

Abstract
The gold standard to characterize idiopathic inflammatory myopathies is the morphological, immunohistochemical and immunopathological analysis of muscle biopsy. Mononuclear cell infiltrates and muscle fiber necrosis are commonly shared histopathological features. Inflammatory cells that surround, invade and destroy healthy muscle fibers expressing MHC class I antigen are the typical pathological finding of polymyositis. Perifascicular atrophy and microangiopathy strongly support a diagnosis of dermatomyositis. Randomly distributed necrotic muscle fibers without mononuclear cell infiltrates represent the histopathological hallmark of immune-mediated necrotizing myopathy; meanwhile, endomysial inflammation and muscle fiber degeneration are the two main pathological features in sporadic inclusion body myositis. A correct differential diagnosis requires immunopathological analysis of the muscle biopsy and has important clinical implications for therapeutic approach. In particular, unnecessary, potentially harmful, immune-suppressive therapy should be avoided alike in dystrophic myopathies with secondary inflammation.
Keywords: Inflammatory myopathy, Autoimmune myositis, Histopathology, Differential diagnosis
Introduction
Idiopathic inflammatory myopathies (IIM) are a heterogeneous group of acquired muscle diseases, which have distinct clinical, pathological and histological features [1, 2]. The most common IIM seen in clinical practice can be separated into four categories including polymyositis (PM), dermatomyositis (DM), immune-mediated necrotizing myopathy (NM) and sporadic inclusion body myositis (sIBM) [1, 3].
In the diagnostic workup of an inflammatory myopathy, muscle biopsy is an indispensable and sensitive tool for establishing the diagnosis. Although all inflammatory myopathies have in common the presence of mononuclear cell infiltrates and muscle fiber necrosis, certain pathologic findings are unique for each subset.
Histopathology of polymyositis
The presence of mononuclear inflammatory cells that surround, invade and destroy healthy muscle fibers is the typical pathological finding of PM (Fig. 1a) [4]. These inflammatory cells consist predominantly of cytotoxic CD8+ T cells along with a lesser number of macrophages, only a few CD4+ T cells occur among the invading cells (Fig. 2) [4]. Muscle fibers surrounded by inflammatory cells express on their sarcolemma, the major histocompatibility complex (MHC) class I antigen, which is not constitutively expressed under normal conditions, but is ubiquitously upregulated on the surface of most fibers in PM (Fig. 1c) [4]. In addition, the cytotoxic CD8+ T cells invading morphologically normal-appearing muscle fibers have granules containing cytotoxic molecules including perforin and granzymes [4]. In PM, mononuclear cellular infiltrates may also occur in the perimysium and rarely at perivascular sites [4]. Nonspecific histological abnormalities consist of increased variation in fiber diameter, scattered necrotic and regenerating muscle fibers and increase in perimysial and/or endomysial connective tissue (Fig. 1b) [4]. Taken together these features strongly suggest that a cell-mediated cytotoxicity plays a key role in the pathogenesis of PM; according with this interpretation, clonally expanded CD8+ T cells invade muscle fibers expressing antigen-presenting MHC class I molecules and release cytotoxic granules, thus leading to myofiber necrosis [5].
Fig. 1.
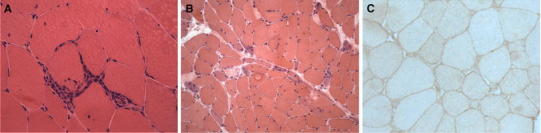
Polymyositis (muscle biopsy, hematoxylin and eosin staining). a A small endomysial inflammatory infiltrate surrounding and invading a healthy muscle fiber. b Scattered necrotic and regenerating muscle fibers. c MHC class I antigen is expressed on the sarcolemma of almost all muscle fibers
Fig. 2.
