

Significance
Biomineralization is an important and widespread phenomenon by which living systems produce solid materials from soluble metal ions and has key implications with regard to environmental and health processes. In this work, we report studies of redox and structural components that control mineralization events in the production of iron nanoparticles in directional-sensing bacteria.
Keywords: biomineralization, iron, magnetosome, cytochrome, MamP
Abstract
Magnetotactic bacteria have evolved complex subcellular machinery to construct linear chains of magnetite nanocrystals that allow the host cell to sense direction. Each mixed-valent iron nanoparticle is mineralized from soluble iron within a membrane-encapsulated vesicle termed the magnetosome, which serves as a specialized compartment that regulates the iron, redox, and pH environment of the growing mineral. To dissect the biological components that control this process, we have carried out a genetic and biochemical study of proteins proposed to function in iron mineralization. In this study, we show that the redox sites of c-type cytochromes of the Magnetospirillum magneticum AMB-1 magnetosome island, MamP and MamT, are essential to their physiological function and that ablation of one or both heme motifs leads to loss of function, suggesting that their ability to carry out redox chemistry in vivo is important. We also develop a method to heterologously express fully heme-loaded MamP from AMB-1 for in vitro biochemical studies, which show that its Fe(III)–Fe(II) redox couple is set at an unusual potential (−89 ± 11 mV) compared with other related cytochromes involved in iron reduction or oxidation. Despite its low reduction potential, it remains competent to oxidize Fe(II) to Fe(III) and mineralize iron to produce mixed-valent iron oxides. Finally, in vitro mineralization experiments suggest that Mms mineral-templating peptides from AMB-1 can modulate the iron redox chemistry of MamP.
The unparalleled ability of living systems to evolve new chemistry taps into both organic and inorganic reaction space as well as homogeneous and heterogeneous catalysis. Indeed, the transformation of soluble metal ions to form various biological nanostructures, from the intricately patterned diatom frustule to the unusually strong mollusk shell, provides important function at the organism level (1, 2) and has inspired new methods for the preparation of synthetic materials (3). The mineralization of calcium is the most well understood as it produces perhaps the largest class of biominerals (1, 2). However, the controlled mineralization of elements such as silicon (4), iron (5, 6), copper (7), and manganese (8) is also observed, although less broadly distributed. Of the latter group, iron biomineralization by magnetotactic bacteria represents a particularly interesting case for understanding how the production of nanomaterials can be programmed at the genetic level.
Magnetotactic bacteria house a sophisticated system to sense the earth’s magnetic field for magnetoaerotaxis using linear chains of biological magnets, consisting of membrane-encapsulated magnetite [Fe(II)Fe(III)2O4] nanoparticles of various sizes (35–120 nm) and shapes with several unusual properties (9–11). First, these nanoparticles are generated as a single magnetic domain, suggesting that they have naturally evolved to maximize the magnetic contribution of each iron atom. In addition, the magnetite is synthesized as an internally pure material with surface-passivating proteins rather than as a composite material with organic layers, as is often observed in calcium-based biominerals. Also in contrast with the structural biomaterials built from calcium and silicon, the production of magnetite requires redox chemistry to produce the mixed-valent Fe(II)–Fe(III) product and could allow us to access large classes of functional transition metal-based materials using synthetic biology approaches.
The magnetite nanocrystals are grown by the cell under genetically controlled conditions within a subcellular vesicle, called the magnetosome, to produce defectless crystals with a narrow size and shape distribution. Based on its Pourbaix diagram, magnetite remains stable only at select redox potentials and pHs, implying that the magnetosome forms a compartment that has been optimized for synthesis of these materials (11). Interestingly, an analysis of the magnetosome island (MAI) of Magnetospirillum magneticum AMB-1, which houses a large part of the genetic material encoding this process, reveals that several essential genes are annotated as potential iron transporters, proton transporters, and redox proteins and could serve to tune these different parameters (10, 11). Our group is interested in identifying and characterizing the components involved directly in mineral synthesis to facilitate efforts to engineer the magnetosome for new synthetic functions. In this study, we focus on MamP and MamT, which are predicted c-type cytochromes in the MAI proposed to participate in redox reactions of iron in the magnetosome (12, 13). A recent crystal structure of the MamP ortholog from the Marine magnetotactic ovoid bacterium MO-1 shows that it comprises a novel “magnetochrome” fold involving the minimal association of 23 amino acids around each c-type heme cofactor (13). In this work, we demonstrate that the double CXXCH heme motifs of MamP and MamT are essential for magnetotaxis function, which supports a physiological role in redox chemistry. In vitro biochemical experiments on purified MamP demonstrate that it is competent to catalytically oxidize Fe(II) to Fe(III) despite its relatively unusual reduction potential compared with other iron-metabolizing cytochromes. Finally, in vitro iron mineralization experiments show that MamP is capable of producing mixed-valent iron oxides from soluble Fe(II) species under several different pH and redox environments and that its behavior can be modulated by a putative lattice-templating peptide from the Mms family (Mms7) to control both mineral structure and redox state.
Results
Genetic Characterization of the Physiological Function of mamP and mamT in AMB-1.
