

Abstract
Among the many neuromodulators used by the mammalian brain to regulate circuit function and plasticity, dopamine (DA) stands out as one of the most behaviorally powerful. For example, release of DA within nucleus accumbens signals reward and rapidly modifies brain function to drive repetition of motor action in search of further reward. The control that DA exerts over behavior is also clear in humans, as mimicry of dopaminergic reward signals underlies addiction to drugs of abuse, whereas death of midbrain dopaminergic neurons causes Parkinson’s disease (PD). In addition, perturbations of DA are implicated in the pathogenesis, or exploited in the treatment of many neuropsychiatric diseases including schizophrenia, obsessive compulsive disorder, and Tourette’s syndrome. Although the precise mechanisms employed by DA to exert its control over behavior are not fully understood, DA is known to regulate many electrical and biochemical aspects of neuronal function including excitability, synaptic transmission, integration and plasticity, protein trafficking and gene transcription. In this review, we discuss the actions of DA on ionic and synaptic signaling in neurons of the prefrontal cortex and striatum, brain areas in which dopaminergic dysfunction is thought to be central to the above-mentioned diseases. We focus on actions of DA on the pre- and postsynaptic terminals and restrict our discussion to studies in which the site of action or the molecular target of DA is clearly identified.
Introduction
DA is a catecholamine (CA) that was initially identified as the metabolic precursor of the neurotransmitter norepinephrine (NE). Pioneering studies by Arvid Carlsson in the late 1950s first lent support to the idea that DA does not merely serve as an intermediate for NE biosynthesis, but rather functions as a transmitter in the mammalian central nervous system (CNS) in its own right. Specifically, he noted that the striatum – a large subcortical forebrain structure known at the time to form part of the extrapyramidal system that controls motor activity – contains high levels of DA and very little NE, and that systemic administration of the DA precursor 3,4-dihydroxyphenylalanine restored motor function and striatal DA levels in rabbits whose monoamines had been pharmacologically depleted with the vesicular monoamine transporter blocker reserpine (Carlsson et al., 1957; 1958; Carlsson, 1959). Importantly, the experimental akinesia produced by reserpine resembled parkinsonian motor deficits, and the subsequent replication of Carlsson’s observations by clinicians in the early 1960s paved the way for the utilization to this day of (S) 3,4-dihydroxyphenylalanine – better known as L-DOPA – as a major component of standard pharmacological treatments for PD.
These discoveries naturally prompted the search for neurons that produce DA. This quest started in the early 1960s, following the remarkable finding that CA-containing neurons could be visualized in tissue following chemical conversion of CAs into fluorescent molecules with formaldehyde (Carlsson et al., 1962; Falck et al., 1962). Using this method, seventeen groups of CA cells (designated A1–A17) were identified in the CNS. The connectivity of these cell groups was established in the early 1970s with the introduction of axonal tracing techniques and the use of stereotaxic lesions. The development of immunohistochemical techniques in the 1980s to label CAs and CA-synthesizing enzymes offered the possibility to distinguish more accurately different transmitters. DA is produced via hydroxylation and decarboxylation of tyrosine by tyrosine hydroxylase (TH) and aromatic amino acid decarboxylase (AADC), respectively, and subsequent decarboxylation of DA by dopamine-β-hydroxylase (DBH) yields NE. TH and AADC are therefore expressed by both NE and DA-producing cells and cannot be used as the sole markers to identify dopaminergic centers. Similarly, packaging into synaptic vesicles and degradation of CAs and other monoamines, such as serotonin, rely on a common set of enzymes: vesicular monoamine transporters are responsible for packaging all monoaminergic transmitters into synaptic vesicles, whereas monoamine oxidases catalyze the breakdown of many neurotransmitters. Thus, the specific identification of DA-producing cells is complex even with modern techniques. The absence of DBH and the presence of enzymes that act specifically on DA, such as the plasma membrane DA transporter, which is exclusively expressed in the presynaptic terminals of DA-releasing terminals (Ciliax et al., 1995) have proven particularly useful in helping determine the dopaminergic phenotype of CA cell groups.
Collectively, these approaches have largely confirmed the existence of ten DA-producing nuclei in the mammalian brain (A8–A17). Midbrain DA neurons in the substantia nigra pars compacta (SNc; field A9) and ventral tegmental area (VTA; field A10) are perhaps the best studied of these because of their central roles, respectively, in the pathology of PD and in reward signaling and reinforcement. These two centers provide the bulk of DA to the basal ganglia and forebrain and contain the vast majority of DA neurons in the CNS. In the rat, VTA and SNc each contain ~20,000 neurons bilaterally, approximately 10 fold more than the smaller retrorubral A8 nucleus (German and Manaye, 1993). Given their small numbers and powerful impact on many aspects of behavior, each midbrain DA neuron must exert influence over large brain areas and many cells. Indeed, individual SNc/VTA neurons extend impressive axons of half a meter in total length that densely ramify throughout up to 1 mm3 of tissue (Matsuda et al., 2009). Furthermore, midbrain DA neurons are spontaneously active at low frequencies, suggesting that each neuron provides a basal DA tone to many target neurons that is rapidly adjusted by either phasic bursts or transients pauses of activity. Additional and yet smaller groups of putative DA neurons are found in the diencephalic A11–A15 fields (~60–400 cells per nucleus) (Qu et al., 2006; Skagerberg and Lindvall, 1985) as well as dispersed within the glomerular layer of the olfactory blub (field A16) and amacrine cell population of the retina (field A17). Interestingly, it has become increasingly evident in recent years that neurons within each field can differ significantly with respect to axonal projection areas, electrophysiological properties and the expression of synthetic enzymes, membrane and vesicular transporters, neuropeptides and other amino-acid transmitters (Bjorklund and Dunnett, 2007; Hnasko et al., 2010; Lammel et al., 2011). Therefore, firmly establishing a dopaminergic identify necessitates the analysis of multiple cellular markers and ideally the demonstration of stimulus-evoked DA release from genetically-defined neurons such as by combining optogenetics and carbon fiber voltammetry (e.g. Stuber et al., 2010; Tecuapetla et al., 2010).
Since the identification of DA as a neurotransmitter over 50 years ago, neuroscientists have sought to elucidate the influence that DA exerts on behavior and neural circuits, and to uncover the underlying cellular and molecular underpinnings of such effects. Interest in the actions of this molecule is further stimulated by the recognition of its involvement in several neurological and psychiatric disorders, including PD, addiction, schizophrenia, obsessive compulsive disorder and Tourette’s syndrome. DA plays an important role in the control of fine motor actions and higher cognitive functions such as learning, working memory, attention, decision-making and appetitive and consummatory aspects of reward. However, the precise mechanisms employed by DA to mediate these effects remain largely unknown owing to the multiplicity and complexity of its actions. Unlike classical amino-acid neurotransmitters such as glutamate and γ-aminobutyric acid (GABA), which signal for the most part by directly gating ionotropic receptors to rapidly and briefly alter membrane potential, DA activates G protein-coupled receptors (GPCRs) that modulate the biochemical and electrical state of a cell via a plethora of signaling elements including kinases, phosphatases, transcription factors, ion channels and membrane receptors. Moreover, DA’s actions have largely defied interpretation because they vary greatly between cell types, depend on the strength and duration of receptor stimulation, are influenced by current and past cellular states, and compete with other neuromodulatory systems impinging on similar pathways. Thus, despite extensive investigation, there is no unified view of DA’s actions in the CNS, and many studies have yielded contradictory conclusions.
One of the principal actions of DA in the CNS is to modulate electrical communication between neurons. DA clearly promotes persistent changes in the efficacy of synapses and in the structural organization of neural circuits, and influences the moment-to-moment flow of electrical signals across synapses. Here we review the latter, namely DA’s ability to rapidly influence synaptic transmission, dendritic integration and membrane excitability. Many recent reviews cover the longer term effects of DA signaling on synaptic and circuit plasticity from the perspective of normal brain function, PD and drug addiction – these topics are therefore not covered here.
Some of the first electrophysiological investigations of DA’s influence in the 1970s and 1980s utilized in vivo and in vitro extracellular and intracellular recordings and examined the effects of electrical stimulation of DA centers or local ionotophoretic/pressure application of exogenous DA. These studies invariably reported complex, variable and often contradictory findings (see Nicola et al., 2000; Seamans and Yang, 2004 for review). Some of these disparities likely arose because, as discussed below, DA activates multiple classes of receptors that are heterogeneously distributed and engage different intracellular signaling cascades.
In addition, neuromodulators such as DA can affect several distinct steps of synaptic transmission, including the probability of neurotransmitter release, the postsynaptic sensitivity to neurotransmitter, and the membrane excitability of the pre- and postsynaptic cells (Figure 1). These neuromodulatory targets are expected to alter synaptic communication in different ways and should be considered separately. First, the excitability of presynaptic neurons directly determines the frequency of activation of synapses by controlling the rate of action potential invasion of presynaptic boutons. Many ion channels, including those controlling resting membrane potential and resistance as well as action potential generation and waveform, are regulated by neuromodulators. Such changes may fall under the general category of ‘gain-control’ mechanisms, which transform the input-output relationship of a circuit. Modulation of the excitability of interneurons that mediate feedback and feed forward inhibition can additionally introduce time-dependent transformations that alter circuit activity in complex ways. Second, neuromodulators directly regulate the probability of action potential-evoked vesicular neurotransmitter release (Prelease) from presynaptic boutons by altering the size and properties of the vesicle pool or of the state of active zone proteins. In addition, Prelease is indirectly controlled by ion channels that determine action potential-evoked Ca2+ influx. Importantly, neurotransmitter release is generally facilitated during repetitive activation of synapses with low basal Prelease whereas it generally depresses at synapses with high basal Prelease. For this reason, changes to Prelease result in complex alterations in the time-dependence of neurotransmitter release that can profoundly alter the dynamics of action potential firing. Third, neuromodulators control the number, classes, and properties of neurotransmitter receptors in the synapse, thereby regulating the postsynaptic and biochemical electrical response. In the simplest cases, changing the number of synaptic ionotropic receptors is analogous to gain control – e.g., increasing the number of synaptic AMPA-type glutamate receptors enlarges the excitatory postsynaptic potential (EPSP) thus altering the gain in the transformation from pre- to postsynaptic activity. However, more subtle modes of regulation are possible with specific changes on subsets of neurotransmitter receptors. Finally, regulation of postsynaptic ion channels that open secondarily as a consequence of synaptic activity can have profound effects on the generation of synaptic potentials, Ca2+ influx, synaptic integration, plasticity and action potential firing. This review will therefore dissect the reported effects of DA on each of three steps that broadly define synaptic transmission: presynaptic neurotransmitter release, postsynaptic neurotransmitter detection and membrane excitability and synaptic integration.