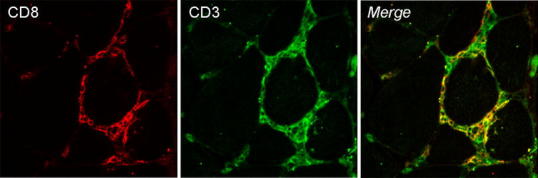
Confocal fluorescence microscopical images of muscle biopsy from a PM patient. CD3+ T cells (green) surrounding two nonnecrotic muscle fibers are mainly CD8+ T cells (red)
Histopathology of dermatomyositis
In DM, inflammatory infiltrates occur predominantly at perivascular sites or within the interfascicular septae (Fig. 3a), rarely in the endomysium, and are composed largely of B cells accompanied by CD4+ T helper cells, plasmacytoid dendritic cells and macrophages; CD8+ T cells are rare and NK cells sparse [4]. Single muscle fibers or clusters of muscle fibers in various stages of necrosis and/or regeneration are observed frequently [4]. The main pathological hallmark of DM is the distribution of atrophic, degenerating and regenerating fibers at the periphery of the fascicle (Fig. 3b); perifascicular atrophy involves both type 1 and type 2 muscle fibers, may affect two to ten layers and strongly support a diagnosis of DM, even in the absence of inflammation [4]. An early histological feature is the involvement of intramuscular blood vessels; the angiopathy is characterized by: (a) the deposition of immunoglobulins and complement, including the C5b-C9 membrane attack complex (MAC), on endomysial capillaries and small blood vessels and (b) the reduction in number of capillaries with endothelial hyperplasia and enlargement of the lumen of the remaining capillaries [6]. On electron microscopy, a unique ultrastructural alteration is the presence of tubuloreticular inclusions within the endothelial cytoplasm of arterioles and capillaries [7]. Therefore, DM is primarily considered a complement-mediated microangiopathy leading to capillary drop-out, necrosis of muscle fibers and perifascicular atrophy [5].
Fig. 3.
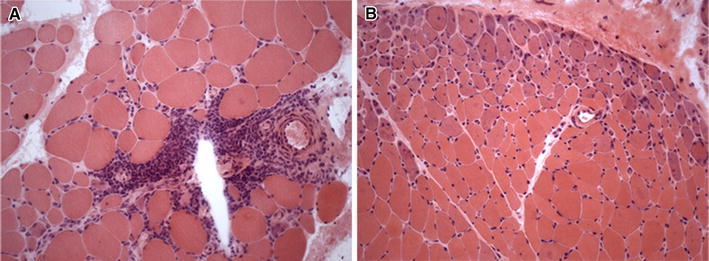
Dermatomyositis (muscle biopsy, hematoxylin and eosin staining). a A large inflammatory infiltrate at perivascular site. b Prominent perifascicular atrophy
Histopathology of immune-mediated necrotizing myopathy
Immune-mediated NM is an emerging entity characterized by a specific histological pattern [1–3, 5, 8]. The histopathological hallmark is the presence of randomly distributed necrotic muscle fibers along with fibers in various stages of regeneration, but in the absence of or sparse mononuclear cell infiltrates (Fig. 4) [1, 2, 5, 8]. Necrotic fibers can be invaded by macrophages which are the predominant mononuclear cell type, whereas T and B cells are virtually absent [1, 2, 5, 8]. MHC class I antigen is usually not expressed on the sarcolemma of muscle fibers and, if present, its expression is weak and focal [1, 2, 5, 8]. Deposition of MAC on intramuscular blood vessels has been reported only in a few cases [1, 2, 5, 8]. Therefore, in immune-mediated NM, muscle biopsy lacks the typical diagnostic features of DM and of PM. Immune-mediated NM is found in association with viral infections, connective tissue diseases, malignancy and statin therapy [5, 8]. Although the pathogenesis still remains elusive, the identification of specific antibodies in some patients with NM emphasizes an antibody-mediated mechanism underlying this disorder [5, 8].
Fig. 4.