The genetic analysis of the AMB-1 MAI implicates both mamP and mamT as essential but distinct components involved in the mineralization of iron based on differences in iron nanoparticle morphology that arise upon genetic deletion (12). To further explore their physiological function, we used low-resolution transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) to probe mineralization defects in genetic knockouts of mamP and mamT. The mamT deletion was found to be unstable in a wild-type strain background and was thus generated in the ΔR9 strain, in which a repeated sequence is eliminated to increase genetic stability with no apparent effect on iron mineralization (12). Wild-type AMB-1 produces 15–25 cubooctahedral magnetite nanoparticles per cell with a size distribution centered around 40–60 nm (Fig. 1 and SI Appendix, Figs. S1 and S2). In contrast, the ΔmamP strain produces mostly small, flake-like crystals along with 1–2 wild-type or larger-sized crystals per cell. HRTEM analysis of these small flakes proved to be difficult, due to either their poor crystallinity or their small size. Crystals observed in the ΔmamTΔR9 strain are small, elongated, and dumbbell-shaped.
Fig. 1.

TEM images and statistical analysis of nanoparticles produced by AMB-1 MamP and MamT heme mutants in vivo. For each strain, >300 particles from >20 cells from three different growths were analyzed. (A) Representative images of wild-type and ΔmamP strains compared with complemented strains. (B) Histograms of crystal sizes observed in the MamP strains. (C) Representative image of HRTEM image and derived fast Fourier transform (FFT) lattice for ΔmamP crystals. (D) Representative images of ΔR9 and ΔmamTΔR9 strains compared with complemented strains. (E) Histograms of crystal sizes observed in the MamT strains. (F) Representative image of HRTEM image and derived FFT lattice for ΔmamTΔR9 crystals. (ΔP, ΔmamP; ΔTΔR9, ΔmamTΔR9; h1, heme1; h2, heme2; h, heme).
Interestingly, HRTEM shows that all flakes and particles with resolved 2D lattices produced in the ΔmamP, ΔmamTΔR9, and wild-type strains are consistent with a magnetite structure (Fig. 1 and SI Appendix, Fig. S3), indicating that mamP and mamT are not required for magnetite production although they are needed for magnetotaxis. The ability of the ΔmamP strain to produce a limited number of wild-type crystals per cell may be surprising but could arise from redundant control of the iron biomineralization process, both with respect to iron redox chemistry itself or other magnetosome properties such as pH. Indeed, magnetite nanoparticles can also be produced abiotically by simple mixing of Fe(II) and Fe(III) at the appropriate pH and it is unusual to observe the formation of iron oxides other than magnetite in magnetotactic bacteria, even with severe cell and magnetite growth disadvantages from deleterious genetic knockouts or incubation with toxic concentrations of metal ions (14).
We sought to further probe the in vivo role of the heme-binding sites of MamP and MamT, which are proposed to enable the transfer of redox equivalents to or from iron species within the magnetosome (13). Therefore, each of the two CXXCH c-type cytochrome motifs found within both MamP and MamT was either individually or doubly mutated to AXXAA to remove the coordinating histidine and two cysteines for covalent heme attachment and expressed from a constitutive tac promoter in the appropriate ΔmamP or ΔmamTΔR9 background (SI Appendix, Table S1). Initial characterization of the mamP and mamT heme mutant strains was carried out using a qualitative assay for cellular magnetization (Cmag), which measures the ability of cells to turn in the presence of an external magnetic field. The parental ΔmamP and ΔmamTΔR9 strains exhibit a strong magnetic defect with Cmag measurements of 1.2 ± 0.1 and 1.2 ± 0.05, respectively, where 1.0 indicates no cellular magnetization and 2.0 is the characteristic Cmag observed for wild-type AMB-1 (12) (Fig. 2). Complementation of the chromosomal deletion of mamP or mamT with the corresponding plasmid-borne wild-type gene rescues Cmag to wild-type levels. In contrast, we found that deletion of one or both of the hemes was sufficient to impair magnetite formation with little to no recovery of magnetic response (Fig. 2). TEM was then used to analyze the size and shape of iron oxide particles produced by these mutants. Ablation of one or more of the heme sites of either MamP or MamT yields a phenotype indistinguishable from the parental ΔmamP and ΔmamTΔR9 strains based on the nanoparticle histograms (Fig. 1 and SI Appendix, Fig. S1). Overall, these results are consistent with a model where the heme redox sites of both MamP and MamT are critical for magnetite growth as the mutation of a single site yields a phenotype that cannot be differentiated from the deletion strain. The difference in the crystal morphologies between the MamP and MamT heme mutants further suggests that the changes in mineralization behavior related to the ablation of the heme sites are related to the loss of MamP or MamT function rather than a more general defect in magnetosome biogenesis.
Fig. 2.

Magnetic response of AMB-1 MamP and MamT heme mutants in vivo. (A) Wild-type and ΔmamP strains compared with ΔmamP complemented with MamP heme variants. (B) ΔR9 and ΔmamTΔR9 strains compared with ΔmamTΔR9 complemented with MamT heme variants. All data are reported as the mean ± SD of three biological replicates with technical duplicates (n = 6). (Δh1, mamPΔheme1; Δh2, mamPΔheme2; Δheme, mamPΔheme.)
