

Abstract
Autoantibodies are powerful diagnostic tools in idiopathic inflammatory myopathies, especially for confirming the diagnosis and contributing to the definition of disease subsets. They are present in over 80 % of patients with immuno-mediated myositis and directed towards ubiquitously expressed intracellular complexes. Most of these autoantibodies are reported also in other autoimmune diseases, while some are considered myositis-specific. Myositis autoantibodies are traditionally categorized in two groups, based on their diagnostic accuracy: myositis-specific antibodies (MSA) and myositis-associated antibodies (MAA), the latter mostly occurring in myositis-overlap syndromes. Besides the so-called traditional MSA, including anti-synthetases, anti-SRP and anti-Mi-2 antibodies, additional newly conceived immune targets have been recently identified, mostly in patients with severe forms of dermatomyositis or necrotizing myopathy. They mainly encompass enzymatic proteins essentially involved in the regulation of gene transcription or post-translational modifications, i.e., TIF1-γ, NXP-2, MDA5, SAE and HMGCR. Among the MAA, anti-PM/Scl and anti-Ku characterize an overlap polydermatomyositis/systemic sclerosis syndrome with severe interstitial lung involvement.
Keywords: Autoimmune myositis, Autoantibodies, Myositis-specific antibodies, Overlap syndrome
Introduction
Serum autoantibodies towards ubiquitary intracellular constituents are found in more than 80 % of patients with polymyositis (PM) or dermatomyositis (DM). Some autoantibody specificities are shared with other connective tissue diseases (CTD), others are almost exclusive of patients with autoimmune myositis. As well as in other CTD, the identification of autoantibodies in serum has become an important milestone in the diagnosis of idiopathic inflammatory myopathies (IIM).
The clinically important autoantibodies are classically categorized in two groups based on their diagnostic accuracy: the myositis-specific antibodies (MSA) and the myositis-associated antibodies (MAA) (Table 1). By definition, MSA are specific of autoimmune myositis, diagnostic specificity exceeding 90 %, and target cytoplasmic or nuclear ribonucleoproteins involved in key processes of cell biology such as gene transcription, protein synthesis and translocation, and innate antiviral immune response (Table 2). They are mutually exclusive and closely associated with distinct disease subsets differing in clinical involvement and prognosis [1]. On the other hand MAA, even though present in up to 50 % of myositis patients, are not disease-specific, frequently associated with MSA, and mostly found in myositis-overlap syndrome, primarily myositis-systemic sclerosis (PDM/SSc) [2].
Table 1.
Autoantibodies in poly/dermatomyositis

Table 2.
Myositis-specific antibodies: target antigens and clinical associations in adult myositis patients
| Autoantibody | Immune target | Function of autoantigen | Clinical associations |
|---|---|---|---|
| Anti-ARS (Jo-1, PL-7, PL-12, EJ, OJ, KS, Ha, Zo) | tRNA synthetases | Aminoacylation of tRNAs | PM Anti-synthetase syndrome |
| Anti-Mi-2 | NuRD subunit | Gene transcription Nucleosome remodeling |
“Classic DM” Mild disease |
| Anti-TIF1-γ | Transcriptional intermediary factor 1γ | Ubiquitination Gene transcription |
Severe DM Cancer-associated DM |
| Anti-NXP-2 | Nuclear matrix protein 2 | Gene transcription | Severe DM Cancer-associated DM |
| Anti-MDA5 | Melanoma differentiation-associated protein 5 | Innate antiviral response | Amyopathic DM ILD Poor prognosis |
| Anti-SAE | SUMO-1 activating enzyme | Protein sumoylation Gene transcription |
DM Initially amyopathic DM |
| Anti-SRP | Signal recognition particle | Protein translocation across the ER | Necrotizing myopathy |
| Anti-HMGCR | 3-Hydroxy-3-methylglutaryl-CoA reductase | Cholesterol biosynthesis | Necrotizing myopathy Prior statin use |
tRNAs transfer RNAs, PM polymyositis, NuRD nucleosome remodeling-histone deacetylase, DM dermatomyositis, ILD interstitial lung disease, SUMO-1 small ubiquitin-like modifier 1, ER rough endoplasmic reticulum
Myositis-specific autoantigens are largely heterogeneous in functional and biochemical characteristics: the classic ones are the aminoacyl-tRNA synthetases (ARS), the Mi-2 helicase/histone deacetylase protein complex, and the signal recognition particle (SRP) [3–5]. During the last decade, new putative myositis autoantigens have been identified, including TIF1-γ, NXP-2, MDA5, SAE, and HMGCR [6–11]. The diagnostic accuracy of newly conceived autoantibodies is far from being ascertained; however, they are promising and in the future they could expand the MSA spectrum improving our ability in diagnosis and classification of myositis.