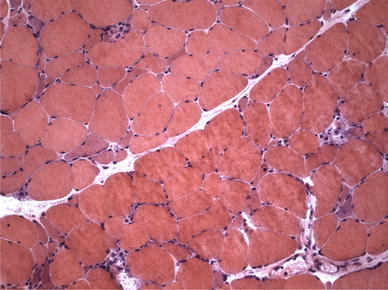
Immune-mediated necrotizing myopathy (muscle biopsy, hematoxylin and eosin staining): a few necrotic muscle fibers invaded by macrophages
Histopathology of sporadic inclusion body myositis
In sIBM, the distribution and the immunophenotypic profile of the inflammatory cells are similar to those seen in PM, consisting predominantly of macrophages and CD8+ T cells which invade nonnecrotic muscle fibers that express MHC class I antigen on the sarcolemma [1, 5, 9, 10]. Besides endomysial inflammation, muscle biopsies from patients with sIBM show distinct light microscopic findings which include the following: (a) nonnecrotic muscle fibers containing one or more, irregular and various-sized vacuoles, most of them with basophilic granular deposits around the edges (rimmed vacuoles) (Fig. 5a, b); (b) eosinophilic cytoplasmic inclusions, usually near or in the vacuoles (Fig. 5a, b); (c) single or multiple intracellular amyloid deposits, usually in non-vacuolated areas and rarely within vacuoles, identified with fluorescence-enhanced Congo red staining (Fig. 5c); (d) mitochondrial abnormalities consisting of increased number of ragged red fibers (muscle fibers containing excessive mitochondrial proliferation) and of cytochrome c oxidase (COX)-negative fibers; (e) small angulated muscle fibers, which are considered indicative of a neurogenic process [1, 5, 9, 10]. Ultrastructural examination documents tubulofilamentous inclusions of 15–21 nm in the cytoplasm and/or in the nucleus of muscle fibers (Fig. 5d), referred to as paired helical filaments (PHFs), cytoplasmic 6–10 nm amyloid-like filaments and deposits of flocculomembranous and amorphous material [9, 10]. In addition, several proteins accumulate in muscle fibers of sIBM patients, including amyloid-β and its precursor protein AβPP, phosphorylated tau, ubiquitin, TDP-43, p62 and others [5, 9, 10]. However, none of these proteins can be considered a specific marker for a diagnosis of sIBM. To date, sIBM remains a complex and enigmatic muscle disorder, in which endomysial inflammation and muscle fiber degeneration are the two main pathological features, but the contribution of each to the pathogenesis is unknown [5, 9, 10].
Fig. 5.
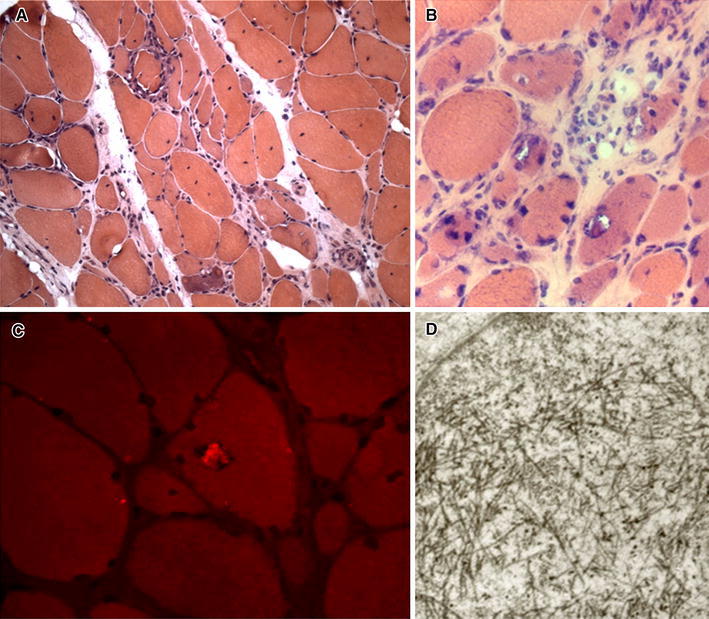
Sporadic inclusion body myositis (muscle biopsy, hematoxylin and eosin staining). a, b Rimmed vacuoles and eosinophilic inclusions in the cytoplasm of some muscle fibers. c Fluorescence-enhanced Congo red staining shows amyloid deposit within a vacuole. d (electron microscopy): several filamentous inclusions in the nucleus of muscle fiber
Differential diagnosis of idiopathic inflammatory myopathies
