

Abstract
Almost all eukaryotic mRNAs must be polyadenylated at their 3′ ends to function in protein synthesis. This modification occurs via a large nuclear complex that recognizes signal sequences surrounding a poly(A) site on mRNA precursor, cleaves at that site, and adds a poly(A) tail. While the composition of this complex is known, the functions of some subunits remain unclear. One of these is a multidomain protein called Mpe1 in the yeast Saccharomyces cerevisiae and RBBP6 in metazoans. The three conserved domains of Mpe1 are a ubiquitin-like (UBL) domain, a zinc knuckle, and a RING finger domain characteristic of some ubiquitin ligases. We show that mRNA 3′-end processing requires all three domains of Mpe1 and that more than one region of Mpe1 is involved in contact with the cleavage/polyadenylation factor in which Mpe1 resides. Surprisingly, both the zinc knuckle and the RING finger are needed for RNA-binding activity. Consistent with a role for Mpe1 in ubiquitination, mutation of Mpe1 decreases the association of ubiquitin with Pap1, the poly(A) polymerase, and suppressors of mpe1 mutants are linked to ubiquitin ligases. Furthermore, an inhibitor of ubiquitin-mediated interactions blocks cleavage, demonstrating for the first time a direct role for ubiquitination in mRNA 3′-end processing.
INTRODUCTION
Polyadenylation is an essential step in the production of functional eukaryotic mRNA that will be efficiently utilized in translation. It is a nuclear processing event that involves the cleavage of mRNA precursor followed by the addition of a poly(A) tail and is carefully coordinated with other events involved in mRNA synthesis and utilization, such as transcription, splicing, assembly of mRNA into ribonucleoprotein complexes, and mRNA export. Many recent studies have also revealed how selection of the cleavage site can globally affect the type of mRNA produced by cells (1–3). Polyadenylation requires a suite of multiple factors whose subunits are conserved across eukaryotic species (4–6). Significant progress has been made in understanding the contributions of each factor to the recognition of the poly(A) site, the execution of the processing steps, and regulation. However, the precise role of each subunit in this complex process and the impact of posttranslational modifications on the regulation of polyadenylation have not been completely defined.
In Saccharomyces cerevisiae, the factors needed for mRNA 3′-end processing are the RNA-binding protein Hrp1 and two multisubunit complexes called cleavage/polyadenylation factor (CPF) and cleavage factor IA (CF IA). One poorly characterized but essential subunit of CPF identified more than 10 years ago is Mpe1, which is needed for both processing steps (7). The mammalian Mpe1 homolog, RBBP6, interacts with the tumor suppressor proteins p53 and pRb (8, 9), possibly linking polyadenylation to regulation of cell growth. Even though RBBP6 is found in the mammalian mRNA 3′-end processing complex (10), it has not been shown to function in polyadenylation.
Mpe1 is a particularly interesting subunit because of its evolutionarily conserved tripartite domain structure, yet it is not clear how this protein contributes to a fully functional polyadenylation complex. Mpe1 contains three domains separated by linker regions (Fig. 1A). At its N terminus is a ubiquitin -like (UBL) domain. UBL domains have been shown to interact with ubiquitin-binding domains (UBDs) found on proteins involved in a wide spectrum of cellular activities (11–13). The middle domain of Mpe1 is a zinc knuckle. This motif can mediate both RNA and protein interactions (14–20).
FIG 1.

MPE1 mutants show growth defects. (A) Schematic of mutations analyzed in this study, with conserved domains and their amino acid boundaries indicated. Amino acids 2 to 81 were deleted in the mpe1-ΔUBL construct, and amino acids 283 to 441 were deleted in the mpe1-ΔRING construct. UBL, ubiquitin-like domain. (B) Relative growth rates of mpe1 mutants at different temperatures. Strains were grown in liquid YPD, 10-fold serially diluted, spotted onto YPD plates, and incubated for 3 to 7 days at the indicated temperatures. (C) In vivo expression of the mpe1 mutant proteins. Cells containing a chromosomal deletion of the MPE1 gene but expressing Myc-tagged forms of wt MPE1 or mpe1 mutants on plasmids were first grown at 30°C and then, as indicated, shifted to 37°C for 2 h (mpe1-F9A) or to 16°C for 4 h (mpe1-C182G,C185G and mpe1-L337A). Extracts from these cells were analyzed by Western blotting using an antibody against the Myc epitope or against actin. For the nonviable mpe1-ΔUBL mutation, an extract was made from cells coexpressing an untagged wild-type Mpe1 protein, and only the truncated protein was detected by the anti-Myc antibody. The asterisk indicates a nonspecific band sometimes seen with the anti-Myc antibody. Numbers on the left of the bottom panel indicate the positions of molecular mass markers (in kilodaltons). The amount of mutant protein relative to the amount of wild-type Mpe1 was determined by using actin as a loading control and is indicated under each lane.
The third Mpe1 domain is a RING finger domain found in some E3 and E4 enzymes (21–23). RING-type E3 ubiquitin ligases directly interact both with target substrates and with E2 ubiquitin-conjugating enzymes, thereby facilitating the transfer of the ubiquitin from E2 to the target protein (24). E4 enzymes help to elongate short ubiquitin chains (25). RBBP6 has been shown to enhance the polyubiquitination of p53 by the Mdm2 E3 ligase and to act directly as a ubiquitin ligase on the oncogenic transcription factor YB-1 (26, 27). In addition to targeting proteins for degradation by the proteasome, the reversible addition of ubiquitin can alter protein-protein interactions, and examples of this can be found in aspects of mRNA biology such as transcription, splicing, and nuclear export (28–31). However, no direct role for ubiquitin in mRNA 3′-end processing has been demonstrated.
To understand the precise roles of the individual domains of Mpe1 in mRNA polyadenylation, we examined the consequences of mutating or removing each domain on Mpe1 function and on the abilities of these regions to facilitate interaction with the polyadenylation complex or its RNA substrate. This analysis has revealed critical functions for the UBL domain in mediating protein-protein contacts within the processing complex and an unexpected requirement of the RING finger for RNA binding. We have also uncovered a previously unknown role for ubiquitination in modulating the efficiency of mRNA 3′-end processing.
MATERIALS AND METHODS
Yeast strains and culture.
The S. cerevisiae strains used in this study are as follows: SDL1 (MATa leu2Δ1 ura3-52 trp1Δ63 mpe1Δ [YCp50-URA-MPE1]), SDL2 (MATa leu2Δ1 ura3-52 trp1Δ63 mpe1Δ [pRS315-LEU-Myc-MPE1-His6]), SDL3 (MATa leu2Δ1 ura3-52 trp1Δ63 mpe1Δ [pRS315-LEU-Myc-mpe1-F9A-His6]), SDL4 (MATa leu2Δ1 ura3-52 trp1Δ63 mpe1Δ [pRS315-LEU-Myc-mpe1-C182G,C185G-His6]) (where mpe1-C182G,C185G indicates the mpe1 gene with glycine substituted for cysteine at amino acids 182 and 185), SDL5 (MATa leu2Δ1 ura3-52 trp1Δ63 mpe1Δ [pRS315-LEU-Myc-mpe1-L337A-His6]), SDL6 (MATa leu2Δ1 ura3-52 trp1Δ63 mpe1Δ [YCp50-URA-MPE1] [pRS315-LEU-Myc-mpe1-ΔUBL-His6]), SDL7 (MATa leu2Δ1 ura3-52 trp1Δ63 mpe1Δ [pRS315-LEU-Myc-mpe1-ΔRING-His6]), SDL8 (MATa leu2Δ1 ura3-52 trp1Δ63 mpe1Δ [pRS315-leu::URA3-Myc-MPE1-His6]), SDL9 (MATa leu2Δ1 ura3-52 trp1Δ63 mpe1Δ [pRS315-leu::URA3-Myc-mpe1-F9A-His6]), SDL10 (MATa leu2Δ1 ura3-52 trp1Δ63 mpe1Δ [pRS315-leu::URA3-Myc-mpe1-C182G,C185G-His6]), SDL11 (MATa leu2Δ1 ura3-52 trp1Δ63 mpe1Δ [pRS315-leu::URA3-Myc-mpe1-L337A-His6]), W303 (MATa ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112), SDL13 (MATa ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 mpe1-1) (7), SDL70 (MATa his3-Δ200 leu2-3,112 lys2-801 trp1Δ63 ura3-52) (32), SDL72 (MATa ump1-Δ1::HIS3 his3-Δ200 leu2-3,112 lys2-801 trp1Δ63 ura3-52) (32), SDL68 (MATa leu2-3,112 ura3-52 his3-11) (33), SDL69 (pre1-1 pre4-1 leu2-3,112 ura3-52 his3-11) (33), CM351 (MATa PTA1-TAP::TRP1 leu2Δ1 ura3-52 trp1Δ63) (34), SDL60 (MATa PTA1-TAP::TRP1 leu2Δ1 ura3-52 trp1Δ63 mpe1Δ [pRS315-LEU-Myc-MPE1-His6]), and PJ69-4A (MATa trp1-901 leu2-3,112 ura3-52 his3Δ200 gal4Δ gal80Δ LYS2::GAL1-HIS3 GAL2-ADE2 met2::GAL7-lacZ) (35).
FY23 was used as the host for the construction of strains SDL1 to SDL11. Strains SDL8 to SDL11, which were used for the high-copy-number suppressor screen, were made from strains SDL2 to SDL5 by using “marker swap” plasmids (36). SDL60 was constructed as described previously (34). To determine the effect of mpe1 mutations on yeast cell growth, the plasmid shuffle complementation assay was used (37). The pRS315 plasmids containing mpe1 mutations were analyzed in the MPE1 plasmid shuffle strain SDL1. Growth properties were analyzed by growing the strains in liquid yeast extract-peptone-dextrose (YPD) at room temperature to an optical density at 600 nm (OD600) of 1.0, spotting 5 μl of 10-fold serial dilutions onto YPD plates, and incubating the plates for 3 to 7 days at 12, 16, 24, 30, or 37°C.
MPE1 plasmids and mutant construction.
For pRS315-MPE1, the MPE1 sequence is flanked by a Myc epitope tag at the N terminus and by a 6× histidine tag at the C terminus and is under the control of the MPE1 promoter and terminator sequences (900 bp upstream and 900 bp downstream of the coding region of the MPE1 gene). The coding sequence of MPE1 was amplified from genomic DNA by PCR using the upstream primer NdeI-Myc-MPE1(5′ ORF) and the downstream primer MPE1(3′ ORF)-His6-NotI. The PCR product was digested with NdeI and NotI and was cloned into the pRS315 (LEU2) vector flanked by the 900-bp regions upstream and downstream of the coding region of the MPE1 gene. MPE1 point mutations and truncation mutations were made by the overlap PCR method (38), and pRS315-mpe1 derivatives were constructed by inserting these PCR-amplified fragments into the NdeI and NotI sites of pRS315-MPE1. Amino acids 2 to 81 were deleted in the mpe1-ΔUBL construct, and amino acids 283 to 441 were deleted in the mpe1-ΔRING construct. Mutations were verified by sequencing.
