, and DCP1A.
Fig. 1.

GR associates with UPF1 and DCP1A via PNRC2. (A) IP experiments with endogenous GR. HEK293T cells were transiently cotransfected with plasmids expressing MYC-PNRC2 and FLAG-UPF1. Two days later, the cells were either treated or not treated with Dex for 3 h. Cell extracts were prepared and were either not treated or treated with RNase A for 15 min before IP. The IP experiments were performed using either α-GR antibody or a nonspecific mouse IgG (mIgG, control). The protein fractions before and after IPs were analyzed using Western blotting with the indicated antibodies. The levels of coimmunopurified MYC-PNRC2, FLAG-UPF1, and endogenous DCP1A were normalized to the level of immunopurified GR. The normalized levels obtained from IPs from the extracts of cells not treated with Dex were arbitrarily set to 1.0. For quantitative analysis, threefold serial dilutions of total cell extracts that were obtained before IP were loaded in the four leftmost lanes (Upper). Efficient digestion of cellular transcripts by RNase A treatment was demonstrated by the complete absence of cellular GAPDH mRNA according to RT-PCR using α-[32P]-dATP and specific oligonucleotides. For quantitative analysis, total cell RNA samples obtained before IP were serially diluted twofold and analyzed using RT-PCR. The RT-PCR products were loaded in the four leftmost lanes (Lower). (B and C) IP experiments of endogenous GR from the extracts of cells depleted of PNRC2. The cells were transiently transfected with either PNRC2 siRNA or a nonspecific control siRNA. Two days later, the cells were retransfected with a plasmid expressing FLAG-UPF1. One day later, the cells were treated with Dex for 3 h before IP. After that, cell extracts were prepared and treated with RNase A before IP. (B) A Western blot of PNRC2 demonstrating specific down-regulation by siRNA. (C) IP experiments using either the α-GR antibody or mIgG (control). The levels of coimmunopurified FLAG-UPF1 and endogenous DCP1A were normalized to the level of immunopurified GR. The normalized levels obtained from IPs from the extracts of undepleted cells were arbitrarily set to 1.0. All data represent at least two independently performed transfection experiments, IPs, and RT-PCRs.
Next, we evaluated the role of PNRC2 in the formation of the GR-containing complex. To this end, we conducted IP experiments using α-GR antibody and the extracts of either undepleted or PNRC2-depleted cells. The cells were pretreated with Dex before IP to stimulate formation of the GR-containing complex. Transfection of PNRC2 siRNA selectively reduced the level of endogenous PNRC2, 2% of the normal level (Fig. 1B). The IP results showed that after the PNRC2 down-regulation, FLAG-UPF1 and endogenous DCP1A coimmunopurified with GR in 5- and 10-fold smaller amounts, respectively (Fig. 1C), suggesting that PNRC2 stabilizes or serves as a link for the associations between GR and UPF1, and between GR and DCP1A. Because GR directly binds to the 5′UTR of specific mRNAs even in the absence of a ligand (21, 22), it is likely that mRNA-bound GR recruits PNRC2 or the PNRC2/UPF1/DCP1A complex to mRNA in the presence of a ligand.
GR-Mediated mRNA Decay Targets a Subset of Cellular mRNAs in a Manner Requiring PNRC2, UPF1, and GR.
The results shown in Fig. 1 led us to hypothesize that the recruitment of UPF1 and DCP1A to GR-bound mRNA through PNRC2 may elicit rapid mRNA degradation, which we termed GR-mediated mRNA decay. In support of this idea, it was reported that GR binds directly to the 5′UTR of chemokine (C-C motif) ligand 2 (CCL2) mRNA and CCL7 mRNA, and destabilizes these mRNAs in the presence of Dex (21, 22). To test the above hypothesis, we first searched for natural GMD substrates by performing microarray analysis using 48K human gene chips and total transcripts purified from HeLa cells that were either treated or not treated with Dex. The microarray analysis revealed that 147 and 77 transcripts were up-regulated and down-regulated, respectively, by at least 1.5-fold after Dex treatment (Dataset S1). The GMD substrates are expected to be down-regulated after Dex treatment. We next compared the list of transcripts down-regulated after Dex treatment with previously reported microarray data on transcripts coimmunoprecipitated with GR (22), and finally obtained four natural transcripts that were putative GMD substrates. Interestingly, among them was CCL2 mRNA, which is known to be rapidly degraded as a result of Dex treatment. The others were transcripts encoding B-Cell CLL/lymphoma 3 (BCL3), zinc finger SWIM-type containing 4 (ZSWIM4), and pleckstrin homology-like domain, family A, member 1 (PHLDA1).
We next tested whether the putative GMD substrates require PNRC2, UPF1, and GR for efficient decay. To this end, endogenous PNRC2, UPF1, and GR were down-regulated to 15%, 8%, and 2% of the normal level, respectively, using specific siRNA (Fig. S2 A and B). After that process, the levels and half-lives of all of the putative GMD substrates including CCL7 mRNA, whose expression is known to be reduced by Dex treatment (21, 22), were analyzed using quantitative RT-PCR (RT-qPCR). The results showed that the levels and half-lives of all of the putative GMD substrates were drastically decreased after Dex treatment and were significantly restored via the down-regulation of PNRC2, UPF1, or GR (Fig. 2 and Fig. S2 C and D). In contrast, the level and half-life of endogenous CCL5 mRNA, which lacks a GR-binding site in its 5′UTR (22), were not significantly affected by Dex treatment and by the down-regulation of PNRC2, UPF1, or GR. All these data indicate that these transcripts are bona fide substrates of GMD, which requires PNRC2, UPF1, and GR.
Fig. 2.

The half-lives of newly identified GMD substrates are increased after a down-regulation of PNRC2 or UPF1. HeLa cells were transiently transfected with the indicated siRNAs. Three days after the transfection, the cells were treated with DRB, which is a potent transcription inhibitor, and then treated with Dex. After that, total cell RNA and protein were prepared at the indicated time points after Dex treatment in the presence of DRB. Specific down-regulation by siRNAs was demonstrated by Western blotting (Fig. S2A). The levels of the mRNAs tested, which were normalized to endogenous GAPDH mRNA, were plotted as a function of time after Dex treatment. The normalized levels of mRNAs at 0 h were arbitrarily set to 100%. The dots and bars represent the mean and SD of two independent biological replicates.
GMD Requires Binding of GR to a Target mRNA in a Position-Independent Manner.
To gain molecular insight into GMD, we next tested the positional effect of GR binding to mRNA. For this purpose, we designed several reporter constructs in which the 5′UTR, 3′UTR, or both, of CCL2 mRNA, BCL3 mRNA, or PHLDA1 mRNA were inserted immediately upstream or downstream of the ORF of Renilla luciferase (RLuc) cDNA (Fig. 3A). The levels of reporter mRNAs were monitored using RT-qPCR. The results showed that Dex treatment reduced the levels of C5′-RLuc mRNA, B5′-RLuc mRNA, and P5′-RLuc mRNA, which contain the CCL2 5′UTR (C5′), BCL3 5′UTR (B5′), and PHLDA1 5′UTR (P5′), respectively, upstream of the RLuc ORF, to ∼40% of untreated levels (Fig. 3 B–E). The reduction was almost completely reversed after a down-regulation of GR, PNRC2, or UPF1 (Fig. 3 B–E). The level of C5′-RLuc-C3′ mRNA, which contains both the CCL2 5′UTR and CCL2 3′UTR (C3′) upstream and downstream, respectively, of the RLuc ORF, was also reduced after Dex treatment and restored by a down-regulation of PNRC2 or UPF1 (Fig. 3E). On the other hand, the level of RLuc-C3′ mRNA, which contains the CCL2 3′UTR downstream of the RLuc ORF, was not affected by Dex treatment and by down-regulation of PNRC2 or UPF1 (Fig. 3E), suggesting that the 5′UTR of GMD substrates is sufficient to trigger GMD.