Figure 1. Potential sites of modulation of synaptic transmission by DA.
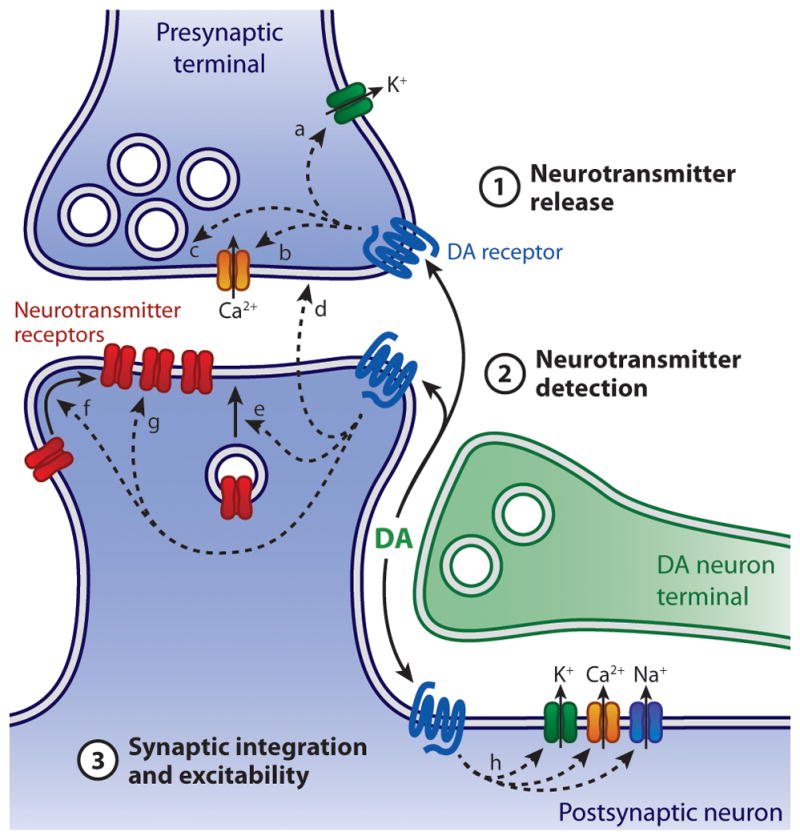
DA may affect Prelease by modulating axon terminal excitability (a), Ca2+ influx (b) or vesicular release machinery (c). This can occur directly, through activation of presynaptic DA receptors, or indirectly, following the recruitment of postsynaptic DA receptors and liberation of retrograde signaling molecules (d). Postsynaptic DA receptors may influence neurotransmitter detection by modulating the membrane insertion (e), synaptic recruitment (f) or properties (g) of neurotransmitter receptors. In addition, DA alters synaptic integration and the excitability of pre- and postsynaptic membranes by modulating ion channels that control resting potential, Ca2+ influx, and action potential threshold and waveform (h).
Our initial intent was to cover dopaminergic modulation of synaptic transmission at all central synapses. However, the available information could not reasonably be contained within the allotted space. Thus, we restricted our analysis to prefrontal cortex (PFC) and striatum, as they are the major targets of the largest group of DA neurons in the mammalian brain and perturbations of DA in these brain regions are implicated in the pathogenesis or exploited in the treatment of numerous neuropsychiatric diseases. No mention will be made of the important modulatory functions of DA in the retina, olfactory bulb, hypothalamus and neuroendocrine system, or of the autoregulatory effects of DA on its synthesis and release. In an attempt to further simplify and reduce this complex field, we limited our presentation to studies in which pharmacological, biochemical, or electrophysiological assays were used to specifically assign (to the extent possible) the regulatory targets of DA to each of the three synaptic transmission steps mentioned above. This largely precludes the discussion of in vivo studies or in vitro electrophysiological investigations that rely exclusively on field or whole-cell current-clamp recordings since effects on synaptic potentials can arise via the modulation of many pre- and postsynaptic targets. Finally, we narrowed our discussion to studies of rodents because they constitute the model of choice for the majority of in vitro electrophysiological studies and have significantly contributed to our understanding of DA signaling in recent years with the application of molecular, genetic and optogenetic techniques.
Intracellular signaling by DA receptors
Once released from presynaptic terminals, DA mediates its effects by interacting with members of a family of GPCRs termed D1 through D5. These distinct but closely-related DA receptors are commonly segregated in two major classes based on their structural, pharmacological and signaling properties: D1 and D5 receptors belong to the subfamily of D1-like receptors, whereas D2, D3 and D4 receptors are grouped into the D2-like receptor class (Table 1). The genes encoding D2-like receptors (Drd2, Drd3 and Drd4) contain several exons that are alternatively spliced, giving rise to isoforms with distinct physiological properties and subcellular localization. The best characterized of these isoforms are the short and long variants of D2 receptors (D2S and D2L, respectively), although several variants of D3 and D4 receptors have also been described (Callier et al., 2003; Rankin et al., 2010). By contrast, the genes encoding D1-like receptors (Drd1a and Drd5) consist of a single exon and therefore do not generate splice variants. At the protein level, receptors within the D1-like and D2-like receptor classes share a high level of homology with each other and consequently display similar pharmacological properties. The affinity of D2-like receptors for DA is generally reported to be 10 to 100-fold greater than that of D1-like receptors, with D3 and D4 receptors displaying the highest sensitivity for DA, and D1 receptors the lowest (Beaulieu and Gainetdinov, 2011). However, it is difficult to infer from these measures, which rely on displacement of radiolabeled antagonists from heterologously-expressed DA receptors and do not capture the coupling efficacy to downstream signaling cascades, whether D2-like receptors are preferentially activated by basal extracellular levels of DA in vivo. Moreover, D1 and D2 receptors can exist in both high and low affinity states, and have similar nanomolar affinities for DA in their high affinity states (reviewed in Wickens and Arbuthnott, 2005). Pharmacological agonists and antagonists of DA receptors can readily distinguish between receptor families, but less so between individual subtypes within a family. Finally, the D1-like and D2-like receptor classes differ functionally in the intracellular signaling pathways they modulate. As GPCRs, all DA receptors activate heterotrimeric G proteins, but the second messenger pathways and effector proteins activated by both receptor classes vary greatly and often mediate opposite effects (Figure 2). These signaling cascades are described in detail elsewhere (see Beaulieu and Gainetdinov, 2011; Fisone, 2010; Neve et al., 2004 and references within); only a brief overview is presented here.
Table 1.
Basic characteristics of DA receptors
| DA receptor subtype | D1-like family
|
D2-like family
|
|||
|---|---|---|---|---|---|
| D1 | D5 | D2 | D3 | D4 | |
| Gene name | Drd1a | Drd5 | Drd2 | Drd3 | Drd4 |
| Number of introns | 0 | 0 | 6 | 5 | 3 |
| Splice variants | No | No | Yes (D2S, D2L) | Yes | Yes |
| Affinity for DA (uM)* | 1.0–5.0 | 0.2–2.0 | 0.2–2.0 | 0.02–0.2 | 0.01–0.1 |
| G protein coupling | Gαs, Gαolf | Gαs, Gαq | Gαi, Gα0 | Gαi, Gα0 | Gαi, Gα0 |
| Common family-specific agonists | SKF-38393, SKF-81297 | (−)Quinpirole, Cabergoline | |||
| Common family-specific antagonists | SCH-23390, SKF-83566 | (−)Sulpiride, Spiperone, Nemonapride | |||
Ki ranges for cloned human DA receptors obtained from the NIMH Psychoactive Drug
Screening Program database (http://pdsp.med.unc.edu) and the International Union of Basic and Clinical Pharmacology database (http://www.iuphar-db.org)
Figure 2. Intracellular DA signaling pathways.

Schematic of cAMP/PKA-dependent (A) and independent (B) pathways recruited by DA receptors. D1- and D2-like receptors are depicted in the same cell for illustrative purposes. Note that some of the targets of Gβγ are ion channels (Kir3, CaV1 and CaV2.2). Black and red arrows depict activation and inhibition, respectively. IP3, inositol triphosphate; DAG, diacylglycerol.
D1-like receptors stimulate the heterotrimeric G proteins Gαs and Gαolf, which are positively coupled to adenylyl cyclase (AC), leading to the production of cyclic adenosine monophosphate (cAMP) and the activation of protein kinase A (PKA). By contrast, D2-like receptors activate Gαi and Gαo proteins, which inhibit AC and limit PKA activation. PKA mediates most of the effects of D1-like receptors by phosphorylating and regulating the function of a wide array of cellular substrates such as voltage-gated K+, Na+ and Ca2+ channels, ionotropic glutamate and GABA receptors and transcription factors. One of the major targets of PKA is the 32-kilodalton DA and cAMP-regulated phosphoprotein DARPP-32, which is highly expressed in DA-responsive striatal and cortical neurons and plays a critical role in the regulation of downstream signal transduction pathways. DARPP-32 integrates signals from several neurotransmitters to bi-directionally modulate PKA activity. When phosphorylated by PKA, DARPP-32 amplifies PKA signaling by inhibiting protein phosphatase 1 (PP1), which counteracts PKA’s actions. By contrast, dephosphorylation by the calmodulin-dependent protein phosphatase 2B (PP2B) upon D2-like receptor stimulation helps convert DARPP-32 into a potent inhibitor of PKA signaling.
DA receptors can also signal independently of cAMP/PKA to modulate intracellular Ca2+ levels and regulate ligand- and voltage-gated ion channels. This is particularly true for Gαi/0-coupled receptors, such as members of the D2-like family, which target several effector proteins through liberation of the Gβγ subunit of heterotrimeric G proteins upon receptor activation. Membrane-bound Gβγ subunits can diffuse along the plasma membrane to directly activate ion channels or second messengers. The best example is the gating of G protein-activated inward-rectifier K+ channels (Kir3) in D2 receptor-expressing midbrain DA neurons (Beckstead et al., 2004). Release of Gβγ subunits following D2-like receptor stimulation can also decrease CaV2.2 (N-type) and CaV1 (L-type) Ca2+ currents directly or indirectly via activation of phospholipase C (PLC). There is also evidence that D1-class receptors can activate PLC by coupling to Gαq heterotrimeric G proteins; but this property may be limited to cells expressing D5 receptors or D1/D2 heterodimers (Lee et al., 2004; Sahu et al., 2009). In addition to their effects on G protein-regulated pathways, D1 and D2 receptors can alter membrane trafficking of CaV2.2 channels as well as NMDA and GABAA receptors through direct protein-protein interactions or following the activation of intracellular tyrosine kinases.