Pulldown assays.
Yeast processing extracts (1 mg) prepared as described previously (39) were incubated with Ni-nitrilotriacetic acid (NTA) resins (Qiagen) overnight at 4°C in 500 μl of buffer IP-150 (10 mM Tris-HCl at pH 7.9, 150 mM NaCl, 1 mM EDTA, 10% glycerol, 1 mM dithiothreitol [DTT], 1 mM phenylmethylsulfonyl fluoride [PMSF], 2 μM pepstatin A, 0.6 μM leupeptin, 0.01% NP-40). In some cases, after two washes with 1 ml of buffer IP-150 containing 5 mM MgCl2–2 mM CaCl2, the beads were treated with an RNase cocktail (15 μg/ml RNase A, 38 U/ml RNase T1; Thermo Scientific) for 40 min at 30°C in the same buffer, followed by three washes with 1 ml of buffer IP-150. Samples were centrifuged, and the pellets were washed five times with 1 ml of buffer IP-150. The pellets were resuspended in 100 μl of IP-150 containing 500 mM imidazole. The eluates were resuspended with SDS sample buffer, boiled for 3 min, and analyzed in an SDS-8% polyacrylamide gel. Western blotting was performed according to standard procedures. Polyclonal antibodies against Rna14, Rna15, and Ysh1 were a gift from H. Domdey. The monoclonal antibodies against Pta1 and Pap1 have been described previously (39). Antibodies against ubiquitin and actin were purchased from Santa Cruz Biotechnology, Santa Cruz, CA (sc-8017), and Abcam, Cambridge, MA (ab8224), respectively. Western blots were quantitated by ImageJ software.
For the detection of ubiquitination associated with Pap1, cells expressing hemagglutinin (HA)-tagged ubiquitin under the control of a copper-inducible promoter (40) were grown in 1 liter of selective medium containing 0.15 mM CuSO4 to an OD600 of 1.5. The cells were resuspended in 5 ml of buffer C (10 mM HEPES-KOH at pH 7.0, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT, 1 mM PMSF, 2 μM pepstatin A, 0.6 μM leupeptin, 5 mM 1,10-phenanthroline monohydrate [Sigma], 20 mM N-ethylmaleimide [NEM; Fisher Scientific], 5 μM MG132 [Sigma], and 8 μg ubiquitin-aldehyde [Boston Biochem]). Extracts were prepared as described previously (41) and were incubated with an anti-Pap1 antibody overnight. Protein A beads (Santa Cruz) were then added and were rotated for 2 h. Beads were washed twice with IP-150 buffer (10 mM Tris-HCl at pH 7.9, 150 mM NaCl, 1 mM EDTA, 10% glycerol, 1 mM DTT, 1 mM PMSF, 2 μM pepstatin A, 0.6 μM leupeptin, 5 mM 1,10-phenanthroline monohydrate, and 20 mM NEM), washed twice with IP-150 buffer containing 0.1% SDS, resuspended in SDS sample buffer, boiled for 3 min, and analyzed in an SDS-8% polyacrylamide gel. Western blotting was performed according to standard procedures.
For glutathione S-transferase (GST) pulldown assays, plasmid GST-MPE1 was generated by inserting the MPE1 open reading frame (ORF) into the SmaI/XhoI sites of pGEX-6P. The expression and purification of GST-Mpe1 from Escherichia coli Rosetta (DE3) cells, as well as GST pulldown assays, were carried out as described previously (41, 42). Radiolabeled Cft1 protein was translated in vitro in the presence of [35S]methionine with a TNT rabbit reticulocyte lysate system (Promega). GST-Mpe1 or GST was incubated with a 30-μl bed volume of glutathione-Sepharose beads in 200 μl of buffer IP-150 for 1 h at 4°C. After washing with buffer IP-150, beads were incubated with radiolabeled Cft1 (10 μl of the in vitro-translated reaction product) for 2 h at 4°C. The beads were centrifuged, washed five times with 1 ml of buffer IP-150 containing 0.05% NP-40, and boiled in SDS-PAGE sample buffer. The eluted sample was then resolved by SDS-6% polyacrylamide gel electrophoresis and was visualized by a Storm 960 PhosphorImager.
Protein purification.
Derivatives of plasmids pET21b-MPE1 and pET21b-mpe1 (tagged with Myc at the N terminus and with 6× histidine at the C terminus) were generated by digesting NdeI and NotI fragments from pRS315-MPE1 and pRS315-mpe1 and inserting these fragments into pET21b digested with the same enzymes. Recombinant proteins were expressed and purified from Escherichia coli Rosetta (DE3) cells as described previously (41, 42).
In vitro RNA processing assay.
Preparation of yeast cell extracts, transcription of [α-32P]UTP-labeled GAL7-1 RNA or precleaved GAL7-9, and processing assays were performed as described previously (39). The template for CYC1 precursor was pGYC1 digested with PvuII (43). Ubistatin A (NSC665534) was generously provided by the Drug Synthesis and Chemistry Branch of the National Cancer Institute, and MG132 was obtained from Sigma. For the RNA-processing rescue experiment with K48-linked tetraubiquitin (Boston Biochem), 70 μM ubistatin A was preincubated with 100 μM K48-linked tetraubiquitin for 10 min at 24°C and then incubated with cell extracts for an additional 10 min at 24°C before the addition of radiolabeled GAL7-1.
RNA binding assays.
RNA binding was carried out in a volume of 20 μl containing [α-32P]UTP-labeled GAL7-1, with 0.6 to 4 μM recombinant Mpe1, 20 mM HEPES-KOH (pH 7.9), 50 mM KCl, 1 mM DTT, 0.1 mM ZnSO4, and 25 μg/ml tRNA. After a 15-min incubation at 30°C, the reaction mixture was loaded onto a 4% polyacrylamide gel with 1× Tris-borate-EDTA (TBE) buffer and was electrophoresed. The gels were dried and were visualized by a Storm 960 PhosphorImager. RNase H protection experiments were performed as described previously (44).
To examine the binding of Mpe1 to RNA when it is incorporated into the CPF complex, CPF was purified from yeast expressing TAP-tagged Pta1. Extracts were prepared from 2 liters of cells grown in YPD at 30°C to an OD600 of 1.5 as described previously (41), except that the ammonium sulfate precipitation step was omitted. Proteins were purified according to the standard TAP procedure (45), separated on an SDS-10% polyacrylamide gel, and detected by silver staining. The known CPF subunits were confirmed by Western blotting. For UV cross-linking experiments, reaction mixtures were assembled in a final volume of 200 μl containing CPF purified from Pta1-TAP or from Pta1-TAP containing pRS315-Myc-Mpe1-His6, 2,800,000 cpm of [α-32P]UTP-labeled GAL7-1 prepared as described above, 50 μg/ml tRNA, 1 mM magnesium acetate, 75 mM potassium acetate, 10 mM Tris-HCl (pH 7.5), and 1.5 mM ATP. After incubation for 15 min at 30°C, samples were returned to ice and were irradiated with 1.2 mJ from a Stratalinker UV cross-linker, model 2400 (Stratagene). One-tenth of the sample was then digested with 2 μg of RNase A and 40 U of RNase T1 for 1 h at 37°C and was resolved on an SDS-10% polyacrylamide gel. The gel was dried and was visualized with a Storm 960 PhosphorImager. Mpe1-His6 from the rest of the sample was then captured by incubation with 80 μl of Ni-NTA (Qiagen) in 600 μl of buffer (150 mM NaCl, 20 mM Tris-HCl [pH 7.9]) for 4 h at 4°C. Unbound proteins were removed by five washes in 600 μl of buffer (150 mM NaCl, 20 mM Tris-HCl [pH 7.9]), followed by two washes in 600 μl of the same buffer containing 0.1% SDS. Bound proteins were eluted in 150 μl of elution buffer (100 mM EDTA, 0.1% SDS, 150 mM NaCl, 20 mM Tris-HCl [pH 7.9]), transferred to another tube, and digested with 20 μg of proteinase K. The eluted sample was then resolved on a 5% acrylamide–8.3 M urea gel and was visualized with a Storm 960 PhosphorImager.
Yeast two-hybrid assay.
pGBD-MPE1, pGBD-UBL, pGBD-ZK, and pGBD-RING were constructed by inserting full-length MPE1, the UBL (amino acids 1 to 173), the zinc knuckle domain and second linker region (amino acids 174 to 282), or the RING finger domain (amino acids 283 to 441) into vector pGBD-C2. Each of these constructs was transformed into the yeast two-hybrid selection strain pJ69-4A along with plasmid pGAD-CFT1. Transformants were selected on complete medium lacking leucine and tryptophan (CM-LT) to select for both plasmids pGBD and pGAD. Positive interactions were determined by the ability of the transformed yeast cells to grow on complete medium lacking leucine, tryptophan, and histidine (CM-LTH). Cells were plated on CM-LT as a growth control or on CM-LTH for the determination of positive interactions. Plates were incubated at 30°C for 3 days.
High-copy-number suppressor screen.
High-copy-number suppressors of the mpe1-F9A, mpe1-C182G,C185G, and mpe1-L337A growth defects were isolated by transforming the mutant strain with a 2μm yeast genomic library (46). Plasmid DNA was transformed into the mutant strains, and transformants were selected on minimal medium lacking leucine. After 3 days at 30°C, transformants were replica plated onto minimal medium lacking leucine and were incubated at 37°C for 3 days (mpe1-F9A) or at 12°C for 18 days (mpe1-C182G,C185G and mpe1-L337A). For the mpe1-F9A screen, 3 suppressors were identified among ∼18,000 Leu+ transformants. DNA sequence analysis revealed that plasmids from these 3 transformants contained MPE1. For the mpe1-C182G,C185G screen, 5 suppressors were isolated from ∼20,000 Leu+ transformants. DNA sequence analysis revealed that 2 plasmids contained MPE1 and the other 3 contained the same region of genomic DNA, which included PEX10, HEL2, CIA1, MSW1, and CCC2. This region was subcloned by cutting out an insert fragment with restriction enzymes MscI, located at the CIA1 coding sequence, and SmaI, located at the multiple cloning site of pGP564, and religating the backbone fragment to produce a construct containing the PEX10 and HEL2 genes that suppressed the mutant phenotype. The PEX10 gene (including 900-bp regions upstream and downstream of the coding sequence) was further cloned into pGP564 by PCR. However, PEX10 was unable to suppress the mutant phenotype, and thus, it was concluded that the HEL2 gene was responsible for the suppression. For the mpe1-L337A screen, 6 suppressors were isolated among ∼23,000 Leu+ transformants. DNA sequence analysis revealed that 2 of the plasmids contained MPE1 and 4 contained the same region of DNA, which included SAY1, MES1, YGR266W, FOL2, and HUA1. The subcloning of this region by cutting out a fragment with restriction enzyme HindIII, located at the multiple cloning site of pGP564 and at the FOL2 coding sequence, and religating the backbone fragment resulted in a construct containing only the HUA1 gene, which was sufficient to suppress the mutant phenotype.
