

Abstract
Airway hyperresponsiveness (AHR) has long been considered a cardinal feature of asthma. The development of the measurement of AHR forty years ago initiated many important contributions to our understanding of asthma and other airway diseases. However, our understanding of AHR in asthma remains complicated by the multitude of potential underlying mechanisms which in reality are likely to have different contributions amongst individual patients. Therefore the present review will discuss the current state of understanding of the major mechanisms proposed to contribute to AHR and highlight the way in which AHR testing is beginning to highlight distinct abnormalities associated with clinically relevant patient populations. In doing so we aim to provide a foundation by which future research can begin to ascribe certain mechanisms to specific patterns of bronchoconstriction and subsequently match phenotypes of bronchoconstriction with clinical phenotypes. We believe that this approach is not only within our grasp but will lead to improved mechanistic understanding of asthma phenotypes and hopefully better inform the development of phenotype-targeted therapy.
1. Introduction
Airway hyperresponsiveness (AHR) is defined as the predisposition of the airways of patients to narrow excessively in response to stimuli that would produce little or no effect in healthy subjects (Figure 1). Cockcroft et al (1) are largely credited with popularising the non-specific test of AHR almost forty years ago; however, the abnormal responses of asthmatics to non-specific stimuli were first described by Tiffeneau and Beauvallet in 1945 (2) and later developed during the 1960s in both Europe (3) and the United States (4). AHR has long been considered a cardinal feature of asthma and its measurement has provided profound insights into the underlying pathophysiology of the disease. Our view of AHR has greatly matured, and because of recent findings it is important to re-assess the current knowledge of AHR particularly in our understanding of the underlying mechanisms. While we did not embark upon a systematical evaluation of all literature regarding mechanisms of AHR in asthma we have endeavoured to provide an extensive update on the most recent findings. We will first introduce a few key clinical studies to provide a foundation for discussing the way in which future research of the mechanisms of AHR may contribute significantly to clinical practice.
Figure 1. Representative dose response curves (DRC) to methacholine in a healthy and a severely asthmatic subject.
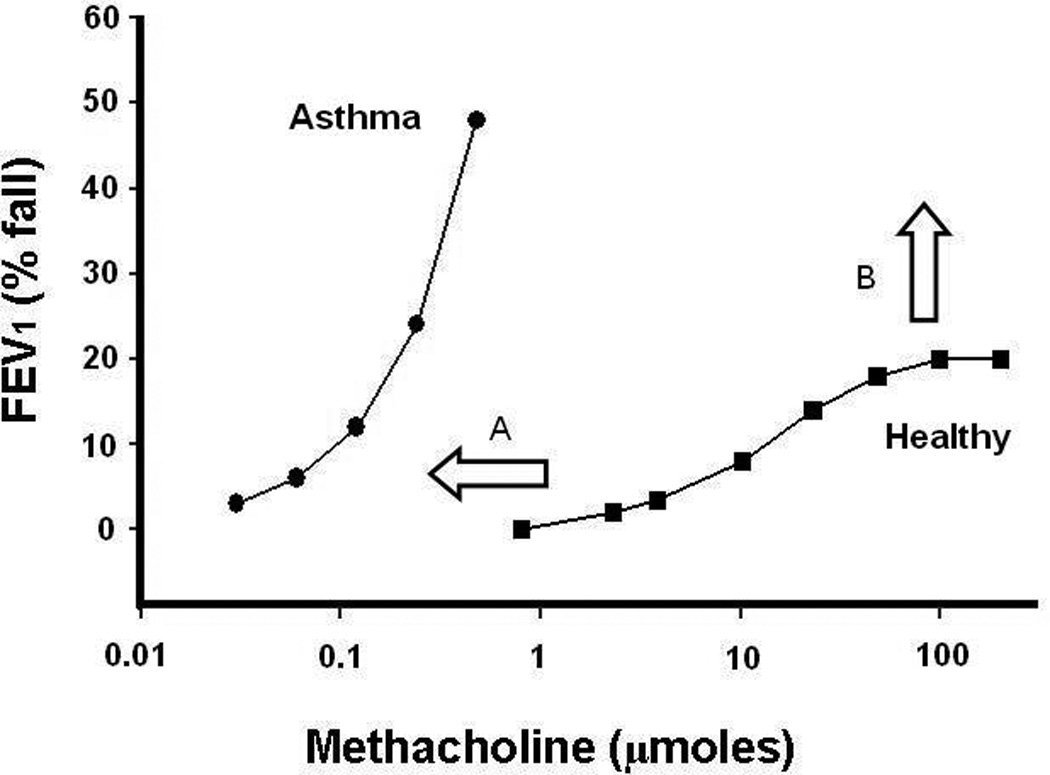
Airway hyperresponsiveness is characterised by both an increased sensitivity, seen as the leftward shift in the DRC of the asthmatic patient (A), and excessive bronchoconstriction, resulting in the loss/increase in the maximal response plateau (B).
But let’s start with a pair of case studies. Both authors have at one time responded positively to bronchial challenge. One (DC) had AHR to methacholine despite being concurrently negative to mannitol challenge. Following treatment, he is now in “AHR remission”. The other (CI) responded positively in the past to exercise, methacholine and histamine when inhaled but negatively during systemic administration. Currently he does not respond to methacholine even up to high doses. Despite this small sample size, a few key concepts emerge. Firstly, AHR is a moving target, in that its severity and presence are dependent upon many factors, including the modality of agonist chosen and level of treatment. Secondly, AHR is likely due to a plethora of underlying mechanisms that will have greater or lesser contributions in individual patients. This heterogeneity is not always well appreciated. While the clinical utility of AHR has been extensively reviewed elsewhere (5–7) we believe that understanding the heterogeneity of the mechanisms underlying AHR is an often over-looked yet important piece of the puzzle. Therefore we propose that future research into the mechanisms of AHR should aim to can move our understanding from a “one size fits all” approach to ascribing specific mechanisms of AHR to distinct patient populations. In this way we believe that it will provide much needed insight into the ever-developing recognition of distinct clinical phenotypes of asthma.
2. Clinical Importance
Airway hyperresponsiveness is used as a tool in the diagnosis, classification of severity (8) and management (9, 10) of asthma. AHR is useful in those who report symptoms (9), particularly in those with normal baseline lung function as measured by spirometry (11). The presence of AHR is associated with increased decline in lung function (12), even in those with asymptomatic AHR (13), increased risk for the development of asthma (12) and increased likelihood of the persistence of wheeze from childhood to adulthood (14). Furthermore, the severity of AHR is associated with an increased risk of exacerbation (15), increased asthma severity as measured by symptoms (16) and an increased level of treatment required to control symptoms (1). While the clinical implications relating specifically to the loss of the maximal response plateau in asthma are unclear, an increased or absent plateau represents uninhibited airway narrowing or closure that has the potential for life-threatening exacerbations (17). Understanding the factors contributing to the presence and severity of AHR therefore provides an important component for improving asthma control and reducing disease progression.
Although AHR is considered a hallmark of asthma, it is important to recognise that the severity, and even presence, of AHR is not stable. AHR to non-specific stimuli, such as histamine and methacholine, is increased in some, but not all, subjects following allergen challenge (18). This increase in AHR occurs most frequently in those subjects with a late asthmatic response and its persistence can be short-lived or remain for up to several months from exposure (19). It is not surprising, then, that seasonal allergen exposure alters the severity of AHR (20). In addition, anti-inflammatory therapy profoundly improves AHR and since its widespread introduction many patients on appropriate treatment regimens may not respond positively to bronchial challenge in the range associated with asthma. For example, in a population of poorly controlled, chronically undertreated asthmatics, Reddel et al (21) reported that 16 weeks of high dose inhaled corticosteroid (ICS) followed by dose titration led to a 4.0 doubling dose increase in PD20FEV1 (reduction in AHR). After 72 weeks, 40% of subjects had responses to methacholine challenge within the normal range. Consistent with this, in asthmatic subjects on regular controller therapy, the sensitivity of a positive methacholine challenge for the diagnosis of asthma is only 77% (22). This sensitivity is further reduced in Caucasian and non-atopic patients. Furthermore, the measurement of AHR is confounded by its moderate repeatability, with estimates of within-subject repeatability ranging from 1–3 doubling doses (reviewed in (23)). Variability of AHR is further increased in those with non-atopic disease and those over 50 years of age (24). It is important to acknowledge that the variability of AHR is not only due to variability in the underlying mechanisms but also due to the imprecision of the measurement itself. Although it is clear that a negative challenge does not exclude the presence of asthma, interpretation of a negative bronchial challenge must consider the presence or absence of current symptoms. In a patient with current symptoms a negative challenge may suggest that diagnoses other than asthma should be considered. However, a negative challenge in a period without symptoms does not preclude asthma and in consideration of history it may be more appropriate to label such a patient as “currently negative AHR”.