DA receptor distribution in forebrain
DA receptors are broadly expressed in the CNS, with their distribution and expression levels largely mirroring the density of innervating DA fibers (see Bentivoglio and Morelli, 2005; Callier et al., 2003 and references within). D1 and D2 receptors are the two most abundant receptor subtypes expressed in the brain, with D1 receptors displaying the most widespread distribution and highest expression levels. D1 and D2 receptors are most prominently found in dorsal striatum, ventral striatum (nucleus accumbens) and olfactory tubercle, which collectively constitute the principal recipient structures of midbrain DA axons. Significant levels of D1 and D2 receptor mRNA are also found in other forebrain structures, including cortex. The expression of D3, D4 and D5 receptors in the brain is considerably more restricted and weaker than of D1 and D2 receptors. D1-like and D2-like receptors are expressed in both striatal projection neurons (SPNs) and interneurons, as well as in subpopulations of pyramidal neurons, interneurons and glial cells in cortex (Table 2). In these brain regions and others, D1- and D2-like receptors are localized presynaptically in nerve terminals and axonal varicosities, as well as postsynaptically in dendritic shafts and spines (Bentivoglio and Morelli, 2005). Thus, no simple and general division of labor exists between D1 and D2 receptor families with respect to receptor distribution in projection vs. locally-projecting neurons, or pre- vs. postsynaptic membrane specializations.
Table 2. Cellular distribution of DA receptors in the cortex and striatum of rodents.
This table reports semi-quantitative expression levels of and their relative cellular distribution (in parentheses) within defined cortical and striatal neuronal populations.
| D1 | D2 | D3 | D4 | D5 | |
|---|---|---|---|---|---|
| Striatum | |||||
| dSPNs | +++ (>90%) | – | + (50%) | + (<10%) | – |
| iSPNs | + (<10%) | +++ (>90%) | + (<10%) | + (<10%) | + (<10%) |
| Cholinergic interneurons | + (<20%) | ++ (>80%) | – | – | ++ (>80%) |
| PV+ interneurons | + (>70%) | ||||
| NPY/SOM/NO+ interneuron | + (<10%) | + (>70%) | |||
| CR+ interneurons | + (50%) | ||||
| Cortex | |||||
| L2/3 pyramidal neurons | ++ (<20%) | + (<10%) | + | + | |
| L5/6 pyramidal neurons | ++ (20–40%) | + (25%) | + | + | |
| L2-6 interneurons | ++ (30–60%)* | + (20%)* | + | + | + |
For the most part PV+ interneurons
Striatum is almost entirely populated by two equally-sized groups of GABAergic SPNs that extend axons either to basal ganglia output nuclei (the striatonigral or so-called direct pathway SPNs, denoted dSPNs) or to the external segment of the globus pallidus (GPe) (the striatopallidal or indirect pathway SPNs, denoted iSPNs). Anatomical, pharmacological and single-cell reverse transcription-polymerase chain reaction (RT-PCR) studies determined that dSPNs express high levels of D1 receptors along with the peptides neurotransmitters substance P and dynorphin, whereas iSPNs express D2 receptors as well as the neurotransmitter enkephalin (Gerfen, 1992; Gerfen and Surmeier, 2011). This dichotomy was recapitulated in transgenic mice using bacterial artificial chromosomes (BACs) that express Cre recombinase or gene reporters such as enhanced green fluorescent protein (EGFP) or tdTomato under control of the promoter region for D1 or D2 receptor genes (Ade et al., 2011; Gong et al., 2003; Gong et al., 2007). In these mice, transgenes driven by D1 and D2 receptor promoters are almost exclusively segregated into striatonigral and striatopallidal populations, respectively, although a small number of D1 receptor-containing dSPNs additionally extend axon collaterals in GPe (Valjent et al., 2009), consistent with earlier anatomical studies of single biocytin-filled cells. Moreover, molecular profiling studies employing fluorescence activated cell sorting or translating ribosomal affinity purification of striatal tissue from BAC transgenic mice to quantitatively profile mRNAs differentially expressed by iSPNs and dSPNs support the selective enrichment (by several hundred folds) of D1 and D2 receptors in distinct SPN populations (Heiman et al., 2008; Lobo et al., 2010). However, some controversy persists as to whether the segregation of DA receptor families in SPNs is absolute and whether subpopulations of SPNs potentially coexpressing both receptor types underlie the synergistic actions of D1 and D2 receptor agonists observed in some experimental preparations (Perreault et al., 2011). Indeed, in situ hybridization and single-cell RT-PCR experiments have revealed that D1 and D2 receptors can both be detected in a subset of SPNs in striatum and that dSPNs and iSPNs also express low levels of D3, D4 and D5 receptor mRNA (Lester et al., 1993; Surmeier et al., 1992; Surmeier et al., 1996). It is unclear whether these low-abundance transcripts significantly contribute to SPN function and whether the apparent cooperative effects of D1- and D2-like receptors observed in some studies instead arise from complex network interactions.
By virtue of the fact that dSPNs and iSPNs share largely similar morphological and physiological properties, they represent an ideal system to compare the differential neuromodulatory effects of D1 and D2 receptors on synaptic transmission and intrinsic excitability. However, despite this seeming simplicity, electrophysiological characterizations DA’s actions have been complicated by the fact that striatal interneurons also express DA receptors, as do the synaptic terminals of striatal afferents. In dorsal striatum, there are at least five distinct subtypes of GABAergic interneurons (Tepper et al., 2011) and one population of large aspiny cholinergic interneurons. Although these interneurons collectively account for only 5–10% of all striatal neurons, they exert a powerful influence on behavior (Gittis et al., 2011; Witten et al., 2010). Striatal GABAergic interneurons can be distinguished based on the expression of neuropeptides, synthetic enzymes and calcium binding proteins: they include parvalbumin (PV)-expressing fast-spiking (FS) interneurons, neuropeptide Y (NPY)/somatostatin (SOM)/nitric oxide synthase (NOS)-coexpressing low threshold spiking (LTS) interneurons, NPY only-expressing neurogliaform, TH-expressing interneurons and calretinin (CR)-expressing interneurons. Cholinergic interneurons mainly coexpress D2 and D5 receptors, whereas PV+, CR+ and NPY/SOM/NOS+ interneurons express D5 receptors (Rivera et al., 2002; Yan and Surmeier, 1997). It is currently unknown whether NPY-neurogliaform and TH+ interneurons express DA receptors. In addition, D2 receptors adorn the presynaptic terminals of DA afferents (Sesack et al., 1994), glutamatergic cortical and thalamic afferents that innervate SPNs and interneurons (Wang and Pickel, 2002), as well as GABAergic pallidostriatal neurons (Hoover and Marshall, 2004), which mostly terminate on PV+ interneurons and SPNs (Mallet et al., 2012). D1 receptors have also been observed in a small number of presynaptic glutamatergic terminals in striatum (Dumartin et al., 2007). Lastly, SPNs provide lateral inhibition onto each other through recurrent axon collaterals that contain D1 or D2 receptors, depending on SPN subtype (Guzman et al., 2003; Taverna et al., 2005; Tecuapetla et al., 2009). Thus, DA likely initiates a complex cascade of modulatory events in striatum that has the potential to vary dynamically depending on the recruitment of distinct striatal circuits.
In cerebral cortex, the cellular distribution of DA receptors is not as well delineated. The distribution and density of mesocortical DA fibers and cortical DA receptors varies between species, as well as between and within cortical areas in a given species (Bentivoglio and Morelli, 2005), limiting the ability to extract general DA signaling principles. Most studies have focused on PFC, which is the principal cortical recipient of DA afferents. During the past two decades, a large number of histological studies have confirmed that D1 receptors are the most widespread and strongly expressed DA receptors in PFC. D1 and D2 receptors distribute to both pyramidal neurons and interneurons throughout layers (L) 2 to 6, but most prominently in deep cortical layers (Bentivoglio and Morelli, 2005; Santana et al., 2009), where DA innervation is densest. In PFC pyramidal neurons, D1 receptor mRNA is expressed in approximately 20% of layer L2/3 and L5, and in 40% of L6 pyramidal cells (Table 2). By contrast, D2 receptor mRNA is only sparsely detected in superficial layer pyramidal neurons (5% in L2/3), and in 25% and 13 % of L5 and L6 pyramidal cells, respectively (Santana et al., 2009). The cellular distribution of D5 receptors in pyramidal neurons overlaps with that of D1 receptors (Bergson et al., 1995), and D3 and D4 receptors mostly distribute to GABAergic interneurons (Khan et al., 1998). Therefore, unlike striatum, DA receptors in PFC may only be expressed in a fraction of projection neurons, indicating that a considerable number of pyramidal cells may not be subject to direct modulation by DA. Moreover, DA receptor expression in PFC pyramidal neurons does not delineate a functionally-homogeneous group of cells, as only a small proportion of corticostriatal (6–11%), corticothalamic (~25%) and corticocortical neurons (4–10%) expressed D1 or D2 receptors (Gaspar et al., 1995).
Although the total number of DA receptor-expressing pyramidal neurons exceeds that of interneurons, DA receptors are proportionally more widespread and homogeneously expressed within local interneuron populations. D1 receptor mRNA is present in 30–60% of all GABA-containing cortical interneurons across cortical layers in rat (Le Moine and Gaspar, 1998; Santana et al., 2009). The vast majority of these cells are PV+ FS interneurons and calbindin (CB)-expressing LTS interneurons, but only rarely CR+ interneurons; it is estimated that up to 60% of PV+ 25% of CB+ and < 10% of CR+ interneurons express D1 receptors (Le Moine and Gaspar, 1998). The fraction of interneurons expressing D1-like receptors may be larger as D5 receptors complement the expression pattern of D1 receptors, labeling mostly CR+ interneurons, and less so PV+ interneurons (Glausier et al., 2009). By contrast, D2 receptors distribute to a comparatively smaller fraction of cortical GABAergic interneurons: only 5–17% of interneurons contain D2 receptor mRNA (Santana et al., 2009), the majority of which consist of PV+ interneurons (Le Moine and Gaspar, 1998). Although D3 and D4 receptors may complement the expression of D2 receptors in cortical interneurons, their overall distribution is limited (Khan et al., 1998), indicating that D2-like receptors are unlikely to distribute to a large proportion of GABAergic interneurons.