RESULTS
The three conserved domains of Mpe1 are important for normal growth and mRNA 3′-end processing.
In order to investigate the role of Mpe1 in mRNA 3′-end processing, we carried out site-directed mutagenesis on amino acid residues that show high conservation among Mpe1 homologs. We generated 14 single alanine substitution mutations across the three conserved domains (F9A, K31A, K39A, L48A, P78A, T212A, Q268A, C290A, V301A, F311A, L316A, D324A, P328A, and L337A), one double glycine substitution mutation located in the zinc knuckle domain (C182G C185G), and two deletion mutations (ΔUBL and ΔRING). The mpe1-ΔUBL mutant lacks amino acids 2 to 81, and the mpe1-ΔRING mutant lacks amino acids 283 to 441. A wild-type (wt) copy of the MPE1 gene was replaced with the mutant mpe1 gene by plasmid shuffling (see Materials and Methods), and strains were tested for viability and conditional growth defects. Of the single alanine substitution mutants, one UBL domain mutant (mpe1-F9A) caused poor growth at 30 and 37°C, and one RING finger domain mutant (mpe1-L337A) grew poorly at 16°C (Fig. 1B). No growth defects were observed with the remaining 12 single amino acid replacements (see Fig. S1A in the supplemental material). The double glycine substitution mutation located in the zinc knuckle also resulted in cold-sensitive growth (Fig. 1B). Deletion of the UBL domain was lethal (see Fig. S1B in the supplemental material), while deletion of the C-terminal region containing the RING finger domain caused slow growth at 30 and 37°C and severely compromised growth at 16°C (Fig. 1B).
To check for the expression levels of these mpe1 mutants, extracts were made from wild-type or mutant cells grown at 30°C or from cultures shifted for 4 h to 16°C (mpe1-C182G,C185G and mpe1-L337A) or for 2 h to 37°C (mpe1-F9A) before harvesting. Western blotting using these extracts showed that the mutant mpe1-C182G,C185G and mpe1-L337A proteins were expressed similarly to wild-type mpe1 at the restrictive temperatures (Fig. 1C). The level of the mpe1-F9A mutant was about half that of the wild type at 37°C, and the level of mpe1-ΔRING was equivalent to that of the wild type at 30°C. The mpe1 mutant with a lethal deletion of the UBL domain was expressed well in the presence of the wild-type MPE1 gene (Fig. 1C). This analysis shows that all of the conserved domains of Mpe1 are important for growth.
To see whether the conditional growth defects conferred by the mpe1-F9A, mpe1-C182G,C185G, and mpe1-L337A mutations might be due to defects in mRNA 3′-end processing, we performed in vitro 3′-end-processing assays using extracts made from each of these mutants. When a radiolabeled RNA substrate containing the GAL7 poly(A) site was incubated with the wild-type extract and ATP, the RNA was efficiently cleaved and polyadenylated regardless of the temperature at which the reactions were performed (Fig. 2A, lanes 2, 3, 8, and 10). However, in the mpe1-C182G,C185G and mpe1-L337A extracts, this activity was lower than that in wild-type extracts at 30°C and dropped further at 18°C (Fig. 2A, lanes 4 to 7). The defect was most severe for the mpe1-C182G,C185G mutant, a finding consistent with its strong cold-sensitive growth phenotype. The activity of the mpe1-F9A extract was comparable to that of the wild-type extract when reactions were conducted at 30°C but about half that of the wild-type extract at 37°C (Fig. 2A, lanes 8 to 11). The cleavage step of 3′-end processing can be studied separately from poly(A) addition by substituting 3′-dATP for ATP, and the poly(A) addition step can be isolated from cleavage by using a substrate that ends at the cleavage site. The activities of the mpe1-C182G,C185G and mpe1-L337A extracts were compromised for both steps (Fig. 2B and C, lanes 4 to 7), while the mpe1-F9A mutant was mostly impaired in poly(A) addition, especially at 37°C (Fig. 2B and C, lanes 9 and 11).
FIG 2.
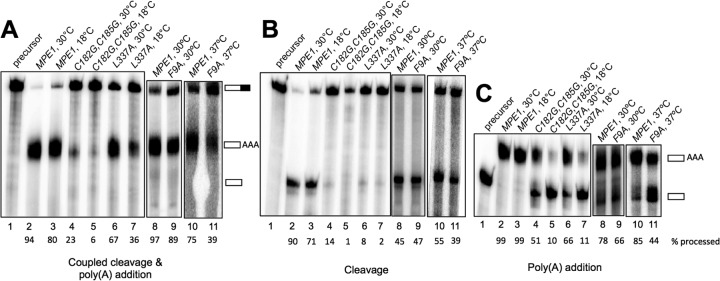
MPE1 mutants show impairment of in vitro processing. Processing extracts were prepared from the indicated strains grown at 30°C and then shifted to 37°C for 2 h (F9A) or to 16°C for 4 h (C182G,C185G and L337A). Assays were conducted for 20 min at the indicated temperatures. (A) For the coupled cleavage-polyadenylation assays, extracts were incubated with ATP and full-length 32P-labeled GAL7-1 RNA. (B) For cleavage-only assays, the reactions were carried out as described above except that 3′-dATP was used instead of ATP. (C) For poly(A) addition assays, the reactions were carried out as described above except that precleaved GAL7-9 RNA, ending at the poly(A) site, was used as the precursor. RNA products were resolved on a denaturing 5% polyacrylamide gel and were visualized with a phosphorimager. The positions of the substrate and products are indicated to the right of panels A and C (partially filled rectangle, full-length GAL7-1 RNA; open rectangles labeled AAA, polyadenylated product; open rectangles, cleaved product). The lane marked “precursor” indicates the unreacted substrate for each reaction. The percentage of processed RNA is given under each lane number.
The original mpe1-1 mutant identified by Vo et al. (7) showed defective polyadenylation in vitro, grew slowly at 24 and 30°C, and was nonviable at 37°C. It contains four mutations: F9S in the UBL domain, Q268K in the linker region between the zinc knuckle and the RING finger domain, L337F in the RING finger domain, and a stop codon created at L354 just outside the RING finger domain. A strain with a TAP tag at L354 was viable and gave intact CPF (7). However, the other mutations were not examined separately. Our results with the F9A, C182G C185G, and L337A mutants show that each of three conserved domains of Mpe1 is needed for optimal cell growth and function in mRNA 3′-end processing.
Interactions with CPF require more than one region of Mpe1.
MPE1 was originally discovered as a suppressor of a mutation in PCF11, which encodes a component of CF IA (7). To test whether Mpe1 functions as a bridge between CPF and CF IA, we performed pulldown assays from cell extracts by using nickel affinity beads to capture versions of Mpe1 that had been doubly tagged with the His6 and Myc epitopes. Extracts were prepared from wild-type (MPE1) cells and from the mpe1-ΔUBL or mpe1-ΔRING mutant. For the mpe1-ΔUBL mutant, which is not viable, the cells were grown in the presence of a plasmid containing untagged wild-type MPE1. The composition of proteins in the pulldown assays was assessed by Western blotting with antibodies against the Rna14 and Rna15 subunits of CF IA (Fig. 3A). Both Rna14 and Rna15 associated with Mpe1 in wild-type and mpe1-ΔRING extracts but were not pulled down in mpe1-ΔUBL extracts, where only background levels were detectable (Fig. 3A).
FIG 3.

Interactions with CPF involve more than one region of Mpe1. (A) Pulldown assays with extracts prepared from cells expressing Myc-His6-tagged forms of mpe1, mpe1-ΔRING, or mpe1-ΔUBL. Cells were grown at 30°C. Pulldown assays were performed using nickel affinity beads, and the eluates were analyzed by Western blotting with antibodies against CF IA and CPF subunits. Mpe1 was detected with the anti-Myc antibody. The control lane shows a pulldown with an extract from cells expressing only untagged Mpe1. The asterisk indicates a nonspecific band detected with the anti-Myc antibody. The amount of protein pulled down in the mutant relative to wild-type Mpe1 is given under each lane. (B) Yeast two-hybrid analysis reveals an interaction between GBD-Mpe1 and GAD-Cft1. A positive two-hybrid interaction was detected by transcription activation of the HIS3 reporter gene, which allowed for growth on a medium lacking histidine. Pta1 and Ssu72, which have been shown previously to interact with each other (47), were used as a positive control. The pairings of two-hybrid constructs are as follows: lane 1, pGBD-PTA1 plus pGAD-SSU72; lane 2, pGBD-MPE1 plus pGAD; lane 3, pGBD plus pGAD-CFT1; lane 4, pGBD-MPE1 plus pGAD-CFT1. (C) Schematic representation of the different Mpe1 regions encoded by pGBD plasmids. Numbers indicate the amino acid boundaries of each Mpe1 fragment. UBL, ubiquitin-like; ZK, zinc knuckle. (D) The region containing the zinc knuckle domain of Mpe1 and the second linker mediates the two-hybrid interaction with Cft1. Yeast two-hybrid analyses were performed as described for panel B. The pairings of two-hybrid constructs are as follows: lane 1, pGBD-PTA1 plus pGAD-SSU72; lane 2, pGBD-ZK plus pGAD; lane 3, pGBD plus pGAD-CFT1; lane 4, pGBD-UBL plus pGAD-CFT1; lane 5, pGBD-ZK plus pGAD-CFT1; lane 6, pGBD-RING plus pGAD-CFT1. (E) Mpe1 interacts with Cft1 in vitro. GST pulldown assays were performed with purified recombinant GST or GST-Mpe1 and 35S-labeled in vitro-translated Cft1. The input lane represents 1/5 of the in vitro-translated Cft1 used for binding.
To sample the association of the mpe1 mutants with CPF subunits, the pulled-down proteins were also probed with antibodies against Pap1, Pta1, and Ysh1. These subunits could not be pulled down with mpe1-ΔUBL, and these interactions were weakened but still present upon deletion of the RING finger domain (Fig. 3A). Pulldowns of Mpe1 in the presence of RNase did not change the amounts of associated proteins (see Fig. S2A in the supplemental material), indicating that the interactions observed are mediated by protein-protein contacts and not by RNA binding.