As the loss of MamP and MamT function could lead to a variety of complex biological outcomes, we carried out additional control experiments to check for other large-scale changes in magnetosome function. For example, the deletion of certain MAI genes, such as mamE, can result in the mislocalization of other proteins and downstream magnetic defects in the deletion strains (15). To test whether the mamP or mamT deletions cause a similar mislocalization of key magnetosome proteins, C-terminal GFP fusions of MamC, MmsF, MamF, and MamI were expressed in ΔmamP and ΔmamTΔR9 deletion strains and control strains. Fluorescence microscopy studies showed that all GFP fusions localized as a filament at midcell in ΔmamP and ΔmamTΔR9 as consistent with wild-type AMB-1, suggesting that localization of these proteins to the magnetosome is not altered (SI Appendix, Fig. S4). Because variation in MamP expression levels has been observed to lead to differences in iron mineralization (16), antibodies to MamP were raised for immunostaining experiments, which indicated that MamP mutants are expressed at levels consistent with or higher than wild-type protein (SI Appendix, Fig. S5). Although we cannot eliminate the possibility that removal of the heme sites of MamP and MamT results in misprocessing, mislocalization, or structural destabilization of MamP, MamT, or other essential magnetosome components, we hypothesize that the CXXCH motifs serve a critical function in the mineralization process of AMB-1.
Heterologous Expression and Isolation of MamP.
We then turned our attention to in vitro characterization of MamP, which is proposed to be localized to the magnetosome and to directly interact with iron (13, 16). A series of plasmids for the expression of MamP in Escherichia coli was constructed that contained the native mamP gene encoding the predicted mature sequence with an N-terminal OmpA tag for periplasmic localization as needed for the maturation of c-type cytochromes. These constructs also included various solubilization tags flanked by an N-terminal His6-affinity tag and protease cleavage site (SI Appendix, Table S1). MamP expression was initially screened in a pCWori backbone, which has been found to frequently increase the functional expression of heme proteins (17). Overall, we found that proteolysis of MamP was problematic in many of these variants, which could possibly be related to the accompanying observation that the second heme site was often unoccupied. After significant screening, the optimal system was identified to be E. coli C43(DE3) coexpressing the pEC86 cytochrome c maturation plasmid with pET29a-OmpA.His6.MBPtev.MamP (18) with low inducer concentration (SI Appendix, Fig. S6).
The His6.MBPtev.MamP fusion protein was isolated from the periplasmic lysate using a nickel-nitrilotriacetic acid (Ni-NTA) affinity column. After proteolytic digestion, the sample was subjected to a second passage over a Ni-NTA column and an amylose column to remove His6.TEV protease, His6.MBP, and other cellular proteins enriched by Ni-NTA chromatography. The lingering minor contaminants were then removed by size-exclusion chromatography, yielding MamP which is >95% pure as judged by an overloaded reducing gel (SI Appendix, Fig. S7). Furthermore, MamP was found to elute in a single asymmetric peak with fractions exhibiting Reinheitzahl ratios (Rz = ASoret, 407 nm/A280 nm) ranging between 2 and 8. The fractions at the leading edge to the center of the peak exhibited the highest heme content (Rz ∼ 7.0–7.2) and were then collected and pooled for downstream biochemical experiments (Fig. 3A).
Fig. 3.
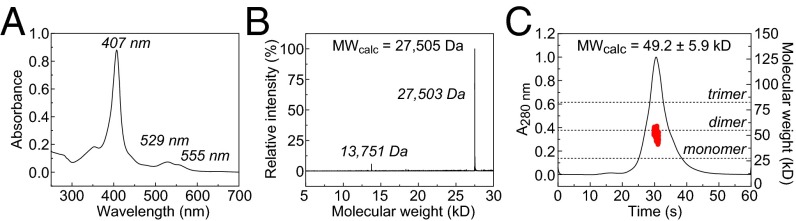
Characterization of heterologously expressed MamP. (A) UV-visible spectrum of MamP. (B) ESI mass spectrum of MamP showing covalent modification by two heme prosthetic groups. The minor peak in the spectrum is an artifact of the deconvolution process. (C) Size-exclusion chromatogram coupled to multiangle light scattering yields a derived molecular weight of 49.2 ± 5.9 kDa, consistent with a dimer (MWmonomer = 27.5 kDa).
The heme content of heterologously expressed MamP was characterized by a combination of protein acidolysis, inductively coupled plasma atomic emission spectroscopy (ICP-AES), and electrospray ionization mass spectrometry (ESI-MS). Based on the low expected molar extinction coefficient of MamP and overlap with the heme absorption bands at 280 nm (Fig. 3A), protein acidolysis was used to measure MamP concentration and determine ε280 nm after amino acid analysis (SI Appendix, Table S2). MamP was then subjected to iron analysis by ICP-AES, which revealed that each MamP monomer contained two equivalents of iron as expected for diheme-loaded MamP (2.0 ± 0.4; SI Appendix, Fig. S8). The molar extinction coefficients calculated by protein and metal content are in good agreement with each other, indicating that both methods provided fairly accurate quantification (SI Appendix, Table S3). The major peak in the ESI mass spectrum of MamP is centered at 27,503 Da, also as expected for the diheme-loaded protein (Fig. 3B). Finally, size-exclusion chromatography coupled to multiangle light scattering and dynamic light scattering suggest that MamP preferentially forms a dimer in solution, which is consistent with the crystal structure of the ortholog from MO-1 (Fig. 3C and SI Appendix, Figs. S9–S11) (13).