Transgenic mice such as D1- and D2-receptor BACs have the potential to help identify cortical cells with transcriptionally-active DA receptor genes. However, currently-available transgenic lines for D1 and D2 receptors were selected based the fidelity of transgene expression in striatal neurons (Valjent et al., 2009). Comparatively little is known in cortex regarding the penetrance and specificity of these transgenes in D1 and D2 receptor-expressing neurons. A recent study by Zhang and colleagues (2010) determined that Drd2-EGFP BAC transgenic mice express EGFP in over 90% of PFC pyramidal neurons, while Drd1a-tdTomato mice label 16–25% of pyramidal cells, most of which coexpress EGFP, without any region or layer-specific differences. This distribution stands in stark contrast to that described previously (Bentivoglio and Morelli, 2005). In another recent study (Gee et al., 2012), PFC pyramidal neurons identified in Drd2-EGFP and Drd2-Cre BAC transgenic mice were found to project to thalamus but not contralateral cortex, unlike previous descriptions using in situ hybridization (Gaspar et al., 1995). These discrepancies likely speak to the weaknesses of both histological and transgenic approaches. BAC transgenes are generated by nonspecific integration into the target genome and are not immune to positional effects, requiring phenotypic characterization of several transgenic lines before identifying the ones that most closely recapitulate endogenous gene expression patterns. Moreover, transgenic reporter and effector proteins are not subject to the same post-transcriptional and homeostatic regulatory mechanisms that control GPCR expression and may therefore highlight cells that do not functionally detect DA under normal conditions. Conversely, low-abundance GPCR transcripts may be functionally relevant but below the detection limit of conventional histological methods. Therefore, more functional studies like the one recently performed on L5 PFC pyramidal neurons in Drd1a-TdTomato mice (Seong and Carter, 2012) are needed to determine whether BAC transgene expression in brain areas other than striatum accurately identifies neurons that are directly modulated by DA.
DA modulation of neurotransmitter release
One of the many ways neuromodulators influence synaptic transmission is by regulating release of neurotransmitters. This is typically accomplished either by altering ionic conductances that control the excitability of the presynaptic membrane or by acting directly on the vesicular release machinery. Neuromodulators can initiate changes in Prelease either by activating presynaptic receptors or by eliciting the liberation of retrograde signaling molecules from the postsynaptic membrane. Thus, modulation of Prelease by DA cannot simply be inferred based on presynaptic localization of DA receptors, nor can it be excluded in their absence. For the purposes of this review, we focus on electrophysiological studies in acute slice that clearly identify a presynaptic modulatory effect of DA either through analysis of tetrodotoxin (TTX)-resistant ‘miniature’ excitatory or inhibitory postsynaptic currents (mEPSCs or mIPSCs), paired-pulse ratios, or evoked excitatory or inhibitory postsynaptic currents (EPSCs or IPSCs) when postsynaptic changes in neurotransmitter receptor composition have been excluded.
DA acting through both D1 and D2 receptor families has been implicated in heterosynaptic regulation of Prelease at glutamatergic, GABAergic and cholinergic terminals (Figure 3). Specifically, D2-like receptor activation decrease release of glutamate onto SPNs in dorsal and ventral striatum (Bamford et al., 2004; Higley and Sabatini, 2010; Salgado et al., 2005; Wang et al., 2012). D2-like receptors also decrease Prelease of GABA onto PFC pyramidal neurons (Chiu et al., 2010; Seamans et al., 2001b; Xu and Yao, 2010), SPNs (Delgado et al., 2000; Guzman et al., 2003; Kohnomi et al., 2011; Taverna et al., 2005; Tecuapetla et al., 2009) and striatal interneurons (Bracci et al., 2002; Centonze et al., 2003; Momiyama and Koga, 2001; Pisani et al., 2000). In addition, D2-like receptors depress release of acetylcholine (Ach) onto striatal cholinergic interneurons (Pisani et al., 2000). D1-like receptor stimulation decreases release of glutamate onto L5 pyramidal cells in PFC (Gao et al., 2001; Gao and Goldman-Rakic, 2003; Gonzalez-Islas and Hablitz, 2003; Seamans et al., 2001a) and SPNs in ventral striatum (Harvey and Lacey, 1997; Nicola and Malenka, 1997; Pennartz et al., 1992; Wang et al., 2012) but not dorsal striatum (Nicola and Malenka, 1998). Moreover, DA-mediated activation of D1-like receptors reduces GABA release onto cortical FS interneurons (Towers and Hestrin, 2008), L2–5 PFC pyramidal neurons (Gao et al., 2003; Gonzalez-Islas and Hablitz, 2001), and SPNs in ventral striatum only (Nicola and Malenka, 1997, 1998; Pennartz et al., 1992; Taverna et al., 2005). Thus, at synapses responsive to DA modulation, DA typically acts to decrease Prelease.
Figure 3. DA modulation of neurotransmitter release.

Summary of modulatory effects of DA on transmitter release (small circled arrows) in striatum (A) and cortex (B). Principal cells are depicted in blue, interneurons in green. Glutamatergic, GABAergic and cholinergic synaptic inputs are represented as triangles, circles and squares shaded to reflect modulation by D1- (black) or D2-class receptors (white). Lack of presynaptic modulation by DA shown in gray. The identity of the presynaptic cell (inferred or deducted from paired recordings) is indicated where possible. Note that some modulatory changes only apply to striatal subdivisions (dorsal vs. ventral) and that inconsistencies exist (e.g. DA modulation of GABAergic inputs onto L5 PCs). CIN, cholinergic interneuron; PC, pyramidal cell.
There are, however, some notable exceptions to this simple view. DA and D1-like receptor agonists enhance GABA release from dSPN axon collaterals (Guzman et al., 2003). Such D1-like receptor-induced facilitation of transmitter release is consistent with the previously-reported presynaptic enhancement of neurotransmission by cAMP and PKA at hippocampal and cerebellar synapses (Chen and Regehr, 1997; Trudeau et al., 1996). In addition, the D2-like receptor agonist quinpirole was reported to increase GABA release in a third of synaptic connections formed by FS interneurons onto SPNs in nucleus accumbens, and decrease it in another third (Kohnomi et al., 2011). The variable or inconsistent nature of some of these observations may arise from cell type or synaptic heterogeneity, or from the recruitment of other neuromodulatory systems that in turn influence release probability. In cortex, DA differentially influences GABAergic transmission from FS and non-FS interneurons onto pyramidal neurons: it depresses GABA release from FS interneurons and potentiates inhibitory postsynaptic potentials initiated by non-FS cells without affecting electrophysiological measures of Prelease (Gao et al., 2003). In striatum, anatomical studies indicate that presynaptic D1 and D2 receptors are only expressed in a small fraction of glutamatergic synapses (Dumartin et al., 2007; Wang and Pickel, 2002), in agreement with reports of sparse DA receptor expression in a subset of striatum-projecting L5 pyramidal neurons (Gaspar et al., 1995). This observation is corroborated by functional imaging studies of vesicular release from corticostriatal afferents, in which DA modulation is limited to only a small number of terminals (Bamford et al., 2004; Wang et al., 2012). Moreover, DA modulates the activity of cholinergic interneurons (Aosaki et al., 1998; Pisani et al., 2000) and can promote the postsynaptic liberation of adenosine and endocannabinoids from SPNs, which independently influence transmitter exocytosis through the activation of presynaptic GPCRs (Harvey and Lacey, 1997; Oldenburg and Ding, 2011; Wang et al., 2012).
Molecular substrates of presynaptic DA modulation
The molecular mechanisms of DA’s action on presynaptic terminals remain poorly understood due to technical difficulties associated with probing presynaptic intercellular signal cascades. D1- and D2-like receptor agonists inhibit somatic CaV2.1 (P/Q-type) and CaV2.2 channels (Salgado et al., 2005; Surmeier et al., 1995; Yan et al., 1997), which are primarily responsible for initiating neurotransmission in the CNS. These Ca2+ channels therefore constitute a likely substrate for the presynaptic modulatory effect of DA. Indeed, inhibition of CaV2.2 underlies the D2 receptor-induced reduction of GABA release onto striatal cholinergic interneurons (Momiyama and Koga, 2001; Pisani et al., 2000), and the D2 receptor-evoked depression of GABA release from SPN axon collaterals depends on modulation of CaV2.1 or CaV2.2 depending on age (Salgado et al., 2005). However, DA’s effects on neurotransmitter release are not exclusively mediated through presynaptic modulation of Ca2+ channels; in ventral striatum, DA acting on D1-like receptors depresses excitatory transmission independently of presynaptic Ca2+ influx (Nicola and Malenka, 1997).
Pre- and postsynaptic regulation of transmitter release by DA
While there are good reasons to believe that DA modulates transmitter release by directly activating presynaptic DA receptors, experimental evidence formally excluding the involvement of postsynaptic receptors is rare, especially at synapses in which DA receptors are expressed both pre- and postsynaptically. Using paired recordings from synaptically-connected SPNs, Tecuapetla and colleagues (2009) showed that DA acting on D2 but not D1 receptors depresses GABA release from iSPNs (expressing D2 receptors) onto dSPNs (expressing D1 receptors), providing compelling evidence for a direct presynaptic locus of action. In striatum, activation of D2 receptors diminishes presynaptic release of glutamate from corticostriatal afferents (Bamford et al., 2004; Higley and Sabatini, 2010; Salgado et al., 2005; Wang et al., 2012). Although commonly attributed to activation of presynaptic D2 receptors, DA and D2 receptor agonists have small (Wang et al., 2012) or negligible effects on mEPSCs (Andre et al., 2010; Nicola and Malenka, 1998), the reduction in evoked glutamate release scales with afferent stimulation frequency (Bamford et al., 2004; Yin and Lovinger, 2006) and is prevented by postsynaptic Ca2+ buffering as well as pharmacological and genetic blockade of metabotropic glutamate and endocannabinoid receptors (Tozzi et al., 2011; Wang et al., 2012; Yin and Lovinger, 2006). While these studies do not exclude a role for presynaptic D2 receptors, they suggest that under conditions of elevated synaptic activity, DA and glutamate interact postsynaptically to decrease synaptic drive through the synthesis of endocannabinoid retrograde messengers. A similar inhibitory feedback pathway relying on postsynaptic release of adenosine has been proposed downstream of D1-like and NMDA receptors in ventral striatum (Harvey and Lacey, 1997; Wang et al., 2012), though this has not been universally observed (Nicola and Malenka, 1997).