To further investigate Mpe1 interactions, we performed yeast two-hybrid analysis using MPE1 fused to the GAL4 DNA binding domain (GBD) and paired with GAL4 activation domain (GAD) fusion constructs, which included CFT1, PTA1, PCF11, SSU72, and HRP1. These fusion constructs represented each of the known polyadenylation factors: Cft1, Pta1, and Ssu72 are subunits of CPF; Pcf11 is a subunit of CF IA; and Hrp1 is CF IB. These GBD and GAD fusion plasmids were cotransformed into a reporter strain containing a copy of HIS3 controlled by the GAL1 promoter. Positive two-hybrid interactions activate the GAL1-HIS3 reporter gene and enable yeast to grow on a medium lacking histidine. Little growth was observed when GBD-MPE1 was combined with the GAD vector or with the GAD fused to PTA1, PCF11, SSU72, or HRP1 (Fig. 3B; see also Fig. S2B in the supplemental material). However, when GBD-MPE1 was paired with GAD-CFT1, cells were able to grow in the absence of histidine at a level comparable to that seen with the known two-hybrid interaction of PTA1 and SSU72 (47) (Fig. 3B). Little growth was observed when GAD-CFT1 was combined with the GBD vector, indicating that the two-hybrid signal required MPE1. This interaction may be direct, since a recombinant GST-Mpe1 fusion protein pulled down twice as much in vitro-translated Cft1 as did GST alone (Fig. 3E).
Given the pulldown analysis shown in Fig. 3A, it was surprising not to see a two-hybrid interaction between Mpe1 and the CPF subunit Pta1 or Ssu72 or between Mpe1 and the CF IA subunit Pcf11. However, as we have observed previously (48), two-hybrid analysis will not always detect interactions that have been demonstrated biochemically, perhaps because of conformational constraints when the fusions are incorporated into the complexes being tested. Nevertheless, two-hybrid signals, when detected, can be helpful in further dissection of critical interaction domains. With this goal in mind, we tested the GAD-CFT1 construct against GBD constructs fused to the different Mpe1 regions diagramed in Fig. 3C. By this in vivo assay, only the fragment containing the zinc knuckle domain and the linker between the zinc knuckle and the RING finger scored positive (Fig. 3D).
In conclusion, the pulldown experiments show that the UBL domain of Mpe1 is essential for the association of Mpe1 with CPF and CF IA and that the RING finger contributes to the interaction with CPF but not to that with CF IA. The two-hybrid analysis suggests that the zinc knuckle of Mpe1 or the second linker region also makes contact with other CPF subunits, perhaps through a direct Cft1 interaction.
Mpe1 binds to RNA.
Mpe1 contains a zinc knuckle domain (CX2CX4HX4C) that in other proteins has been implicated in protein-protein interactions and RNA binding (14–20, 49). To test whether Mpe1 binds directly to RNA, we performed an RNA gel mobility shift assay with purified recombinant wild-type protein (Fig. 4A). Radiolabeled GAL7-1 precursor RNA was incubated with increasing concentrations of Mpe1, and the samples were subjected to gel electrophoresis under native conditions. Mpe1 shifted the position of RNA in a concentration-dependent manner (Fig. 4B, lanes 2 to 4). Higher concentrations of Mpe1 caused a supershift of the Mpe1-RNA complexes, perhaps due to the presence of more than one binding site on the GAL7 RNA.
FIG 4.

Mpe1 binds to RNA with sequence specificity. (A) Coomassie blue-stained gel of purified recombinant Mpe1 proteins used for RNA gel mobility shift assays. (B) RNA gel mobility shift assays were performed with the indicated recombinant proteins as described in Materials and Methods. Concentrations of 300 nM, 600 nM, and 1.8 μM were used for each protein and are indicated by the wedges above the gel. (C) Mapping the binding site of Mpe1 on CYC1 precursor. (Top) Sequence of the CYC1 transcript and positions of signal sequences and the poly(A) site. The locations of sequences complementary to DNA oligonucleotides 1 to 8 are indicated. (Bottom) RNase H protection profile of Mpe1 on 32P-labeled CYC1 RNA, which was first incubated with binding buffer (no protein [lanes 1 to 9]) or Mpe1 (600 nM [lanes 10 to 18] or 1.8 μM [lanes 19 to 27]) and then cleaved by the addition of RNase H and oligonucleotides as indicated. As a control, oligonucleotides were omitted in lanes 1, 10, and 19. The amounts of digested product were determined for lanes using DNA oligonucleotides 6 to 8 and are indicated under the lane numbers.
To determine which domains of Mpe1 are involved in binding to RNA, purified recombinant mpe1-ΔUBL, mpe1-C182G,C185G, and mpe1-ΔRING proteins (Fig. 4A) were used in the mobility shift assay. mpe1-ΔUBL behaved like wt protein in this assay (Fig. 4B, lanes 5 to 7). In contrast, the mpe1-C182G,C185G mutant showed a lower affinity for the GAL7 RNA, with protein-RNA complexes observed only at the highest concentration (Fig. 4B, lanes 8 to 10). The RING finger domain deletion mutant failed to interact with RNA (Fig. 4B, lanes 11 to 13). These results show that both the zinc knuckle and RING finger domains are essential for efficient binding of Mpe1 to RNA.
To investigate whether Mpe1 had any sequence specificity in its interaction with RNA, we employed a method previously used by the Keller laboratory to examine the specific binding of CPF (44, 50, 51). Radioactive RNA containing the CYC1 poly(A) site and flanking sequences is challenged with the indicated cDNA oligonucleotides (Fig. 4C, top) and RNase H, with or without preincubation with recombinant Mpe1. Binding of Mpe1 to specific regions would block hybridization of the oligonucleotide and prevent digestion of the RNA, which is detected by the appearance of smaller RNA fragments. We used the two concentrations of Mpe1 (600 nM and 1.8 μM) that gave complexes in the gel mobility shift assay for which results are shown in Fig. 4B. Protection was observed in the regions corresponding to oligonucleotides 7 and 8 at both Mpe1 concentrations (Fig. 4C, bottom). At the higher concentration of Mpe1, there was protection in the region of oligonucleotide 6. Although cleavage in the region of oligonucleotide 2 in the absence of Mpe1 was very low, there may also have been protection in this area. The section of the transcript complementary to oligonucleotides 7 and 8 matches the cleavage site and the U-rich tracts that flank this site, which are known to constitute part of the signal sequences that define yeast poly(A) sites (52). This observation suggests that Mpe1 contributes to the recognition of this region of the polyadenylation precursor.
Although the mobility shift and RNase H protection assays showed that recombinant Mpe1 binds directly to RNA, the question remained whether Mpe1 binds to RNA when it is part of CPF. To address this issue, we used UV light to photochemically cross-link proteins that are in proximity to nucleotides (53). Extracts were prepared from yeast expressing a TAP-tagged version of the Pta1 subunit (54) and either a His6-tagged or an untagged version of Mpe1, and CPF was purified from extracts by using the TAP tag (Fig. 5A). CPF was mixed with radiolabeled GAL7 RNA precursor, and the mixture was subjected to photochemical cross-linking. Subsequently, RNA was digested, and cross-linked proteins were resolved by SDS-PAGE and were identified by the radioactive RNA tag. Several proteins in CPF are cross-linked to the RNA (Fig. 5B). To see if one of the cross-linked proteins is Mpe1, RNA was cross-linked to CPF, but the RNase digestion step was omitted. His6-Mpe1 was then purified from cross-linked CPF by nickel affinity chromatography, using a bead wash buffer containing 0.1% SDS to disrupt the CPF complex. The samples were then digested with proteinase K, and the released RNA was resolved on a denaturing gel. The RNA signal was significantly increased above background only if the CPF was obtained from the strain expressing His6-Mpe1, not if CPF came from the strain lacking this tagged Mpe1 (Fig. 5C). This experiment demonstrates that Mpe1 can contact polyadenylation precursor RNA when it is part of CPF.
FIG 5.
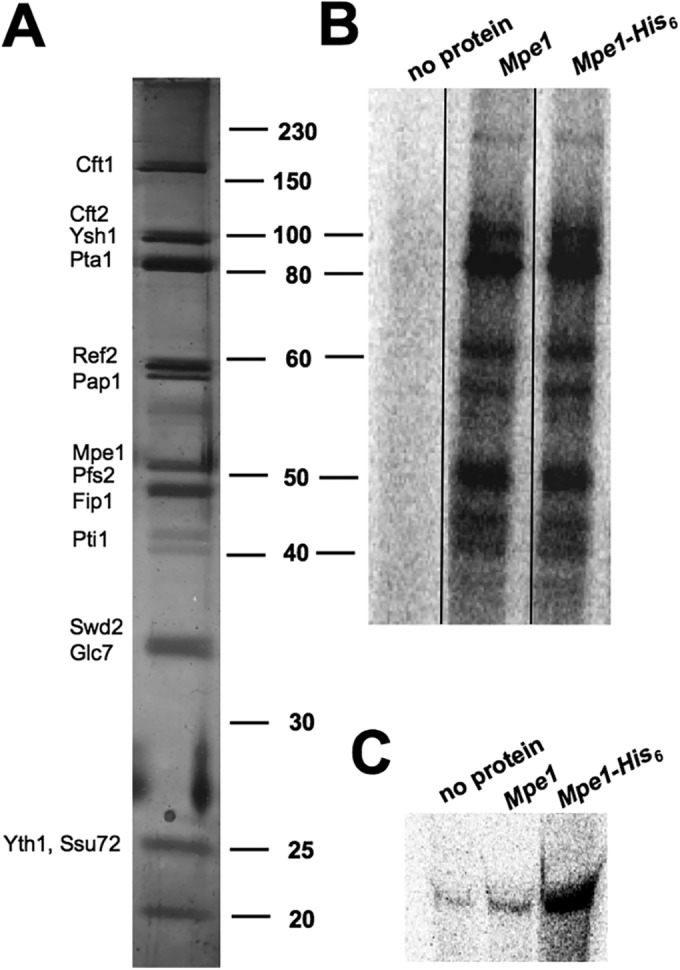
Mpe1 binds RNA when it is a subunit of CPF. (A) Silver-stained gel of CPF TAP-purified from a strain expressing His6-Mpe1, showing the expected subunits (54). The positions of size markers (in kilodaltons) are indicated on the right, and the positions of CPF subunits are noted on the left. (B) Cross-linking of CPF proteins to RNA, using CPF from a strain expressing His6-Mpe1 or from a strain expressing untagged Mpe1. The two CPF preparations were cross-linked to GAL7-1 polyadenylation precursor. After digestion of the RNA, proteins were resolved by SDS-PAGE (10% acrylamide), and radioactively tagged proteins were visualized by phosphorimager analysis. The two CPF preparations gave identical cross-linking patterns. The samples were loaded onto the same gel, and the intervening lanes were removed as demarked by the line between Mpe1 and the Mpe1-His6. (C) Mpe1 binds to RNA when it is part of the CPF complex. After cross-linking of CPF and RNA, His6-tagged Mpe1 was purified by Ni-NTA, followed by digestion with proteinase K. The samples were then resolved on a 5% acrylamide–8.3 M urea gel, and RNAs were visualized by phosphorimager analysis. The “no protein” lanes in panels B and C show results for samples that included RNA but no CPF.