3. Measurement of AHR: To what do we respond and how do we measure it?
Traditionally measurements of airway responsiveness have been presented using two different, yet qualitatively similarly, calculations. The provocative concentration (or dose) causing a 20% fall in FEV1 (PC20FEV1) is calculated by interpolation from the dose causing ≥20% fall in FEV1 and the penultimate dose on a semi-log scale. It is maybe not surprising that determining the actual dose delivered (PD20) appears to be a more robust measure than simply using the concentration of agonist (PC20) (25). The dose response slope (or DRS), also referred to as the response dose ratio (RDR), is calculated as the slope of the dose response curve plotted with a linear dose axis. The advantage of the DRS is that it provides a continuous measure of airway responsiveness allowing inclusion of subjects who do not reach a 20% fall in FEV1. In a more philosophical sense, the DRS more accurately reflects that airway responsiveness is a continuous variable in which AHR merely describes those people at one extreme. As both calculations provide similar information studies providing either calculation will be subsequently mentioned without differentiation.
a. Methacholine vs Mannitol
There are two groups of stimuli utilised in the measurement of AHR; those which allegedly act directly on the airway smooth muscle to induce bronchoconstriction, such as methacholine and histamine, and those which indirectly cause bronchoconstriction through the release of upstream mediators. Indirect challenge tests include exercise, eucapnic voluntary hypernea (EVH), hypertonic saline, mannitol, adenosine 5’monophosphate (AMP) and various allergens. Although extensive review of the clinical application (6) and mechanisms (5) of these various stimuli have been provided elsewhere, the increasing interest in mannitol is worth considering as an example of an indirect challenge. Airway eosinophilia, measured by both sputum eosinophils and exhaled NO (eNO), is more strongly associated with AHR to mannitol than to methacholine (26, 27). Although this suggests that mannitol may provide a marginally better reflection of airway eosinophilia, the clinical utility of this finding is unclear. In addition, mannitol is more variable than methacholine which probably reflects variability in underlying inflammation. Therefore mannitol might be useful in predicting those patients who respond best to anti-IL-13 or anti-IL-5 monoclonal antibody treatment given the projected high cost of these treatments. Compared to treatment guidelines based upon symptoms and lung function, treatment strategies targeting reductions in AHR to mannitol (STAMINA trial) (28) or methacholine (AMPUL trial) (9) both lead to improvements in the number of mild exacerbations. Mannitol appears more specific in detecting a diagnosis of asthma although it is less sensitive than methacholine (27, 29, 30). Interestingly, the biggest distinction may be in the effect of allergen exposure with increased responsiveness to methacholine but reduced responsiveness to mannitol three hours after allergen challenge (31). This highlights the distinct underlying mechanisms between the two tests and suggests that comparison of the two methods may be useful in identifying phenotypes of AHR, and the underlying mechanisms, in subpopulations of patients. Indeed, one recent study reported 15% of asthmatics in primary care had AHR only to methacholine while another 15% had AHR only to mannitol (32). Similar variability in response has been reported when comparing exercise, EVH and methacholine challenge (33). Further research is required to determine the clinical importance of these distinct phenotypes of AHR.
b. What lung function measurement to use?
Traditionally AHR has been measured as reductions in spirometric parameters, most particularly FEV1. However, spirometry is highly effort dependent and therefore requires considerable subject co-operation which is impossible for children under five and difficult for the elderly and those with increased disease severity. In contrast, the forced oscillation technique (FOT) is a measure of the mechanics of the respiratory system which can be acquired without special breathing manoeuvers. The FOT imposes oscillations over tidal breathing with the subsequent changes in pressure and flow analysed to provide measures of respiratory system resistance (Rrs) or its inverse conductance (Grs), as measures of airway calibre, and reactance (Xrs), as a measure of elastance. Recent advancements have allowed the FOT to cross the divide into clinical practice. FOT is capable of detecting patients with AHR, as assessed by spirometry, during methacholine (34), mannitol (35) and carbachol challenges (36). Furthermore, the repeatability of Grs and Xrs is not different to that of FEV1 (2.0 and 1.95 vs 1.67 doubling doses, respectively, (35)). However, it should be noted that the aforementioned comparative studies included deep inspirations (inherent in spirometry) which provide beneficial effects in non-asthmatic but not moderate to severe asthmatic subjects (37). More importantly, removing deep inspirations during FOT measurement may also alter responsiveness in asthmatics with mild AHR since reduced responses are observed in mild disease when challenge inhalation is performed with the deep inspiration method compared to the tidal breathing method (38). Therefore, measuring responses to bronchial challenge during only tidal breathing with FOT may reduce the ability to discriminate borderline AHR from normal responsiveness. Furthermore, decades of research indicates the clinical utility of AHR measured by spirometry, leaving us with a scenario where the use of FOT during bronchial challenge may best be suited as an adjunct to spirometry. Comparison of spirometry and FOT responses to bronchial challenge, as well as comparisons between FOT variables, may provide important clues as to the underlying pathophysiology. Lastly, FOT measurements may be capable of detecting differences in the pattern of response between stimuli (39) which may further aid in assigning phenotypes of AHR to distinct clinical populations.
4. Mechanisms of Airway Hyperresponsiveness
Despite decades of research, there is still little consensus as to the mechanisms underlying AHR in asthma. This is most likely due to the numerous pathophysiological abnormalities associated with asthma and the likely reality that different mechanisms or a combination of these gives rise to AHR in different patient populations. The definition of asthma as an inflammatory airways disease characterised by exaggerated airway narrowing immediately brings attention to the role of airway inflammation and the airway smooth muscle (ASM) in the manifestation of AHR. In addition, the structural remodelling reported in many patients with asthma is also likely to contribute in some, but presently unclear, way to the severity of AHR. Lastly, there is currently renewed interest in airway closure as a cause of AHR rather than merely a consequence.
a. Genetics
The role of familial inheritance in asthma was formally acknowledged by Coca and Cooke in 1923 (40) and the heritability of AHR has since been reported to be approximately 30% (41). However, the mechanisms linking genetics and AHR remain to be defined. Levitt and Mitzner (42) showed the large genetic contribution to AHR in mice by demonstrating that airway responsiveness to acetylcholine was controlled by a single autosomal recessive gene. Genome-wide association studies have revealed a substantial number of genes associated with susceptibility to asthma (recently reviewed in (43)), with genes corresponding to inflammatory pathways, airway epithelial function and ASM function likely contributing to AHR (mechanisms discussed below). Indeed, β2-adrenergic receptor genotype appears to partially determine the improvements in AHR to methacholine following salmeterol/ICS therapy (44). Although not substantiated, this may represent distinct genotype-dependent mechanisms of AHR rather than differences in general treatment efficacy since improvements in baseline lung function, eNO and bronchodilator responsiveness were not affected by genotype. Similarly, it has been reported that allergen exposure in mice induces epigenetic changes in the transforming growth factor-β signaling pathway which are associated with development of AHR (45). Despite considerable advancement of our understanding of genetics in asthma future research is required to determine if and how genetic/epigenetic alterations are causally linked to the development and severity of AHR in human asthma.
b. Airway Inflammation
Asthma is a disease associated with chronic inflammation and the influx of inflammatory proteins likely contributes to AHR. Although allergic asthma has long been known to be associated with increased eosinophils in the airways, recent research suggests that subsets of asthmatic patients have elevated neutrophils with or without increased eosinophils (46). In asthmatic subjects there is a positive correlation between the severity of AHR and the number of eosinophils and metachromic cells in sputum (47, 48), as well as the number of mast cells in the airways (49). Furthermore, the level of exhaled nitric oxide (eNO), considered a biomarker for eosinophilic inflammation (50), correlates with the severity of AHR to methacholine in asthmatic subjects (51, 52). Interestingly, the link between eNO and AHR appears driven by airway narrowing, but not airway closure (53). In a small study of mild asthmatics, Brusasco et al (54) found no relationship between baseline AHR and inflammatory cells in bronchoalveolar lavage. However, after allergen challenge a strong correlation was reported between the increase in AHR and increase in eosinophils, further supporting the role of eosinophils in AHR. In contrast, there is little evidence that airway neutrophilia contributes to the severity of AHR in asthma. Indeed, Porsbjerg et al (48) were unable to show any correlation between neutrophils or neutrophil mediators and AHR. However, sputum neutrophils were correlated with an increased contribution of airway closure to the overall level of bronchoconstriction. Taken together, these findings suggest that eosinophillic airway inflammation may contribute to the severity of AHR whereas airway neutrophilia may be associated (causally or coincidentally) with an alteration in the type of bronchoconstriction towards predominance of airway closure.
c. Airway Smooth Muscle
Bronchoconstriction is due, at least in part, to constriction of the airway smooth muscle (ASM) surrounding the airway. Therefore it is not surprising that increased contractility of the ASM has long been touted as a principal cause of AHR. Abnormal ASM function could be due to intrinsic abnormalities of the ASM itself or to the effects of the asthmatic environment in which it resides.
i. Intrinsic factors
Despite considerable research it is still unclear as to whether asthmatic ASM is intrinsically hyper-contractile, and if so, what factors are mechanistically involved. Recent gene expression profiling of ASM revealed four novel genes that not only differentiated asthmatic and non-asthmatic patients, but were related to the severity of AHR (55). Furthermore, the expression of contractile proteins α-smooth muscle actin and desmin in ASM from asthmatics correlates with the severity of AHR (56) suggesting a role of intrinsic ASM dysfunction. Some in vitro studies have reported increased force generation of ASM from asthmatic patients (57, 58), while others have reported no difference when compared to healthy controls (59–61). However, increased airway narrowing could be due to an increase in the shortening velocity of ASM despite normal force generation. This would theoretically occur because a muscle that shortens quickly would produce greater airway narrowing during expiration before the dilatory effect of the proceeding inspiration (62). Indeed, in vivo findings support an effect of ASM shortening velocity on the magnitude of ASM shortening (63).
ASM contraction involves the formation of actin-myosin cross-bridges with the rate of formation dependent upon the activity of myosin light chain kinase (MLCK) and myosin light chain phosphatase (MLCP). An increase in the activity of either MLCK or MLCP would lead to increased shortening velocity of ASM. Indeed, both an increased expression of MLCK and increased shortening velocity of ASM have been reported in asthma (64). Furthermore, the fast myosin heavy chain isoform is also increased in patients with asthma which murine models suggest would also increase cross-bridge cycling and AHR (65). Functionally, this may not be important as increased shortening velocity in asthma is not a consistent finding (61). Asthmatic ASM is also more sensitive to oxidative stress with the extent of oxidative damage within the ASM bundle correlated with the severity of AHR (66). This relationship is in part mediated by increased NOX4 expression since siRNA knockdown of NOX4 attenuates in vitro ASM contractility. In contrast to dysregulation of the molecular pathways controlling ASM contraction, subcellular structure of the ASM appears similar between asthmatic and non-asthmatic subjects (67).
ii. Extrinsic factors
The asthmatic airway resides in a pro-inflammatory environment which likely contributes to ASM dysfunction independent of any intrinsic abnormalities. Pro-inflammatory cytokines such as IL-4, IL-13 and tumour necrosis factor-α (TNFα) increase ASM responsiveness in vitro, possibly via effects on calcium signaling (68). Proteases, such as matrix metalloproteinase-1 (MMP-1), are increased within ASM bundles of asthmatics and also regulate in vitro ASM contractility (69) and structural integrity. Additionally, the number of mast cells within the ASM correlates with the severity of AHR in asthma (70). Although the mechanisms are not yet clear, mast cell mediators such as histamine, leukotriene D4 (71) and prostaglandin D2 (72) may contribute to increased basal ASM tone. Bossé and colleagues (73) reported that ovine tracheal ASM adapts to increased basal tone so that subsequent ASM shortening is synergistically amplified. Computational modelling suggested that this synergistic effect on ASM shortening, termed force adaptation, would translate to an increase in airway narrowing as high as 48% and increase in airflow resistance up to 274% for a prototypic ninth generation airway (74). Although force adaptation has recently been demonstrated in mice in vivo (75), it is unknown whether force adaptation occurs in humans in vivo and to what extent, if any, that would contribute to AHR measured by spirometry.