Myositis-specific antibodies (MSA) and disease subsets
Anti-ARS antibodies
ARS are cytoplasmic enzymes that catalyze the binding of each amino acid to its cognate tRNA during protein synthesis. As immunological targets, eight tRNA synthetases have been identified so far: histidyl (Jo-1), threonyl (PL-7), alanyl (PL-12), glycyl (EJ), isoleucyl (PL-12), asparaginyl (KS), tyrosyl (Ha), and phenylalanyl (Zo) synthetases. Antibodies to ARS overall occur in 25–35 % of IIM patients [1]. Anti-Jo-1 is the most common, found in 20–30 % of patients with PM and 60–70 % of myositis patients with interstitial lung disease (ILD). Anti-PL-7, PL-12, are found in less than 5 %, and anti-KS, -OJ, -EJ, -Zo, -Ha, in less than 2 % of PM or DM. Taken together, anti-ARS are associated with a clinical syndrome featured by myositis and high occurrence of ILD, arthritis, Raynaud’s phenomenon and “mechanic’s hands”, named anti-ARS syndrome. Lungs and joints are the major organs involved, and disease prognosis is strictly related to pulmonary involvement. Even if anti-ARS syndrome was described as a clinical variant of PM or DM, it is now considered a true overlap syndrome [12].
Anti-Mi-2 antibody
Mi-2/nucleosome remodeling and histone-deacetylase complex participates in the regulation of gene expression via chromatin modifications. Anti-Mi-2 antibodies are found in 10–30 % of patients with IIM, especially DM, being associated with specific skin involvement, like Gottron’s sign or papules and heliotrope rash, lung sparing and good response to steroids [4]. Moreover, anti-Mi-2 antibody seems to be associated with a lower risk of paraneoplastic myositis, thus being considered a good prognostic factor [4, 13].
Anti-SRP antibody
SRP is a highly conserved cytoplasmic multimeric ribonucleoprotein, consisting of six polypeptides complexed with one 7SL RNA, that is involved in secretory protein recognition and translocation across the rough endoplasmic reticulum. Anti-SRP antibodies are specific for IIM, being found in 4–8 % of PM patients [1, 5]. They are strictly associated with the anti-SRP syndrome, a severe necrotizing myopathy, histologically characterized by abundant myofiber necrosis and regeneration, with scarce inflammation. Rapidly progressive muscle weakness and poor response to standard treatment are the characteristic prognostic features [5].
Anti-TIF1-γ antibody
This novel putative MSA, firstly described few years ago, is exclusively found in 20–30 % of adult as well as juvenile DM. It targets nuclear transcription factors belonging to the human transcriptional intermediatory factor (TIF-1) family, primarily TIF1-γ. Anti-TIF1-γ antibody is significantly associated with aggressive skin lesions both in adult and juvenile DM, whereas the established association with paraneoplastic DM is largely confined to adult patients, especially the older ones (>50 years). Intriguingly, TIF-1 proteins are over-expressed in solid tumors, i.e., adenocarcinoma, and implicated in the regulation of p53 oncogene suppressor. The diagnostic value of anti-TIF1-γ antibody testing has been recently assessed: high antibody levels anti-TIF1-γ increase the risk of cancer-associated DM in adults, whereas negative antibody prompts to exclude concurrent malignancy, due to its very high negative predictive value [6].