Ubiquitination plays a role in mRNA 3′-end processing.
One role of ubiquitination is to target proteins for degradation by the proteasome (55). To determine if inactivation of the proteasome might affect mRNA 3′-end processing, extracts were prepared from the proteasome-defective Δump1 and pre1-1 pre4-1 strains (33, 56) grown at the nonpermissive temperature of 37°C. These extracts were deficient in polyadenylation in vitro (Fig. 6A), suggesting that failure to degrade certain proteins is detrimental to processing.
FIG 6.

Role of ubiquitination in mRNA 3′-end processing. (A) Processing extracts were prepared from wild-type, Δump1, and pre1-1 pre4-1 strains that had been grown at 30°C and then shifted to 37°C for 2 h. Processing reactions used GAL7-1 RNA, and RNAs were detected as described for Fig. 2. (B) Disruption of ubiquitin interactions inhibits 3′-end processing activity and, in particular, the cleavage step, in vitro. Whole-cell extracts were incubated with increasing amounts of ubistatin A in DMSO (5 μM, 20 μM, 100 μM, and 1 mM; indicated by the wedges above lanes 4 to 7, 10 to 12, and 17 to 20) or with (lanes 3, 9, and 16) or without (lanes 2, 8, and 15) DMSO for 10 min at room temperature before the addition of GAL7-1 RNA precursor to initiate the 3′-end processing assay, using the reaction conditions and the procedure for detection and quantitation of RNAs that are described in the legend to Fig. 2. (C) Ubistatin A inhibits the processing of the precursor containing the CYC1 poly(A) site. The coupled cleavage and poly(A) addition reaction was performed using ATP and radioactive precursor containing the CYC1 poly(A) site. (D) Ubistatin A inhibition is rescued by polyubiquitin. Seventy micromolar ubistatin A was preincubated with 100 μM K48-linked tetraubiquitin for 10 min at room temperature and then incubated with the whole-cell extract for an additional 10 min before the addition of the GAL7-1 RNA. (E) MG132 inhibits 3′-end processing activity. The processing extract was incubated with MG132 in DMSO (1 μM, 10 μM, 100 μM, and 1 mM) or with DMSO alone for 10 min at room temperature before the addition of GAL7-1 RNA precursor to initiate the 3′-end processing assay, using the reaction conditions and the procedure for detection and quantitation of RNAs that are described in the legend to Fig. 2. The samples were loaded onto the same gel, and the intervening lane was removed as demarked by the line between lanes 1 and 2.
However, because of the pleiotropic influence of ubiquitin on many cellular activities, the defects in processing observed with the proteasome mutants may be indirect. To address this concern, we next asked whether ubiquitin modifications already present in the extract are important for mRNA 3′-end processing. In addition to directing proteins for degradation, ubiquitination can modulate a protein's activity through conformational changes or by creating or obstructing binding sites for interacting partners. This type of regulation often involves interactions of ubiquitinated proteins with other proteins harboring ubiquitin-binding domains (UBDs) (11, 57). The consequences of blocking such interactions can be assessed in vitro by using ubistatin A, an inhibitor that binds in the vicinity of the hydrophobic patch on the surface of ubiquitin that interacts with UBDs (28, 58). When ubistatin A was included in an assay that measures coupled cleavage and poly(A) addition, 3′-end processing in vitro was inhibited (Fig. 6B, lanes 4 to 7). Similar concentrations of ubistatin A inhibited mRNA splicing in vitro (28). When the two steps of processing were examined separately, we observed that ubistatin A inhibited only the cleavage step and did not affect poly(A) addition when this reaction was uncoupled from cleavage (Fig. 6B, lanes 10 to 13 and 17 to 20). Ubistatin A also inhibited the processing of RNA containing the CYC1 poly(A) site (Fig. 6C), which differs from the GAL7 site by having longer U-rich tracts flanking the cleavage site but a poorer match to the UA-rich efficiency element (44), suggesting that ubistatin A inhibition is not specific to the GAL7 precursor.
To confirm that the inhibitory effect of ubistatin A on in vitro processing is due to the drug itself and not to any contaminants in drug preparation, ubistatin A was preincubated with an excess of K48-linked polyubiquitin chains before it was added to the in vitro processing reaction mixture. Because of the high affinity of ubistatin A for K48-linked polyubiquitin chains (58), this treatment should block ubistatin A from interacting with ubiquitins in the extract. The inclusion of this step completely restored in vitro processing activity (Fig. 6D), suggesting that the inhibitory effect was indeed due to ubistatin A itself.
Ubistatins also block the binding of ubiquitinated proteins to the proteasome (58). While the inhibitory effect of ubistatin A on mRNA 3′ processing could reflect a nonproteolytic function of ubiquitin, it could also indicate that ubistatin A is preventing the proteasomal degradation of an inhibitor of mRNA 3′ processing that is present in yeast extract. To test more directly whether proteasomal activity is important for processing in vitro, we added the proteasomal inhibitor MG132 to our processing reaction mixtures. Approximately half as much precursor was processed in the presence of 100 μM and 1 mM MG132 as in the presence of the dimethyl sulfoxide (DMSO) control (Fig. 6E). The effective concentration of MG132 is similar to the concentration applied to yeast cells to inhibit the proteasome in vivo (50 μM) (59) or to cell extracts to inhibit the proteasome in vitro (132 μM) (60) but is much higher than that reported to inhibit purified proteasomes (5 μM) (61). The effect of MG132 on polyadenylation suggests that active protein degradation contributes directly to efficient processing in vitro.
Mpe1 is needed for the association of ubiquitin with Pap1.
Taken together, our findings support a role for ubiquitination in modulating mRNA 3′-end processing. We have previously shown evidence that the yeast poly(A) polymerase Pap1 could be ubiquitinated (62), and we looked for effects of Mpe1 mutation on this modification. For this purpose, a plasmid harboring HA-tagged ubiquitin under the control of a copper-inducible promoter was used to overexpress ubiquitin (40). After extract preparation, immunoprecipitation was performed using an antibody against Pap1 and 0.1% SDS in the wash buffer to remove Pap1-interacting proteins. We have shown previously that this treatment disrupts protein-protein interactions within the Pap1-containing CPF complex (34). The immunoprecipitated Pap1 was then subjected to Western blotting using an antibody against ubiquitin to detect ubiquitinated species. This revealed a smear of high-molecular-weight bands, which were not observed in the control pulldown assay with no antibody and thus could correspond to polyubiquitinated forms of Pap1 (Fig. 7A, left). However, these high-molecular-weight bands were not detected when the precipitate was blotted with an anti-Pap1 antibody (Fig. 7A, right), suggesting that they are not abundant Pap1 species but are seen with the antibody against ubiquitin because of the multiple ubiquitin moieties in these forms of Pap1. Alternatively, the ubiquitinated species could represent a protein that is so tightly associated with Pap1 that it is retained in the immunoprecipitate even after the 0.1% SDS washes.
FIG 7.
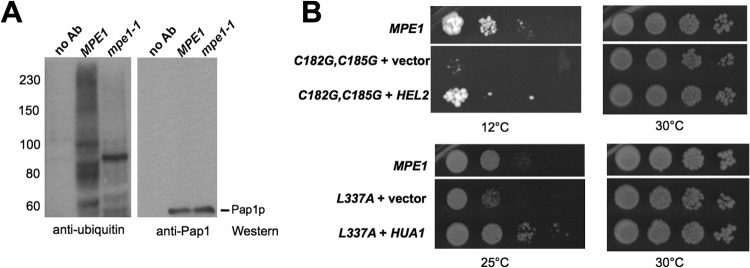
Mpe1 and ubiquitination. (A) Association of ubiquitinated species with Pap1 requires Mpe1. A plasmid harboring HA-tagged ubiquitin under the control of a copper-inducible promoter was introduced into the wild-type and mpe1-1 strains. Yeast cells were grown at 30°C and were shifted to 37°C for 4 h in the presence of 0.15 mM CuSO4. Whole-cell extracts were used for immunoprecipitation with an anti-Pap1 antibody or a no-antibody (no Ab) control, and the precipitated proteins were subjected to Western blotting against the indicated antibodies. The positions of molecular mass markers (in kilodaltons) are indicated on the left. (B) Suppressors of mpe1 mutants have links to ubiquitination. (Top) HEL2 is a high-copy-number suppressor of the cold sensitivity of mpe1-C182G,C185G. (Bottom) HUA1 is a suppressor of the mpe1-L337A growth defect. Yeast cells were grown on a YPD plate and were incubated for 20 days at 12°C or for 4 days at 25 or 30°C.
The experiments described above show that ubiquitin-mediated interactions are important for mRNA 3′-end processing and that one recipient of this modification is likely to be Pap1. Given that the mammalian homolog of Mpe1, RBBP6, promotes ubiquitination (26, 27), we next asked whether the appearance of the ubiquitinated proteins in the Pap1 immunoprecipitate was dependent on Mpe1. These species disappeared in the mpe1-1 strain (Fig. 7A, left), suggesting a role for Mpe1 in their formation. Curiously, a single ubiquitin-containing band of ∼90 kDa persisted in the Pap1 immunoprecipitated from the mpe1-1 extract (Fig. 7A). Pap1 migrates at around 60 kDa, suggesting that this species might contain 1 to 2 ubiquitin moieties whose addition to Pap1 would not be dependent on Mpe1.