IIM include, in addition to the four main idiopathic forms (PM, DM, NM and sIBM), various diseases characterized by skeletal muscle inflammation, either primary or secondary to other causes. Main goal of IIM differential diagnosis is to accurately specify myositis type, ruling out mimics with secondary inflammatory changes (muscular dystrophies, metabolic, infectious, toxic or drug-induced myopathies) as well other neurological diseases (Table 1). Focal invasion by CD8+ T cells of nonnecrotic muscle fibers expressing MHC-I complex is the main diagnostic feature, since the CD8/MHC-1 complex is distinctive of IIM (Figs. 6, 7), differently from degenerative, toxic, necrotizing myopathies with secondary macrophagic infiltration. Diagnostic workup of myositis includes a comprehensive screening for medical conditions that may confound the diagnosis: anamnesis for iatrogenic myopathies, tests for autoimmunity, connective tissue diseases (CTD), systemic, endocrine and electrolytic abnormalities, electrophysiology and imaging studies [11].
Table 1.
Main differential diagnosis of idiopathic myositis
| Muscular dystrophies | Metabolic myopathies |
| Facioscapulohumeral dystrophy (FSHD) | Myophosphorylase deficiency |
| Dysferlinopathies (LGMD2B, Miyoshi type) | Adult-onset acid maltase deficiency |
| Congenital dystrophies (merosin deficiency) | Lipid storage myopathies |
| Dystrophinopathies (Becker) | Carnitine deficiency |
| Mitochondrial myopathies | |
| Endocrine myopathies | Toxic/drug-induced myopathies |
| Hypercorticosurrenalism | Statin-induced myopathies |
| Hyper/hypothyroidism | Antiretroviral drug myopathy |
| Hyper/hypoparathyroidism | Steroid myopathy |
| Others: antibiotics, chinidine, D-penicillamine, procainamide, FANS, TNF and H+ pump inhibitors | |
| Non-muscular diseases | Systemic disorder-associated myopathies |
| Guillain–Barrè syndrome | Nutritional deficits associated myopathy (alcoholism, malabsorption) |
| Transverse myelitis | Electrolyte imbalance myopathies (hypophosphatemia, hypomagnesemia, hypokalemia) |
| Neuromuscular junction pathology (myasthenia gravis, Lambert–Eaton syndrome) | Critical illness myopathy (CIM) |
| Rheumatic diseases, fibromyalgia | Cancer-associated myopathies (disuse, cachexia, drug-induced, paraneoplastic neuromyopathy) |
| Periodic paralysis |
Fig. 6.
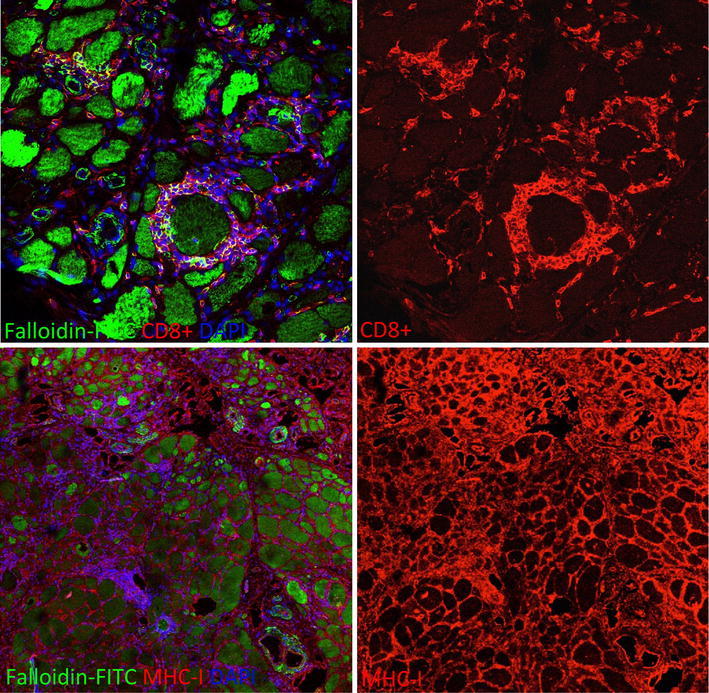
Endomysial CD8+ T lymphocytes surrounding partially invaded and apparently not invaded muscle fibers in a muscle biopsy of a patient with sporadic inclusion body myositis (IBM). All muscle fibers and inflammatory cells display a strong MHC-I immunoreactivity
Fig. 7.