Anti-NXP-2 antibody
This autoantibody targets a 140-kDa nuclear protein, named nuclear matrix protein 2 (NXP2), which plays a role in the regulation of p53-induced apoptosis after oncogenic stimuli. To date, anti-NXP-2 antibodies have been reported in about 25 % of juvenile or adult DM, and rarely in PM. Joung-onset DM, severe cutaneous lesions, including calcinosis, and muscle contractures are the prominent features [14]. An association with cancer has been observed in adults, especially in males. Recently, Fiorentino et al. [7] assessed the diagnostic value of anti-TIF1-γ and anti-NXP-2 antibody testing for cancer-associated DM, confirming that they overall occur in more than 50 % of otherwise “antibody negative” patients, and are independently associated with cancer. Moreover, the authors provided evidence that accurate laboratory testing for such antibodies should be included in the diagnostic workup in order to identify the vast majority of patients with cancer-associated DM.
Anti-MDA5 antibody
This novel MSA has been firstly described in 20–30 % of Asian populations, as specific of DM and associated with a clinical subset characterized by clinically amyopathic DM and rapidly progressive ILD [8]. IFN-induced melanoma differentiation-associated protein 5 (MDA5) is as the identified autoantigen, which regulates the innate antiviral immune response against cytoplasmic viral RNA. The clinical relevance of anti-MDA5 antibodies seems to be confirmed also in Caucasian IIM cohorts [9], but it is still a matter of research.
Anti-SAE antibody
Strictly related to a specific HLA haplotype, anti-small ubiquitin-like modifier activating enzyme (SAE) antibodies are found in 8 % of adult DM patients, presenting with severe skin disease, dysphagia and systemic features; prognosis is favorable [10]. They target the SUMO-1 activating enzyme heterodimer, a small nuclear protein structurally similar to ubiquitin, which leads to a post-translational enzymatic aggregation/conjugation of proteins, called “protein sumoylation”.
Anti-HMGCR antibody
A putative MSA was discovered in 3–8 % of adult patients with IIM, as directed against 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR), a key enzyme of the cholesterol biosynthesis, specifically inhibited by statins. Anti-HMGCR antibody has been proposed as a serological marker of immune-mediated necrotizing myopathy, often but not exclusively induced by statin exposure [11]. Clinical phenotype of anti-HMGCR-positive patients resembled that observed in other forms of IIM: proximal muscle weakness, markedly elevated creatine kinase levels, myopathic features on electromyography, and response to immunosuppressive therapy. Myofiber necrosis and degeneration is the characteristic histopathologic feature, occasionally accompanied by aspecific inflammatory cell infiltration (Fig. 1).
Fig. 1.
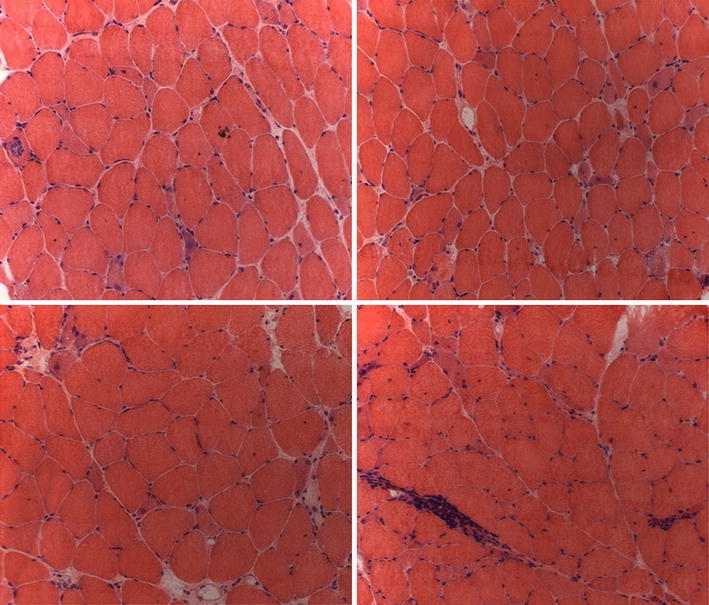
Muscle biopsy from a patient with polymyositis and anti-HMGCR autoantibodies (hematoxylin–eosin stain, original magnification ×20). The biopsy shows scattered necrotic fibers, some of which invaded by mononuclear cells, basophilic regenerating fibers and an inflammatory infiltrate in the perimysium. (Courtesy of Dr. Vattemi G.)