We reasoned that if Mpe1 is involved in ubiquitination, then the suppressors of the growth defects of mpe1 mutants might be other yeast genes involved in this posttranslational modification. To identify such genes, we searched for high-copy-number suppressors of mpe1 conditional mutants. We transformed each of three mpe1 mutants (mpe1-F9A, mpe1-C182G,C185G, and mpe1-L337A) with a high-copy-number yeast genomic library and screened for genes that suppressed the growth defects of these mutants at nonpermissive temperatures. For each mutant, three to six suppressors were isolated. When plasmids were recovered from yeast and were reintroduced into the mutant strains, the growth defects were again suppressed. DNA sequencing revealed that at least one of the plasmids recovered from each suppressor screen contained MPE1. In addition to MPE1, the HUA1 and HEL2 genes were able to suppress mpe1-L337A and mpe1-C182G,C185G, respectively (Fig. 7B). Interestingly, HEL2 encodes a RING finger ubiquitin ligase that functions in ubiquitination and degradation of excess histones (63). Hua1 has been shown to physically interact with the Rsp5 E3 ubiquitin ligase and with ubiquitin (64–67). The identification of these two suppressors suggests that the growth defects of the mpe1 mutants may be due to defective ubiquitination.
DISCUSSION
Mpe1 is an essential subunit of CPF needed for both the cleavage and poly(A) addition steps of mRNA 3′-end processing (7). It possesses a tripartite domain structure consisting of a UBL, a zinc knuckle, and a RING finger. In this study, we have investigated the role of each of these highly conserved domains in promoting efficient 3′-end processing of mRNA precursor. As might be expected from this domain architecture, this work has revealed that Mpe1 is a multifunctional protein, contributing to protein-protein interactions within the complex, to interaction with the RNA substrate, and to possible modification of the poly(A) polymerase by ubiquitination.
A role for ubiquitination in mRNA polyadenylation.
Several studies have indicated that posttranslational modification by SUMO, a ubiquitin-like molecule, can modulate the activity of the polyadenylation machinery (68–70). Other studies, described in more detail below, have implicated ubiquitination and proteolytic degradation in regulating mRNA 3′-end processing, especially under conditions in which mRNA synthesis or mRNP assembly is perturbed (2, 71). However, no direct role for ubiquitination in the mRNA 3′-end processing reaction has been demonstrated. Our work reported here indicates that this modification has important functions in polyadenylation.
Examples of ubiquitin-regulated assembly of protein complexes have been documented in events closely linked to polyadenylation, such as the formation of gene loops between the 5′ and 3′ ends of genes, mRNP assembly, and mRNA export. The release of export-competent mRNPs from transcription sites is facilitated not only by polyadenylation but also by polyubiquitination and proteasomal degradation of Hpr1, a subunit of the THO transcription complex (72, 73). Cotranscriptional recruitment of the Yra1 protein is an important part of this maturation process and may facilitate the assembly of the 3′-end processing complex at the poly(A) site (74). Yra1 must then be ubiquitinated to remove it from the mRNP before export (75). A recent study has added a further dimension to this ubiquitin-regulated network (76), showing that histone H2B ubiquitination stimulates the ubiquitination of the Swd2 subunit of CPF. This Swd2 modification, in turn, facilitates the recruitment of Yra1 and the assembly of gene loops, thus helping to coordinate events at the beginning and ends of genes.
The studies described above show that ubiquitin is clearly a key player in coordinating mRNA transactions that are coupled with polyadenylation. We have found that ubiquitination also has a direct impact on the polyadenylation reaction (Fig. 6). Blocking of the interactions between ubiquitin and ubiquitin-binding proteins by ubistatin A inhibits the cleavage step of mRNA 3′-end processing. Prevention of proteasomal activity by mutation in vivo or by the inclusion of a proteasomal inhibitor, MG132, in processing reactions also leads to defective processing. Mpe1 may play a role in this ubiquitination, since the mpe1-1 mutation prevents the association of ubiquitinated species with Pap1. The identification of HUA1 and HEL2 as suppressors of mpe1 mutants suggests that Mpe1 may function as an E3 ligase, an E4 conjugating factor (25) that helps the E3 ligase polyubiquitinate its substrates, or a scaffold protein that helps an E3 ligase identify its substrates.
Ubiquitination could promote important shifts in protein-protein interactions as the processing complex proceeds through a cycle of assembly, cleavage, poly(A) addition, and release from the mature mRNA. Alternatively, ubiquitin-mediated degradation could reduce the level of an inhibitor of processing or help maintain appropriate stoichiometry of the subunits that make up the processing complex. For example, our earlier work has indicated that the cell carefully balances the levels of Pta1 and Ssu72 (34, 54). The binding of Ssu72 to the N-terminal domain of Pta1 appears to block a negative effect of this domain on cleavage (47). Our studies have also shown that certain types of stress can cause a decline in the level of specific subunits of the processing complex. In one case, accumulation of immature mRNPs in the nucleus caused by defects in the THO complex induces degradation of Fip1, a protein needed to recruit Pap1 to CPF (71). Loss of Fip1 is proposed to enhance cell survival by allowing more-rapid exosome-mediated destruction of poorly formed mRNPs because they are not polyadenylated. In another recent example, UV-type DNA damage inhibits mRNA 3′-end processing and causes the depletion of several CPF subunits but not of the mRNAs encoding them, suggesting that targeted protein degradation could limit the polyadenylation of truncated or damaged RNAs (2). A low level of polyubiquitination of Pap1 may occur as cells move from G2 to M in the cell cycle (62). However, the significance of this timing is unknown, and details about the contribution of ubiquitination to the events described above remain to be established.
Protein-protein interactions made by Mpe1.
The work presented here indicates that at least three regions of Mpe1 contribute to interactions within the cleavage/polyadenylation complex. The UBL domain appears most critical for the association of Mpe1 with CPF (Fig. 3A), which may explain the lethal phenotype of mpe1-ΔUBL. The RING finger also contributes to interaction between Mpe1 and CPF (Fig. 3A), and our two-hybrid analysis suggests that the region containing the zinc knuckle and second linker makes contacts with Cft1, one of the CPF subunits. In agreement with our findings, Vo et al. (7) showed that the mpe1-1 mutant, which contains the F9S mutation in the UBL domain and the L337F mutation in the RING finger domain, is not stably incorporated into CPF. Moreover, they found that otherwise intact CPF can be purified from this mutant, indicating that Mpe1 itself is not important for CPF integrity.
The genetic interaction between Mpe1 and the Pcf11 subunit of CF IA (7) suggests that Mpe1 may contact CF IA directly instead of through other CPF subunits. In our study, the RING finger deletion mutant was able to pull down an amount of CF IA comparable to that seen with wild-type Mpe1, even though this mutant showed reduced interaction with CPF. This supports the idea of a direct contact with CF IA mediated by a region different from the RING finger. The UBL deletion mutant showed defective interaction with both CPF and CF IA, suggesting that the UBL domain is important for the interaction with both complexes.
Interaction of Mpe1 with RNA.
We have found that recombinant Mpe1 binds to polyadenylation precursor in gel shift assays (Fig. 4B) and prefers to bind in the region around the poly(A) site of CYC1 RNA (Fig. 4C). Mpe1 also interacts with RNA precursor when it is part of the CPF complex (Fig. 5), suggesting that Mpe1 cooperates with other subunits of CPF to define where processing should occur on the RNA substrate. Previous studies have shown that the Cft1, Cft2, Ysh1, and Yth1 components of CPF also contact the RNA in the region directly around the cleavage site, while the CF IA and Hrp1 factors recognize elements farther upstream (reviewed in references 6 and 77). This contact point for CPF around the cleavage site makes sense given that the endonuclease Ysh1 and the poly(A) polymerase Pap1 are part of CPF and need to act at this site on the RNA.
Interestingly, two domains of Mpe1, the zinc knuckle and the RING finger, are needed for efficient interaction with RNA. Several RING-type E3 ubiquitin ligases bind RNA, or are predicted to bind RNA because of canonical RNA-binding domains (RBDs) (78, 79). Some have been implicated in regulating the translation or stability of mRNAs, but the role of ubiquitination in this process remains to be elucidated. The zinc knuckle of Mpe1 may be equivalent to the RBD on the ubiquitin ligases described above. Zinc knuckles are known to bind RNA and are found in proteins with diverse functions in RNA transactions, such as the Air1/Air2 subunits of the TRAMP RNA quality control complex, the Lin28 regulator of microRNA (miRNA) biogenesis, and two splicing factors, 9G8 and the branch point-binding protein BBP (14–19). Mutations of a zinc knuckle cysteine reduced RNA binding in Lin28 (16, 19), a finding consistent with the defect we have observed upon the mutation of comparable residues in Mpe1.
The RING finger could facilitate RNA interaction in two ways. The RING finger in the Mpe1 homolog RBBP6 promotes protein dimerization (23), and bringing together two zinc knuckles in an Mpe1 homodimer may be critical for stable RNA interaction. Alternatively, the Mpe1 RING finger could bind RNA directly. In support of the latter possibility, the Mdm2 E3 ligase, a major regulator of the p53 tumor suppressor, stimulates the translation or stability of p53 and some other mRNAs by binding to these RNAs through its RING finger domain, an interaction that can also suppress the known E3 ligase activity of Mdm2 (80, 81). Like Mdm2, the RING finger of Mpe1 may provide both RNA binding and E3 ligase functions.
In summary, we have shown that all three domains of Mpe1 contribute to efficient mRNA polyadenylation. The UBL domain is most important for interaction with the rest of CPF but is aided by the regions containing the zinc knuckle and the RING finger. In contrast, the UBL domain is not needed for RNA interaction, a function determined by the RING finger and the zinc knuckle. Our findings also suggest that like its mammalian homolog RBBP6, Mpe1 functions in ubiquitination. Consistent with such an activity residing in the 3′-end processing complex, ubiquitin-mediated interactions facilitate polyadenylation. Global analysis of ubiquitinated proteins in yeast has suggested the presence of ubiquitin on the Cft1, Hrp1, Pta1, Rna14, and Pap1 subunits of the polyadenylation machinery (65, 82, 83). It will be important in future work to identify proteins whose interactions or degradation might be affected by ubiquitination and the biochemical contribution of Mpe1 to such modifications. We also do not know if Mpe1, acting as a mediator of ubiquitination, a structural protein, or an RNA-binding protein, is involved in the coordination of cotranscriptional events, and this will be another avenue for further research.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the laboratories of C. Moore and A. Bohm for critical input regarding our experiments and the manuscript.
This work was supported by NIH grant GM68887 and NSF grant MCB-1244043 to C. L. Moore.
Footnotes
Published ahead of print 18 August 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00077-14.