Alternatively, the inflammatory milieu may induce the transition of asthmatic ASM from a contractile to a “synthetic” phenotype (reviewed in (76)). This synthetic ASM phenotype is characterised by reduced contractile-associated proteins but increased proliferation and chemokine secretion. In vitro stimulation of human ASM with TNFα or IL-1β induces the secretion of chemokines such as regulated on activation, normal T cells expressed and secreted (RANTES), interleukin-6 (77, 78) and IL-8 (79, 80). This proliferative/secretory phenotype is associated with reduced expression of contractile proteins such smooth muscle myosin heavy chain, smooth muscle α-actin, myosin light chain kinase (81). It is presently unclear whether the transition of ASM to the synthetic phenotype confers protection against, or further contributes to, AHR.
Damage to the airway epithelium, which provides an initial barrier for inhaled spasmogens, also likely contributes to AHR. Disruption of the airway epithelium would increase the amount of stimulus interacting with the ASM and thus potentiate bronchoconstriction. In addition, epithelial damage or dysregulation likely reduces the ability of the epithelium to maintain relaxation of ASM via release of epithelial-derived relaxing factor(s) (82). For example, intratracheal administration of cationic proteins reduces both the barrier effect and control of ASM relaxation by the airway epithelium, and results in AHR in animal models (83). Additionally, damage to the epithelium may also directly contribute to airway narrowing. Recent murine in vitro findings suggest that rupture of small airway epithelial cells induce intracellular [Ca2+] waves and subsequent contraction in neighboring ASM (84). The contribution of damage of the airway epithelium is most likely to be highly relevant to AHR following exposure of noxious inhalants, such as in occupational asthma.
d. Structural airway remodelling
The lung of the asthmatic exhibits a gamut of structural pathologies that are collectively termed airway remodelling. Remodelling here merely means that the structure is no longer normal with the implication that the change is permanent. These changes include subepitheial fibrosis (85), ASM hypertrophy/hyperplasia (86), angiogensis (87) and changes in extracellular matrix composition (88). AHR correlates with airway wall thickening (89, 90) , reticular basement membrane thickness (91) and components of the extracellular matrix (56, 92), although others have been unable to replicate these findings (70).
Thickening of the airway wall could contribute to excessive bronchoconstriction in two ways. Airway resistance is inversely related to airway radius such that an increase in the submucosal area would amplify the reduction in airway calibre for any given degree of ASM shortening. Although an attractively simple explanation, it remains unclear whether reduced airway calibre is causally associated with increased severity of AHR. On one hand, several studies report a correlation between baseline airway calibre and AHR, measured by FEV1/FVC (53, 93) and FEF25–75/FVC (94, 95). On the other hand, improvements in airway calibre appear dissociated from improvements in AHR. Salome et al (96) administered fenoterol prior to histamine challenge and reported that although baseline FEV1 and AHR both improved, AHR returned to control levels much more rapidly than FEV1. This disconnect was later strengthened by Britton et al (97) who reported that ipratropium did not alter AHR despite increasing baseline FEV1 and sGaw. However, determining whether reductions in airway calibre are similarly disassociated from AHR is confounded by an inability to reduce airway calibre without confounding effects, such as those related to transpulmonary pressure or ASM tone.
An increase in the thickness of the adventitial layer has the potential to uncouple the ASM layer from the surrounding parenchyma. Under this condition, the ASM is essentially untethered from the lung parenchyma, reducing the load against which the ASM shortens. This would allow for increased ASM shortening. Increased ASM mass, due to either hypertrophy or hyperplasia, is thought to increase the total force generated by ASM and thus exaggerate airway narrowing without any alteration in ASM contractile function (98). Indeed, increased ASM area in explanted bronchial segments from asthmatics correlates with increased in vitro airway narrowing (99). However, airway remodelling may protect against AHR. Should the remodelling processes increase airway wall stiffness, it would in fact oppose, and therefore limit, airway narrowing during bronchoconstriction (100). On the other hand, some features of airway remodeling may be a consequence, rather than a cause, of AHR since bronchoconstriction itself is sufficient to induce subepithelial fibrosis and mucous metaplasia without affecting AHR (101).
e. Airway Closure
It is not well recognized that bronchoconstriction is also associated with increased airway closure. Moreover, it is important to determine whether increased airway closure is merely the consequence of exaggerated airway narrowing (102) or whether asthma is associated with a predisposition to airway closure. Increased airway closure assessed by bronchial challenge is associated with increased disease severity (103), oral steroid use (104) and a history of exacerbations requiring intubation (103). Irvin and Bates (105) reviewed the literature that supports the notion that bronchoncostriction in asthma is not due to central airway narrow as commonly assumed, but rather due to peripheral airway closure. Consistent with this hypothesis, AHR in allergically sensitised mice can be fully attributed to an increased susceptibility to small airway closure (106, 107). The importance of airway closure to human asthma was validated by the finding that the extent of airway closure during methacholine challenge was a significant determinant of the severity of AHR, independent of airway narrowing (53). However, unlike in the allergically sensitised mouse, there is great variability in the contribution of airway closure to bronchoconstriction in human asthma. As shown in Figure 2A and B, asthmatic patients can respond to bronchial challenge through predominantly airway narrowing or airway closure. It is important to highlight that these examples are the extremes of a continuum with the majority of subjects falling in between. Hence, the severity of AHR in specific phenotypes of asthmatic patients is likely due in large part to airway closure. However, the clinical features or underlying mechanisms of patients who respond predominantly due to airway closure require further investigation.
Figure 2. The contribution of airway narrowing and airway closure to the fall in FEV1 during bronchial challenge.

FEV1 is reduced by airway narrowing because a narrowed airway loses some capacity to transmit flow. However, FEV1 is also determined by the number of parallel airways contributing to flow and is thus reduced by functional airway closure (both true airway closure and severe airway narrowing). By contrast, FVC is determined by the volume of air in communication with the mouth and is reduced by functional airway closure but not by airway narrowing. Air narrowing, per se, is thus reflected in the ratio FEV1/FVC. There is substantial variation in the contribution of airway narrowing and airway closure to the fall in FEV1 amongst patients with asthma. Shown are dose response curves for an asthmatic subject with predominantly airway narrowing (A: 24 years old, baseline FEV1 122%pred, PC20FEV1 0.23µmol) and one with predominantly airway closure (B: 24 years old, baseline FEV1 78%pred, PC20FEV1 0.28µmol). These examples represent extremes of a continuum of responses, with the majority of subjects falling in between. The extent of airway narrowing is expected to contribute to airway closure so to determine excessive airway closure we have analysed the relationship between %fall FVC and FEV1/FVC (C). A steeper slope represents greater airway closure for a given level of airway narrowing. Absolute FEV1/FVC, rather than % fall FEV1/FVC, maintains the contribution of baseline airway calibre. Representative regression lines were calculated from the mean baseline FEV1/FVC, mean fall in FEV1/FVC and mean % fall FVC for lean non-asthmatics (blue), lean asthmatics (red), obese non-asthmatics (green), non-allergic obese asthmatics prior to bariatric surgery (purple) and the same subjects 12 months following bariatric surgery (dashed purple). Data were adapted from two of our previous studies (53, 130). Important to note is the increased slope in asthmatics compared to non-asthmatics, and in all obese groups compared to the two lean groups. Following weight loss, the slope of the obese non-allergic asthmatics decreased suggesting reduced predisposition to airway closure. Interestingly, the position and slope of obese non-allergic asthmatics post-surgery is almost identical to obese non-asthmatics suggesting that the effect of obesity on airway closure is dependent upon the level of adiposity in non-allergic subjects.
There are several mechanisms by which asthma pathophysiology may lead to increased airway closure during bronchoconstriction. Firstly, mucous plugging would obviously induce airway closure so it is not surprising that pharmacologically blocking the release of mucous protects against AHR in allergically inflamed mice (108). Secondly, increased airway closure may be due to surfactant dysfunction caused by inflammation (109, 110). Similarly, fibrin is known to inactivate surfactant (111), accumulate in the airways of asthmatics and is associated with AHR (107). A role of surfactant dysfunction in AHR is consistent with the protective effect of inhaled surfactant against allergen-induced bronchoconstriction (112). On the other hand, recent computational and physiological evidence suggests that increased baseline ventilation heterogeneity may promote increased airway closure during bronchoconstriction. Venegas et al (113) developed a highly advanced lung model that takes account of the effects of the parenchymal tethering forces, the intra- and extra-luminal pressures and ASM forces. The model predicted that uniform ASM contraction with the addition of small, random heterogeneities in airway calibre would lead to the abrupt development of airway closure when ASM contraction reached a critical level of instability. The validity of the model predictions have been strengthened by subsequent findings that the severity of AHR in asthma strongly correlates with the degree of baseline ventilation heterogeneity (52, 114, 115). This has recently been extended by the report that baseline ventilation heterogeneity correlates with the increase in airway closure during methacholine challenge (116). Importantly, the association between baseline ventilation heterogeneity and AHR remained following three months of ICS treatment suggesting that it is independent of (steroid-responsive) airway inflammation (52). Further research is needed to ascertain the causes of the baseline ventilation heterogeneity and whether they can be targeted to treat AHR, which may provide more effective treatment strategies for asthma.
5. Can AHR contribute to our understanding of asthma phenotypes?
Asthma is not a single disease but a combination of many pathophysiological features culminating in the clinical presentation of asthma symptoms. This underlies the importance of personalised medicine, in which the foundation has been built on improved phenotyping of asthmatic patients utilising a variety of clinical, inflammatory and physiological features (117, 118). However, these approaches are yet to include charateristics of AHR, such as differences between modalities or the pattern of bronchostriction. Below we highlight three phenotypes associated with worse asthma control and discuss the current understanding of AHR in each group (Table 1).