Biochemical Characterization of MamP from AMB-1.
To investigate the electron transfer capabilities of MamP, spectroelectrochemical titrations were performed to measure the Fe(III)–Fe(II) redox couple in both the oxidative and reductive directions. The Fe(III)–MamP resting state was reduced with sodium dithionite and then titrated with an oxidant while quantifying the fraction of reduced and oxidized MamP (Fig. 4A). The oxidized MamP was subsequently rereduced to collect the reductive curve, with MamP demonstrating reversible, Nernstian behavior in both directions. The average Fe(III)–Fe(II) midpoint reduction potential was fit to a value of −89 ± 11 mV at pH 7.5, where the width of the transition predicts a one-electron redox process with very similar if not identical redox potentials for each of the heme sites within the MamP monomer (Fig. 4B). This observation is consistent with the crystal structure of the MO-1 ortholog, which shows that the cofactors are exposed, likely enabling rapid and reversible electron transfer to and from the iron center without significant perturbation from the protein environment. Furthermore, the redox potentials of the MamP orthologs fall within a similar range despite their divergence in sequence (SI Appendix, Fig. S11).
Fig. 4.

Characterization of MamP redox behavior. (A) Changes in the electronic absorption spectrum showing the stepwise oxidation of Fe(II)–MamP (red) to Fe(III)–MamP (black). (B) Three individual redox titrations of MamP monitored by ∆A551 nm with the curve fit for a one-electron (red line) or two-electron process (black line). (C) Redox cycle of MamP-catalyzed oxidation of Fe(II) showing reduction of Fe(III)–MamP by ammonium iron(II) sulfate followed by reoxidation of Fe(II)–MamP by titration of 1 eq of O2.
Interestingly, the measured midpoint reduction potential for MamP is quite different from other iron oxidoreductases and falls between the ranges of the measured potentials for iron-reducing and iron-oxidizing cytochromes known to be involved in iron transformations. For example, the Fe(III)–Fe(II) couple for iron oxidases from either Rhodobacter ferrooxidans SW2 (19) or Ferrobacillus ferrooxidans (20, 21) was reported to be in the +200 to +300-mV range. In contrast, iron-reducing organisms, such as Shewanella oneidensis, contain c-type cytochromes such as OmcA with reduction potentials ranging from −234 mV to −324 mV (22). However, some c-type cytochromes do have more positive reduction potentials, such as an extracellular electron carrier found in Geobacter sulfurreducins (−167 mV) (23). Although different, the MamP Fe(III)–Fe(II) couple remains slightly higher than the reduction potentials for various reactions between insoluble iron oxides and soluble, complexed Fe(II) (24). For example, values for ferrihydrite and the Fe(III)(OH)3–Fe(II) couple are between −200 mV and +100 mV, whereas the potentials for crystalline iron oxides reside between −88 mV (lepidocrocite) and −314 mV (magnetite). The standard reduction potential of the Fe(III)–Fe(II) couple (+770 mV) applies only to strongly acidic solutions in which both oxidation states have high solubility (25). Indeed, the reducing power of ferrous iron increases dramatically at neutral pH due to the formation of insoluble iron oxides and oxyhydroxides. Therefore, the measurement of reduction potentials of insoluble iron minerals is quite complex, as chelated iron oxides have higher potentials than ferric iron oxides and are dependent on pH and even particle size (26). Thus, at circumneutral pH, reduction potentials for these species can range from –300 mV to +400 mV and the MamP couple is found in the middle of this range. This could suggest that MamP is tuned to transform very specific iron species at controlled pH values.
As the reduction potential of MamP falls directly in between characterized iron-reducing and iron-oxidizing cytochromes, we decided to investigate its chemical competence to carry out redox chemistry with iron. In the absence of additional mediators, we found that the resting Fe(III)–MamP could be reduced by the addition of iron(II) salts (Fig. 4C). Reduced Fe(II)–MamP could further be rapidly and stoichiometrically oxidized by the titration of one equivalent of molecular oxygen (Fig. 4C). These results demonstrate that Fe(II) and O2 enable the formation of a closed catalytic cycle for overall oxidation of Fe(II) to Fe(III) catalyzed by MamP.
Mineralization of Iron Oxides from Fe(II) with Magnetosome Proteins.