REFERENCES
- 1.Elkon R, Ugalde AP, Agami R. 2013. Alternative cleavage and polyadenylation: extent, regulation and function. Nat. Rev. Genet. 14:496–506. 10.1038/nrg3482. [DOI] [PubMed] [Google Scholar]
- 2.Graber JH, Nazeer FI, Yeh PC, Kuehner JN, Borikar S, Hoskinson D, Moore CL. 2013. DNA damage induces targeted, genome-wide variation of poly(A) sites in budding yeast. Genome Res. 23:1690–1703. 10.1101/gr.144964.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tian B, Manley JL. 2013. Alternative cleavage and polyadenylation: the long and short of it. Trends Biochem. Sci. 38:312–320. 10.1016/j.tibs.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan S, Choi EA, Shi Y. 2011. Pre-mRNA 3′-end processing complex assembly and function. Wiley Interdiscip. Rev. RNA 2:321–335. 10.1002/wrna.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Darmon SK, Lutz CS. 2012. mRNA 3′ end processing factors: a phylogenetic comparison. Comp. Funct. Genomics 2012:876893. 10.1155/2012/876893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Millevoi S, Vagner S. 2010. Molecular mechanisms of eukaryotic pre-mRNA 3′ end processing regulation. Nucleic Acids Res. 38:2757–2774. 10.1093/nar/gkp1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vo LT, Minet M, Schmitter JM, Lacroute F, Wyers F. 2001. Mpe1, a zinc knuckle protein, is an essential component of yeast cleavage and polyadenylation factor required for the cleavage and polyadenylation of mRNA. Mol. Cell. Biol. 21:8346–8356. 10.1128/MCB.21.24.8346-8356.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pugh DJ, Eiso AB, Faro A, Lutya PT, Hoffmann E, Rees DJ. 2006. DWNN, a novel ubiquitin-like domain, implicates RBBP6 in mRNA processing and ubiquitin-like pathways. BMC Struct. Biol. 6:1. 10.1186/1472-6807-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simons A, Melamed-Bessudo C, Wolkowicz R, Sperling J, Sperling R, Eisenbach L, Rotter V. 1997. PACT: cloning and characterization of a cellular p53 binding protein that interacts with Rb. Oncogene 14:145–155. 10.1038/sj.onc.1200825. [DOI] [PubMed] [Google Scholar]
- 10.Shi Y, Di Giammartino DC, Taylor D, Sarkeshik A, Rice WJ, Yates JR, III, Frank J, Manley JL. 2009. Molecular architecture of the human pre-mRNA 3′ processing complex. Mol. Cell 33:365–376. 10.1016/j.molcel.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hicke L, Schubert HL, Hill CP. 2005. Ubiquitin-binding domains. Nat. Rev. Mol. Cell Biol. 6:610–621. 10.1038/nrm1701. [DOI] [PubMed] [Google Scholar]
- 12.Kiel C, Serrano L. 2006. The ubiquitin domain superfold: structure-based sequence alignments and characterization of binding epitopes. J. Mol. Biol. 355:821–844. 10.1016/j.jmb.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 13.Su V, Lau AF. 2009. Ubiquitin-like and ubiquitin-associated domain proteins: significance in proteasomal degradation. Cell. Mol. Life Sci. 66:2819–2833. 10.1007/s00018-009-0048-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ali PS, Ghoshdastider U, Hoffmann J, Brutschy B, Filipek S. 2012. Recognition of the let-7g miRNA precursor by human Lin28B. FEBS Lett. 586:3986–3990. 10.1016/j.febslet.2012.09.034. [DOI] [PubMed] [Google Scholar]
- 15.Cavaloc Y, Bourgeois CF, Kister L, Stevenin J. 1999. The splicing factors 9G8 and SRp20 transactivate splicing through different and specific enhancers. RNA 5:468–483. 10.1017/S1355838299981967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fasken MB, Leung SW, Banerjee A, Kodani MO, Chavez R, Bowman EA, Purohit MK, Rubinson ME, Rubinson EH, Corbett AH. 2011. Air1 zinc knuckles 4 and 5 and a conserved IWRXY motif are critical for the function and integrity of the Trf4/5-Air1/2-Mtr4 polyadenylation (TRAMP) RNA quality control complex. J. Biol. Chem. 286:37429–37445. 10.1074/jbc.M111.271494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garrey SM, Voelker R, Berglund JA. 2006. An extended RNA binding site for the yeast branch point-binding protein and the role of its zinc knuckle domains in RNA binding. J. Biol. Chem. 281:27443–27453. 10.1074/jbc.M603137200. [DOI] [PubMed] [Google Scholar]
- 18.Holub P, Lalakova J, Cerna H, Pasulka J, Sarazova M, Hrazdilova K, Arce MS, Hobor F, Stefl R, Vanacova S. 2012. Air2p is critical for the assembly and RNA-binding of the TRAMP complex and the KOW domain of Mtr4p is crucial for exosome activation. Nucleic Acids Res. 40:5679–5693. 10.1093/nar/gks223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loughlin FE, Gebert LF, Towbin H, Brunschweiger A, Hall J, Allain FH. 2012. Structural basis of pre-let-7 miRNA recognition by the zinc knuckles of pluripotency factor Lin28. Nat. Struct. Mol. Biol. 19:84–89. 10.1038/nsmb.2202. [DOI] [PubMed] [Google Scholar]
- 20.Lu K, Heng X, Summers MF. 2011. Structural determinants and mechanism of HIV-1 genome packaging. J. Mol. Biol. 410:609–633. 10.1016/j.jmb.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deshaies RJ, Joazeiro CA. 2009. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78:399–434. 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 22.Hatakeyama S, Nakayama KI. 2003. U-box proteins as a new family of ubiquitin ligases. Biochem. Biophys. Res. Commun. 302:635–645. 10.1016/S0006-291X(03)00245-6. [DOI] [PubMed] [Google Scholar]
- 23.Kappo MA, Eiso AB, Hassem F, Atkinson RA, Faro A, Muleya V, Mulaudzi T, Poole JO, McKenzie JM, Chibi M, Moolman-Smook JC, Rees DJ, Pugh DJ. 2012. Solution structure of RING finger-like domain of retinoblastoma-binding protein-6 (RBBP6) suggests it functions as a U-box. J. Biol. Chem. 287:7146–7158. 10.1074/jbc.M110.217059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Metzger MB, Pruneda JN, Klevit RE, Weissman AM. 2014. RING-type E3 ligases: master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim. Biophys. Acta 1843:47–60. 10.1016/j.bbamcr.2013.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoppe T. 2005. Multiubiquitylation by E4 enzymes: ‘one size' doesn't fit all. Trends Biochem. Sci. 30:183–187. 10.1016/j.tibs.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 26.Chibi M, Meyer M, Skepu A, Rees DJG, Moolman-Smook JC, Pugh DJR. 2008. RBBP6 interacts with multifunctional protein YB-1 through its RING finger domain, leading to ubiquitination and proteosomal degradation of YB-1. J. Mol. Biol. 384:908–916. 10.1016/j.jmb.2008.09.060. [DOI] [PubMed] [Google Scholar]
- 27.Li L, Deng B, Xing G, Teng Y, Tian C, Cheng X, Yin X, Yang J, Gao X, Zhu Y, Sun Q, Zhang L, Yang X, He F. 2007. PACT is a negative regulator of p53 and essential for cell growth and embryonic development. Proc. Natl. Acad. Sci. U. S. A. 104:7951–7956. 10.1073/pnas.0701916104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bellare P, Small EC, Huang X, Wohlschlegel JA, Staley JP, Sontheimer EJ. 2008. A role for ubiquitin in the spliceosome assembly pathway. Nat. Struct. Mol. Biol. 15:444–451. 10.1038/nsmb.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Geng F, Wenzel S, Tansey WP. 2012. Ubiquitin and proteasomes in transcription. Annu. Rev. Biochem. 81:177–201. 10.1146/annurev-biochem-052110-120012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song EJ, Werner SL, Neubauer J, Stegmeier F, Aspden J, Rio D, Harper JW, Elledge SJ, Kirschner MW, Rape M. 2010. The Prp19 complex and the Usp4Sart3 deubiquitinating enzyme control reversible ubiquitination at the spliceosome. Genes Dev. 24:1434–1447. 10.1101/gad.1925010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tutucci E, Stutz F. 2011. Keeping mRNPs in check during assembly and nuclear export. Nat. Rev. Mol. Cell Biol. 12:377–384. 10.1038/nrm3119. [DOI] [PubMed] [Google Scholar]
- 32.Ramos PC, Hockendorff J, Johnson ES, Varshavsky A, Dohmen RJ. 1998. Ump1p is required for proper maturation of the 20S proteasome and becomes its substrate upon completion of the assembly. Cell 92:489–499. 10.1016/S0092-8674(00)80942-3. [DOI] [PubMed] [Google Scholar]
- 33.Hilt W, Enenkel C, Gruhler A, Singer T, Wolf DH. 1993. The PRE4 gene codes for a subunit of the yeast proteasome necessary for peptidylglutamyl-peptide-hydrolyzing activity. Mutations link the proteasome to stress- and ubiquitin-dependent proteolysis. J. Biol. Chem. 268:3479–3486. [PubMed] [Google Scholar]
- 34.He X, Moore C. 2005. Regulation of yeast mRNA 3′ end processing by phosphorylation. Mol. Cell 19:619–629. 10.1016/j.molcel.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 35.James P, Halladay J, Craig EA. 1996. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics 144:1425–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cross FR. 1997. ‘Marker swap' plasmids: convenient tools for budding yeast molecular genetics. Yeast 13:647–653. . [DOI] [PubMed] [Google Scholar]
- 37.Boeke JD, Trueheart J, Natsoulis G, Fink GR. 1987. 5-Fluoroorotic acid as a selective agent in yeast molecular genetics. Methods Enzymol. 154:164–175. 10.1016/0076-6879(87)54076-9. [DOI] [PubMed] [Google Scholar]
- 38.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59. 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 39.Zhao J, Kessler M, Helmling S, O'Connor JP, Moore C. 1999. Pta1, a component of yeast CF II, is required for both cleavage and poly(A) addition of mRNA precursor. Mol. Cell. Biol. 19:7733–7740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hochstrasser M, Ellison MJ, Chau V, Varshavsky A. 1991. The short-lived MATα2 transcriptional regulator is ubiquitinated in vivo. Proc. Natl. Acad. Sci. U. S. A. 88:4606–4610. 10.1073/pnas.88.11.4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gross S, Moore C. 2001. Five subunits are required for reconstitution of the cleavage and polyadenylation activities of Saccharomyces cerevisiae cleavage factor I. Proc. Natl. Acad. Sci. U. S. A. 98:6080–6085. 10.1073/pnas.101046598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Helmling S, Zhelkovsky A, Moore CL. 2001. Fip1 regulates the activity of poly(A) polymerase through multiple interactions. Mol. Cell. Biol. 21:2026–2037. 10.1128/MCB.21.6.2026-2037.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Butler JS, Sadhale PP, Platt T. 1990. RNA processing in vitro produces mature 3′ ends of a variety of Saccharomyces cerevisiae mRNAs. Mol. Cell. Biol. 10:2599–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dichtl B, Keller W. 2001. Recognition of polyadenylation sites in yeast pre-mRNAs by cleavage and polyadenylation factor. EMBO J. 20:3197–3209. 10.1093/emboj/20.12.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M, Seraphin B. 2001. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods 24:218–229. 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- 46.Jones GM, Stalker J, Humphray S, West A, Cox T, Rogers J, Dunham I, Prelich G. 2008. A systematic library for comprehensive overexpression screens in Saccharomyces cerevisiae. Nat. Methods 5:239–241. 10.1038/nmeth.1181. [DOI] [PubMed] [Google Scholar]