Table 1.
Current understanding of AHR in asthma phenotypes associated with worse control
| Asthma Phenotype | Severity of AHR | Pattern of AHR (Closure vs narrowing) |
Modality | Associated pathophysiology |
|---|---|---|---|---|
| Elderly | Increased | Closure | Methacholine | ↑ Neutrophils |
| ↓ Elastic recoil | ||||
| Obese | ||||
| • non-allergic | Increased | Closure | Methacholine | ↓ FRC volume |
| ↑ airway compliance | ||||
| ↓ surfactant (?) | ||||
| • allergic | Unaltered | Closure | Methacholine | ↓ FRC volume |
| Smoking | Increased | Closure | AMP | Inflammation/ Acutely reversible |
| Increased | ~ Equal | Methacholine | Structural | |
FRC = functional residual capacity
a. Asthma in the elderly
Asthma control worsens with age (119) with one recent study reporting that 25% of asthma patients over 65 years experienced at least one severe exacerbation in the preceding year (120). Hardaker et al (114) recently compared the physiological determinants of AHR in young and elderly asthmatics. In those below 55 years, the severity of AHR was predicted by increased eosinophilic airway inflammation (exhaled NO) and baseline ventilation heterogeneity in conducting airways. In contrast, AHR in the elderly was associated with baseline gas trapping and ventilation heterogeneity in acinar airways. This suggests that AHR in the elderly is associated with more peripheral disease. This is consistent with previous reports that AHR in the elderly is associated with increased airway closure during methacholine challenge (121). This may be due to the increase in neutrophilia with age, since increased airway closure during bronchoconstriction in elderly asthmatics correlates with sputum neutrophil levels (48). Alternatively the distinct AHR of the elderly may be due to ural change of the loss of elastic recoil due to emphysema-like changes associated with aging (122). Bronchial challenge with AMP has been suggested to induce a more peripheral response than that due to methacholine (123) and therefore age may have greater effects on AHR to AMP than to methacholine challenge. However, it is unclear whether the more peripheral disease is due to the additive effects of age on asthma or a synergistic effect of disease duration.
b. Asthma in the obese
Cross-sectional studies report an increased prevalence of asthma in the obese (124), while obesity and weight gain appear to precede the development of asthma (125). Obesity appears to worsen asthma control (126) while weight loss leads to an improvement in asthma symptoms (127). There is growing recognition that obese asthmatics comprise two distinct clinical populations; those with high IgE and early-onset asthma (allergic), and those with late-onset disease and low serum IgE (non-allergic). Following weight loss, only the non-allergic obese asthmatics had an improvement in methacholine responsiveness suggesting that obesity negatively impacts AHR only in those with non-allergic disease (128). This effect of obesity on AHR is due to increased collapsibility of peripheral airways that predisposes to increased airway closure during methacholine challenge (129, 130). This is illustrated in Figure 2C, in which the extent of airway closure, adjusted for the level of airway narrowing, is substantially greater in non-allergic obese asthmatics compared to non-obese asthmatics (ie significantly steeper slope). Interestingly, following weight loss, the response to methacholine appears identical to that of non-asthmatic obese subjects (ie position and slope of regression). These data suggest that allergic obese asthmatics may have asthma that is complicated by obesity, whereas non-allergic obese asthmatics have asthma secondary to obesity. Since the severity of AMP appears closely associated with atopy and IgE levels (131, 132), it is possible that AMP challenge may be better able to differentiate these two phenotypes of obese asthma.
c. Asthma in smokers
Asthmatics who smoke report worse asthma control (133) and smoking history is associated with the severity of AHR (134). Airway closure during AMP challenge is increased in asthmatic smokers compared to non-smokers, but not during methacholine challenge (135). This is consistent with a more peripheral response during AMP than methacholine challenge (123). Interestingly, improvements in AHR to AMP following smoking cessation occur earlier than improvements in AHR to methacholine (136). This may reflect a greater sensitivity of AMP to smoking-related pathophysiology. On the other hand, methacholine challenge may better reflect underlying structural changes than AMP (131) such that differences between the two stimuli may reflect distinct mechanisms underlying the severity of AHR in smoking asthmatics. Further research will determine whether these differences can be used to detect early smoking-related disease or determine those smokers who may respond to asthma guideline therapy and those unlikely to benefit.
6. Looking through the crystal ball: the future of AHR testing
We must ensure that our view of the measurement of AHR does not remain as a “one size fits all” approach. On one hand we have many different stimuli for bronchial challenge testing and on the other, an extensive list of potential mechanisms underlying AHR. Currently we do not completely understand whether specific mechanisms play a greater role in AHR assessed by one challenge test over another. Similarly, we do not know whether differences between challenge modalities provide a better assessment of the various clinical asthma phenotypes. Physiologists have long known that FEV1 is a polyvalent measure of lung function that provides little information about the precise pattern of bronchoconstriction. Understanding the pattern of bronchoconstriction in an individual patient, whether through comparison of spirometric variables or from measurements such as the FOT and inert gas washout, may allow us to ascribe certain phenotypes of bronchoconstriction to specific clinical phenotypes. This, too, would help elucidate the underlying mechanisms and may contribute to more targeted therapies. As discussed above, a combination of these two approaches has already been applied to asthma in the obese, elderly and those who smoke. While only in its infancy, the evidence to date suggests that phenotyping AHR may help to uncover the pathophysiology contributing to poor asthma control in numerous distinct subsets of patients with asthma.
7. Conclusion
The development of the measurement of AHR forty years ago sparked many important contributions to our understanding of asthma and other airway diseases. However, it is time to re-evaluate our assumptions of AHR in light of the current population of asthmatic patients. We must look towards the future, embracing the technological advancements which provide potentially complimentary techniques to measure the response to bronchial challenge. These complementary measurements may lead us to better partition global bronchoconstriction into its components of airway narrowing and airway closure as well as proximal and distal airway effects. This enhancement has to the potential of allowing us to assign certain mechanisms to specific patterns of bronchoconstriction, opening the door for matching phenotypes of bronchoconstriction with clinical phenotypes. In doing so we are likely to gain improved mechanistic understanding of asthma phenotypes, and help better focus as well as better assess the development of phenotype-targeted therapy.
Acknowledgments
Grant support: DGC is a recipient of a CJ Martin Fellowship from the National Health and Medical Research Council of Australia (1053790). CGI is supported by the American Lung Association and the National Institutes of Health (P30 GM103532).
References
- 1.Cockcroft DW, Killian DN, Mellon JJ, Hargreave FE. Bronchial reactivity to inhaled histamine: a method and clinical survey. Clin Allergy. 1977;7(3):235–243. doi: 10.1111/j.1365-2222.1977.tb01448.x. [DOI] [PubMed] [Google Scholar]
- 2.Tiffeneau R, Beauvallet M. Epreuve de bronchoconstriction et de bronchodilation par aerosols. Bull Acad Med. 1945;129:165–168. [Google Scholar]
- 3.de Vries, Tammeling GJ, Orie NG. [Hyperreactivity of the bronchi in bronchial asthma and chronic bronchitis] Ned Tijdschr Geneeskd. 1962;106:2295–2296. [PubMed] [Google Scholar]
- 4.Parker CD, Bilbo RE, Reed CE. METHACHOLINE AEROSOL AS TEST FOR BRONCHIAL ASTHMA. Arch Intern Med. 1965;115:452–458. doi: 10.1001/archinte.1965.03860160078013. [DOI] [PubMed] [Google Scholar]
- 5.Brannan JD, Lougheed MD. Airway hyperresponsiveness in asthma: mechanisms, clinical significance, and treatment. Front Physiol. 2012;3:460. doi: 10.3389/fphys.2012.00460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tepper RS, Wise RS, Covar R, Irvin CG, Kercsmar CM, Kraft M, Liu MC, O'Connor GT, Peters SP, Sorkness R, Togias A. Asthma outcomes: pulmonary physiology. J Allergy Clin Immunol. 2012;129(3 Suppl):S65–S87. doi: 10.1016/j.jaci.2011.12.986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davis BE, Cockcroft DW. Past, present and future uses of methacholine testing. Expert Rev Respir Med. 2012;6(3):321–329. doi: 10.1586/ers.12.29. [DOI] [PubMed] [Google Scholar]