The ability of MamP to rapidly generate Fe(III) from Fe(II) should also enable catalysis of the formation of mixed-valent iron oxides related to magnetite. Indeed, we observed that reaction of Fe(II) with resting Fe(III)–MamP, while introducing oxygen as the terminal oxidant, led to the formation of a green mineral within 10 min and reached high levels within 20 min at pH 8.0 (Fig. 5A). In comparison, little to no mineralization takes place above background in the control reaction with no MamP, as it was not observed to significantly increase within the timeframe of the reaction (30 min; Fig. 5A). Furthermore, the mineralization reaction catalyzed by MamP is significantly faster than the formation of iron(III) oxides or hydroxides in the reaction with no MamP, even with the introduction of oxygen (Fig. 5A). Studies of the pH dependence of this reaction show that orange rust or fully oxidized ferrihydrite is formed at pH 6.0, where magnetite is thermodynamically unstable, and that green rust formation appears to be accelerated when the pH is increased to 9.0, which accesses the middle of pH range of magnetite stability (SI Appendix, Fig. S12). The formation of a green rust-like mineral in the MamP reaction at pH >7.5 is consistent with a mixed Fe(II)–Fe(III) oxyhydroxide green rust [2–4 Fe(II):1 Fe(III)], which is a possible precursor to magnetite [1 Fe(II):2 Fe(III)] that is incompletely oxidized. Given the variation of the reduction potential of green rust with Fe(II):Fe(III) ratio (−210 mV to +270 mV) (27), the rates of mineralization and Fe(II) oxidation can affect the identity of the mineral formed, as MamP would be competent to further oxidize some of these species. Indeed, magnetic materials are formed with longer incubation (>60 min) at pH >7.5 but could be caused by chemical oxidation rather than a MamP-catalyzed process (SI Appendix, Fig. S12). Thus, the bulk formation of green rust rather than magnetite under these in vitro conditions may indicate that the stoichiometry of Fe(II) to MamP may be controlled in vivo such that Fe(II) and Fe(III) are formed in the appropriate ratio for direct formation of magnetite, or that other components could be involved in the oxidation of green rust to magnetite. In addition to possible functional redundancy in the redox chain, it is also possible that control over pH via predicted proton transporters may play a role in modulating this behavior given the relationship between pH and iron oxide stability as well as Fe(II) and Fe(III) solubility and stability.
Fig. 5.

In vitro mineralization of iron by MamP. (A) Reactions containing ammonium Fe(II) sulfate (40 mM) in 50 mM Hepes pH 8.0, 100 mM NaCl with (+) and without (−) 2.5 μM MamP. (B) Reactions containing ammonium Fe(II) sulfate (20 mM) and MamP (1.2 μM) in 50 mM Hepes pH 7.5, 100 mM NaCl with (+) and without (–) 45 μM Mms7ct. After the end of the mineralization reaction, reaction vials were opened to air for 20 min.
Given the competence of MamP to transform soluble iron(II) species to mixed-valent Fe(II)–Fe(III) oxide minerals, we were interested to see how other magnetosome proteins might interact with the growing material. In addition to redox partners that are necessary to generate both Fe(II) and Fe(III), there also exist small magnetosome proteins proposed to template mineralization by direct binding to the material (28) analogous to the more well-understood mineralization of calcium. These Mms proteins were identified by their tight association with the magnetite nanoparticles isolated from magnetotactic bacteria and have been shown experimentally to be able to control mineral shape in vitro through their highly acidic C-terminal sequences (29, 30). Similar to other magnetotactic bacteria, the MAI of AMB-1 contains multiple mms genes, mms5 (mamG), mms6, and mms7 (mamD), which have been previously shown to possess overlapping genetic function as all genes must be deleted to observe the resulting biomineralization defect (12). Consequently, we cloned, expressed, and purified the acidic C terminus of the Mms7 protein (Mms7ct) from AMB-1 as an N-terminal fusion with MBP. The Mms7ct peptide resulting from cleavage of the fusion with TEV protease was purified by reverse-phase HPLC and characterized by analytical HPLC and MALDI-TOF MS (SI Appendix, Fig. S13).
Using our in vitro mineralization assay, we set out to explore the relationship between redox catalysis and structural templating, respectively, provided by MamP and Mms7. In the absence of MamP, the addition of Mms7Ct to soluble Fe(II) and introduction of oxygen did not lead to immediate mineralization (SI Appendix, Fig. S14). Furthermore, reactions containing both MamP and Mms7ct proceeded similarly to those with MamP alone, with rapid formation of green rust on the same timescale. However, an interesting change in behavior was induced by Mms7ct after mineralization. If the reactions were opened to air, those that did not contain Mms7ct quickly turned orange, which signifies the formation of fully oxidized Fe(III) oxide red rusts and represents the typical endpoint for reactions of Fe(II) with oxygen (Fig. 5B). In contrast, the green rust remained mostly intact in reactions containing Mms7ct after direct exposure to air, indicating that the mixed-valent Fe(II)–Fe(III) material was somehow being protected from further oxidation (Fig. 5B). This behavior is sensitive to the Fe(II):Mms7ct ratio based on the observation that full protection can be lost at large excess of Fe(II) (>1:1,000). These results suggest that both the redox setpoint of MamP as well as the structural templating of the Mms proteins control the identity of the mineral being formed.