- 47.Ghazy MA, He X, Singh BN, Hampsey M, Moore C. 2009. The essential N terminus of the Pta1 scaffold protein is required for snoRNA transcription termination and Ssu72 function but is dispensable for pre-mRNA 3′-end processing. Mol. Cell. Biol. 29:2296–2307. 10.1128/MCB.01514-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ezeokonkwo C, Ghazy MA, Zhelkovsky A, Yeh PC, Moore C. 2012. Novel interactions at the essential N-terminus of poly(A) polymerase that could regulate poly(A) addition in Saccharomyces cerevisiae. FEBS Lett. 586:1173–1178. 10.1016/j.febslet.2012.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Guzman RN, Wu ZR, Stalling CC, Pappalardo L, Borer PN, Summers MF. 1998. Structure of the HIV-1 nucleocapsid protein bound to the SL3 psi-RNA recognition element. Science 279:384–388. 10.1126/science.279.5349.384. [DOI] [PubMed] [Google Scholar]
- 50.Barabino SM, Ohnacker M, Keller W. 2000. Distinct roles of two Yth1p domains in 3′-end cleavage and polyadenylation of yeast pre-mRNAs. EMBO J. 19:3778–3787. 10.1093/emboj/19.14.3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dichtl B, Blank D, Sadowski M, Hubner W, Weiser S, Keller W. 2002. Yhh1p/Cft1p directly links poly(A) site recognition and RNA polymerase II transcription termination. EMBO J. 21:4125–4135. 10.1093/emboj/cdf390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Graber JH, McAllister GD, Smith TF. 2002. Probabilistic prediction of Saccharomyces cerevisiae mRNA 3′-processing sites. Nucleic Acids Res. 30:1851–1858. 10.1093/nar/30.8.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bucheli ME, He X, Kaplan CD, Moore CL, Buratowski S. 2007. Polyadenylation site choice in yeast is affected by competition between Npl3 and polyadenylation factor CFI. RNA 13:1756–1764. 10.1261/rna.607207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.He X, Khan AU, Cheng H, Pappas DL, Jr, Hampsey M, Moore CL. 2003. Functional interactions between the transcription and mRNA 3′ end processing machineries mediated by Ssu72 and Sub1. Genes Dev. 17:1030–1042. 10.1101/gad.1075203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Finley D. 2009. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 78:477–513. 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramos PC, Marques AJ, London MK, Dohmen RJ. 2004. Role of C-terminal extensions of subunits β2 and β7 in assembly and activity of eukaryotic proteasomes. J. Biol. Chem. 279:14323–14330. 10.1074/jbc.M308757200. [DOI] [PubMed] [Google Scholar]
- 57.Dikic I, Wakatsuki S, Walters KJ. 2009. Ubiquitin-binding domains—from structures to functions. Nat. Rev. Mol. Cell Biol. 10:659–671. 10.1038/nrm2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Verma R, Peters NR, D'Onofrio M, Tochtrop GP, Sakamoto KM, Varadan R, Zhang M, Coffino P, Fushman D, Deshaies RJ, King RW. 2004. Ubistatins inhibit proteasome-dependent degradation by binding the ubiquitin chain. Science 306:117–120. 10.1126/science.1100946. [DOI] [PubMed] [Google Scholar]
- 59.Lee DH, Goldberg AL. 1998. Proteasome inhibitors cause induction of heat shock proteins and trehalose, which together confer thermotolerance in Saccharomyces cerevisiae. Mol. Cell. Biol. 18:30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Adachi K, Lakka V, Zhao Y, Surrey S. 2004. Ubiquitylation of nascent globin chains in a cell-free system. J. Biol. Chem. 279:41767–41774. 10.1074/jbc.M405059200. [DOI] [PubMed] [Google Scholar]
- 61.Lavelin I, Beer A, Kam Z, Rotter V, Oren M, Navon A, Geiger B. 2009. Discovery of novel proteasome inhibitors using a high-content cell-based screening system. PLoS One 4:e8503. 10.1371/journal.pone.0008503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mizrahi N, Moore C. 2000. Posttranslational phosphorylation and ubiquitination of the Saccharomyces cerevisiae poly(A) polymerase at the S/G2 stage of the cell cycle. Mol. Cell. Biol. 20:2794–2802. 10.1128/MCB.20.8.2794-2802.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Singh RK, Gonzalez M, Kabbaj MH, Gunjan A. 2012. Novel E3 ubiquitin ligases that regulate histone protein levels in the budding yeast Saccharomyces cerevisiae. PLoS One 7:e36295. 10.1371/journal.pone.0036295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y. 2001. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl. Acad. Sci. U. S. A. 98:4569–4574. 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Peng J, Schwartz D, Elias JE, Thoreen CC, Cheng D, Marsischky G, Roelofs J, Finley D, Gygi SP. 2003. A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 21:921–926. 10.1038/nbt849. [DOI] [PubMed] [Google Scholar]
- 66.Ren J, Kee Y, Huibregtse JM, Piper RC. 2007. Hse1, a component of the yeast Hrs-STAM ubiquitin-sorting complex, associates with ubiquitin peptidases and a ligase to control sorting efficiency into multivesicular bodies. Mol. Biol. Cell 18:324–335. 10.1091/mbc.E06-06-0557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu H, Braun P, Yildirim MA, Lemmens I, Venkatesan K, Sahalie J, Hirozane-Kishikawa T, Gebreab F, Li N, Simonis N, Hao T, Rual JF, Dricot A, Vazquez A, Murray RR, Simon C, Tardivo L, Tam S, Svrzikapa N, Fan C, de Smet AS, Motyl A, Hudson ME, Park J, Xin X, Cusick ME, Moore T, Boone C, Snyder M, Roth FP, Barabasi AL, Tavernier J, Hill DE, Vidal M. 2008. High-quality binary protein interaction map of the yeast interactome network. Science 322:104–110. 10.1126/science.1158684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.del Olmo M, Mizrahi N, Gross S, Moore CL. 1997. The Uba2 and Ufd1 proteins of Saccharomyces cerevisiae interact with poly(A) polymerase and affect the polyadenylation activity of cell extracts. Mol. Gen. Genet. 255:209–218. 10.1007/s004380050491. [DOI] [PubMed] [Google Scholar]
- 69.Vethantham V, Rao N, Manley JL. 2007. Sumoylation modulates the assembly and activity of the pre-mRNA 3′ processing complex. Mol. Cell. Biol. 27:8848–8858. 10.1128/MCB.01186-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vethantham V, Rao N, Manley JL. 2008. Sumoylation regulates multiple aspects of mammalian poly(A) polymerase function. Genes Dev. 22:499–511. 10.1101/gad.1628208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Saguez C, Schmid M, Olesen JR, Ghazy MA, Qu X, Poulsen MB, Nasser T, Moore C, Jensen TH. 2008. Nuclear mRNA surveillance in THO/sub2 mutants is triggered by inefficient polyadenylation. Mol. Cell 31:91–103. 10.1016/j.molcel.2008.04.030. [DOI] [PubMed] [Google Scholar]
- 72.Gwizdek C, Iglesias N, Rodriguez MS, Ossareh-Nazari B, Hobeika M, Divita G, Stutz F, Dargemont C. 2006. Ubiquitin-associated domain of Mex67 synchronizes recruitment of the mRNA export machinery with transcription. Proc. Natl. Acad. Sci. U. S. A. 103:16376–16381. 10.1073/pnas.0607941103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Libri D, Dower K, Boulay J, Thomsen R, Rosbash M, Jensen TH. 2002. Interactions between mRNA export commitment, 3′-end quality control, and nuclear degradation. Mol. Cell. Biol. 22:8254–8266. 10.1128/MCB.22.23.8254-8266.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Johnson SA, Kim H, Erickson B, Bentley DL. 2011. The export factor Yra1 modulates mRNA 3′ end processing. Nat. Struct. Mol. Biol. 18:1164–1171. 10.1038/nsmb.2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Iglesias N, Tutucci E, Gwizdek C, Vinciguerra P, Von Dach E, Corbett AH, Dargemont C, Stutz F. 2010. Ubiquitin-mediated mRNP dynamics and surveillance prior to budding yeast mRNA export. Genes Dev. 24:1927–1938. 10.1101/gad.583310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vitaliano-Prunier A, Babour A, Herissant L, Apponi L, Margaritis T, Holstege FC, Corbett AH, Gwizdek C, Dargemont C. 2012. H2B ubiquitylation controls the formation of export-competent mRNP. Mol. Cell 45:132–139. 10.1016/j.molcel.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mandel CR, Bai Y, Tong L. 2008. Protein factors in pre-mRNA 3′-end processing. Cell. Mol. Life Sci. 65:1099–1122. 10.1007/s00018-007-7474-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cano F, Bye H, Duncan LM, Buchet-Poyau K, Billaud M, Wills MR, Lehner PJ. 2012. The RNA-binding E3 ubiquitin ligase MEX-3C links ubiquitination with MHC-I mRNA degradation. EMBO J. 31:3596–3606. 10.1038/emboj.2012.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cano F, Miranda-Saavedra D, Lehner PJ. 2010. RNA-binding E3 ubiquitin ligases: novel players in nucleic acid regulation. Biochem. Soc. Trans. 38:1621–1626. 10.1042/BST0381621. [DOI] [PubMed] [Google Scholar]
- 80.Biderman L, Manley JL, Prives C. 2012. Mdm2 and MdmX as regulators of gene expression. Genes Cancer 3:264–273. 10.1177/1947601912455331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Naski N, Gajjar M, Bourougaa K, Malbert-Colas L, Fahraeus R, Candeias MM. 2009. The p53 mRNA-Mdm2 interaction. Cell Cycle 8:31–34. 10.4161/cc.8.1.7326. [DOI] [PubMed] [Google Scholar]
- 82.Seyfried NT, Xu P, Duong DM, Cheng D, Hanfelt J, Peng J. 2008. Systematic approach for validating the ubiquitinated proteome. Anal. Chem. 80:4161–4169. 10.1021/ac702516a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Swaney DL, Beltrao P, Starita L, Guo A, Rush J, Fields S, Krogan NJ, Villen J. 2013. Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nat. Methods 10:676–682. 10.1038/nmeth.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.