Fig. 3.

Artificial GMD reporter mRNA harboring a GR-binding site is subject to efficient GMD in a way that depends on GR, PNRC2, and UPF1. (A) A schematic of GMD reporter RLuc constructs used in this study. See Results for details. A plasmid expressing FLuc served as a control of variation in transfection efficiency. C5′, CCL2 5′UTR; C3′, CCL2 3′UTR; B5′, BCL3 5′UTR; P5′, PHLDA1 5′UTR; SL, stem-loop structure; and Δ, a deletion of the GR-binding site. (B–E) The effects of a down-regulation of GR, UPF1, or PNRC2 on GMD reporter RLuc mRNAs. HeLa cells were transfected with the indicated siRNAs. One day later, the cells were transfected with a GMD reporter RLuc plasmid and a FLuc control plasmid. Two days later, the cells were either treated or not treated with Dex for 12 h before harvesting. (B and D) Western blots demonstrating specific down-regulation of GR, UPF1, or PNRC2. (C and E) RT-qPCR analysis of reporter RLuc mRNAs. The levels of GMD reporter RLuc mRNAs were normalized to the levels of FLuc mRNAs. The normalized levels of GMD reporter RLuc mRNAs without Dex treatment were arbitrarily set to 100%. (F) The effect of deletion of a GR-binding site and a positional effect of the GR-binding site. HeLa cells were transfected with the indicated GMD reporter RLuc plasmid and a FLuc control plasmid. Two days later, the cells were either treated or not treated with Dex for 12 h before harvesting. The levels of GMD reporter RLuc mRNAs were normalized as described in Fig. 3C. The columns and bars represent the mean and SD of three independent biological replicates. Two-tailed, equal-sample variance Student′s t tests were used to calculate the P values. **P < 0.01.
We further analyzed molecular features of GR binding to the 5′UTR of a GMD substrate. It is known that GR directly binds to the region spanning nucleotides 44–60 within the 5′UTR of CCL2 mRNA (22). Deletion of this region from C5′-RLuc mRNA significantly inhibited GMD [C5′(Δ)-RLuc mRNA] (Fig. 3F). We also designed RLuc-C5′ mRNA, which contains the CCL2 5′UTR downstream, rather than upstream, of the RLuc ORF. Intriguingly, the level of RLuc-C5′ mRNA was significantly reduced after Dex treatment to the extent observed in C5′-RLuc mRNA (Fig. 3F). The reduction was reversed by a down-regulation of GR, PNRC2, or UPF1, but not by a down-regulation of an NMD-specific factor, UPF2 (Fig. S3). These results suggest that loading of GR onto a GR-binding site within target mRNA is sufficient for efficient GMD and that the position of the GR-binding site relative to the ORF is not critical for GMD.
The Loading of GR onto Target mRNA and Binding of GR to a Ligand Are Crucial for GMD.
Previous UV cross-linking studies using in vitro-synthesized CCL2 mRNA and the extracts of cells either treated or not treated with Dex revealed that GR directly binds to a GR-binding site within CCL2 mRNA (21, 22). It may be possible, however, that Dex-induced signaling molecules or GR-related proteins other than GR itself are involved in the recognition of a GR-binding site and induce GMD within the cells. To determine whether the loading of GR alone onto a target mRNA is sufficient for GMD, we used an artificial tethering system, the bacteriophage λN-5BoxB system (Fig. 4A). The results showed that artificial tethering of λN-HA-GR, but not of λN-HA-GFP, to the 5′UTR of a reporter mRNA containing five tandem repeats of the bacteriophage BoxB sequence (5BoxB) triggered rapid degradation of the reporter mRNA in the presence of Dex, in a way that was completely reversed after down-regulation of PNRC2 and UPF1, but not of UPF2 (Fig. 4B). The Dex-induced rapid degradation of the reporter mRNA elicited by tethered λN-HA-GR was almost completely blocked by either a single amino acid substitution, V729I, which reduces the ligand-binding ability of GR (25), or by deletion of the ligand-binding domain (LBD) of GR (Fig. 4C). A specific down-regulation by siRNA and proper expression of the effectors were demonstrated by means of Western blotting (Fig. S4 A–C). These results suggest that the binding of GR to the 5′UTR is sufficient for eliciting GMD and that direct interaction between GR and its ligand is necessary for GMD.
Fig. 4.
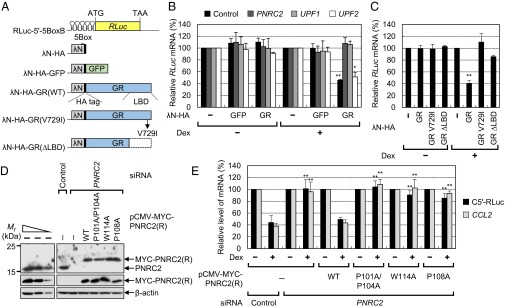
GMD requires a ligand-binding ability of GR and GR- and DCP1A-binding abilities of PNRC2. (A) A schematic diagram of (i) the tethering reporter construct, RLuc-5′-5BoxB, which contains five tandem repeats of 5BoxB in the 5′UTR, and (ii) the effector construct, which expresses a C-terminally HA-tagged bacteriophage λN polypeptide (λN-HA), λN-HA-GFP, λN-HA-GR, or λN-HA-GR variants. (B) Analysis of rapid mRNA degradation elicited by GR artificially tethered to the 5′UTR. HeLa cells were transiently transfected with the indicated siRNAs and, 1 d later, transfected with a tethering reporter plasmid, an effector plasmid, and pCI-F, which encodes FLuc cDNA and serves as a control for variation in transfection. Two days later, the cells were either treated or not treated with Dex for 30 min before harvesting. Specific down-regulation of PNRC2, UPF1, and UPF2 (Fig. S4A), and comparable expressions of λN-HA-GFP and λN-HA-GR (Fig. S4B) were validated by Western blotting. The levels of RLuc-5′-5BoxB mRNAs were normalized to the levels of FLuc mRNAs. Normalized levels of RLuc-5′-5BoxB mRNAs in the presence of λN-HA were set to 100%. (C) Effects of GR variants on mRNA degradation. Comparable expression of GR variants were determined by Western blotting (Fig. S4C). (D and E) Complementation experiments using siRNA-resistant PNRC2 variants. HEK293T cells were transiently transfected with either PNRC2 siRNA or a nonspecific control siRNA. One day after the transfection, the cells were cotransfected with three plasmids: (i) a plasmid expressing siRNA-resistant MYC-PNRC2(R) either WT or its variant (Fig. S4D), (ii) a GMD reporter plasmid expressing C5′-RLuc mRNA, and (iii) a reference plasmid, pCI-F. Two days later, the cells were either treated or not treated with Dex for 12 h before harvesting. (D) Western blotting of endogenous PNRC2 and MYC-PNRC2(R) with either WT or its variants. (E) RT-qPCR analysis of C5′-RLuc mRNA and endogenous CCL2 mRNA. The levels of C5′-RLuc mRNA and endogenous CCL2 mRNA were normalized to the levels of FLuc mRNAs and GAPDH mRNAs, respectively. The normalized levels of C5′-RLuc mRNA and endogenous CCL2 mRNA in the cells not treated with Dex were arbitrarily set to 100%. The columns and bars represent the mean and SD of three independent biological replicates. **P < 0.01, *P < 0.05.