Discussion
Magnetotactic bacteria have evolved a remarkable organelle that serves to genetically control the size, shape, and structure of the iron oxide nanoparticles that are used for passive sensing of direction. As the production of magnetite requires both the formation of Fe(II) and Fe(III), the redox components of the magnetosome play an essential role in this process. Using genetic complementation studies, we have shown that the redox cofactors or heme sites of the two putative redox partners, MamP and MamT, are required for this process in vivo and that removal of one or both sites leads to defects in mineralization that are nearly indistinguishable from the phenotypes of the mamP and mamT deletion strains. Although we cannot rule out other effects caused by mutation of the heme sites, TEM studies show the heme mutants retain the crystallization phenotypes that are a signature of either mamP or mamT loss of function, whereas controls indicate both that the processed forms of the mutant proteins are present in the complementation strains and that key magnetosome proteins retain their proper localization. The difference in the defects does suggest that MamP and MamT do not have redundant functions in magnetite formation. The ability of MamP- and MamT-deficient strains to produce magnetite may further imply that they are more important in mineral growth rather than nucleation, although it is also likely that the magnetosome possesses several complementary mechanisms, including pH and redox control and structural templating, which help to maintain magnetite formation should a defect arise in an individual pathway.
To further explore MamP function, we have developed and optimized a heterologous expression method to cleanly isolate fully heme-loaded MamP for biochemical studies as shown by amino acid analysis, iron analysis, and MS. Spectrochemical redox titrations show that the reduction potential of MamP lies in a different range than other c-type cytochromes involved in either Fe(III) reduction or Fe(II) oxidation. Nonetheless, Fe(III)–MamP remains competent to oxidize soluble Fe(II) species and can be reoxidized to the Fe(III) resting state to form a closed catalytic cycle leading to overall formation of Fe(III) from Fe(II). Indeed, the ability of MamP to oxidize Fe(II) at a lower overpotential provides a mechanism for the generation of Fe(II) and Fe(III) within the magnetosome for magnetite formation while preventing the rereduction of Fe(III) to Fe(II). In this manner, MamP could be optimized for controlling the stoichiometry of Fe(II) and Fe(III) so that the magnetite nanoparticle can be grown without defects.
In vitro mineralization studies with MamP and Fe(II) show that MamP is able to catalyze the formation of mixed-valent Fe(II)–Fe(III) oxides at neutral pH within a short timescale that are competent to be oxidized further to magnetic iron oxides. Whereas we use oxygen as the terminal oxidant for this reaction, other small-molecule oxidants, such as nitrate or perchlorate, could also be used for anaerobic mineralization based on the MamP reduction potential. The formation of the incompletely oxidized mineral under these conditions is likely a result of kinetic competition between the rates of mineralization versus Fe(II) oxidation and could be controlled in vivo by local Fe(II) concentration, iron:MamP stoichiometry, or pH inside the magnetosome. Further studies with Mms7ct indicate that it and other Mms proteins may play a more significant role in controlling magnetite mineral structure than previously hypothesized. Beyond simple control of size and shape of magnetite (29, 30), they may also template the crystal lattice of the mineral itself similar to what has been observed with calcium biomineralization, where unstable crystal forms and phases of the mineral are stabilized by interaction with peptides and other macromolecules (1, 2). In the specific case of iron minerals, lattice stabilization could also affect the redox potential of individual Fe atoms within the mineral.
Taken together, our results suggest that the biological mechanism for iron mineralization in AMB-1 involves the transport of iron into the magnetosome in the Fe(II) oxidation state and its subsequent oxidation to Fe(III) to form magnetite in a process that is controlled by magnetosome redox (MamP and MamT) and templating (Mms proteins) components (Fig. 6). In this regard, biomineralization in magnetotactic bacteria may be directly related to Fe(II) oxidation by anaerobic nitrate-dependent bacteria and, by analogy, ultimately coupled to the respiratory chain. In this regard, it has been postulated that periplasmic components of these nitrate-reducing bacteria enable Fe(II) oxidation to overcome toxicity of iron and radical byproducts (31) based on the observation that several strains can precipitate iron minerals within the periplasm. Furthermore, nitrate reduction has been shown to be linked to biomineralization in other species of magnetotactic bacteria and is mediated by key periplasmic proteins (32). Because the magnetosome is ultimately derived from the periplasmic compartment of the host, it would be interesting to hypothesize that magnetite nanoparticle formation represents an inside-out respiratory process, in which the electron transport chain provides not only energy to the cell but also oxidizing equivalents to the tightly controlled biomineralization process.
Fig. 6.

Model for redox-controlled magnetite formation in AMB-1. Soluble Fe(II) is oxidized to Fe(III) either in the periplasm or magnetosome through a redox chain involving magnetosome-specific cytochromes, such as MamP and MamT, as well as other general electron transport proteins in the periplasmic compartment. The overall oxidation of Fe(II) leads to the formation of 4 reducing eq per MamP dimer, which could potentially be passed to the respiratory chain for energy metabolism. The localization of MamP and MamT to either the periplasm or magnetosome have yet to be determined, but their respective sequences are predicted to contain a signal sequence and transmembrane segment. A mixture of Fe(II) and Fe(III) is generated in the magnetosome either by direct oxidation with a magnetosome-localized MamP or MamT or through transport from the periplasm [Fe(II) or Fe(III)] or cytoplasm [Fe(II)] via predicted cation diffusion facilitors (MamB/MamM). The Fe(II) and Fe(III) ions are templated by Mms proteins to form the Fe(II)Fe(III)2O4 mineral lattice with oxide bridges derived from H2O. We hypothesize that the Fe(II):Fe(III) ratio in the magnetosome is set by MamP, whereas the Fe(II):Fe(III) ratio in the mineral lattice may be further controlled by Mms proteins. The in vivo product of MamP could possibly be magnetite itself or a Fe(II)–Fe(III) oxide precursor that is further oxidized by other redox components while being stabilized and protected by Mms family from overoxidation to Fe(III) oxides. The protons lost during mineralization from the bridging waters are transported out of the magnetosome by a predicted Na+/H+ antiporter (MamN) to maintain the pH required for stable magnetite formation.