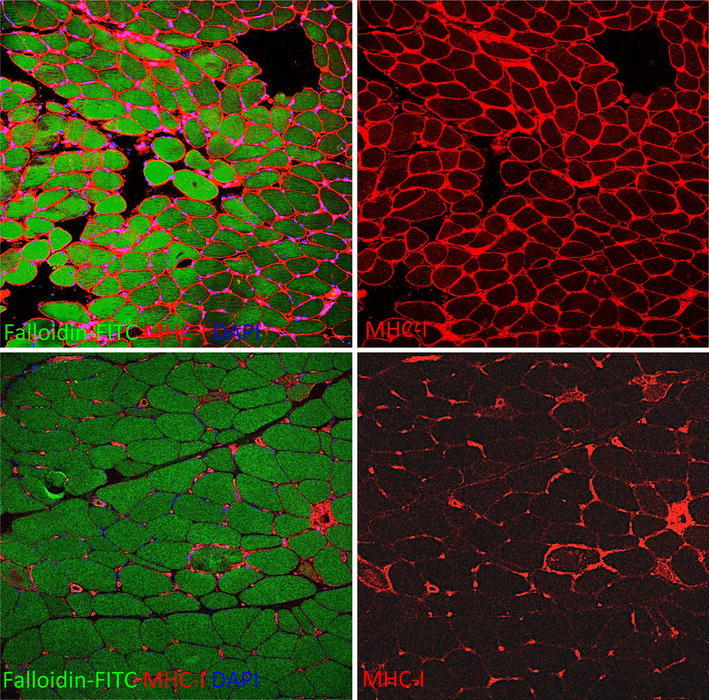
Diffuse MHC-I sarcolemmal immunoreactivity in a muscle biopsy of a patient with polymyositis (PM) (upper panels). On the contrary in necrotizing autoimmune myopathy (NAM), MHC-I is expressed only on membrane and/or sarcoplasm of necrotic fibers, by endomysial macrophages besides, as normal finding, in muscle blood vessels (lower panels)
Immune-mediated NM, besides to be associated with autoantibodies such as anti-SRP or anti-HMGCoA reductase [12], is often related to drug exposition (statins), CTD or neoplastic diseases [13].
Brachio-cervical myositis (BCM) is a rare form of IIM, responsive to steroids, clinically characterized by proximal muscle weakness mainly involving cervical and shoulder girdle muscles with dysphagia. Histologically BCM presents typical perivascular inflammatory infiltrates largely composed by B lymphocytes [14] (Fig. 8).
Fig. 8.