An Interaction Between GR and DCP1A via PNRC2 Is Important for Efficient GMD.
Because the ligand-dependent interaction between GR and PNRC2 occurs via an SH3-binding motif of PNRC2 (7, 8), we further analyzed the role of the Dex-dependent GR–PNRC2 interaction in GMD using a complementation approach. First, we generated siRNA-resistant PNRC2 constructs [PNRC2(R)] (Fig. S4D) that expressed: (i) MYC-tagged PNRC2 WT; (ii) a double-mutant P101A/P104A, which has amino acid substitutions from proline to alanine at positions 101 and 104 located within the SH3-binding motif and renders PNRC2 unable to interact with GR (8) without affecting its interactions with UPF1 (10); (iii) W114A; and (iv) P108A, both of which lack the DCP1A-binding ability of PNRC2 (12). In this study, we also confirmed that the P101A/P104A mutant failed to interact with endogenous GR even in the presence of Dex, without affecting its interactions with endogenous DCP1A (Fig. S4 E and F). In addition, the W114A and P108A mutants failed to interact with endogenous DCP1A, without affecting the ability of PNRC2 to interact with FLAG-GR and FLAG-UPF1 (Fig. S4G). Furthermore, these mutants failed to associate with FLAG-tagged DCP2 (Fig. S4H), which is known to interact with DCP1A, suggesting that PNRC2 indirectly interacts with DCP2 via DCP1A. All these data demonstrate the binding specificity of PNRC2 variants.
Next, we conducted complementation experiments using HEK293T cells, PNRC2 siRNA, and the PNRC2(R) constructs (Fig. 4 D and E). When HeLa cells were transiently transfected with an excess amount of a plasmid expressing MYC-PNRC2(R), the expression level of MYC-PNRC2(R) was too low for a complementation experiment. Therefore, we used HEK293T instead of HeLa cells to achieve a sufficient level of MYC-PNRC2(R) expression that corresponded to the endogenous level of the PNRC2 protein.
Western blotting confirmed that transfection of PNRC2 siRNA reduced the level of endogenous PNRC2 to 4% of the level in untransfected cells, and that MYC-PNRC2(R)-WT, -P101A/P104A, -W114A, and -P108A were expressed at a level comparable to the endogenous PNRC2 level (Fig. 4D). Under the same conditions, the levels of C5′-RLuc reporter mRNA and endogenous CCL2 mRNA were analyzed using RT-qPCR (Fig. 4E). The results showed that Dex treatment reduced the levels of C5′-RLuc mRNA and endogenous CCL2 mRNA to 44% and 38%, respectively, of untreated levels and that the reductions were almost completely reversed by a PNRC2 down-regulation. Intriguingly, when MYC-PNRC2(R)-WT was expressed, efficient GMD of both C5′-RLuc mRNA and endogenous CCL2 mRNA was restored. On the other hand, comparable expression of MYC-PNRC2(R)-P101A/P104A, -W114A, or -P108A failed to restore GMD. These results suggest that the abilities of PNRC2 to interact with GR and DCP1A are necessary for efficient GMD.
GMD Is Mechanistically Distinct from NMD and SMD.
Because PNRC2 and UPF1 are commonly involved in NMD and SMD (10, 11) as well as in GMD (present study), it is possible that there is a cross-talk between NMD/SMD and Dex-mediated signaling or Dex-induced GMD. To first determine whether NMD and SMD are affected by Dex treatment, we assessed the changes in expression of NMD and SMD factors and measured the efficiencies of NMD and SMD before and after Dex treatment (Fig. S5). The results revealed that the abundances of all tested NMD and SMD factors were not significantly changed after Dex treatment. In addition, NMD and SMD efficiencies were not significantly affected by Dex treatment (see SI Results for more details).
Conversely, we also tested the effect of an NMD-specific factor on GMD. To this end, UPF1 (a common factor for GMD and NMD), GR (a GMD-specific factor), and UPF2 and UPF3X (both of which are known NMD factors) were down-regulated using specific siRNA (Fig. 5 A and B). The results of RT-qPCR revealed that the levels of all tested endogenous GMD substrates, but not of endogenous CCL5 mRNA, which lacks a GR-binding site (22), were drastically reduced upon Dex treatment and restored by a down-regulation of GR or UPF1, but not of UPF2 or UPF3X (Fig. 5 A and B).
Fig. 5.

GMD is mechanistically distinct from NMD. (A and B) Effects of NMD factors on GMD. HeLa cells were transfected with the indicated siRNAs, and endogenous GMD substrates were analyzed using RT-qPCR. The levels of endogenous GMD substrates were normalized to levels of endogenous GAPDH mRNA. The normalized levels of endogenous GMD substrates in Dex-untreated cells were arbitrarily set to 100%. Specific down-regulation by siRNAs was demonstrated by Western blotting (Fig. S5 G and H). (C and D) A test of translation independence of GMD. HeLa cells were transfected with the indicated GMD reporter RLuc plasmid and the FLuc control plasmid. Two days later, the cells were either treated or not treated with Dex for 12 h before harvesting. (C) Translation efficiency of GMD reporter RLuc mRNAs. The protein activities of RLuc translated from GMD reporter mRNAs were normalized to the levels of the GMD reporter RLuc mRNAs. The normalized activities of RLuc translated from C5′-RLuc mRNA or RLuc-C5′ mRNA were arbitrarily set to 100%. (D) RT-qPCR analysis of GMD reporter RLuc mRNAs. The levels of RLuc mRNA were normalized to the levels of FLuc mRNAs. The normalized levels of RLuc mRNA in Dex-untreated cells were arbitrarily set to 100%. The columns and bars represent the mean and SD of three independent biological replicates. **P < 0.01, *P < 0.05.
It is known that NMD and SMD are coupled to translation (17, 26). Given that GMD shares common factors UPF1 and PNRC2 with NMD and SMD, it is likely that GMD is also dependent on translation. To test this possibility, we designed SL-C5′-RLuc and SL-RLuc-C5′ reporter constructs that harbor a stem-loop (SL) structure with ΔG = −87.8 kcal/mol (Fig. 3A). It is known that an insertion of a stable SL structure (ΔG = −75 kcal/mol) into the 5′UTR blocks ribosome scanning and causes drastic inhibition of translation (27–29). Consistent with previous studies, translation efficiencies of SL-C5′-RLuc mRNA and SL-RLuc-C5′ mRNA were drastically inhibited compared with translation efficiencies of C5′-RLuc mRNA and RLuc-C5′ mRNA (Fig. 5C). Nevertheless, the GMD efficiencies of SL-C5′-RLuc mRNA and SL-RLuc-C5′ mRNA were comparable to those of C5′-RLuc mRNA and RLuc-C5′ mRNA, respectively (Fig. 5D). It should be noted that when the same SL structure is inserted into the 5′UTR of NMD reporter mRNA, NMD and translation of the reporter mRNA are inhibited by ∼3- and 10-fold, respectively (29). These unexpected results suggest that GMD is independent of a translation event. All of the data shown in Fig. 5 indicate that although all three pathways share common factors UPF1 and PNRC2, GMD is mechanistically distinct from NMD and SMD.