Methods
Detailed procedures for AMB-1 transformation, cell culture, and characterization are provided in SI Appendix along with those for plasmid construction, protein purification, and biochemical assays.
Supplementary Material
Acknowledgments
We thank Prof. Peidong Yang for assistance with HRTEM using the National Center for Electron Microscopy at Lawrence Berkeley National Laboratory, supported by the Office of Basic Energy Sciences of the Department of Energy (DE-AC02-05CH11231). This work was funded by generous support from the Defense Advanced Research Projects Agency (N66001-12-1-4230). S.R.J. and L.L.F. also acknowledge the support of NIH Training Grant (1 T32 GMO66698) and NSF Graduate Research Fellowships. A.K. was supported by grants from NIH (R01GM084122) and the Office of Naval Research (N000141310421). This work also used the Vincent J. Coates Proteomics/Mass Spectrometry Laboratory, supported in part by NIH S10RR025622.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1417614112/-/DCSupplemental.
References
- 1.Mann S. Biomineralization: Principles and Concepts in Bioinorganic Materials Chemistry. Oxford Univ Press; Oxford: 2001. [Google Scholar]
- 2.Weiner S, Addadi L. Crystallization pathways in biomineralization. Annu Rev Mater Res. 2011;41:21–40. [Google Scholar]
- 3.Zhang S. Fabrication of novel biomaterials through molecular self-assembly. Nat Biotechnol. 2003;21(10):1171–1178. doi: 10.1038/nbt874. [DOI] [PubMed] [Google Scholar]
- 4.Hildebrand M. Diatoms, biomineralization processes, and genomics. Chem Rev. 2008;108(11):4855–4874. doi: 10.1021/cr078253z. [DOI] [PubMed] [Google Scholar]
- 5.Banfield JF, Welch SA, Zhang H, Ebert TT, Penn RL. Aggregation-based crystal growth and microstructure development in natural iron oxyhydroxide biomineralization products. Science. 2000;289(5480):751–754. doi: 10.1126/science.289.5480.751. [DOI] [PubMed] [Google Scholar]
- 6.Theil EC. Ferritin: The protein nanocage and iron biomineral in health and in disease. Inorg Chem. 2013;52(21):12223–12233. doi: 10.1021/ic400484n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lichtenegger HC, Schöberl T, Bartl MH, Waite H, Stucky GD. High abrasion resistance with sparse mineralization: Copper biomineral in worm jaws. Science. 2002;298(5592):389–392. doi: 10.1126/science.1075433. [DOI] [PubMed] [Google Scholar]
- 8.Tebo BM, Johnson HA, McCarthy JK, Templeton AS. Geomicrobiology of manganese(II) oxidation. Trends Microbiol. 2005;13(9):421–428. doi: 10.1016/j.tim.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 9.Bazylinski DA, Frankel RB. Magnetosome formation in prokaryotes. Nat Rev Microbiol. 2004;2(3):217–230. doi: 10.1038/nrmicro842. [DOI] [PubMed] [Google Scholar]
- 10.Komeili A. Molecular mechanisms of magnetosome formation. Annu Rev Biochem. 2007;76(1):351–366. doi: 10.1146/annurev.biochem.74.082803.133444. [DOI] [PubMed] [Google Scholar]
- 11.Faivre D, Schüler D. Magnetotactic bacteria and magnetosomes. Chem Rev. 2008;108(11):4875–4898. doi: 10.1021/cr078258w. [DOI] [PubMed] [Google Scholar]
- 12.Murat D, Quinlan A, Vali H, Komeili A. Comprehensive genetic dissection of the magnetosome gene island reveals the step-wise assembly of a prokaryotic organelle. Proc Natl Acad Sci USA. 2010;107(12):5593–5598. doi: 10.1073/pnas.0914439107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siponen MI, et al. Structural insight into magnetochrome-mediated magnetite biomineralization. Nature. 2013;502(7473):681–684. doi: 10.1038/nature12573. [DOI] [PubMed] [Google Scholar]
- 14.Raschdorf O, Müller FD, Pósfai M, Plitzko JM, Schüler D. The magnetosome proteins MamX, MamZ and MamH are involved in redox control of magnetite biomineralization in Magnetospirillum gryphiswaldense. Mol Microbiol. 2013;89(5):872–886. doi: 10.1111/mmi.12317. [DOI] [PubMed] [Google Scholar]
- 15.Quinlan A, Murat D, Vali H, Komeili A. The HtrA/DegP family protease MamE is a bifunctional protein with roles in magnetosome protein localization and magnetite biomineralization. Mol Microbiol. 2011;80(4):1075–1087. doi: 10.1111/j.1365-2958.2011.07631.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taoka A, et al. A magnetosome-associated cytochrome MamP is critical for magnetite crystal growth during the exponential growth phase. FEMS Microbiol Lett. 2014;358(1):21–29. doi: 10.1111/1574-6968.12541. [DOI] [PubMed] [Google Scholar]