- 8.Fowler SJ, Dempsey OJ, Sims EJ, Lipworth BJ. Screening for bronchial hyperresponsiveness using methacholine and adenosine monophosphate. Relationship to asthma severity and beta(2)-receptor genotype. Am J Respir Crit Care Med. 2000;162(4 Pt 1):1318–1322. doi: 10.1164/ajrccm.162.4.9912103. [DOI] [PubMed] [Google Scholar]
- 9.Sont JK, Willems LN, Bel EH, van Krieken JH, Vandenbroucke JP, Sterk PJ. Clinical control and histopathologic outcome of asthma when using airway hyperresponsiveness as an additional guide to long-term treatment. The AMPUL Study Group. Am J Respir Crit Care Med. 1999;159(4 Pt 1):1043–1051. doi: 10.1164/ajrccm.159.4.9806052. [DOI] [PubMed] [Google Scholar]
- 10.Woolcock AJ, Yan K, Salome CM, Sedgwick CJ, Peat JK. What determines the severity of asthma? Chest. 1985;87(suppl):209S–213S. [Google Scholar]
- 11.Barisione G, Crimi E, Bartolini S, Saporiti R, Copello F, Pellegrino R, Brusasco V. How to interpret reduced forced expiratory volume in 1 s (FEV1)/vital capacity ratio with normal FEV1. Eur Respir J. 2009;33(6):1396–1402. doi: 10.1183/09031936.00183708. [DOI] [PubMed] [Google Scholar]
- 12.Brutsche MH, Downs SH, Schindler C, Gerbase MW, Schwartz J, Frey M, Russi EW, Ackermann-Liebrich U, Leuenberger P. Bronchial hyperresponsiveness and the development of asthma and COPD in asymptomatic individuals: SAPALDIA cohort study. Thorax. 2006;61(8):671–677. doi: 10.1136/thx.2005.052241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parker DR, O'Connor GT, Sparrow D, Segal MR, Weiss ST. The relationship of nonspecific airway responsiveness and atopy to the rate of decline of lung function. The Normative Aging Study. Am Rev Respir Dis. 1990;141(3):589–594. doi: 10.1164/ajrccm/141.3.589. [DOI] [PubMed] [Google Scholar]
- 14.Sears MR, Greene JM, Willan AR, Wiecek EM, Taylor DR, Flannery EM, Cowan JO, Herbison GP, Silva PA, Poulton R. A longitudinal, population-based, cohort study of childhood asthma followed to adulthood. N Engl J Med. 2003;349(15):1414–1422. doi: 10.1056/NEJMoa022363. [DOI] [PubMed] [Google Scholar]
- 15.Leuppi JD, Salome CM, Jenkins CR, Anderson SD, Xuan W, Marks GB, Koskela H, Brannan JD, Freed R, Andersson M, Chan HK, Woolcock AJ. Predictive markers of asthma exacerbation during stepwise dose reduction of inhaled corticosteroids. Am J Respir Crit Care Med. 2001;163(2):406–412. doi: 10.1164/ajrccm.163.2.9912091. [DOI] [PubMed] [Google Scholar]
- 16.Yan K, Salome C, Woolcock AJ. Rapid method for measurement of bronchial responsiveness. Thorax. 1983;38(10):760–765. doi: 10.1136/thx.38.10.760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Macklem PT. The clinical relevance of respiratory muscle research. J. Burns Amberson lecture. Am Rev Respir Dis. 1986;134(4):812–815. doi: 10.1164/arrd.1986.134.4.812. [DOI] [PubMed] [Google Scholar]
- 18.Cockcroft DW, Ruffin RE, Dolovich J, Hargreave FE. Allergen-induced increase in non-allergic bronchial reactivity. Clin Allergy. 1977;7(6):503–513. doi: 10.1111/j.1365-2222.1977.tb01481.x. [DOI] [PubMed] [Google Scholar]
- 19.Cartier A, Thomson NC, Frith PA, Roberts R, Hargreave FE. Allergen-induced increase in bronchial responsiveness to histamine: relationship to the late asthmatic response and change in airway caliber. J Allergy Clin Immunol. 1982;70(3):170–177. doi: 10.1016/0091-6749(82)90038-0. [DOI] [PubMed] [Google Scholar]
- 20.Boulet LP, Cartier A, Thomson NC, Roberts RS, Dolovich J, Hargreave FE. Asthma and increases in nonallergic bronchial responsiveness from seasonal pollen exposure. J Allergy Clin Immunol. 1983;71(4):399–406. doi: 10.1016/0091-6749(83)90069-6. [DOI] [PubMed] [Google Scholar]
- 21.Reddel HK, Jenkins CR, Marks GB, Ware SI, Xuan W, Salome CM, Badcock CA, Woolcock AJ. Optimal asthma control, starting with high doses of inhaled budesonide. Eur Respir J. 2000;16(2):226–235. doi: 10.1034/j.1399-3003.2000.16b08.x. [DOI] [PubMed] [Google Scholar]
- 22.Sumino K, Sugar EA, Irvin CG, Kaminsky DA, Shade D, Wei CY, Holbrook JT, Wise RA, Castro M. Methacholine challenge test: diagnostic characteristics in asthmatic patients receiving controller medications. J Allergy Clin Immunol. 2012;130(1):69–75. doi: 10.1016/j.jaci.2012.02.025. e6. [DOI] [PubMed] [Google Scholar]
- 23.Chinn S, Schouten JP. Reproducibility of non-specific bronchial challenge in adults: implications for design, analysis and interpretation of clinical and epidemiological studies. Thorax. 2005;60(5):395–400. doi: 10.1136/thx.2004.039230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sumino K, Sugar EA, Irvin CG, Kaminsky DA, Shade D, Wei CY, Holbrook JT, Wise RA, Castro M. Variability of methacholine bronchoprovocation and the effect of inhaled corticosteroids in mild asthma. Ann Allergy Asthma Immunol. 2014;112(4):354–360. doi: 10.1016/j.anai.2014.01.013. e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dell SD, Bola SS, Foty RG, Marshall LC, Nelligan KA, Coates AL. PD20 Should be Used to Interpret Methacholine Challenge Tests with Modern Nebulizers. Annals of the American Thoracic Society. 2015 doi: 10.1513/AnnalsATS.201409-433OC. [DOI] [PubMed] [Google Scholar]
- 26.Porsbjerg C, Brannan JD, Anderson SD, Backer V. Relationship between airway responsiveness to mannitol and to methacholine and markers of airway inflammation, peak flow variability and quality of life in asthma patients. Clin Exp Allergy. 2008;38(1):43–50. doi: 10.1111/j.1365-2222.2007.02878.x. [DOI] [PubMed] [Google Scholar]
- 27.Sverrild A, Porsbjerg C, Thomsen SF, Backer V. Airway hyperresponsiveness to mannitol and methacholine and exhaled nitric oxide: a random-sample population study. J Allergy Clin Immunol. 2010;126(5):952–958. doi: 10.1016/j.jaci.2010.08.028. [DOI] [PubMed] [Google Scholar]
- 28.Lipworth BJ, Short PM, Williamson PA, Clearie KL, Fardon TC, Jackson CM. A randomized primary care trial of steroid titration against mannitol in persistent asthma: STAMINA trial. Chest. 2012;141(3):607–615. doi: 10.1378/chest.11-1748. [DOI] [PubMed] [Google Scholar]
- 29.Anderson SD, Charlton B, Weiler JM, Nichols S, Spector SL, Pearlman DS. Comparison of mannitol and methacholine to predict exercise-induced bronchoconstriction and a clinical diagnosis of asthma. Respir Res. 2009;10:4. doi: 10.1186/1465-9921-10-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim MH, Song WJ, Kim TW, Jin HJ, Sin YS, Ye YM, Kim SH, Park HW, Lee BJ, Park HS, Yoon HJ, Choi DC, Min KU, Cho SH. Diagnostic properties of the methacholine and mannitol bronchial challenge tests: a comparison study. Respirology. 2014;19(6):852–856. doi: 10.1111/resp.12334. [DOI] [PubMed] [Google Scholar]
- 31.Amakye DO, Davis BE, Martin AL, Peters GE, Cockcroft DW. Refractoriness to inhaled mannitol 3 hours after allergen challenge. Ann Allergy Asthma Immunol. 2013;111(3):182–184. doi: 10.1016/j.anai.2013.06.011. [DOI] [PubMed] [Google Scholar]
- 32.Manoharan A, Lipworth B, Craig E, Jackson C. The potential role of direct and indirect bronchial challenge testing to identify overtreatment of community managed asthma. Clin Exp Allergy. 2014 doi: 10.1111/cea.12352. [DOI] [PubMed] [Google Scholar]
- 33.Eliasson AH, Phillips YY, Rajagopal KR, Howard RS. Sensitivity and specificity of bronchial provocation testing. An evaluation of four techniques in exercise-induced bronchospasm. Chest. 1992;102(2):347–355. doi: 10.1378/chest.102.2.347. [DOI] [PubMed] [Google Scholar]
- 34.Broeders ME, Molema J, Hop WC, Folgering HT. Bronchial challenge, assessed with forced expiratory manoeuvres and airway impedance. Respir Med. 2005;99(8):1046–1052. doi: 10.1016/j.rmed.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 35.McClean MA, Htun C, King GG, Berend N, Salome CM. Cut-points for response to mannitol challenges using the forced oscillation technique. Respir Med. 2011;105(4):533–540. doi: 10.1016/j.rmed.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 36.Bohadana AB, Peslin R, Megherbi SE, Teculescu D, Sauleau EA, Wild P, Pham QT. Dose-response slope of forced oscillation and forced expiratory parameters in bronchial challenge testing. Eur Respir J. 1999;13(2):295–300. doi: 10.1034/j.1399-3003.1999.13b13.x. [DOI] [PubMed] [Google Scholar]
- 37.Chapman DG, King GG, Berend N, Diba C, Salome CM. Avoiding deep inspirations increases the maximal response to methacholine without altering sensitivity in non-asthmatics. Respir Physiol Neurobiol. 2010;173(2):157–163. doi: 10.1016/j.resp.2010.07.011. [DOI] [PubMed] [Google Scholar]
- 38.Cockcroft DW. Methacholine challenge methods. Chest. 2008;134(4):678–680. doi: 10.1378/chest.08-1306. [DOI] [PubMed] [Google Scholar]