GR Bound to a Target mRNA Recruits PNRC2 to the Target mRNA in a Ligand-Dependent Manner to Elicit GMD.
We next characterized the molecular hierarchy involved in the recruitment of GMD factors to mRNA. We performed IPs using antibody against a GMD factor and the extracts of cells either depleted or not depleted of GR, PNRC2, or UPF1 (Fig. 6 and Fig. S6). After that, the levels of coimmunoprecipitated endogenous GMD substrates were analyzed using RT-qPCR. Specific IPs and selective down-regulations by siRNAs were demonstrated by Western blotting (Fig. S6). The results showed that in IP of GR, endogenous GMD substrates (CCL2 mRNA, PHLDA1 mRNA, and BCL3 mRNA) were enriched approximately twofold in a way that was unaffected by Dex treatment (Fig. 6A), indicating that GR is loaded onto GMD substrates independently of a ligand. On the other hand, lower amounts of the endogenous GMD substrates coimmunopurified with either MYC-PNRC2 or FLAG-UPF1 relative to GR, when the cells were not treated with Dex (Fig. 6A). Dex treatment, however, significantly increased the amounts of the coimmunopurified GMD substrates in the IPs of MYC-PNRC2 or FLAG-UPF1 (Fig. 6A). The enrichments of GMD substrates in the IPs of FLAG-UPF1 and MYC-PNRC2 were significantly reversed by down-regulation of GR or PNRC2 (Fig. 6B) and by down-regulation of GR or UPF1 (Fig. 6C), respectively. Consistent with a recent report showing that UPF1 promiscuously binds to mRNA (30), the CCL5 mRNA and N-acetyltransferase 9 (NAT9) mRNA, both of which are not targeted for GMD, were also enriched in the IP of FLAG-UPF1, but not in the IP of GR or MYC-PNRC2 (Fig. 6A). However, this enrichment was not affected by Dex treatment. All data suggest that ligand-free GR loaded onto a GR-binding site within mRNA binds to a ligand and recruits PNRC2 and UPF1 to the mRNA.
Fig. 6.

GR bound to mRNA recruits PNRC2 and UPF1 to the target mRNA and the GR-PNRC2 association is stabilized by UPF1. (A) RT-qPCR of coimmunoprecipitated endogenous GMD substrates in IPs of endogenous GR, MYC-PNRC2, and FLAG-UPF1. HeLa Cells were either transfected or not transfected with plasmid expressing MYC-PNRC2 or FLAG-UPF1. Two days later, the cells were either treated or not treated with Dex for 3 h. IP experiments were performed using α-GR, α-MYC, α-FLAG antibody, or a nonspecific mouse IgG (mIgG, control). IP specificity was demonstrated by Western blotting (Fig. S6 A–C). The levels of coimmunoprecipitated GMD substrates relative to GAPDH mRNA were normalized to the levels of the input amount (GMD substrates relative to GAPDH mRNA before IP). The normalized levels of GMD substrates obtained in IPs using mIgG without Dex treatment were arbitrarily set to 1.0. (B) RT-qPCR of coimmunoprecipitated endogenous CCL2 mRNA in IPs of FLAG-UPF1. As performed in A, except that HeLa cells were depleted or not depleted of GR or PNRC2. The cells were treated with Dex for 3 h before IP. Specific down-regulation (Fig. S6D) and IP specificity (Fig. S6E) were demonstrated by Western blotting. (C) RT-qPCR of coimmunoprecipitated endogenous CCL2 mRNA in IPs of MYC-PNRC2. As performed in A, except that HeLa cells were depleted or not depleted of GR or UPF1. The cells were treated with Dex for 3 h before IP. Specific down-regulation (Fig. S6 F and H) and IP specificity (Fig. S6 G and I) were demonstrated by Western blotting. (D and E) IP of endogenous GR using the extracts of cells depleted or not depleted of UPF1. HeLa cells, either depleted or not depleted of UPF1, were transiently transfected with the plasmid expressing MYC-PNRC2. The cells were treated with Dex for 3 h before IP. After that, the cell extracts were treated with RNase A. IPs were performed using α-GR antibody or mIgG. (D) A Western blot demonstrating specific down-regulation of UPF1. (E) IPs using either an antibody against endogenous GR or nonspecific mIgG. The columns and bars represent the mean and SD of two or three independent biological replicates. **P < 0.01; *P < 0.05.
UPF1 Facilitates the Interaction Between GR and PNRC2.
With regard to mRNA degradation, the recruitment of PNRC2 to the GR-binding site via GR may be sufficient for mRNA degradation because PNRC2 interacts directly with DCP1A (11) and stabilizes the interaction between DCP1A and DCP2 (12). If so, why does GMD require UPF1? To address this question, we performed IPs using extracts of the cells either depleted or not depleted of UPF1 (Fig. 6D). The cells were pretreated with Dex before IP to induce efficient formation of the GR-containing complex. The results of IP using α-GR antibody showed that MYC-PNRC2 coimmunopurified with GR in 10-fold smaller amounts when UPF1 was down-regulated (Fig. 6E). The reciprocal IP using MYC-PNRC2 also showed decreased association between GR and MYC-PNRC2 after UPF1 down-regulation (Fig. S6 J and K). All of these results indicate that UPF1 promotes an interaction between GR and PNRC2, which leads to more efficient GMD.
Helicase Activity of UPF1, but Not Phosphorylation of UPF1, Is Required for Efficient GMD.
The above findings that UPF1 facilitates the interaction between GR and PNRC2 led us to examine which abilities of UPF1 are involved in GMD. First, to test a role of helicase activity of UPF1 in GMD, we performed the complementation experiments using UPF1 siRNA and siRNA-resistant MYC-UPF1-WT or -R843C, which lacks helicase activity (31, 32), under the modified conditions (Fig. 7 A and B). In Fig. 2, we observed that the half-lives of endogenous CCL2 and CCL7 mRNAs were increased to almost normal levels upon UPF1 down-regulation. On the other hand, steady-state levels of endogenous CCL2 and CCL7 mRNAs were moderately increased by UPF1 down-regulation (Fig. 5A). One critical difference between the half-life and steady-state experiments was the treatment with a transcription inhibitor 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (DRB) in the half-life experiment. The simple interpretation for the difference in the extent of GMD inhibition by UPF1 down-regulation is that Dex may be involved in the transcription of the CCL2 gene, as well as GMD of CCL2 mRNA. In support of this idea, down-regulation of UPF1 almost completely inhibited GMDs of artificially designed reporter mRNAs, the transcription of which is driven by the CMV promoter (Figs. 3 C and E and 4B). Therefore, to minimize the possible effect of Dex on transcription of CCL2 gene although GMD efficiency is reduced as well, the incubation time with Dex was reduced to 1 h in complementation experiments.
Fig. 7.