- 17.Barnes HJ, Arlotto MP, Waterman MR. Expression and enzymatic activity of recombinant cytochrome P450 17 α-hydroxylase in Escherichia coli. Proc Natl Acad Sci USA. 1991;88(13):5597–5601. doi: 10.1073/pnas.88.13.5597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arslan E, Schulz H, Zufferey R, Künzler P, Thöny-Meyer L. Overproduction of the Bradyrhizobium japonicum c-type cytochrome subunits of the cbb3 oxidase in Escherichia coli. Biochem Biophys Res Commun. 1998;251(3):744–747. doi: 10.1006/bbrc.1998.9549. [DOI] [PubMed] [Google Scholar]
- 19.Saraiva IH, Newman DK, Louro RO. Functional characterization of the FoxE iron oxidoreductase from the photoferrotroph Rhodobacter ferrooxidans SW2. J Biol Chem. 2012;287(30):25541–25548. doi: 10.1074/jbc.M112.360636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vernon LP, Mangum JH, Beck JV, Shafia FM. Studies on a ferrous-ion-oxidizing bacterium. II. Cytochrome composition. Arch Biochem Biophys. 1960;88(2):227–231. doi: 10.1016/0003-9861(60)90227-7. [DOI] [PubMed] [Google Scholar]
- 21.Blaylock BA, Nason A. Electron transport systems of the chemoautotroph Ferrobacillus ferrooxidans. I. Cytochrome c-containing iron oxidase. J Biol Chem. 1963;238(10):3453–3462. [PubMed] [Google Scholar]
- 22.Firer-Sherwood M, Pulcu GS, Elliott SJ. Electrochemical interrogations of the Mtr cytochromes from Shewanella: Opening a potential window. J Biol Inorg Chem. 2008;13(6):849–854. doi: 10.1007/s00775-008-0398-z. [DOI] [PubMed] [Google Scholar]
- 23.Seeliger S, Cord-Ruwisch R, Schink B. A periplasmic and extracellular c-type cytochrome of Geobacter sulfurreducens acts as a ferric iron reductase and as an electron carrier to other acceptors or to partner bacteria. J Bacteriol. 1998;180(14):3686–3691. doi: 10.1128/jb.180.14.3686-3691.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Straub KL, Benz M, Schink B. Iron metabolism in anoxic environments at near neutral pH. FEMS Microbiol Ecol. 2001;34(3):181–186. doi: 10.1111/j.1574-6941.2001.tb00768.x. [DOI] [PubMed] [Google Scholar]
- 25.Stumm W, Lee GF. The chemistry of aqueous iron. Schweiz Z Hydrol. 1960;22(1):295–319. [Google Scholar]
- 26.Thamdrup B. Bacterial manganese and iron reduction in aquatic sediments. Adv Microb Ecol. 2000;16:41–84. [Google Scholar]
- 27.Ruby C, Upadhyay C, Géhin A, Ona-Nguema G, Génin JM. In situ redox flexibility of FeII-III Oxyhydroxycarbonate green rust and fougerite. Environ Sci Technol. 2006;40(15):4696–4702. doi: 10.1021/es0606834. [DOI] [PubMed] [Google Scholar]
- 28.Arakaki A, Webb J, Matsunaga T. A novel protein tightly bound to bacterial magnetic particles in Magnetospirillum magneticum strain AMB-1. J Biol Chem. 2003;278(10):8745–8750. doi: 10.1074/jbc.M211729200. [DOI] [PubMed] [Google Scholar]
- 29.Amemiya Y, Arakaki A, Staniland SS, Tanaka T, Matsunaga T. Controlled formation of magnetite crystal by partial oxidation of ferrous hydroxide in the presence of recombinant magnetotactic bacterial protein Mms6. Biomaterials. 2007;28(35):5381–5389. doi: 10.1016/j.biomaterials.2007.07.051. [DOI] [PubMed] [Google Scholar]
- 30.Baumgartner J, Carillo MA, Eckes KM, Werner P, Faivre D. Biomimetic magnetite formation: From biocombinatorial approaches to mineralization effects. Langmuir. 2014;30(8):2129–2136. doi: 10.1021/la404290c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carlson HK, Clark IC, Blazewicz SJ, Iavarone AT, Coates JD. Fe(II) oxidation is an innate capability of nitrate-reducing bacteria that involves abiotic and biotic reactions. J Bacteriol. 2013;195(14):3260–3268. doi: 10.1128/JB.00058-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, Katzmann E, Borg S, Schüler D. The periplasmic nitrate reductase Nap is required for anaerobic growth and involved in redox control of magnetite biomineralization in Magnetospirillum gryphiswaldense. J Bacteriol. 2012;194(18):4847–4856. doi: 10.1128/JB.00903-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.