Myositis-associated antibodies (MAA) and overlap syndromes
Major MAA include anti-Ro/SSA, anti-PM/Scl, and anti-Ku and anti-U1RNP antibodies. MAA are often found when PM or DM occurs as a component of another CTD, and thus contributing to the clinical spectrum of an overlap syndrome, though MAA may also be present in patients without overlap CTD. The overlap syndrome between PM/DM and systemic sclerosis (PDM/SSc), also called scleromyositis, is the most common and according to a recent review of the literature it appears to represent more than 44 % of all scleroderma overlap syndromes [2].
Anti-Ro/SSA antibody, mostly directed against the Ro52 subunit, is the most prevalent MAA in myositis, found in more than 30 % of patients, frequently concomitant with anti-ARS antibodies or other MAA [15]. It has been recently reported that anti-Ro52 antibody could have a prognostic value in anti-Jo-1 positive patients, being associated with a higher risk of severe ILD, myositis, joint involvement, and cancer compared with anti-Jo1-positive patients without anti-Ro52 antibodies [16]. In addition, anti-Ro/SSA and/or anti-La/SSB antibodies are frequently encountered in PDM/Sjögren (SS) overlap syndrome, which is reported in 5.3 % of PDM patients, while myositis in primary SS is variably reported in 1–14 % of cases [17].
Anti-PM/Scl and anti-Ku antibodies can be somehow considered markers of PDM/SSc. Antibodies to PM/Scl are generally found in PM, DM or SSc patients, with the highest occurrence in PDM/SSc. A meta-analysis of the studies on anti-PM/Scl antibodies showed that anti-PM/Scl was found in 31 % of patients with PDM/SSc, compared to 8 % of PM, 11 % of DM and 2 % of SSc. On the other hand, PDM/SSc is diagnosed in 59 % of patients with anti-PM/Scl [18]. Apart from myositis, the most frequent clinical manifestations reported in anti-PM/Scl-positive PDM/SSc patients consist of Raynaud’s phenomenon, ILD, arthritis and skin involvement, including “mechanic’s hands”. While muscle involvement is often subclinical in PDM/SSc with PM/Scl antibodies, ILD has the same clinical and pathological features and severity observed in patients with anti-ARS syndrome [19].
Even though detected in a wide spectrum of CTD, anti-Ku antibody should be considered as a marker of PDM/SSc, either because it was first described in patients with PDM/SSc, which was reported in about 25 % of anti-Ku positive patients, or because the clinical manifestations are the same as in anti-ARS and anti-PM/Scl overlap syndrome [20]. Moreover, while the inflammatory myopathy is usually mild and responds to corticosteroid treatment, lung involvement is severe and refractory to corticosteroids, as in both anti-ARS and anti-PM-Scl overlap syndrome [21]. Anti-PM/Scl and anti-Ku positive PDM/SSc are preferentially associated with the limited cutaneous variant of SSc [20, 22], presenting less digital ulcers and much more myositis, arthritis and ILD, compared with anti-centromere antibody positive limited SSc [20]. Notably, both anti-PM/Scl antibody and the associated PDM/SSc recognize an immunogenetic background characterized by the haplotype HLA-DQA1*0501, DQB1*02, DRB1*0301 [22].