- 39.Weersink EJ, vd Elshout FJ, van Herwaarden CV, Folgering H. Bronchial responsiveness to histamine and methacholine measured with forced expirations and with the forced oscillation technique. Respir Med. 1995;89(5):351–356. doi: 10.1016/0954-6111(95)90007-1. [DOI] [PubMed] [Google Scholar]
- 40.Coca FAC RA. On the Classification of the Phenomena of Hypersensitiveness. J Immunol. 1923;8:163–182. [Google Scholar]
- 41.Palmer LJ, Burton PR, James AL, Musk AW, Cookson WO. Familial aggregation and heritability of asthma-associated quantitative traits in a population-based sample of nuclear families. Eur J Hum Genet. 2000;8(11):853–860. doi: 10.1038/sj.ejhg.5200551. [DOI] [PubMed] [Google Scholar]
- 42.Levitt RC, Mitzner W. Expression of airway hyperreactivity to acetylcholine as a simple autosomal recessive trait in mice. FASEB J. 1988;2(10):2605–2608. doi: 10.1096/fasebj.2.10.3384240. [DOI] [PubMed] [Google Scholar]
- 43.Portelli MA, Hodge E, Sayers I. Genetic risk factors for the development of allergic disease identified by genome-wide association. Clin Exp Allergy. 2015;45(1):21–31. doi: 10.1111/cea.12327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wechsler ME, Kunselman SJ, Chinchilli VM, Bleecker E, Boushey HA, Calhoun WJ, Ameredes BT, Castro M, Craig TJ, Denlinger L, Fahy JV, Jarjour N, Kazani S, Kim S, Kraft M, Lazarus SC, Lemanske RF, Jr., Markezich A, Martin RJ, Permaul P, Peters SP, Ramsdell J, Sorkness CA, Sutherland ER, Szefler SJ, Walter MJ, Wasserman SI, Israel E. Effect of beta2-adrenergic receptor polymorphism on response to longacting beta2 agonist in asthma (LARGE trial): a genotype-stratified, randomised, placebo-controlled, crossover trial. Lancet. 2009;374(9703):1754–1764. doi: 10.1016/S0140-6736(09)61492-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheng RY, Shang Y, Limjunyawong N, Dao T, Das S, Rabold R, Sham JS, Mitzner W, Tang WY. Alterations of the lung methylome in allergic airway hyper-responsiveness. Environ Mol Mutagen. 2014;55(3):244–255. doi: 10.1002/em.21851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simpson JL, Scott R, Boyle MJ, Gibson PG. Inflammatory subtypes in asthma: assessment and identification using induced sputum. Respirology. 2006;11(1):54–61. doi: 10.1111/j.1440-1843.2006.00784.x. [DOI] [PubMed] [Google Scholar]
- 47.Kirby JG, Hargreave FE, Gleich GJ, O'Byrne PM. Bronchoalveolar cell profiles of asthmatic and nonasthmatic subjects. Am Rev Respir Dis. 1987;136(2):379–383. doi: 10.1164/ajrccm/136.2.379. [DOI] [PubMed] [Google Scholar]
- 48.Porsbjerg CM, Gibson PG, Pretto JJ, Salome CM, Brown NJ, Berend N, King GG. Relationship between airway pathophysiology and airway inflammation in older asthmatics. Respirology. 2013;18(7):1128–1134. doi: 10.1111/resp.12142. [DOI] [PubMed] [Google Scholar]
- 49.Brightling CE, Bradding P, Symon FA, Holgate ST, Wardlaw AJ, Pavord ID. Mast-cell infiltration of airway smooth muscle in asthma. N Engl J Med. 2002;346(22):1699–1705. doi: 10.1056/NEJMoa012705. [DOI] [PubMed] [Google Scholar]
- 50.Payne DN, Adcock IM, Wilson NM, Oates T, Scallan M, Bush A. Relationship between exhaled nitric oxide and mucosal eosinophilic inflammation in children with difficult asthma, after treatment with oral prednisolone. Am J Respir Crit Care Med. 2001;164(8 Pt 1):1376–1381. doi: 10.1164/ajrccm.164.8.2101145. [DOI] [PubMed] [Google Scholar]
- 51.Salome CM, Roberts AM, Brown NJ, Dermand J, Marks GB, Woolcock AJ. Exhaled nitric oxide measurements in a population sample of young adults. Am J Respir Crit Care Med. 1999;159(3):911–916. doi: 10.1164/ajrccm.159.3.9802108. [DOI] [PubMed] [Google Scholar]
- 52.Downie SR, Salome CM, Verbanck S, Thompson B, Berend N, King GG. Ventilation heterogeneity is a major determinant of airway hyperresponsiveness in asthma, independent of airway inflammation. Thorax. 2007;62(8):684–689. doi: 10.1136/thx.2006.069682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chapman DG, Berend N, King GG, Salome CM. Increased airway closure is a determinant of airway hyperresponsiveness. Eur Respir J. 2008;32(6):1563–1569. doi: 10.1183/09031936.00114007. [DOI] [PubMed] [Google Scholar]
- 54.Brusasco V, Crimi E, Gianiorio P, Lantero S, Rossi GA. Allergen-induced increase in airway responsiveness and inflammation in mild asthma. J Appl Physiol (1985) 1990;69(6):2209–2214. doi: 10.1152/jappl.1990.69.6.2209. [DOI] [PubMed] [Google Scholar]
- 55.Yick CY, Zwinderman AH, Kunst PW, Grunberg K, Mauad T, Chowdhury S, Bel EH, Baas F, Lutter R, Sterk PJ. Gene expression profiling of laser microdissected airway smooth muscle tissue in asthma and atopy. Allergy. 2014;69(9):1233–1240. doi: 10.1111/all.12452. [DOI] [PubMed] [Google Scholar]
- 56.Slats AM, Janssen K, van Schadewijk A, van der Plas DT, Schot R, van den Aardweg JG, de Jongste JC, Hiemstra PS, Mauad T, Rabe KF, Sterk PJ. Expression of smooth muscle and extracellular matrix proteins in relation to airway function in asthma. J Allergy Clin Immunol. 2008;121(5):1196–1202. doi: 10.1016/j.jaci.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 57.Bai TR. Abnormalities in airway smooth muscle in fatal asthma. A comparison between trachea and bronchus. Am Rev Respir Dis. 1991;143(2):441–443. doi: 10.1164/ajrccm/143.2.441. [DOI] [PubMed] [Google Scholar]
- 58.Bjorck T, Gustafsson LE, Dahlen SE. Isolated bronchi from asthmatics are hyperresponsive to adenosine, which apparently acts indirectly by liberation of leukotrienes and histamine. Am Rev Respir Dis. 1992;145(5):1087–1091. doi: 10.1164/ajrccm/145.5.1087. [DOI] [PubMed] [Google Scholar]
- 59.Goldie RG, Spina D, Henry PJ, Lulich KM, Paterson JW. In vitro responsiveness of human asthmatic bronchus to carbachol, histamine, beta-adrenoceptor agonists and theophylline. Br J Clin Pharmacol. 1986;22(6):669–676. doi: 10.1111/j.1365-2125.1986.tb02956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Whicker SD, Armour CL, Black JL. Responsiveness of bronchial smooth muscle from asthmatic patients to relaxant and contractile agonists. Pulm Pharmacol. 1988;1(1):25–31. doi: 10.1016/0952-0600(88)90007-5. [DOI] [PubMed] [Google Scholar]
- 61.Chin LY, Bosse Y, Pascoe C, Hackett TL, Seow CY, Pare PD. Mechanical properties of asthmatic airway smooth muscle. Eur Respir J. 2012;40(1):45–54. doi: 10.1183/09031936.00065411. [DOI] [PubMed] [Google Scholar]
- 62.Solway J, Fredberg JJ. Perhaps airway smooth muscle dysfunction contributes to asthmatic bronchial hyperresponsiveness after all. Am J Respir Cell Mol Biol. 1997;17(2):144–146. doi: 10.1165/ajrcmb.17.2.f137. [DOI] [PubMed] [Google Scholar]
- 63.Bullimore SR, Siddiqui S, Donovan GM, Martin JG, Sneyd J, Bates JH, Lauzon AM. Could an increase in airway smooth muscle shortening velocity cause airway hyperresponsiveness? Am J Physiol Lung Cell Mol Physiol. 2011;300(1):L121–L131. doi: 10.1152/ajplung.00228.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ma X, Cheng Z, Kong H, Wang Y, Unruh H, Stephens NL, Laviolette M. Changes in biophysical and biochemical properties of single bronchial smooth muscle cells from asthmatic subjects. Am J Physiol Lung Cell Mol Physiol. 2002;283(6):L1181–L1191. doi: 10.1152/ajplung.00389.2001. [DOI] [PubMed] [Google Scholar]
- 65.Leguillette R, Laviolette M, Bergeron C, Zitouni N, Kogut P, Solway J, Kachmar L, Hamid Q, Lauzon AM. Myosin, transgelin, and myosin light chain kinase: expression and function in asthma. Am J Respir Crit Care Med. 2009;179(3):194–204. doi: 10.1164/rccm.200609-1367OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sutcliffe A, Hollins F, Gomez E, Saunders R, Doe C, Cooke M, Challiss RA, Brightling CE. Increased nicotinamide adenine dinucleotide phosphate oxidase 4 expression mediates intrinsic airway smooth muscle hypercontractility in asthma. Am J Respir Crit Care Med. 2012;185(3):267–274. doi: 10.1164/rccm.201107-1281OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Syyong HT, Pascoe CD, Zhang J, Arsenault BA, Solomon D, Elliott WM, Hackett TL, Walker DC, Pare PD, Seow CY. Ultrastructure of Human Tracheal Smooth Muscle from Asthmatic and Non-asthmatic Subjects: Standardized Methods for Comparison. Am J Respir Cell Mol Biol. 2014 doi: 10.1165/rcmb.2014-0176OC. [DOI] [PubMed] [Google Scholar]
- 68.Sieck GC, White TA, Thompson MA, Pabelick CM, Wylam ME, Prakash YS. Regulation of store-operated Ca2+ entry by CD38 in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2008;294(2):L378–L385. doi: 10.1152/ajplung.00394.2007. [DOI] [PubMed] [Google Scholar]
- 69.Rogers NK, Clements D, Dongre A, Harrison TW, Shaw D, Johnson SR. Extra-cellular matrix proteins induce matrix metalloproteinase-1 (MMP-1) activity and increase airway smooth muscle contraction in asthma. PLoS One. 2014;9(2):e90565. doi: 10.1371/journal.pone.0090565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Siddiqui S, Mistry V, Doe C, Roach K, Morgan A, Wardlaw A, Pavord I, Bradding P, Brightling C. Airway hyperresponsiveness is dissociated from airway wall structural remodeling. J Allergy Clin Immunol. 2008;122(2):335–341. doi: 10.1016/j.jaci.2008.05.020. 41 e1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hanna CJ, Bach MK, Pare PD, Schellenberg RR. Slow-reacting substances (leukotrienes) contract human airway and pulmonary vascular smooth muscle in vitro. Nature. 1981;290(5804):343–344. doi: 10.1038/290343a0. [DOI] [PubMed] [Google Scholar]