GMD requires a helicase activity but not phosphorylation of UPF1. (A and B) Complementation experiments using siRNA-resistant UPF1-WT and its R843C variant. HeLa cells were transiently transfected with either UPF1 siRNA or a nonspecific control siRNA. One day after the transfection, the cells were retransfected with a plasmid expressing siRNA-resistant MYC-UPF1(R)-WT or -R843C. Two days later, the cells were either treated or not treated with Dex for 1 h before harvesting. (A) Western blots demonstrating selective down-regulation of UPF1 and comparable levels of exogenously expressed UPF1(R) and endogenous UPF1. (B) RT-qPCR analysis of endogenous CCL2 mRNA. The levels of endogenous CCL2 mRNA were normalized to the levels of endogenous GAPDH mRNAs. The normalized levels of endogenous CCL2 mRNA in the cells not treated with Dex were arbitrarily set to 100%. (C–E) GMD is not dependent on SMG1 activity. HeLa cells were transiently transfected with GR siRNA, SMG1 siRNA, or a nonspecific control siRNA. Three days later, the cells were either treated or not treated with Dex for 3 h before harvesting. (C) Western blots demonstrating selective down-regulation of GR and SMG1 by siRNAs. (D) RT-qPCR analysis of endogenous CCL2, CCL7, and CCL5 mRNAs. The levels of endogenous GMD substrates were normalized to the levels of endogenous GAPDH mRNAs. (E) RT-qPCR analysis of endogenous NAT9 and SC35 (1.6 kb) mRNAs. The columns and bars represent the mean and SD of three independent biological replicates. **P < 0.01; *P < 0.05.
The results of complementation experiments using UPF1 siRNA and siRNA-resistant MYC-UPF1 revealed that Dex treatment for 1 h reduced the level of endogenous CCL2 mRNA to ∼50% of the untreated level, and that down-regulation of UPF1 significantly inhibited the GMD of CCL2 mRNA (Fig. 7 A and B). Such inhibition was significantly reversed by expression of UPF1-WT but not of UPF1-R843C, suggesting that helicase activity of UPF1 is involved in efficient GMD.
Next, we asked if UPF1 phosphorylation contributes to GMD (Fig. 7 C–E). In the cases of NMD and SMD, it is well known that the phosphorylation of UPF1 by SMG1 kinase is critical for target mRNA degradation (33–36). Consistent with previous reports, down-regulation of SMG1 significantly inhibited the NMDs of endogenous NAT9 and SC35 (1.6 kb) mRNAs. On the other hand, down-regulation of SMG1 did not significantly affect the GMDs of endogenous CCL2 and CCL7 mRNAs, suggesting that GMD occurs independently of UPF1 phosphorylation. In contrast, down-regulation of GR selectively inhibited GMD but not NMD. All these results strengthen our conclusion that GMD is mechanistically distinct from NMD and SMD (Fig. 5).
GMD Controls Chemotaxis of Monocytes by Regulating CCL2 mRNA Stability.
It is known that the CCL2 protein belongs to the CC chemokine family and is involved in chemotaxis of various immune cells toward a site of injury or infection (37). Because we found that GMD of CCL2 mRNA is dependent on PNRC2, UPF1, and GR in this study, we tested whether CCL2-mediated chemotaxis is influenced by PNRC2, UPF1, and GR. Accordingly, we first measured the protein levels of secreted CCL2 using an ELISA. Dex treatment reduced the protein levels of secreted CCL2 to ∼50% of the untreated control (Fig. 8B and Fig. S7B). The reduced protein levels were restored when PNRC2, UPF1, and GR were down-regulated using specific siRNAs to 16%, 17%, and 9% of normal levels, respectively (Fig. 8A and Fig. S7A). On the other hand, the reduced protein level of CCL2 was not restored by the UPF2 down-regulation (Fig. S7B).
Fig. 8.

Chemotaxis of monocytes is mediated by GMD. The lower and upper compartments of each well in a chemotaxis microchamber were separated on a polyvinylpyrrolidone-treated membrane with 5-μm pores. The lower compartments of wells were filled with the supernatants of HeLa cells depleted of either PNRC2 or UPF1 and either not treated or treated with Dex for 3 h. The upper compartments were filled with THP-1 cells (a monocyte cell line). After incubation of the microchamber for 3 h at 37 °C, the membranes were removed from the wells. Then, the migrating cells that adhered to the underside of the membranes were stained and counted under a microscope in six randomly selected visual fields in each well. (A) A Western blot of PNRC2 and UPF1, demonstrating specific down-regulation by siRNAs. (B) ELISA for the CCL2 protein concentration in the supernatants of the HeLa cells depleted of either PNRC2 or UPF1. The levels of CCL2 protein were normalized to the levels of total protein in the supernatants. The normalized levels of CCL2 without Dex treatment were arbitrarily set to 100%. (C) Analysis of chemotaxis of THP-1 cells. The migrating cells were stained and images were acquired to demonstrate the effects of either the PNRC2 or UPF1 down-regulation on cell migration. (Scale bars, 20 μm.) (D) The chemotactic index. The stained cells were counted under a microscope in six randomly selected visual fields in each well to determine the chemotactic index. The numbers of stained cells without Dex treatment in each visual field were arbitrarily set to 100%. The columns and bars represent the mean and SD of at least three independent transfection experiments and ELISA or chemotaxis assay. **P < 0.01. (E) A model of GMD-mediated chemotaxis. In the absence of a glucocorticoid, GMD is inactive and its target mRNA (CCL2 mRNA) is stable and abundant. Consequently, the expressed CCL2 protein induces chemoattraction of monocytes (Left). In the presence of a glucocorticoid, GMD is activated, destabilizes CCL2 mRNA, and thereby causes inefficient chemoattraction of monocytes (Right). The details are described in Discussion.
We next assessed the effects of the down-regulation of PNRC2, UPF1, or GR on cell migration in a chemotaxis microchamber that had two compartments separated by a polyvinylpyrrolidone-treated membrane with 5-μm pores. The lower compartments of wells were filled with a supernatant (medium) from cells that were either treated or not treated with Dex, and were either depleted or not depleted of PNRC2, UPF1, or GR. Human acute monocytic leukemia cell line THP-1 (expressing CCL2 receptors on the plasma membrane) was seeded in the upper compartments of wells. When THP-1 cells sense CCL2 that diffuses from the lower compartment, they are expected to migrate toward the lower compartment and to consequently adhere to the membrane. The number of migrating cells (cells adherent to the membrane) was counted to determine the chemotactic index. The results revealed that cell migration was decreased by 50–60% when the lower compartments of wells contained supernatants from the cells treated with Dex (Fig. 8 C and D and Fig. S7 C and D). Intriguingly, the rate of cell migration was restored by a down-regulation of PNRC2, UPF1, or GR, but not by UPF2 (Fig. 8 C and D and Fig. S7 C and D). These results suggest that GMD plays an important role in the chemotaxis of monocytes via regulation of CCL2 mRNA stability.