Using optical imaging of exocytic events and electrophysiological recordings from EGFP-labeled dSPNs and iSPNs in BAC transgenic mice, Wang and colleagues (2012) recently dissected the presumed pre- and postsynaptic effects of D1 and D2 receptors on glutamate release from corticoaccumbal afferents. Under conditions of minimal synaptic activity (in TTX), their studies revealed slight presynaptic excitatory and inhibitory effects of D1- and D2-like receptor agonists on glutamate release, respectively. Under conditions of moderate to high corticoaccumbal activity (spontaneous and evoked EPSCs), stimulation of D1- and D2-like receptors both evoked a more pronounced decrease in glutamate release that originated postsynaptically and occluded presynaptic contributions. The D1 receptor-mediated downregulation of glutamate release required AMPA and NMDA receptors and resulted from activation of presynaptic adenosine A1 receptors following postsynaptic liberation of adenosine. By contrast, D2 receptors interacted postsynaptically with metabotropic glutamate receptors to stimulate endocannabinoid production. This same pathway is known to be required for long-term depression of glutamate release onto iSPNs in dorsal striatum (Kreitzer and Malenka, 2007). The effects of DA on transmitter release are therefore complex, context-dependent and not limited to the action of presynaptically-localized DA receptors. The observation that other neuromodulatory systems can independently be engaged by DA further complicates analyses of the mechanisms employed by endogenous DA to modulate transmitter release.
DA modulation of postsynaptic neurotransmitter receptors
Ionotropic glutamate and GABA receptors mediate the majority of fast synaptic transmission in the CNS. The subunits that compose these receptors as well as the molecular scaffolds that guide their assembly, membrane trafficking and synaptic integration contain PKA and protein kinase C (PKC) phosphorylation sites that control the function of these proteins reviewed in (Gladding and Raymond, 2011; Kittler and Moss, 2003; Shepherd and Huganir, 2007). Thus, postsynaptic neurotransmitter receptors are likely targets for the neuromodulatory effects of DA. During the past two decades, in vitro studies demonstrating rapid DA receptor-mediated modulation of ionotropic receptor function and trafficking have abounded, leaving little doubt as to the ability of DA to regulate them. However, the nature and consequences of these interactions are complex and controversial, owing to differences in the expression of DA’s actions across brain areas, cell types and experimental conditions.
DA modulation of NMDA receptors
It is generally accepted that DA acting on D1-like receptors potentiates currents, membrane depolarization and cytosolic Ca2+ levels evoked by ionotophoretic or bath application of NMDA receptor agonists in acutely-dissociated neurons (Andre et al., 2010; Chen et al., 2004; Flores-Hernandez et al., 2002; Jocoy et al., 2011) or slice preparations from PFC and striatum (Cepeda et al., 1998; Levine et al., 1996a; Tseng and O’Donnell, 2004; Zheng et al., 1999). Neuronal glutamate receptors distribute to both synaptic and non-synaptic membranes, but the receptors that populate these membrane domains are distinct with respect to subunit composition, trafficking regulatory mechanisms and function (Gladding and Raymond, 2011; Shepherd and Huganir, 2007). By virtue of the fact that exogenous application of agonists preferentially targets somatic and extrasynaptic receptors, these studies collectively indicate that D1 receptor stimulation can potentiate extrasynaptic NMDA receptor function. Several mechanisms have been proposed to underlie this potentiation, most of which implicate NMDA receptor phosphorylation and membrane trafficking, although the intracellular effectors involved are a matter of debate (Braithwaite et al., 2006; Flores-Hernandez et al., 2002; Gao and Wolf, 2008; Hallett et al., 2006). Importantly, many of the studies reporting enhancements of NMDA receptor function in slices either measured membrane potential or recorded membrane currents under conditions that do not minimize errors associated with the inability to adequately voltage-clamp distal dendrites. This is particularly problematic when investigating functional contributions of NMDA receptors, for which gating is voltage-dependent. Indeed, DA modulation of dendritic voltage-gated Ca2+ channels has been shown to contribute indirectly to NMDA receptor potentiation, possibly by helping relieve Mg2+ block (Cepeda et al., 1998).
It has been comparatively more difficult to establish whether D1 receptors also affect synaptically-localized NMDA receptors, as synaptic stimulation experiments require conditions that additionally exclude contributions from DA’s actions on local interneurons and presynaptic release. Nevertheless, activation of D1-like receptors potentiates miniature and electrically-evoked NMDA receptor EPSCs through postsynaptic signaling involving PKA and PKC in PFC (Gonzalez-Islas and Hablitz, 2003; Li et al., 2010; Seamans et al., 2001a). In striatum, synaptically-evoked NMDA receptor EPSCs are potentiated by D1-like receptor stimulation in some studies (Jocoy et al., 2011; Levine et al., 1996b) but remain unaffected by DA in others (Beurrier and Malenka, 2002; Nicola and Malenka, 1998).
Several studies have also presented evidence that currents evoked by exogenous NMDA application can be attenuated by stimulation of D1-like (Castro et al., 1999; Lee et al., 2002; Lin et al., 2003; Tong and Gibb, 2008) or D2-like receptors (Andre et al., 2010; Flores-Hernandez et al., 2002; Jocoy et al., 2011; Kotecha et al., 2002; Li et al., 2009; Liu et al., 2006; Wang et al., 2003; Zheng et al., 1999). One concern associated with some electrophysiological experiments showing depressing effects of D1-like receptor agonists is that they may have been confounded by direct, non-specific effects of these agents on NMDA receptors; high concentrations of DA or SKF38393, a D1-like receptor agonist, promote rapid, reversible and voltage-dependent blockade of NMDA receptor currents in cultured hippocampal, striatal and thalamic neurons (Castro et al., 1999; Kotecha et al., 2002). With few exceptions (Wang et al., 2003), most reports of decreased NMDA receptor function by DA point to mechanisms independent of G protein signaling, resulting either from direct protein-protein interactions between NMDA receptors and D1 and D2 receptors (Lee et al., 2002; Liu et al., 2006) or from the activation of intracellular tyrosine kinases (Kotecha et al., 2002; Li et al., 2009; Tong and Gibb, 2008). However, few studies have revealed diminished function of synaptic NMDA receptors following DA application. In striatum, postsynaptic NMDA receptor currents evoked by electrical stimulation or 2-photon glutamate uncaging are unperturbed by D2 receptor agonists (Higley and Sabatini, 2010; Levine et al., 1996b). Interestingly, a PKA-dependent decrease in Ca2+ influx through NMDA receptors was observed in iSPNs upon D2 receptor activation, indicating that while D2 receptors do not directly modulate the depolarizing currents contributed by NMDA receptors, they can limit the activation of Ca2+-dependent pathways downstream of NMDA receptors (Higley and Sabatini, 2010), an effect likely due to the regulation of NMDA receptor Ca2+-permeability by PKA phosphorylation of the channel (Chalifoux and Carter, 2010; Skeberdis et al., 2006). Likewise, the D2-like receptor agonist quinpirole did not significantly affect NMDA receptor EPSCs in L2/3 PFC pyramidal neurons (Gonzalez-Islas and Hablitz, 2003). By contrast, selective pharmacological activation of D4 receptors suppresses synaptically-evoked NMDA receptor EPSCs in cortex through PKA-dependent NMDA receptor internalization (Wang et al., 2003). Thus, DA has the capacity to bi-directionally modulate synaptic NMDA receptors through D1- and D2-class receptors, but the susceptibility of individual synapses across brain areas and the intracellular pathways recruited vary greatly.
DA modulation of AMPA receptors
As for NMDA receptors, there is a large body of evidence showing that DA bi-directionally modulates the function and membrane trafficking of AMPA receptors. Biochemical studies have demonstrated that D1 receptor agonists and D2 receptor antagonists promote PKA-dependent phosphorylation of AMPA receptors, whereas D2 receptor agonists diminish it by favoring PP1 activity (Hakansson et al., 2006; Snyder et al., 2000). PKA phosphorylation increases AMPA receptor peak open probability and extrasynaptic membrane expression (Shepherd and Huganir, 2007). Consistent with this, D1 receptors acting through PKA increase surface AMPA receptors in neuronal cultures prepared from nucleus accumbens (Sun et al., 2008) and PFC (Sun et al., 2005), whereas D2 receptor agonists decreases surface AMPA receptor levels (Sun et al., 2005). Moreover, membrane currents and potentials evoked by local application of AMPA receptor agonists in striatal and cortical neurons are depressed by D2 receptor stimulation (Andre et al., 2010; Hernandez-Echeagaray et al., 2004; Levine et al., 1996a) and are either unaffected (Calabresi et al., 1995; Seamans et al., 2001a; Zheng et al., 1999) or potentiated (Andre et al., 2010; Levine et al., 1996a; Lin et al., 2003; Yan et al., 1999) by D1 receptor agonists. Importantly, DA receptor signaling is not sufficient to recruit AMPA receptors to postsynaptic terminals (Sun et al., 2005; Sun et al., 2008), likely because AMPA receptor surface expression and synaptic targeting by lateral diffusion constitute two independent and separately-regulated trafficking steps (Shepherd and Huganir, 2007). Thus, modifications of AMPA receptor surface expression at extrasynaptic membranes by DA may not necessarily extend to synaptic sites. Indeed, very few studies have reported increased or decreased postsynaptic AMPA receptor currents in response stimulation of D1- or D2-class receptors, respectively (Gonzalez-Islas and Hablitz, 2003; Levine et al., 1996b). In most cases, postsynaptic AMPA receptor function was unaltered by DA or D1 receptor agonists in PFC (Gao et al., 2001; Gao and Goldman-Rakic, 2003; Seamans et al., 2001a; Zhou and Hablitz, 1999) and striatum (Bracci et al., 2002; Levine et al., 1996b; Nicola and Malenka, 1997, 1998). Likewise, D2 receptor agonists do not significantly alter synaptically-evoked AMPA receptor EPSCs in cortex or striatum using either electrical stimulation (Gonzalez-Islas and Hablitz, 2003; Pisani et al., 2000), 2-photon glutamate uncaging (Higley and Sabatini, 2010) or paired recordings (Gao et al., 2001). Together, these studies indicate that activation of DA receptors is not sufficient to modify the number or conductance of synaptic AMPA receptors. Instead, DA might need to work in concert with other signaling molecules to promote synaptic AMPA receptor incorporation.