Anti-U1RNP antibodies are significantly more frequent in PDM/systemic lupus erythematosus (SLE) overlap syndrome than in SLE and they are associated with anti-Jo-1 antibodies in such condition. In addition to myositis, erosive arthritis, alopecia, ILD and oral ulcers are prevalent findings in patients with PDM/SLE overlap syndrome. Conversely, renal disease is very rare. Nevertheless, patients with SLE and myositis were likely to die at a young age. The onset of myositis can antedate, be concomitant or follow the appearance of SLE manifestations. DM is more common than PM [23].
By reviewing a cohort of 110 outpatients with PDM attending Brescia Rheumatology and Clinical Immunology Unit from 1990 to 2013, an overlap syndrome with CTD was diagnosed in 23 patients (21 %): 14 with SSc (61 %), 3 with SLE (13 %), 4 with SS (17 %) and 2 with rheumatoid arthritis (9 %). A similar prevalence of overlap syndrome in PDM was also reported in recent studies [17], where myositis was classified according to the commonly used Bohan and Peter’s criteria. Interestingly, a modified classification of these criteria has been proposed [15], taking into account the presence of extra muscular clinical manifestations and/or autoantibodies other than anti-Mi-2, considered highly specific for pure DM. With such an approach, the majority of inflammatory myositis should be diagnosed as overlap myositis syndrome. Furthermore, combining autoantibody testing to the original classification allows us to better understand the value of autoantibodies in the diagnosis and prognosis definition, and in predicting the response to corticosteroid treatment.
Laboratory detection of myositis autoantibodies
Testing for MSA and MAA is a useful aid for the diagnosis and subset definition of autoimmune myositis. However, many target antigens are poorly expressed in cell/tissue extracts or highly sensitive to degradation/denaturation, thus limiting the standardization of methods for diagnostic purpose. Therefore, most MSA are still detected through different time-consuming non-standardized techniques. Recently, a single multianalytic line blot assay has been validated as screening test for MSA/MAA in clinical practice [24]. On the basis of our experience, we suggest to include line blot as an initial screen in suspected IIM. Patients seronegative by line blot testing should be evaluated by in-house speculative testing.
At present, new antibody specificities are accurately detected by immunoprecipitation of radiolabeled proteins or RNA molecules; however, commercially available ELISA or immunoblot assays will be available in the near future.
Autoantibodies as clues in the pathogenesis of myositis
The aforementioned association of MSA with distinct clinical phenotypes and disease prognosis, and the correlation between serum antibody levels and disease activity, provide clues on the pathogenetic role of autoantibodies in PM/DM. Evidence is increasing that myositis autoantigens are able to drive a B cell antigen-specific immune response in muscles. Intriguingly, after the pioneeristic study by Casciola-Rosen et al. [25], several groups confirmed the aberrant expression of Jo-1, Mi-2, HMGCR and other myositis antigens in regenerating fibers from PM and DM muscle biopsies compared with controls. In addition, overexpression of both classic and novel myositis antigens has been demonstrated in other target organs, such as Jo-1 in the lung, Mi-2 in the epidermal basal membrane, and also TIF1-γ and NXP-2 in adenocarcinoma tissues. In target tissues, myositis antigens show strong adjuvant and chemoattractant properties, acting per se as proinflammatory and immunostimulating agents, either in the muscle or far from it, i.e., in the lung, skin or tumors. Thus, regenerating muscle fibers and tumor cells seem to share similar immunostimulating phenotypes, suggesting a pathogenetic link between cancer and autoimmunity in myositis [26]. Finally, association of most myositis autoantigens with nucleic acids makes them likely to stimulate the IFN type I secretion pathways which in turn can increase antigen availability and propagation of the immune response [27].
Conclusions
MSA are disease serological markers, mutually exclusive and closely associated with distinct disease subsets. Moreover, they seem to be directly involved in the induction and perpetuation of muscle damage. Therefore, their detection in the early phase of the disease might be helpful in the prediction of clinical course and disease prognosis.