Discussion
In this study, we demonstrate a new mRNA decay pathway, GMD, which is induced by a ligand (glucocorticoid) and requires GR, PNRC2, UPF1, and DCP1A. According to our findings, we propose the following molecular details of GMD. GR can bind to a GR-binding site of a target mRNA even in the absence of a ligand (Fig. 8E, Left). In the presence of a ligand (Fig. 8E, Right), on the other hand, the ligand binds to GR, which is preloaded onto a target mRNA, and the resulting ligand-bound GR would recruit PNRC2 and eventually DCP1A and UPF1, both of which directly interact with PNRC2 (11). The resulting complex may be displaced by the scanning ribosome. Nonetheless, the complex may quickly reassociate with 5′UTR because of the intrinsic RNA-binding ability of GR (22). The recruited UPF1 may transiently trigger a remodeling of messenger ribonucleoprotein particle (mRNP) via its helicase activity (Fig. 7 A and B), consequently further stabilizing the GR-PNRC2 interaction (Fig. 6 D and E and Fig. S6 J and K). Then, the resulting complex at the GR-binding site would trigger decapping followed by 5′-to-3′ degradation of the mRNA. The recruited UPF1 may also repress translation initiation before mRNA degradation, as observed in NMD (38).
In the case of NMD, it has been reported that UPF1 helicase activity is promoted by UPF2 (39–41) and that SMG1 provides a binding platform for UPF1 and UPF2, resulting in the activation of UPF1 helicase activity (42). However, we observed that UPF2 and SMG1 are not essential for efficient GMD (Figs. 4B, 5B, and 7 C–E). Therefore, it is most likely that other cellular factors rather than UPF2 may control the helicase ability of UPF1 during GMD. In addition, another kinase rather than SMG1 may act to activate UPF1 in GMD. Indeed, UPF1 is known to be phosphorylated by ATR and DNA-PK during HMD (43, 44).
Recently, genome-wide analysis showed that UPF1 associates with mRNAs throughout the entire sequences and is displaced from mRNA by an elongating ribosome (30). Therefore, it is possible that the ligand-bound GR-PNRC2 complex bound to the GR-binding site of mRNA recruits either a preexisting UPF1 within close proximity of the GR-binding site or mRNA-free UPF1. Our data demonstrate that the latter mechanism is predominant because the amount of coimmunopurified GMD substrate in the IP of UPF1 is significantly increased by Dex treatment (Fig. 6A). The resulting complex elicits efficient GMD, possibly in the 5′-to-3′ direction because of the enhancement of decapping by DCP1A. The possible 3′-to-5′ degradation of a GMD substrate remains to be tested. Consequently, GMD substrates would be down-regulated, affecting various physiological and metabolic pathways, such as chemotaxis of monocytes (Fig. 8).
Other groups as well as ours have shown that the C terminus of PNRC2 commonly interacts with GR, DCP1A, and UPF1, although the critical residues in the C terminus involved in the interactions between PNRC2 and its binding partners are characterized (8, 10, 11, 23). Because of the common binding site, once one or two binding partners interact with PNRC2, the third binding partner may not be easily recruited to the complex because of steric hindrance. In the present study, however, we showed that PNRC2 is complexed with DCP1A, UPF1, and GR. Therefore, one possible explanation is that the steric hindrance would be overcome by PNRC2 dimerization, which may provide a sufficient binding platform for loading of its binding partners. In support of this idea, it has recently been reported that PNRC1, an isoform of PNRC2, forms a dimer (45).
It is generally considered that cytosolic GR enters the nucleus when it binds to a ligand. However, we observed that although most of the cytosolic GR moved to the nuclear fraction upon Dex treatment, a significant amount of GR remained in the cytoplasmic fractions (Fig. S8). Moreover, the intracellular distribution of PNRC2 was only marginally affected by Dex treatment. Therefore, the GR and PNRC2 remaining in the cytoplasm upon Dex treatment might be sufficient for eliciting GMD.
Here, we also demonstrate that GMD is mechanistically distinct from other UPF1-dependent mRNA decay pathways—NMD, SMD, and HMD—although all these pathways share some common factors: UPF1, PNRC2, and DCP1A. One major difference is the method of recruitment of UPF1 to the working site on mRNA and the method of stabilization of the UPF1-containing complex. In the case of NMD, either mRNA-unbound or prebound UPF1 is recruited to a terminating ribosome and is stabilized by an exon junction complex downstream of PTC. In the case of SMD and HMD, UPF1 is also recruited to a terminating ribosome during translation and is stabilized by downstream STAU and SL binding protein, respectively. Alternatively, UPF1 could be recruited to mRNA via direct interaction with STAU or SL binding protein and then could join the termination complex. Therefore, NMD, SMD, and HMD are tightly coupled to translation events, although the exact timing of UPF1 recruitment is not clear at this point. In the case of GMD, however, UPF1 recruitment to the working site on mRNA occurs independently of translation (Fig. 5 C and D). Instead, the UPF1 recruitment is mediated by its direct interaction with PNRC2, which bridges mRNA-bound GR and UPF1 in a Dex-dependent manner. In addition, GMD is not affected by down-regulation of known NMD-specific factors (Fig. 5). Therefore, GMD is a previously unidentified translation-independent and UPF1-dependent mRNA decay pathway.
Another novel feature of GMD is that the presence, rather than a relative position, of the GR-binding site within mRNA is crucial for eliciting GMD after Dex treatment. Nonetheless, all GMD substrates tested in this study were shown to contain a functional GR-binding site in the 5′UTR (Fig. 3). Although more GMD substrates should be tested, it seems that there is an evolutionary selective pressure to maintain the GR-binding site in the 5′UTR. This observation points to the possibility of an additional regulatory mechanism of gene expression through the GR-binding site, aside from GMD. A genome-wide survey should help to uncover a number of additional natural GMD substrates and will identify further molecular details and biological relevance of GMD.
Materials and Methods
Construction of Plasmids.
The details of plasmid construction are provided in SI Materials and Methods.
Cell Culture, Transfection, and the siRNA Sequences.
The details of cell culture, transfection, and the siRNA sequences are provided in SI Materials and Methods.
The Tethering Assay.
HeLa cells were transiently cotransfected with 0.1 μg of pRL-5′-5BoxB, 0.2 μg of an effector plasmid, and 0.01 μg of pCI-F using Lipofectamine 2000 (Invitrogen). Two days later, the cells were treated with 100 nM of Dex for 30 min. The cells were then harvested, and total cell RNA and protein were purified using TRIzol Reagent (Life Technologies).
Quantitative Real-Time RT-PCR and Semiquantitative RT-PCR Using α-[32P]-dATP and Specific Oligonucleotides.
RT-qPCR analyses were performed as described previously (10, 11, 46). The oligonucleotides used in our study are listed in Dataset S2.
RT-PCR using α-[32P]-dATP and specific oligonucleotides was performed as described previously (11, 17). The 32P-labeled PCR products were analyzed using 5% polyacrylamide gel electrophoresis, visualized with PhosphorImaging (BAS-2500; Fuji Photo Film), and then quantitated using MultiGauge (Fuji Photo Film).
Immunoprecipitation.
HeLa cells and HEK293T cells were transiently transfected with the indicated plasmids using Lipofectamine 2000 and the calcium phosphate precipitation method, respectively. The IPs and RNA IPs were performed as described previously (9, 11, 46).
Western Blotting.