DA modulation of GABAA receptors
Despite widespread reports of GABAA receptor phosphorylation and current modulation by PKA and PKC (reviewed in Kittler and Moss, 2003), comparatively few studies have observed DA modulation of GABAA receptor function. In deep layer PFC pyramidal neurons, DA reduces postsynaptic GABAA receptor currents at synaptic and extrasynaptic sites through D4 receptor-mediated downregulation of surface receptors (Graziane et al., 2009; Seamans et al., 2001b; Wang et al., 2002). In striatum, D1 and D5 receptors respectively decrease and enhance GABAA receptor currents evoked by local application of GABA on the somata of acutely-dissociated SPNs (Flores-Hernandez et al., 2000) and cholinergic interneurons (Yan and Surmeier, 1997). Aside from these, most studies investigating DA modulation of synaptic GABAergic transmission either failed to detect changes in postsynaptic inhibitory currents or potentials, or assigned them to presynaptic modifications in GABA release or postsynaptic membrane properties in PFC (Gao et al., 2003; Gonzalez-Islas and Hablitz, 2001; Gulledge and Jaffe, 2001; Kroner et al., 2007; Towers and Hestrin, 2008; Zhou and Hablitz, 1999) and striatum (Bracci et al., 2002; Centonze et al., 2003; Delgado et al., 2000; Kohnomi et al., 2011; Nicola and Malenka, 1997, 1998; Pisani et al., 2000; Taverna et al., 2005; Tecuapetla et al., 2009).
DA modulation of postsynaptic excitability and synaptic integration
Striatal projection neurons
During the past two and a half decades, evidence has accumulated that DA exerts a powerful influence on SPN intrinsic excitability. Early electrophysiological studies in slice indicated that DA can both enhance and reduce SPN spiking evoked by intracellular current injection (reviewed in Nicola et al., 2000). Not surprisingly, the polarity and magnitude of these alterations depended in large part on the type of DA receptor activated. However, the picture that arose initially is opposite of the one that constitutes our current understanding of DA’s effects on intrinsic excitability. It was determined that activation of D1 receptors diminishes SPN excitability, whereas D2 receptor signaling promotes excitation (Nicola et al., 2000). The advent of improved methodologies to identify and record from dSPNs and iSPNs in situ, combined with a greater understanding of the contribution of individual ionic conductances to the overall behavior of SPNs have reversed this view to the one originally advanced by anatomists and pathologists some 20 years ago, namely that DA favors the activation of dSPNs and reduces the excitability of iSPNs (Gerfen and Surmeier, 2011).
Under basal conditions, dSPNs and iSPNs exhibit small but significant differences in dendritic morphology and membrane properties that translate into greater excitability of iSPNs over dSPNs. Although both cell types do not differ with regards to input resistance and resting membrane potential, the action potential discharge rate of iSPNs is twice that of dSPNs in response to somatic current injection (Gertler et al., 2008; Kreitzer and Malenka, 2007). Morphologically, the dendrites of dSPNs and iSPNs are studded with a similarly high density of spines, but iSPNs possess more primary dendrites compared to dSPNs, resulting in a functionally-greater number of excitatory synaptic contacts onto these cells (Day et al., 2006; Gertler et al., 2008; Kreitzer and Malenka, 2007). The dendrites of iSPNs are also more excitable than those of dSPNs (Day et al., 2008). While some of the differential effects of DA on SPN excitability admittedly originate from circuit-level interactions between striatal cells, DA directly influences SPN excitability by modulating ion channels, several of which have been defined. Modulation of any of these channels has the potential to significantly alter SPN excitability, although the relative impact of these changes critically depends on membrane potential, as the array of voltage-gated ion channels engaged at different potentials varies considerably.
DA does not significantly alter SPN excitability by modulating leak conductances as DA receptor agonists exert little or no influence on SPN resting membrane potential or input resistance. Instead, most of DA’s reported effects on intrinsic excitability and synaptic integration involve PKA-dependent modulation of voltage-gated K+, Na+ and Ca2+ channels. In both dorsal and ventral striatum, studies of pharmacologically-isolated currents have revealed that D1 receptors facilitate inward rectifier K+ channels belonging to the Kir2 family (Pacheco-Cano et al., 1996; Uchimura and North, 1990) and decrease slowly inactivating A-type K+ currents attributed to KV4 channels (Kitai and Surmeier, 1993). These changes are predicted to impede synaptically-driven transitions from the hyperpolarized resting potential (so-called down state) to a more depolarized, sustained potential near spike threshold (up state), while enhancing action potential firing during up states (Wickens and Arbuthnott, 2005). In addition, D1 receptor stimulation increases CaV1 currents (Hernandez-Lopez et al., 1997; Song and Surmeier, 1996; Surmeier et al., 1995), which potentiate up state transitions, excitatory synaptic potentials and action potential discharge (Plotkin et al., 2011; Vergara et al., 2003), and suppresses currents carried by CaV2.1 and CaV2.2 (Surmeier et al., 1995; Zhang et al., 2002), which limit repetitive action potential firing by activating small- (SK) and large-conductance (BK) Ca2+-dependent K+ channels (Hopf et al., 2010; Vilchis et al., 2000). Interestingly, activation of D1 receptors was recently shown to prolong ‘up state-like’ potentials evoked by repetitive glutamate uncaging on distal SPN dendrites (Plotkin et al., 2011). Generation of these regenerative plateau potentials required NMDA receptors and low threshold CaV3 channels, which are enriched in distal dendrites and spines (Carter and Sabatini, 2004; Carter et al., 2007; Day et al., 2006), but it is currently unclear whether the D1 receptor-evoked enhancement is mediated by direct modulation of NMDA receptors or dendritic K+ and Ca2+ conductances, or both. Thus, through its actions on voltage-gated K+ and Ca2+ channels, D1 receptors promote synaptic integration and spike discharge during up states while increasing the threshold for upward transitions, effectively acting to enhance contrast between up and down states. However, this relatively simple and consistent view of DA’s action on dSPNs is complicated by the reported effects on voltage-gated Na+ currents, which are reduced in amplitude by DA and D1-like receptor agonists (Cepeda et al., 1995; Schiffmann et al., 1995; Surmeier et al., 1992; Zhang et al., 1998). This observation is largely responsible for the initial conclusion that D1 receptors exert a net inhibitory action on SPN excitability (Nicola et al., 2000). The apparently conflicting actions of DA on various ionic conductances reflect some of the difficulties associated with extrapolating overall neuromodulatory effects of DA from changes in isolated conductances. Given the importance of subthreshold membrane potential fluctuations to SPN function and the inability of somatic current injection protocols to engage distal dendritic conductances (Day et al., 2008) and evoke state transitions in acute slices (Wilson, 2004), analyses of spike discharge modulation upon somatic depolarization may not adequately capture DA’s influence on synaptic integration and intrinsic excitability. Nevertheless, although the spike-promoting effects of D1 receptors on K+ and Ca2+ channel may be moderated by reduced Na+ channel availability, most of the evidence accrued to date favors models in which D1 receptors promote dSPN intrinsic excitability (Gerfen and Surmeier, 2011; Wickens and Arbuthnott, 2005).
The reported effects of D2 receptor activation on isolated ionic conductances and up state potentials in SPNs largely oppose those of D1 receptors. Through their inhibitory action on PKA, D2 receptors suppress currents attributable to Kir2 channels but enhance depolarization-activated and ATP-sensitive K+ channels (Greif et al., 1995; Perez et al., 2006; Sun et al., 2000; Surmeier and Kitai, 1993), indicating that D2 receptor activation may facilitate up state transitions but stunt their duration and the depolarization achieved. D2 receptors further limit somatic excitability by decreasing Ca2+ influx through somatic CaV1 channels (Hernandez-Lopez et al., 2000; Salgado et al., 2005). In dendritic shafts and spines, D2 receptor stimulation mainly decreases Ca2+ currents carried by CaV2.3, which gate dendritic SK channels (Higley and Sabatini, 2010), although synaptic potentials evoked by glutamate uncaging remain unaffected by quinpirole (Higley and Sabatini, 2010), perhaps due to concurrent potentiation of dendritic Kv4 channels (Day et al., 2008). Moreover, D2 receptors shorten regenerative plateau potentials evoked by glutamate uncaging on the distal dendrites of iSPNs (Plotkin et al., 2011), possibly by decreasing Ca2+ influx through NMDA receptors or voltage-gated Ca2+ channels (Day et al., 2008; Higley and Sabatini, 2010). Unlike D1 receptors, the effects of D2 receptors on Na+ channels are inconsistent: they were found to enhance, suppress or have no effects in subpopulations of SPNs in ventral and dorsal striatum (Hu et al., 2005; Surmeier et al., 1992; Zhang et al., 1998). Together, this body of work depicts a relatively coherent model of modulation by D2 receptors, in which DA suppresses iSPN synaptic integration and spiking output by diminishing the potential and duration of up states and by limiting the spread and depolarization of synaptic potentials.
Striatal interneurons
Amongst the six populations of striatal interneurons characterized to date, the modulatory actions of DA have only been investigated in cholinergic, FS and LTS interneurons. The latter possess wide axonal arbors that innervate a large number of SPNs and DA depolarizes these cells by activating D5 receptors (Centonze et al., 2002; Tepper et al., 2011). FS interneurons integrate glutamatergic inputs from cortex and establish strong GABAergic synapses on the somata of surrounding SPNs, forming a potent feed forward inhibitory circuit that preferentially targets dSPNs over iSPNs (Tepper et al., 2011). FS interneurons also receive a GABAergic projection from GPe (Mallet et al., 2012). Studies in striatal slices have revealed that DA directly and dose-dependently depolarizes the membrane of FS interneurons via D5 receptors, possibly by promoting the closure of a K+ conductance (Bracci et al., 2002; Centonze et al., 2003). In combination with the D2 receptor-mediated selective decrease of GABAergic (but not glutamatergic) inputs onto these cells, DA is believed to limit the influence of GPe afferents and local interneurons, while promoting corticostriatal feedforward inhibition of SPNs.