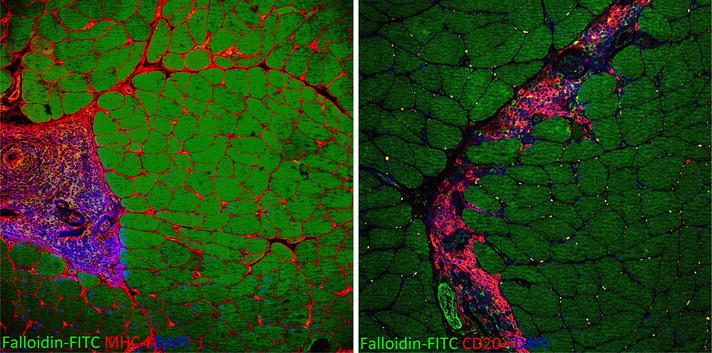
MHC-I is highly expressed on sarcolemmal of all muscle fibers and in mononuclear cells of a large perivascular infiltrate in a muscle biopsy of a patient with brachio-cervical inflammatory myopathy (BCM). The vast majority of the inflammatory cells are CD20+ B lymphocytes
Eosinophilia in peripheral blood or within muscle biopsy, fascial involvement, and muscle pain suggest the presence of eosinophilic myositis. Eosinophilia-associated myopathies include a focal usually benign eosinophilic myositis, eosinophilic perimyositis mostly associated with fasciitis and eosinophilic polymyositis that can be idiopathic (to be distinguished by LMGD2A due to calpain-3 gene mutation) or occurring as a feature of multiorgan hypereosinophilic syndrome (parasitic infections, CTD, hematologic malignancies, drugs, toxics) [15]. In presence of hypereosinophilia, myalgias and focal muscle swelling, parasitic infections by protozoa, cestodes or nematodes (toxoplasmosis, trypanosomiasis, echinococcosis, trichinosis, cysticercosis) must be ruled out by parasite identification on muscle biopsy and serologic findings.
Granulomatous myositis can occur during systemic diseases such as sarcoidosis or as idiopathic form clinically similar to classical IIM but histologically characterized by epithelioid and Langhans giant cells to form epithelioid granulomas [16].
Serum/urine immunofixation may detect a monoclonal gammopathy in patients with a severe sporadic late-onset nemaline bodies’ myopathy (SLONM). SLONM shows clinical and electromyographic features similar to IIM, moderate elevation of creatine phosphokinase (CPK) and fibers containing nemaline bodies visible with Trichrome staining, usually with no inflammation on muscle biopsy [17].
In HIV patients, a zidovudine-induced mitochondrial myopathy may coexist with HIV-induced T cell-mediated myositis. Inflammatory changes may be found on muscle biopsy of patients treated with procainamide, D-penicillamine (DM-like), α-interferon (PM-like), while chloroquine and amiodarone may induce cytoplasmic vacuoles and fiber necrosis. Drugs affecting microtubules (colchicine and vincristine) also provoke myopathic changes with autophagic vacuoles in muscle fibers.
It is important to identify a steroid myopathy presenting with subacute proximal muscle weakness and type II fibers atrophy. When occurring during IIM treatment, it may be mistaken as an exacerbation leading to more aggressive steroid treatment rather than its discontinuation.
Thyroid evaluation is needed because hypothyroidism can cause proximal muscle weakness and enzymes elevation with myoedema and pseudohypertrophy, usually without lymphomonocitic infiltrates and isolated necrosis that may be found instead in hyperthyroidism [18]. Hypercortisolism and hyper/hypoparathyroidism can also induce the onset of a subacute myopathy. Diabetes may sometimes be complicated by skeletal muscle ischemia/infarction, beginning acutely with focal muscle pain, swelling and weakness in proximal lower limb muscles.
Genetic defects in dysferlin gene cause LGMD2B or a distal muscular dystrophy (Miyoshi phenotype): a relatively acute onset of pelvifemoral weakness in early 20 s with elevated CPK levels may suggest a PM. Serum CPK levels may be very high during active phase and biopsy shows significant mononuclear cell infiltration with dystrophic changes and small sarcolemmal defects with thickened basal lamina.
A significant number of muscle biopsies from patients with facioscapulohumeral dystrophy (FSHD) show inflammatory changes. FSHD differentiates from IIM because of typically selective asymmetric muscle involvement of facial and shoulder muscles; weakness and molecular studies will confirm diagnosis.