Antibodies against the following proteins or peptides were used in this study: FLAG and β-actin (Sigma-Aldrich); MYC (Calbiochem), UPF1 (a gift from Lynne E. Maquat, University of Rochester, Rochester, NY), PNRC2 (11), and human STAU1 (47); DCP1A, UPF2, eIF4AIII, phospho-S1078-UPF1, and phospho-S1096-UPF1 (10); UPF3X (48), eIF4E (Cell Signaling Technology), CBP80 (49), CBP80/20-dependent translation initiation factor (CTIF; ref. 46), GR (BD Biosciences and Santa Cruz Biotechnology), snRNP70 (Santa Cruz), and SMG1 (Bethyl). The antibody against UPF1 phosphorylated at threonine 28 was raised in rabbits using the synthetic peptide LGADpTQGSEF (AbClon).
Microarray Analysis.
Microarray analysis was performed by Macrogen, as described previously (9, 10, 50). The microarray data were deposited in the National Center for Biotechnology Information Gene Expression Omnibus web-based data repository (series ID: GSE49591).
CCL2 Measurements and Chemotaxis Assay.
The details of CCL2 measurements and the chemotaxis assay are provided in SI Materials and Methods.
Statistical Analysis.
Two-tailed, equal-sample variance Student’s t-tests were used to calculate the P-values. Differences with P < 0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
We thank Dr. Lynne E. Maquat for providing nonsense-mediated mRNA decay reporter plasmids and the α-UPF1 antibody; Jens Lykke-Andersen for pcDNA3-FLAG-DCP2; and Juan Ortín for α-human STAU1 antibody. This work was supported by the National Research Foundation of Korea grant funded by the Korea government (MSIP) (2012R1A2A1A01002469 and 2014R1A2A1A11050412); O.H.P. was in part supported by the Global PhD Fellowship Program through the National Research Foundation funded by the Korean Government.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. A.J. is a guest editor invited by the Editorial Board.
Data deposition: The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE49591).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1409612112/-/DCSupplemental.
References
- 1.Vandevyver S, Dejager L, Libert C. On the trail of the glucocorticoid receptor: Into the nucleus and back. Traffic. 2012;13(3):364–374. doi: 10.1111/j.1600-0854.2011.01288.x. [DOI] [PubMed] [Google Scholar]
- 2.Oakley RH, Cidlowski JA. Cellular processing of the glucocorticoid receptor gene and protein: new mechanisms for generating tissue-specific actions of glucocorticoids. J Biol Chem. 2011;286(5):3177–3184. doi: 10.1074/jbc.R110.179325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Santos GM, Fairall L, Schwabe JW. Negative regulation by nuclear receptors: A plethora of mechanisms. Trends Endocrinol Metab. 2011;22(3):87–93. doi: 10.1016/j.tem.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lonard DM, O’Malley BW. Nuclear receptor coregulators: Modulators of pathology and therapeutic targets. Nat Rev Endocrinol. 2012;8(10):598–604. doi: 10.1038/nrendo.2012.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kato S, Yokoyama A, Fujiki R. Nuclear receptor coregulators merge transcriptional coregulation with epigenetic regulation. Trends Biochem Sci. 2011;36(5):272–281. doi: 10.1016/j.tibs.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 6.Heery DM, Kalkhoven E, Hoare S, Parker MG. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature. 1997;387(6634):733–736. doi: 10.1038/42750. [DOI] [PubMed] [Google Scholar]
- 7.Zhou D, et al. PNRC: A proline-rich nuclear receptor coregulatory protein that modulates transcriptional activation of multiple nuclear receptors including orphan receptors SF1 (steroidogenic factor 1) and ERRalpha1 (estrogen related receptor alpha-1) Mol Endocrinol. 2000;14(7):986–998. doi: 10.1210/mend.14.7.0480. [DOI] [PubMed] [Google Scholar]
- 8.Zhou D, Chen S. PNRC2 is a 16 kDa coactivator that interacts with nuclear receptors through an SH3-binding motif. Nucleic Acids Res. 2001;29(19):3939–3948. doi: 10.1093/nar/29.19.3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cho H, et al. SMG5-PNRC2 is functionally dominant compared with SMG5-SMG7 in mammalian nonsense-mediated mRNA decay. Nucleic Acids Res. 2013;41(2):1319–1328. doi: 10.1093/nar/gks1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cho H, et al. Staufen1-mediated mRNA decay functions in adipogenesis. Mol Cell. 2012;46(4):495–506. doi: 10.1016/j.molcel.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 11.Cho H, Kim KM, Kim YK. Human proline-rich nuclear receptor coregulatory protein 2 mediates an interaction between mRNA surveillance machinery and decapping complex. Mol Cell. 2009;33(1):75–86. doi: 10.1016/j.molcel.2008.11.022. [DOI] [PubMed] [Google Scholar]
- 12.Lai T, et al. Structural basis of the PNRC2-mediated link between mrna surveillance and decapping. Structure. 2012;20(12):2025–2037. doi: 10.1016/j.str.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 13.Choe J, Ahn SH, Kim YK. The mRNP remodeling mediated by UPF1 promotes rapid degradation of replication-dependent histone mRNA. Nucleic Acids Res. 2014;42(14):9334–9349. doi: 10.1093/nar/gku610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karam R, Wengrod J, Gardner LB, Wilkinson MF. Regulation of nonsense-mediated mRNA decay: implications for physiology and disease. Biochim Biophys Acta. 2013;1829(6-7):624–633. doi: 10.1016/j.bbagrm.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schoenberg DR, Maquat LE. Regulation of cytoplasmic mRNA decay. Nat Rev Genet. 2012;13(4):246–259. doi: 10.1038/nrg3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schweingruber C, Rufener SC, Zünd D, Yamashita A, Mühlemann O. Nonsense-mediated mRNA decay—Mechanisms of substrate mRNA recognition and degradation in mammalian cells. Biochim Biophys Acta. 2013;1829(6-7):612–623. doi: 10.1016/j.bbagrm.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 17.Kim YK, Furic L, Desgroseillers L, Maquat LE. Mammalian Staufen1 recruits Upf1 to specific mRNA 3’UTRs so as to elicit mRNA decay. Cell. 2005;120(2):195–208. doi: 10.1016/j.cell.2004.11.050. [DOI] [PubMed] [Google Scholar]
- 18.Park E, Maquat LE. Staufen-mediated mRNA decay. Wiley Interdiscip Rev RNA. 2013;4(4):423–435. doi: 10.1002/wrna.1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gong C, Maquat LE. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature. 2011;470(7333):284–288. doi: 10.1038/nature09701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marzluff WF, Wagner EJ, Duronio RJ. Metabolism and regulation of canonical histone mRNAs: Life without a poly(A) tail. Nat Rev Genet. 2008;9(11):843–854. doi: 10.1038/nrg2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dhawan L, Liu B, Blaxall BC, Taubman MB. A novel role for the glucocorticoid receptor in the regulation of monocyte chemoattractant protein-1 mRNA stability. J Biol Chem. 2007;282(14):10146–10152. doi: 10.1074/jbc.M605925200. [DOI] [PubMed] [Google Scholar]