The DA and cholinergic systems dynamically and reciprocally influence each other in numerous ways, many of which continue to be unraveled (Cragg, 2006; Threlfell et al., 2012). Cholinergic interneurons are tonically active in vivo and respond to behaviorally-salient stimuli with a brief pause in activity that can be preceded or followed by a transient increase in firing (Cragg, 2006). Experiments in vivo have suggested that this pause is dependent on DA (Aosaki et al., 1994), and several in vitro studies have since revealed potential cellular and molecular substrates for this interaction (reviewed in Goldberg and Reynolds, 2011). Cholinergic interneurons express D2 and D5 receptors (Rivera et al., 2002; Yan and Surmeier, 1997) and pharmacological studies have reported both excitatory and inhibitory modulatory changes by DA. In slice, DA and D5 receptor stimulation depolarize cholinergic interneurons and promotes spiking through cAMP-dependent suppression of a K+ conductance and opening of an undefined cation channel (Aosaki et al., 1998; Centonze et al., 2003; Pisani et al., 2000). By contrast, application of DA together with a D1-like receptor antagonist reveals a hyperpolarizing current (Aosaki et al., 1998), indicating that D5 and D2 receptors both influence the membrane potential of cholinergic interneurons. In agreement with this, direct activation of D2 receptors evokes a dose-dependent depolarization and action potential firing (Maurice et al., 2004; Tozzi et al., 2011) [but see (Pisani et al., 2000; Pisani et al., 2006)]. The effect of D2 receptors has been proposed to be mediated by reductions in a persistent Na+ current and a hyperpolarization-activated, cyclic-nucleotide-gated (HCN) cation current that controls pacemaking (Deng et al., 2007; Maurice et al., 2004). In addition, D2 receptor stimulation reduces CaV2.2 currents in dissociated cholinergic interneurons (Pisani et al., 2006; Yan et al., 1997) and in striatal slices (Ding et al., 2006), which may underlie the D2 receptor-mediated depression of presynaptic Ach release (Pisani et al., 2000). Because CaV2.2 channels are functionally coupled to somatodendritic SK channels, which contribute to spike after hyperpolarizing potentials (Goldberg and Wilson, 2005), downregulation of CaV2.2 by D2 receptors is expected to disrupt autonomous pacemaking and promote a transition to burst firing (Goldberg and Wilson, 2005). Endogenous activation of D5 receptors using electrical stimulation quickly and transiently prolongs interspike intervals by augmenting spike after hyperpolarization (Bennett and Wilson, 1998), which stands in contrast to the increase in firing observed with bath application of DA or D1-like receptor agonists (Aosaki et al., 1998). Thus, although the net effect of DA on the intrinsic excitability of cholinergic interneurons appears to be excitatory (Aosaki et al., 1998; Centonze et al., 2003; Pisani et al., 2000), signaling through D2 and D5 receptors exert opposite effects and the specific conductances that underlie DA’s actions remain to be defined. Given that DA presynaptically inhibits GABAergic but not glutamatergic inputs onto cholinergic interneurons (Momiyama and Koga, 2001; Pisani et al., 2000), and that excitatory inputs precisely regulate spike timing in these cells (Bennett and Wilson, 1998), DA may promote the synchronous activation of cholinergic interneurons, engendering a complex cascade of signaling events resulting in further DA release and inhibition of SPN output (English et al., 2011; Threlfell et al., 2012; Witten et al., 2010).
Prefrontal cortex pyramidal neurons
The prefrontal cortex is the major cortical recipient of DA inputs and DA is believed to play a critical role in several cognitive processes conducted by PFC networks, including working memory, attention and decision making. Although considerable efforts have been invested during the past several decades in elucidating the cellular mechanisms by which DA modulates PFC function, the actions of DA and the underlying receptors and signaling pathways involved remain controversial. What is clear is that DA modulates the intrinsic excitability of both pyramidal neurons and local interneurons, and that DA’s actions on the latter has historically confounded in vivo and in vitro investigations of its effects on the former (reviewed in Seamans and Yang, 2004). In addition, PFC is composed of several functionally distinct pyramidal and non-pyramidal cell types that receive variable dopaminergic innervation along their dendritic trees and express different levels and combinations of DA receptors across cortical layers (Wang et al., 2006). Finally, the functional implications of modulatory effects on isolated currents are often unclear due to the large number of ionic conductances that shape synaptic potentials and spike output, the dependence of these processes on membrane potential, and the complexity of the network these cells are embedded in.
In the majority of in vitro studies in which synaptic contributions are pharmacologically excluded, DA enhances the intrinsic excitability of deep layer PFC pyramidal neurons by elevating the resting membrane potential or promoting a slow but long-lasting increase in the number of action potentials evoked by somatic depolarization (Ceci et al., 1999; Gao and Goldman-Rakic, 2003; Gulledge and Jaffe, 2001; Gulledge and Stuart, 2003; Kroener et al., 2009; Lavin and Grace, 2001; Moore et al., 2011; Penit-Soria et al., 1987; Shi et al., 1997; Wang and Goldman-Rakic, 2004; Yang and Seamans, 1996). In most cases, DA’s actions are selectively abolished by D1-like receptor antagonists and mimicked by D1-like agonists (Chen et al., 2007; Gao and Goldman-Rakic, 2003; Gulledge and Jaffe, 2001; Gulledge and Stuart, 2003; Kroener et al., 2009; Lavin and Grace, 2001; Penit-Soria et al., 1987; Seong and Carter, 2012; Shi et al., 1997; Tseng and O’Donnell, 2004; Witkowski et al., 2008; Yang and Seamans, 1996), implicating signaling through D1-class receptors. Moreover, some studies have indicated that D2-like receptors actively oppose D1 receptor-mediated excitation by directly suppressing intrinsic neuronal excitability (Gulledge and Jaffe, 1998; Tseng and O’Donnell, 2004). However, several other studies have assigned DA-induced increased excitability to D2-class receptors in deep layer pyramidal neurons (Ceci et al., 1999; Gee et al., 2012; Moore et al., 2011; Wang and Goldman-Rakic, 2004) and have reported a net inhibitory effect of D1-class receptors on spike output (Moore et al., 2011; Rotaru et al., 2007). In L2/3 PFC pyramidal neurons, DA was shown to promote (Henze et al., 2000) or leave unaffected action potential firing evoked by somatic current injection (Gonzalez-Islas and Hablitz, 2001; Zhou and Hablitz, 1999).
Regulation of several voltage-gated conductances may contribute to these diverse effects. In PFC pyramidal neurons, activation of D1 receptors reduces K+ currents carried by inward-rectifying (Dong et al., 2004; Witkowski et al., 2008) and voltage-activated K+ channels (Dong and White, 2003; Dong et al., 2004; Dong et al., 2005; Yang and Seamans, 1996), which are respectively expected to facilitate transitions to up states and help sustain them once achieved. D1 receptor activation has been shown to increase (Gorelova and Yang, 2000; Yang and Seamans, 1996), suppress (Geijo-Barrientos and Pastore, 1995; Gulledge and Jaffe, 2001; Rotaru et al., 2007) or exert no effect on (Maurice et al., 2001) the amplitude of persistent voltage-activated Na+ currents. This diversity may result in part from the voltage-dependence of this modulation (Gorelova and Yang, 2000). In addition, D1 receptor agonists inhibit transient voltage-sensitive Na+ currents [(Maurice et al., 2001; Peterson et al., 2006) but see (Gulledge and Jaffe, 2001; Gulledge and Stuart, 2003)]. Some of these effects are consistent with the differential modulation of transient and persistent Na+ currents by PKA and PKC (Chen et al., 2006; Franceschetti et al., 2000), which are both engaged by D1-like receptors in PFC neurons and together exert a net positive influence on membrane excitability (Franceschetti et al., 2000). Modulation of Na+ channels can not only influence action potential initiation and discharge rate, but also the amplitude of synaptic potentials and their active propagation along dendrites (Rotaru et al., 2007). Electrophysiological and Ca2+ imaging experiments in deep layer pyramidal neurons also revealed that D1-like receptor agonists suppress dendritic Ca2+ influx through CaV1, CaV2.2 and possibly CaV2.1 via PKC or direct protein interaction (Kisilevsky et al., 2008; Yang and Seamans, 1996; Young and Yang, 2004; Zhou and Antic, 2012). However, other studies failed to detect any DA modulation of dendritic Ca2+ transients evoked by back-propagating action potentials (Gulledge and Stuart, 2003) or reported PKA-dependent potentiation of CaV1 currents evoked by subthreshold somatic current injection (Young and Yang, 2004). Thus, the reported effects of D1-like receptors on individual ionic conductances in PFC neurons are diverse and a coherent view of the modulatory changes that underlie the excitatory effects of these receptors has yet to emerge.
The ionic conductances that underlie the modulatory effects of D2 receptors in PFC pyramidal neurons have not been investigated as extensively. In instances in which D2-like receptor stimulation promotes the intrinsic excitability of subpopulations of L5 pyramidal cells, the effects have been attributed to suppression of Kir channels (Dong et al., 2004) or potentiation of CaV1 and voltage-gated Na+ channels (Gee et al., 2012; Moore et al., 2011; Wang and Goldman-Rakic, 2004).
Prefrontal cortex interneurons
One of the most consistent and striking effect of DA on PFC pyramidal cells is a selective increase in the frequency of spontaneous (TTX-sensitive), but not miniature (TTX-resistant) IPSCs and IPSPs, reflecting a net enhancement of local GABAergic interneuron spiking activity (Gulledge and Jaffe, 2001; Kroner et al., 2007; Penit-Soria et al., 1987; Seamans et al., 2001b; Zhou and Hablitz, 1999). This effect is largely attributed to PV-expressing FS basket and chandelier cells. Indeed, in vitro studies in PFC slices have repeatedly demonstrated that DA acting on D1-like receptors induces a direct membrane depolarization and increases the input resistance and excitability of the majority of FS interneurons (Gao and Goldman-Rakic, 2003; Gao et al., 2003; Gorelova et al., 2002; Kroner et al., 2007; Towers and Hestrin, 2008; Trantham-Davidson et al., 2008; Zhou and Hablitz, 1999), but exerts variable facilitatory effect on the excitability of other non-FS interneurons (Gao et al., 2003; Gorelova et al., 2002; Kroner et al., 2007). D2 receptor agonists have occasionally been reported to further promote interneuron excitability (Tseng and O’Donnell, 2004; Wu and Hablitz, 2005). In FS interneurons, DA’s actions are mediated by PKA-dependent suppression of leak, inward rectifying and depolarization-activated K+ channels (Gorelova et al., 2002) and amplification of depolarizing currents carried by HCN channels (Gorelova et al., 2002; Trantham-Davidson et al., 2008; Wu and Hablitz, 2005).