Conflict of interest
None.
Human and animal rights
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
The study was approved by the Local Ethics Committee and informed consent was obtained from all patients, in compliance with the Helsinki Declaration.
References
- 1.Ghirardello A, Bassi N, Palma L, et al. Autoantibodies in polymyositis and dermatomyositis. Curr Rheumatol Rep. 2013;15:335. doi: 10.1007/s11926-013-0335-1. [DOI] [PubMed] [Google Scholar]
- 2.Iaccarino L, Gatto M, Bettio S, et al. Overlap connective tissue disease syndromes. Autoimmun Rev. 2012;3:363–373. doi: 10.1016/j.autrev.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 3.Zampieri S, Ghirardello A, Iaccarino L, Tarricone E, Gambari PF, Doria A. Anti-Jo-1 antibodies. Autoimmunity. 2005;38:73–78. doi: 10.1080/08916930400022640. [DOI] [PubMed] [Google Scholar]
- 4.Ghirardello A, Zampieri S, Iaccarino L, et al. Anti-Mi-2 autoantibodies. Autoimmunity. 2005;38:79–83. doi: 10.1080/08916930400022681. [DOI] [PubMed] [Google Scholar]
- 5.Hengstman GJ, ter Laak HJ, Vree Egberts WT, et al. Anti-signal recognition particle autoantibodies: marker of a necrotising myopathy. Ann Rheum Dis. 2006;65:1635–1638. doi: 10.1136/ard.2006.052191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trallero-Araguas E, Rodrigo-Pendas JA, Selva-O’Challagan A, et al. Usefulness of anti-p155 autoantibody for diagnosing cancer-associated dermatomyositis. A systematic review and meta-analysis. Arthritis Rheum. 2012;64:523–532. doi: 10.1002/art.33379. [DOI] [PubMed] [Google Scholar]
- 7.Fiorentino DF, Chung LS, Christopher-Stine L, et al. Most patients with cancer-associated dermatomyositis have antibodies to nuclear matrix protein NXP-2 or transcription intermediary factor 1 γ. Arthritis Rheum. 2013;65:2954–2962. doi: 10.1002/art.38093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao H, Pan M, Kang Y, et al. Clinical manifestations of dermatomyositis and clinically amyopathic dermatomyositis patients with positive expression of anti-melanoma differentiation-associated gene 5 antibody. Arthritis Care Res. 2012;64:1602–1610. doi: 10.1002/acr.21728. [DOI] [PubMed] [Google Scholar]
- 9.Hall JC, Casciola-Rosen L, Samedy LA, et al. Anti-melanoma differentiation-associated protein 5-associated dermatomyositis: expanding the clinical spectrum. Arthritis Care Res. 2013;65:1307–1315. doi: 10.1002/acr.21992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tarricone E, Ghirardello A, Rampudda M, Bassi N, Punzi L, Doria A. Anti-SAE antibodies in autoimmune myositis: identification by unlabelled protein immunoprecipitation in an Italian patient cohort. J Immunol Methods. 2012;384:128–134. doi: 10.1016/j.jim.2012.07.019. [DOI] [PubMed] [Google Scholar]
- 11.Mohassel P, Mammen AL. Statin-associated autoimmune myopathy and anti-HMGCR autoantibodies. Muscle Nerve. 2013;48:477–483. doi: 10.1002/mus.23854. [DOI] [PubMed] [Google Scholar]
- 12.Mahler M, Miller FW, Fritzler MJ. Idiopathic inflammatory myopathies and the anti-synthetase syndrome: a comprehensive review. Autoimmun Rev. 2014;13:367–371. doi: 10.1016/j.autrev.2014.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iaccarino L, Ghirardello A, Bettio S, et al. The clinical features, diagnosis and classification of dermatomyositis. J Autoimmun. 2014;48–49:122–127. doi: 10.1016/j.jaut.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 14.Ceribelli A, Fredi M, Taraborelli M et al (2012) Anti-MJ/NXP-2 autoantibody specificity in a cohort of adult Italian patients with polymyositis/dermatomyositis. Arthritis Res Ther 14:R97. doi:10.1186/ar3822 [DOI] [PMC free article] [PubMed]