- 22.Ishmael FT, et al. The human glucocorticoid receptor as an RNA-binding protein: Global analysis of glucocorticoid receptor-associated transcripts and identification of a target RNA motif. J Immunol. 2011;186(2):1189–1198. doi: 10.4049/jimmunol.1001794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou D, Ye JJ, Li Y, Lui K, Chen S. The molecular basis of the interaction between the proline-rich SH3-binding motif of PNRC and estrogen receptor alpha. Nucleic Acids Res. 2006;34(20):5974–5986. doi: 10.1093/nar/gkl764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schäcke H, Döcke WD, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther. 2002;96(1):23–43. doi: 10.1016/s0163-7258(02)00297-8. [DOI] [PubMed] [Google Scholar]
- 25.Malchoff DM, et al. A mutation of the glucocorticoid receptor in primary cortisol resistance. J Clin Invest. 1993;91(5):1918–1925. doi: 10.1172/JCI116410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ishigaki Y, Li X, Serin G, Maquat LE. Evidence for a pioneer round of mRNA translation: mRNAs subject to nonsense-mediated decay in mammalian cells are bound by CBP80 and CBP20. Cell. 2001;106(5):607–617. doi: 10.1016/s0092-8674(01)00475-5. [DOI] [PubMed] [Google Scholar]
- 27.Kozak M. Influences of mRNA secondary structure on initiation by eukaryotic ribosomes. Proc Natl Acad Sci USA. 1986;83(9):2850–2854. doi: 10.1073/pnas.83.9.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vassilenko KS, Alekhina OM, Dmitriev SE, Shatsky IN, Spirin AS. Unidirectional constant rate motion of the ribosomal scanning particle during eukaryotic translation initiation. Nucleic Acids Res. 2011;39(13):5555–5567. doi: 10.1093/nar/gkr147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choe J, et al. eIF4AIII enhances translation of nuclear cap-binding complex-bound mRNAs by promoting disruption of secondary structures in 5’UTR. Proc Natl Acad Sci USA. 2014;111(43):E4577–E4586. doi: 10.1073/pnas.1409695111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zünd D, Gruber AR, Zavolan M, Mühlemann O. Translation-dependent displacement of UPF1 from coding sequences causes its enrichment in 3′ UTRs. Nat Struct Mol Biol. 2013;20(8):936–943. doi: 10.1038/nsmb.2635. [DOI] [PubMed] [Google Scholar]
- 31.Sun X, Perlick HA, Dietz HC, Maquat LE. A mutated human homologue to yeast Upf1 protein has a dominant-negative effect on the decay of nonsense-containing mRNAs in mammalian cells. Proc Natl Acad Sci USA. 1998;95(17):10009–10014. doi: 10.1073/pnas.95.17.10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kurosaki T, et al. A post-translational regulatory switch on UPF1 controls targeted mRNA degradation. Genes Dev. 2014;28(17):1900–1916. doi: 10.1101/gad.245506.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ohnishi T, et al. Phosphorylation of hUPF1 induces formation of mRNA surveillance complexes containing hSMG-5 and hSMG-7. Mol Cell. 2003;12(5):1187–1200. doi: 10.1016/s1097-2765(03)00443-x. [DOI] [PubMed] [Google Scholar]
- 34.Yamashita A, Kashima I, Ohno S. The role of SMG-1 in nonsense-mediated mRNA decay. Biochim Biophys Acta. 2005;1754(1-2):305–315. doi: 10.1016/j.bbapap.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 35.Yamashita A, Ohnishi T, Kashima I, Taya Y, Ohno S. Human SMG-1, a novel phosphatidylinositol 3-kinase-related protein kinase, associates with components of the mRNA surveillance complex and is involved in the regulation of nonsense-mediated mRNA decay. Genes Dev. 2001;15(17):2215–2228. doi: 10.1101/gad.913001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cho H, Han S, Park OH, Kim YK. SMG1 regulates adipogenesis via targeting of staufen1-mediated mRNA decay. Biochim Biophys Acta. 2013;1829(12):1276–1287. doi: 10.1016/j.bbagrm.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 37.Myers SJ, Wong LM, Charo IF. Signal transduction and ligand specificity of the human monocyte chemoattractant protein-1 receptor in transfected embryonic kidney cells. J Biol Chem. 1995;270(11):5786–5792. doi: 10.1074/jbc.270.11.5786. [DOI] [PubMed] [Google Scholar]
- 38.Isken O, et al. Upf1 phosphorylation triggers translational repression during nonsense-mediated mRNA decay. Cell. 2008;133(2):314–327. doi: 10.1016/j.cell.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clerici M, et al. Unusual bipartite mode of interaction between the nonsense-mediated decay factors, UPF1 and UPF2. EMBO J. 2009;28(15):2293–2306. doi: 10.1038/emboj.2009.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chamieh H, Ballut L, Bonneau F, Le Hir H. NMD factors UPF2 and UPF3 bridge UPF1 to the exon junction complex and stimulate its RNA helicase activity. Nat Struct Mol Biol. 2008;15(1):85–93. doi: 10.1038/nsmb1330. [DOI] [PubMed] [Google Scholar]
- 41.Chakrabarti S, et al. Molecular mechanisms for the RNA-dependent ATPase activity of Upf1 and its regulation by Upf2. Mol Cell. 2011;41(6):693–703. doi: 10.1016/j.molcel.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 42.Melero R, et al. Structures of SMG1-UPFs complexes: SMG1 contributes to regulate UPF2-dependent activation of UPF1 in NMD. Structure. 2014;22(8):1105–1119. doi: 10.1016/j.str.2014.05.015. [DOI] [PubMed] [Google Scholar]
- 43.Kaygun H, Marzluff WF. Regulated degradation of replication-dependent histone mRNAs requires both ATR and Upf1. Nat Struct Mol Biol. 2005;12(9):794–800. doi: 10.1038/nsmb972. [DOI] [PubMed] [Google Scholar]
- 44.Müller B, Blackburn J, Feijoo C, Zhao X, Smythe C. DNA-activated protein kinase functions in a newly observed S phase checkpoint that links histone mRNA abundance with DNA replication. J Cell Biol. 2007;179(7):1385–1398. doi: 10.1083/jcb.200708106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou D, et al. PNRC is a unique nuclear receptor coactivator that stimulates RNA polymerase III-dependent transcription. J Mol Signal. 2007;2:5. doi: 10.1186/1750-2187-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim KM, et al. A new MIF4G domain-containing protein, CTIF, directs nuclear cap-binding protein CBP80/20-dependent translation. Genes Dev. 2009;23(17):2033–2045. doi: 10.1101/gad.1823409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marión RM, Fortes P, Beloso A, Dotti C, Ortín J. A human sequence homologue of Staufen is an RNA-binding protein that is associated with polysomes and localizes to the rough endoplasmic reticulum. Mol Cell Biol. 1999;19(3):2212–2219. doi: 10.1128/mcb.19.3.2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim KM, Cho H, Kim YK. The upstream open reading frame of cyclin-dependent kinase inhibitor 1A mRNA negatively regulates translation of the downstream main open reading frame. Biochem Biophys Res Commun. 2012;424(3):469–475. doi: 10.1016/j.bbrc.2012.06.135. [DOI] [PubMed] [Google Scholar]
- 49.Choe J, et al. Translation initiation on mRNAs bound by nuclear cap-binding protein complex CBP80/20 requires interaction between CBP80/20-dependent translation initiation factor and eukaryotic translation initiation factor 3g. J Biol Chem. 2012;287(22):18500–18509. doi: 10.1074/jbc.M111.327528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cho H, Ahn SH, Kim KM, Kim YK. Non-structural protein 1 of influenza viruses inhibits rapid mRNA degradation mediated by double-stranded RNA-binding protein, staufen1. FEBS Lett. 2013;587(14):2118–2124. doi: 10.1016/j.febslet.2013.05.029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.