- 72.Coleman RA, Sheldrick RL. Prostanoid-induced contraction of human bronchial smooth muscle is mediated by TP-receptors. Br J Pharmacol. 1989;96(3):688–692. doi: 10.1111/j.1476-5381.1989.tb11869.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bosse Y, Chin LY, Pare PD, Seow CY. Adaptation of airway smooth muscle to basal tone: relevance to airway hyperresponsiveness. Am J Respir Cell Mol Biol. 2009;40(1):13–18. doi: 10.1165/rcmb.2008-0150OC. [DOI] [PubMed] [Google Scholar]
- 74.Bosse Y, Chapman DG, Pare PD, King GG, Salome CM. A 'Good' muscle in a 'Bad' environment: the importance of airway smooth muscle force adaptation to airway hyperresponsiveness. Respir Physiol Neurobiol. 2011;179(2–3):269–275. doi: 10.1016/j.resp.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 75.Lee-Gosselin A, Gendron D, Blanchet M-R, Marsolais D, Bossé Y. The gain of smooth muscle's contractile capacity induced by tone on in vivo airway responsiveness in mice. 2015;20(40):07. doi: 10.1152/japplphysiol.00645.2014. 2015-01-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wright DB, Trian T, Siddiqui S, Pascoe CD, Johnson JR, Dekkers BG, Dakshinamurti S, Bagchi R, Burgess JK, Kanabar V, Ojo OO. Phenotype modulation of airway smooth muscle in asthma. Pulm Pharmacol Ther. 2013;26(1):42–49. doi: 10.1016/j.pupt.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 77.Ammit AJ, Hoffman RK, Amrani Y, Lazaar AL, Hay DW, Torphy TJ, Penn RB, Panettieri RA., Jr. Tumor necrosis factor-alpha-induced secretion of RANTES and interleukin-6 from human airway smoothmuscle cells. Modulation by cyclic adenosine monophosphate. Am J Respir Cell Mol Biol. 2000;23(6):794–802. doi: 10.1165/ajrcmb.23.6.4184. [DOI] [PubMed] [Google Scholar]
- 78.Elias JA, Wu Y, Zheng T, Panettieri R. Cytokine- and virus-stimulated airway smooth muscle cells produce IL-11 and other IL-6-type cytokines. Am J Physiol. 1997;273(3 Pt 1):L648–L655. doi: 10.1152/ajplung.1997.273.3.L648. [DOI] [PubMed] [Google Scholar]
- 79.John AE, Zhu YM, Brightling CE, Pang L, Knox AJ. Human airway smooth muscle cells from asthmatic individuals have CXCL8 hypersecretion due to increased NF-kappa B p65, C/EBP beta, and RNA polymerase II binding to the CXCL8 promoter. J Immunol. 2009;183(7):4682–4692. doi: 10.4049/jimmunol.0803832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Perry MM, Hui CK, Whiteman M, Wood ME, Adcock I, Kirkham P, Michaeloudes C, Chung KF. Hydrogen sulfide inhibits proliferation and release of IL-8 from human airway smooth muscle cells. Am J Respir Cell Mol Biol. 2011;45(4):746–752. doi: 10.1165/rcmb.2010-0304OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Halayko AJ, Salari H, Ma X, Stephens NL. Markers of airway smooth muscle cell phenotype. Am J Physiol. 1996;270(6 Pt 1):L1040–L1051. doi: 10.1152/ajplung.1996.270.6.L1040. [DOI] [PubMed] [Google Scholar]
- 82.Vanhoutte PM. Epithelium-derived relaxing factor(s) and bronchial reactivity. J Allergy Clin Immunol. 1989;83(5):855–861. doi: 10.1016/0091-6749(89)90095-x. [DOI] [PubMed] [Google Scholar]
- 83.Coyle AJ, Uchida D, Ackerman SJ, Mitzner W, Irvin CG. Role of cationic proteins in the airway. Hyperresponsiveness due to airway inflammation. Am J Respir Crit Care Med. 1994;150(5 Pt 2):S63–S71. doi: 10.1164/ajrccm/150.5_Pt_2.S63. [DOI] [PubMed] [Google Scholar]
- 84.Zhou J, Alvarez-Elizondo MB, Botvinick E, George SC. Local small airway epithelial injury induces global smooth muscle contraction and airway constriction. J Appl Physiol (1985) 2012;112(4):627–637. doi: 10.1152/japplphysiol.00739.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wilson JW, Li X. The measurement of reticular basement membrane and submucosal collagen in the asthmatic airway. Clin Exp Allergy. 1997;27(4):363–371. [PubMed] [Google Scholar]
- 86.James AL, Elliot JG, Jones RL, Carroll ML, Mauad T, Bai TR, Abramson MJ, McKay KO, Green FH. Airway smooth muscle hypertrophy and hyperplasia in asthma. Am J Respir Crit Care Med. 2012;185(10):1058–1064. doi: 10.1164/rccm.201110-1849OC. [DOI] [PubMed] [Google Scholar]
- 87.Ribatti D, Puxeddu I, Crivellato E, Nico B, Vacca A, Levi-Schaffer F. Angiogenesis in asthma. Clin Exp Allergy. 2009;39(12):1815–1821. doi: 10.1111/j.1365-2222.2009.03385.x. [DOI] [PubMed] [Google Scholar]
- 88.Burgess JK. The role of the extracellular matrix and specific growth factors in the regulation of inflammation and remodelling in asthma. Pharmacol Ther. 2009;122(1):19–29. doi: 10.1016/j.pharmthera.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 89.Niimi A, Matsumoto H, Takemura M, Ueda T, Chin K, Mishima M. Relationship of airway wall thickness to airway sensitivity and airway reactivity in asthma. Am J Respir Crit Care Med. 2003;168(8):983–988. doi: 10.1164/rccm.200211-1268OC. [DOI] [PubMed] [Google Scholar]
- 90.Boulet L, Belanger M, Carrier G. Airway responsiveness and bronchial-wall thickness in asthma with or without fixed airflow obstruction. Am J Respir Crit Care Med. 1995;152(3):865–871. doi: 10.1164/ajrccm.152.3.7663797. [DOI] [PubMed] [Google Scholar]
- 91.Ward C, Pais M, Bish R, Reid D, Feltis B, Johns D, Walters EH. Airway inflammation, basement membrane thickening and bronchial hyperresponsiveness in asthma. Thorax. 2002;57(4):309–316. doi: 10.1136/thorax.57.4.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yick CY, Ferreira DS, Annoni R, von der Thusen JH, Kunst PW, Bel EH, Lutter R, Mauad T, Sterk PJ. Extracellular matrix in airway smooth muscle is associated with dynamics of airway function in asthma. Allergy. 2012;67(4):552–559. doi: 10.1111/j.1398-9995.2011.02773.x. [DOI] [PubMed] [Google Scholar]
- 93.Britton J, Pavord I, Richards K, Knox A, Wisniewski A, Wahedna I, Kinnear W, Tattersfield A, Weiss S. Factors influencing the occurrence of airway hyperreactivity in the general population: the importance of atopy and airway calibre. Eur Respir J. 1994;7(5):881–887. [PubMed] [Google Scholar]
- 94.Litonjua AA, Sparrow D, Weiss ST. The FEF25-75/FVC ratio is associated with methacholine airway responsiveness. The normative aging study. Am J Respir Crit Care Med. 1999;159(5 Pt 1):1574–1579. doi: 10.1164/ajrccm.159.5.9803063. [DOI] [PubMed] [Google Scholar]
- 95.Parker AL, Abu-Hijleh M, McCool FD. Ratio between forced expiratory flow between 25% and 75% of vital capacity and FVC is a determinant of airway reactivity and sensitivity to methacholine. Chest. 2003;124(1):63–69. doi: 10.1378/chest.124.1.63. [DOI] [PubMed] [Google Scholar]
- 96.Salome CM, Schoeffel RE, Yan K, Woolcock AJ. Effect of aerosol fenoterol on the severity of bronchial hyperreactivity in patients with asthma. Thorax. 1983;38(11):854–858. doi: 10.1136/thx.38.11.854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Britton J, Hanley SP, Garrett HV, Hadfield JW, Tattersfield AE. Dose related effects of salbutamol and ipratropium bromide on airway calibre and reactivity in subjects with asthma. Thorax. 1988;43(4):300–305. doi: 10.1136/thx.43.4.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lambert RK, Wiggs BR, Kuwano K, Hogg JC, Pare PD, Lambert RK, Wiggs BR, Kuwano K, Hogg JC, Pare PD. Functional significance of increased airway smooth muscle in asthma and COPD. Journal of Applied Physiology. 1993;74(6):2771–2781. doi: 10.1152/jappl.1993.74.6.2771. [DOI] [PubMed] [Google Scholar]
- 99.Noble PB, Jones RL, Cairncross A, Elliot JG, Mitchell HW, James AL, McFawn PK. Airway narrowing and bronchodilation to deep inspiration in bronchial segments from subjects with and without reported asthma. J Appl Physiol (1985) 2013;114(10):1460–1471. doi: 10.1152/japplphysiol.01489.2012. [DOI] [PubMed] [Google Scholar]
- 100.McParland BE, Macklem PT, Pare PD. Airway wall remodeling: friend or foe? J Appl Physiol (1985) 2003;95(1):426–434. doi: 10.1152/japplphysiol.00159.2003. [DOI] [PubMed] [Google Scholar]
- 101.Grainge CL, Lau LC, Ward JA, Dulay V, Lahiff G, Wilson S, Holgate S, Davies DE, Howarth PH. Effect of bronchoconstriction on airway remodeling in asthma. N Engl J Med. 2011;364(21):2006–2015. doi: 10.1056/NEJMoa1014350. [DOI] [PubMed] [Google Scholar]
- 102.Bates JH, Irvin CG. Time dependence of recruitment and derecruitment in the lung: a theoretical model. J Appl Physiol. 2002;93(2):705–713. doi: 10.1152/japplphysiol.01274.2001. [DOI] [PubMed] [Google Scholar]
- 103.Lee P, Abisheganaden J, Chee CB, Wang YT. A new asthma severity index: a predictor of near-fatal asthma? Eur Respir J. 2001;18(2):272–278. doi: 10.1183/09031936.01.00074401. [DOI] [PubMed] [Google Scholar]
- 104.Gibbons WJ, Sharma A, Lougheed D, Macklem PT. Detection of excessive bronchoconstriction in asthma. Am J Respir Crit Care Med. 1996;153(2):582–589. doi: 10.1164/ajrccm.153.2.8564102. [DOI] [PubMed] [Google Scholar]