Female carrier of dystrophinopathy can present CPK increase, adult-onset proximal muscle weakness with inflammatory alterations on biopsy [19]. A typical mosaic of positive and negative fibers is seen on dystrophin immunostaining. Milder forms of Becker muscular dystrophy with high CPK, myalgias, cramps, exercise intolerance, and mild limb-girdle weakness (quadriceps myopathy) mimic IIM because of endomysial inflammation; Western blot for muscle dystrophin is required because immunohistochemistry may be normal.
Sarcoglycanopathies (LGMD types IIC to IIF) start in childhood as pelvic muscle weakness with high levels of CPK and marked regeneration and necrosis on biopsy. Early evaluation of these patients may raise the suspect of subacute myositis, till immunohistochemical and molecular studies demonstrate the correct diagnosis.
Inherited metabolic myopathies, with intermittent episodes of acute muscle pain and tenderness exertion-induced, such as type V and VII glycogenosis, rarely present as adult-onset muscle weakness with fatigue and exercise intolerance. Forearm lactate test and determination of muscular enzymatic activity are fundamental for diagnosis.
Adult-onset acid maltase deficiency (AMD, type II glycogenosis) presents as progressive myopathy resembling PM with early diaphragmatic involvement. Muscle biopsy often demonstrates a vacuolar myopathy with increased glycogen and lysosomal activation [20].
Mitochondrial myopathies are extremely heterogeneous but may evocate IIM when presenting as symmetric proximal myopathy. Familiar recurrence with maternal inheritance, presence of ophthalmoplegia and systemic involvement helps in diagnosis. Muscle biopsy shows abnormalities of COX/SDH stainings, such as ragged red fibers.
A correct differential diagnosis has important clinical implication: identification of myositis type such as NM, even if steroid resistant, supports the decision to boost immunosuppression, while specific therapies are available for some genetic myopathies (AMD, some lipidosis) and unnecessary, potentially harmful, immune-suppressive therapy should be avoided alike in dystrophic myopathies with secondary inflammation.
Conclusions
Histopathologic features at muscle biopsy, related to the knowledge of disease-specific pathomechanisms, are essential in the diagnostic workup of IIM. The characteristic histopathological features, usually shared by the different forms of immune-mediated IIM, are the presence of mononuclear cell infiltrates and muscle fiber necrosis. Muscle immunohistochemical analysis and cell phenotyping are recommended to achieve correct diagnosis and proper therapeutic intervention.
Conflict of interest
None.
Human and animal rights
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
The study was approved by the Local Ethics Committee and informed consent was obtained from all patients, in compliance with the Helsinki Declaration.
Footnotes
G. Vattemi and M. Mirabella equally contributed to this work.
References
- 1.Dimachkie MM. Idiopathic inflammatory myopathies. J Neuroimmunol. 2011;231:32–42. doi: 10.1016/j.jneuroim.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 2.Luo YB, Mastaglia FL (2014) Dermatomyositis, polymyositis and immune-mediated necrotising myopathies. Biochim Biophys Acta [DOI] [PubMed]
- 3.Hoogendijk JE, Amato AA, Lecky BR, Choy EH, Lundberg IE, Rose MR, et al. 119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis. Neuromuscul Disord. 2004;14:337–345. doi: 10.1016/j.nmd.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Engel AG, Hohlfeld R, Banker BQ. The polymyositis and dermatomyositis syndrome. In: Engel AG, Franzini-Armstrong C, editors. Myology. New York: McGraw-Hill; 2006. pp. 1335–1383. [Google Scholar]