- 15.Troyanov Y, Targoff IN, Tremblay JL, Goulet JR, Raymond Y, Senécal JL. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients. Medicine (Baltimore). 2005;84:231–249. doi: 10.1097/01.md.0000173991.74008.b0. [DOI] [PubMed] [Google Scholar]
- 16.Marie I, Hatron PY, Dominique S, et al. Short-term and long-term outcome of anti-Jo1-positive patients with anti-Ro52 antibody. Semin Arthritis Rheum. 2012;41:890–899. doi: 10.1016/j.semarthrit.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 17.Váncsa A, Gergely L, Ponyi A, et al. Myositis-specific and myositis-associated antibodies in overlap myositis in comparison to primary dermatopolymyositis: Relevance for clinical classification: retrospective study of 169 patients. Jt Bone Spine. 2010;77:125–130. doi: 10.1016/j.jbspin.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 18.Mahler M, Raijmakers R. Novel aspects of antibodies to the PM/Scl complex: clinical, genetics and diagnostic insights. Autoimmun Rev. 2007;6:432–437. doi: 10.1016/j.autrev.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 19.Lega JC, Cottin V, Fabien N, Thivolet-Béjui F, Cordier JF. Interstitial lung disease associated with anti-PM/Scl or anti-aminoacyl-tRNA synthetase autoantibodies: a similar condition? J Rheumatol. 2010;37:1000–1009. doi: 10.3899/jrheum.090652. [DOI] [PubMed] [Google Scholar]
- 20.Cavazzana I, Fredi M, Taraborelli M, Quinzanini M, Tincani A, Franceschini F. A subset of systemic sclerosis but not of systemic lupus erythematosus is defined by isolated anti-Ku autoantibodies. Clin Exp Rheumatol. 2013;31:118–121. [PubMed] [Google Scholar]
- 21.Rigolet A, Musset L, Dubourg O, et al. Inflammatory myopathies with anti-Ku antibodies: a prognosis dependent on associated lung disease. Medicine (Baltimore) 2012;91:95–102. doi: 10.1097/MD.0b013e31824d9cec. [DOI] [PubMed] [Google Scholar]
- 22.Ho KT, Reveille JD. The clinical significance of autoantibodies in scleroderma. Arthritis Res Ther. 2003;5:80–93. doi: 10.1186/ar628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dayal NA, Isenberg DA. SLE/myositis overlap: are the manifestations of SLE different in overlap disease? Lupus. 2002;11:293–298. doi: 10.1191/0961203302lu186oa. [DOI] [PubMed] [Google Scholar]
- 24.Ghirardello A, Rampudda M, Ekholm L, et al. Diagnostic performance and validation of autoantibody testing in myositis by a commercial line blot assay. Rheumatology (Oxford) 2010;49:2370–2374. doi: 10.1093/rheumatology/keq281. [DOI] [PubMed] [Google Scholar]
- 25.Casciola-Rosen L, Nagaraij K, Plots P, et al. Enhanced autoantigen expression in regenerating muscle cells in idiopathic inflammatory myopathy. J Exp Med. 2005;201:591–601. doi: 10.1084/jem.20041367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zampieri S, Doria A, Adami N, et al. Subclinical myopathy in patients affected with early stage colorectal cancer at disease onset: evidence from skeletal muscle biopsies. Neurol Res. 2010;32:20–25. doi: 10.1179/016164110X12556180205997. [DOI] [PubMed] [Google Scholar]
- 27.Lundberg IE, Helmers SB. The type I interferon system in idiopathic inflammatory myopathies. Autoimmunity. 2010;43:239–243. doi: 10.3109/08916930903510955. [DOI] [PubMed] [Google Scholar]