- 105.Irvin CG, Bates JH. Physiologic dysfunction of the asthmatic lung: what's going on down there, anyway? Proc Am Thorac Soc. 2009;6(3):306–311. doi: 10.1513/pats.200808-091RM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lundblad LK, Thompson-Figueroa J, Allen GB, Rinaldi L, Norton RJ, Irvin CG, Bates JH. Airway hyperresponsiveness in allergically inflamed mice: the role of airway closure. Am J Respir Crit Care Med. 2007;175(8):768–774. doi: 10.1164/rccm.200610-1410OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wagers S, Lundblad LK, Ekman M, Irvin CG, Bates JH. The allergic mouse model of asthma: normal smooth muscle in an abnormal lung? J Appl Physiol. 2004;96(6):2019–2027. doi: 10.1152/japplphysiol.00924.2003. [DOI] [PubMed] [Google Scholar]
- 108.Agrawal A, Rengarajan S, Adler KB, Ram A, Ghosh B, Fahim M, Dickey BF. Inhibition of mucin secretion with MARCKS-related peptide improves airway obstruction in a mouse model of asthma. J Appl Physiol (1985) 2007;102(1):399–405. doi: 10.1152/japplphysiol.00630.2006. [DOI] [PubMed] [Google Scholar]
- 109.Hohlfeld JM, Schmiedl A, Erpenbeck VJ, Venge P, Krug N. Eosinophil cationic protein alters pulmonary surfactant structure and function in asthma. J Allergy Clin Immunol. 2004;113(3):496–502. doi: 10.1016/j.jaci.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 110.Jarjour NN, Enhorning G. Antigen-induced airway inflammation in atopic subjects generates dysfunction of pulmonary surfactant. Am J Respir Crit Care Med. 1999;160(1):336–341. doi: 10.1164/ajrccm.160.1.9806155. [DOI] [PubMed] [Google Scholar]
- 111.Seeger W, Stohr G, Wolf HR, Neuhof H. Alteration of surfactant function due to protein leakage: special interaction with fibrin monomer. J Appl Physiol (1985) 1985;58(2):326–338. doi: 10.1152/jappl.1985.58.2.326. [DOI] [PubMed] [Google Scholar]
- 112.Babu KS, Woodcock DA, Smith SE, Staniforth JN, Holgate ST, Conway JH. Inhaled synthetic surfactant abolishes the early allergen-induced response in asthma. Eur Respir J. 2003;21(6):1046–1049. doi: 10.1183/09031936.03.00069202. [DOI] [PubMed] [Google Scholar]
- 113.Venegas JG, Winkler T, Musch G, Vidal Melo MF, Layfield D, Tgavalekos N, Fischman AJ, Callahan RJ, Bellani G, Harris RS. Self-organized patchiness in asthma as a prelude to catastrophic shifts. Nature. 2005;434(7034):777–782. doi: 10.1038/nature03490. [DOI] [PubMed] [Google Scholar]
- 114.Hardaker KM, Downie SR, Kermode JA, Farah CS, Brown NJ, Berend N, King GG, Salome CM. Predictors of airway hyperresponsiveness differ between old and young patients with asthma. Chest. 2011;139(6):1395–1401. doi: 10.1378/chest.10-1839. [DOI] [PubMed] [Google Scholar]
- 115.Kaminsky DA, Daud A, Chapman DG. Relationship between the baseline alveolar volume-to-total lung capacity ratio and airway responsiveness. Respirology. 2014;19(7):1046–1051. doi: 10.1111/resp.12347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Farrow CE, Salome CM, Harris BE, Bailey DL, Bailey E, Berend N, Young IH, King GG. Airway closure on imaging relates to airway hyperresponsiveness and peripheral airway disease in asthma. J Appl Physiol (1985) 2012;113(6):958–966. doi: 10.1152/japplphysiol.01618.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Haldar P, Pavord ID, Shaw DE, Berry MA, Thomas M, Brightling CE, Wardlaw AJ, Green RH. Cluster analysis and clinical asthma phenotypes. Am J Respir Crit Care Med. 2008;178(3):218–224. doi: 10.1164/rccm.200711-1754OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Moore WC, Meyers DA, Wenzel SE, Teague WG, Li H, Li X, D'Agostino R, Jr., Castro M, Curran-Everett D, Fitzpatrick AM, Gaston B, Jarjour NN, Sorkness R, Calhoun WJ, Chung KF, Comhair SA, Dweik RA, Israel E, Peters SP, Busse WW, Erzurum SC, Bleecker ER. Identification of asthma phenotypes using cluster analysis in the Severe Asthma Research Program. Am J Respir Crit Care Med. 2010;181(4):315–323. doi: 10.1164/rccm.200906-0896OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Molimard M, Le Gros V. Impact of patient-related factors on asthma control. J Asthma. 2008;45(2):109–113. doi: 10.1080/02770900701815727. [DOI] [PubMed] [Google Scholar]
- 120.Milanese M, Di Marco F, Corsico AG, Rolla G, Sposato B, Chieco-Bianchi F, Costantino MT, Crivellaro MA, Guarnieri G, Scichilone N. Asthma control in elderly asthmatics. An Italian observational study. Respir Med. 2014;108(8):1091–1099. doi: 10.1016/j.rmed.2014.05.016. [DOI] [PubMed] [Google Scholar]
- 121.Cuttitta G, Cibella F, Bellia V, Grassi V, Cossi S, Bucchieri S, Bonsignore G. Changes in FVC during methacholine-induced bronchoconstriction in elderly patients with asthma: bronchial hyperresponsiveness and aging. Chest. 2001;119(6):1685–1690. doi: 10.1378/chest.119.6.1685. [DOI] [PubMed] [Google Scholar]
- 122.Knudson RJ, Clark DF, Kennedy TC, Knudson DE. Effect of aging alone on mechanical properties of the normal adult human lung. J Appl Physiol Respir Environ Exerc Physiol. 1977;43(6):1054–1062. doi: 10.1152/jappl.1977.43.6.1054. [DOI] [PubMed] [Google Scholar]
- 123.Michils A, Elkrim Y, Haccuria A, Van Muylem A. Adenosine 5'-monophosphate challenge elicits a more peripheral airway response than methacholine challenge. J Appl Physiol (1985) 2011;110(5):1241–1247. doi: 10.1152/japplphysiol.01401.2010. [DOI] [PubMed] [Google Scholar]
- 124.Ford ES. The epidemiology of obesity and asthma. J Allergy Clin Immunol. 2005;115(5):897–909. doi: 10.1016/j.jaci.2004.11.050. quiz 10. [DOI] [PubMed] [Google Scholar]
- 125.Beuther DA, Sutherland ER. Overweight, obesity, and incident asthma: a meta-analysis of prospective epidemiologic studies. Am J Respir Crit Care Med. 2007;175(7):661–666. doi: 10.1164/rccm.200611-1717OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Peters-Golden M, Swern A, Bird SS, Hustad CM, Grant E, Edelman JM. Influence of body mass index on the response to asthma controller agents. Eur Respir J. 2006;27(3):495–503. doi: 10.1183/09031936.06.00077205. [DOI] [PubMed] [Google Scholar]
- 127.Stenius-Aarniala B, Poussa T, Kvarnstrom J, Gronlund EL, Ylikahri M, Mustajoki P. Immediate and long term effects of weight reduction in obese people with asthma: randomised controlled study. Bmj. 2000;320(7238):827–832. doi: 10.1136/bmj.320.7238.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dixon AE, Pratley RE, Forgione PM, Kaminsky DA, Whittaker-Leclair LA, Griffes LA, Garudathri J, Raymond D, Poynter ME, Bunn JY, Irvin CG. Effects of obesity and bariatric surgery on airway hyperresponsiveness, asthma control, and inflammation. J Allergy Clin Immunol. 2011;128(3):508–515. doi: 10.1016/j.jaci.2011.06.009. e1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Al-Alwan A, Bates JH, Chapman DG, Kaminsky DA, DeSarno MJ, Irvin CG, Dixon AE. The nonallergic asthma of obesity. A matter of distal lung compliance. Am J Respir Crit Care Med. 2014;189(12):1494–1502. doi: 10.1164/rccm.201401-0178OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chapman DG, Irvin CG, Kaminsky DA, Forgione PM, Bates JH, Dixon AE. Influence of distinct asthma phenotypes on lung function following weight loss in the obese. Respirology. 2014 doi: 10.1111/resp.12368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Suh DI, Choi SH, Lee JK, Kim JT, Koh YY. Relationships of methacholine and adenosine 5'-monophosphate (AMP) responsiveness to the postbronchodilator FEV(1)/FVC ratio in children with asthma. J Asthma. 2011;48(4):345–350. doi: 10.3109/02770903.2011.563809. [DOI] [PubMed] [Google Scholar]
- 132.De Meer G, Heederik D, Postma DS. Bronchial responsiveness to adenosine 5'-monophosphate (AMP) and methacholine differ in their relationship with airway allergy and baseline FEV(1) Am J Respir Crit Care Med. 2002;165(3):327–331. doi: 10.1164/ajrccm.165.3.2104066. [DOI] [PubMed] [Google Scholar]
- 133.Thomson NC, Chaudhuri R, Heaney LG, Bucknall C, Niven RM, Brightling CE, Menzies-Gow AN, Mansur AH, McSharry C. Clinical outcomes and inflammatory biomarkers in current smokers and exsmokers with severe asthma. J Allergy Clin Immunol. 2013;131(4):1008–1016. doi: 10.1016/j.jaci.2012.12.1574. [DOI] [PubMed] [Google Scholar]
- 134.Juusela M, Pallasaho P, Ronmark E, Sarna S, Sovijarvi A, Lundback B. Dose-dependent association of smoking and bronchial hyperresponsiveness. Eur Respir J. 2013;42(6):1503–1512. doi: 10.1183/09031936.00073712. [DOI] [PubMed] [Google Scholar]
- 135.Prieto L, Palop J, Llusar R, Herrera S, Perez-Frances C, Lanuza A, Aguilar D. Effects of cigarette smoke on methacholine- and AMP-induced air trapping in asthmatics. J Asthma. 2014:1–8. doi: 10.3109/02770903.2014.944981. [DOI] [PubMed] [Google Scholar]
- 136.Piccillo G, Caponnetto P, Barton S, Russo C, Origlio A, Bonaccorsi A, Di Maria A, Oliveri C, Polosa R. Changes in airway hyperresponsiveness following smoking cessation: comparisons between Mch and AMP. Respir Med. 2008;102(2):256–265. doi: 10.1016/j.rmed.2007.09.004. [DOI] [PubMed] [Google Scholar]