- 5.Dalakas MC. An update on inflammatory and autoimmune myopathies. Neuropathol Appl Neurobiol. 2011;37:226–242. doi: 10.1111/j.1365-2990.2010.01153.x. [DOI] [PubMed] [Google Scholar]
- 6.Emslie-Smith AM, Engel AG. Microvascular changes in early and advanced dermatomyositis: a quantitative study. Ann Neurol. 1990;27:343–356. doi: 10.1002/ana.410270402. [DOI] [PubMed] [Google Scholar]
- 7.De Visser M, Emslie-Smith AM, Engel AG. Early ultrastructural alterations in adult dermatomyositis. Capillary abnormalities precede other structural changes in muscle. J Neurol Sci. 1989;94:181–192. doi: 10.1016/0022-510X(89)90228-1. [DOI] [PubMed] [Google Scholar]
- 8.Liang C, Needham M. Necrotizing autoimmune myopathy. Curr Opin Rheumatol. 2011;23:612–619. doi: 10.1097/BOR.0b013e32834b324b. [DOI] [PubMed] [Google Scholar]
- 9.Askanas V, Engel WK. Inclusion-body myositis: muscle-fiber molecular pathology and possible pathogenic significance of its similarity to Alzheimer’s and Parkinson’s disease brains. Acta Neuropathol. 2008;116:583–595. doi: 10.1007/s00401-008-0449-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Askanas V, Engel WK, Nogalska A. Pathogenic considerations in sporadic inclusion-body myositis, a degenerative muscle disease associated with aging and abnormalities of myoproteostasis. J Neuropathol Exp Neurol. 2012;71:680–693. doi: 10.1097/NEN.0b013e31826183c8. [DOI] [PubMed] [Google Scholar]
- 11.Nirmalananthan N, Holton JL, Hanna MG. Is it really myositis? A consideration of the differential diagnosis. Curr Opin Rheumatol. 2004;16:684–691. doi: 10.1097/01.bor.0000143441.27065.bc. [DOI] [PubMed] [Google Scholar]
- 12.Mohassel P, Mammen AL. Statin-associated autoimmune myopathy and anti-HMGCR autoantibodies. Muscle Nerve. 2013;48:477–483. doi: 10.1002/mus.23854. [DOI] [PubMed] [Google Scholar]
- 13.Stenzel W, Goebel HH, Aronica E. Review: immune-mediated necrotizing myopathies–a heterogeneous group of diseases with specific myopathological features. Neuropathol Appl Neurobiol. 2012;38(7):632–646. doi: 10.1111/j.1365-2990.2012.01302.x. [DOI] [PubMed] [Google Scholar]
- 14.Pestronk A, Kos K, Lopate G, Al-Lozi MT. Brachio-cervical inflammatory myopathies: clinical, immune, and myopathologic features. Arthritis Rheum. 2006;54:1687–1696. doi: 10.1002/art.21822. [DOI] [PubMed] [Google Scholar]
- 15.Selva-O’Callaghan A, Trallero-Araguás E, Grau JM. Eosinophilic myositis: An updated review. Autoimmun Rev. 2014;13:375–378. doi: 10.1016/j.autrev.2014.01.018. [DOI] [PubMed] [Google Scholar]
- 16.Prieto-González S, Grau JM. Diagnosis and classification of granulomatous myositis. Autoimmun Rev. 2014;13:372–374. doi: 10.1016/j.autrev.2014.01.017. [DOI] [PubMed] [Google Scholar]
- 17.Chahin N, Selcen D, Engel AG. Sporadic late onset nemaline myopathy. Neurology. 2005;65:1158–1164. doi: 10.1212/01.wnl.0000180362.90078.dc. [DOI] [PubMed] [Google Scholar]
- 18.Madariaga MG. Polymyositis-like syndrome in hypothyroidism: review of cases reported over the past twenty-five years. Thyroid. 2002;12:331–336. doi: 10.1089/10507250252949478. [DOI] [PubMed] [Google Scholar]
- 19.Yoon J, Kim SH, Ki CS, et al. Carrier woman of duchenne muscular dystrophy mimicking inflammatory myositis. J Korean Med Sci. 2011;26:587–591. doi: 10.3346/jkms.2011.26.4.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schüller A, Wenninger S, Strigl-Pill N, Schoser B. Toward deconstructing the phenotype of late-onset pompe disease. Am J Med Genet C Semin Med Genet. 2012;160C(1):80–88. doi: 10.1002/ajmg.c.31322. [DOI] [PubMed] [Google Scholar]