Early studies in which GABAergic signaling is left unperturbed had reported that DA predominantly depresses evoked and spontaneous firing of PFC pyramidal cells in vivo (reviewed in Seamans and Yang, 2004) and in vitro (Geijo-Barrientos and Pastore, 1995; Gulledge and Jaffe, 1998; Zhou and Hablitz, 1999). It is now believed that the reported inhibitory effect of DA on pyramidal neuron excitability was indirectly mediated through GABAergic FS cells, which primarily innervate the cell bodies, initial axon segments, and proximal dendritic shafts of pyramidal cells and exert a powerful influence over action potential initiation and timing. Indeed, bath application of GABAA receptor antagonists reverses the polarity of DA’s influence on pyramidal neuron excitability, from inhibition to facilitation (Gulledge and Jaffe, 2001; Zhou and Hablitz, 1999), stressing the importance of excluding synaptic contributions to investigate modulation of intrinsic excitability. In addition to these changes, DA alters the release of glutamate and GABA onto pyramidal and non-pyramidal neurons differentially based pre- and postsynaptic cell identity through D1- and D2-like receptors (Chiu et al., 2010; Gao et al., 2001; Gao and Goldman-Rakic, 2003; Gao et al., 2003; Gonzalez-Islas and Hablitz, 2001; Penit-Soria et al., 1987; Seamans et al., 2001b; Towers and Hestrin, 2008; Trantham-Davidson et al., 2004), revealing a rich and complex array of modulatory influences that collectively contribute to DA’s important role in PFC function.
Future Directions
As described above, dozens of mechanisms have been identified through which DA receptors alter the properties of neurons and synapses. However, several important challenges remain and it is likely that many of these results will have to be revisited with newer approaches. The studies listed above all use pharmacological activation of DA receptors to probe changes that can occur under strong modulation of DA receptors. In the era of optogenetics, in which light can be used to trigger synaptic DA release directly from dopaminergic axons, it will be necessary to determine which of these effects occur under more natural patterns of DA receptor activation. Early studies using this approach have demonstrated that midbrain DA neurons additionally release glutamate and GABA that act on ionotropic receptors in SPNs to rapidly regulate postsynaptic excitability (Stuber et al., 2010; Tecuapetla et al., 2010; Tritsch et al., 2012), adding another dimension to the consequences of DA neuron firing on downstream targets. Similarly, the effects of DA in cortex will need to be reexamined in transgenic mice expressing genetically-encoded fluorophores or recombinases in specific subsets of DA-sensitive neurons to mitigate the experimental variability that has historically confused this field (e.g. Gee et al., 2012; Seong and Carter, 2012). These approaches continue to transform our understanding of DA action in the striatum, in which decades of studies were plagued by mixing data from two classes of SPNs that express different DA receptors. Lastly, the challenge remains of trying to understand how the results of these largely reductionist studies explain the consequences of DA and DA receptor perturbation on behavior. The hope is that knowledge gained from these studies, combined with more specific and physiological methodologies will permit the elucidation of the cellular and molecular means by which DA influences neural circuits.
Acknowledgments
We apologize to those authors whose work was not cited due to space restrictions. Work in our laboratory on neuromodulation is supported by grants from the National Institute of Health (NS046579 to B.L.S.), the Lefler family fund, and the Howard Hughes Medical Institute. N.X.T. is supported by a fellowship from the Nancy Lurie Marks Family Foundation.
References
- Ade KK, Wan Y, Chen M, Gloss B, Calakos N. An Improved BAC Transgenic Fluorescent Reporter Line for Sensitive and Specific Identification of Striatonigral Medium Spiny Neurons. Front Syst Neurosci. 2011;5:32. doi: 10.3389/fnsys.2011.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andre VM, Cepeda C, Cummings DM, Jocoy EL, Fisher YE, William Yang X, Levine MS. Dopamine modulation of excitatory currents in the striatum is dictated by the expression of D1 or D2 receptors and modified by endocannabinoids. Eur J Neurosci. 2010;31:14–28. doi: 10.1111/j.1460-9568.2009.07047.x. [DOI] [PubMed] [Google Scholar]
- Aosaki T, Graybiel AM, Kimura M. Effect of the nigrostriatal dopamine system on acquired neural responses in the striatum of behaving monkeys. Science. 1994;265:412–415. doi: 10.1126/science.8023166. [DOI] [PubMed] [Google Scholar]
- Aosaki T, Kiuchi K, Kawaguchi Y. Dopamine D1-like receptor activation excites rat striatal large aspiny neurons in vitro. J Neurosci. 1998;18:5180–5190. doi: 10.1523/JNEUROSCI.18-14-05180.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford NS, Zhang H, Schmitz Y, Wu NP, Cepeda C, Levine MS, Schmauss C, Zakharenko SS, Zablow L, Sulzer D. Heterosynaptic dopamine neurotransmission selects sets of corticostriatal terminals. Neuron. 2004;42:653–663. doi: 10.1016/s0896-6273(04)00265-x. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63:182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- Beckstead MJ, Grandy DK, Wickman K, Williams JT. Vesicular dopamine release elicits an inhibitory postsynaptic current in midbrain dopamine neurons. Neuron. 2004;42:939–946. doi: 10.1016/j.neuron.2004.05.019. [DOI] [PubMed] [Google Scholar]
- Bennett BD, Wilson CJ. Synaptic regulation of action potential timing in neostriatal cholinergic interneurons. J Neurosci. 1998;18:8539–8549. doi: 10.1523/JNEUROSCI.18-20-08539.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentivoglio M, Morelli M. The organization and circuits of mesencephalic dopaminergic neurons and the distribution of dopamine receptors in the brain. In: dunnett SB, Bentivoglio M, Bjorklund A, Hokfelt T, editors. Dopamine. San Diego: Elsevier; 2005. pp. 1–107. [Google Scholar]
- Bergson C, Mrzljak L, Smiley JF, Pappy M, Levenson R, Goldman-Rakic PS. Regional, cellular, and subcellular variations in the distribution of D1 and D5 dopamine receptors in primate brain. J Neurosci. 1995;15:7821–7836. doi: 10.1523/JNEUROSCI.15-12-07821.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurrier C, Malenka RC. Enhanced inhibition of synaptic transmission by dopamine in the nucleus accumbens during behavioral sensitization to cocaine. J Neurosci. 2002;22:5817–5822. doi: 10.1523/JNEUROSCI.22-14-05817.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorklund A, Dunnett SB. Dopamine neuron systems in the brain: an update. Trends Neurosci. 2007;30:194–202. doi: 10.1016/j.tins.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Bracci E, Centonze D, Bernardi G, Calabresi P. Dopamine excites fast-spiking interneurons in the striatum. J Neurophysiol. 2002;87:2190–2194. doi: 10.1152/jn.00754.2001. [DOI] [PubMed] [Google Scholar]
- Braithwaite SP, Adkisson M, Leung J, Nava A, Masterson B, Urfer R, Oksenberg D, Nikolich K. Regulation of NMDA receptor trafficking and function by striatal-enriched tyrosine phosphatase (STEP) Eur J Neurosci. 2006;23:2847–2856. doi: 10.1111/j.1460-9568.2006.04837.x. [DOI] [PubMed] [Google Scholar]
- Calabresi P, De Murtas M, Pisani A, Stefani A, Sancesario G, Mercuri NB, Bernardi G. Vulnerability of medium spiny striatal neurons to glutamate: role of Na+/K+ ATPase. Eur J Neurosci. 1995;7:1674–1683. doi: 10.1111/j.1460-9568.1995.tb00689.x. [DOI] [PubMed] [Google Scholar]
- Callier S, Snapyan M, Le Crom S, Prou D, Vincent JD, Vernier P. Evolution and cell biology of dopamine receptors in vertebrates. Biol Cell. 2003;95:489–502. doi: 10.1016/s0248-4900(03)00089-3. [DOI] [PubMed] [Google Scholar]
- Carlsson A, Lindqvist M, Magnusson T. 3,4-Dihydroxyphenylalanine and 5-hydroxytryptophan as reserpine antagonists. Nature. 1957;180:1200. doi: 10.1038/1801200a0. [DOI] [PubMed] [Google Scholar]
- Carlsson A, Lindqvist M, Magnusson T, Waldeck B. On the presence of 3-hydroxytyramine in brain. Science. 1958;127:471. doi: 10.1126/science.127.3296.471. [DOI] [PubMed] [Google Scholar]
- Carlsson A. The occurrence, distribution and physiological role of catecholamines in the nervous system. Pharmacol Rev. 1959;11:490–493. [PubMed] [Google Scholar]
- Carlsson A, Falck B, Hillarp NA. Cellular localization of brain monoamines. Acta Physiol Scand Suppl. 1962;56:1–28. [PubMed] [Google Scholar]
- Carter AG, Sabatini BL. State-dependent calcium signaling in dendritic spines of striatal medium spiny neurons. Neuron. 2004;44:483–493. doi: 10.1016/j.neuron.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Carter AG, Soler-Llavina GJ, Sabatini BL. Timing and location of synaptic inputs determine modes of subthreshold integration in striatal medium spiny neurons. J Neurosci. 2007;27:8967–8977. doi: 10.1523/JNEUROSCI.2798-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro NG, de Mello MC, de Mello FG, Aracava Y. Direct inhibition of the N-methyl-D-aspartate receptor channel by dopamine and (+)-SKF38393. Br J Pharmacol. 1999;126:1847–1855. doi: 10.1038/sj.bjp.0702479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceci A, Brambilla A, Duranti P, Grauert M, Grippa N, Borsini F. Effect of antipsychotic drugs and selective dopaminergic antagonists on dopamine-induced facilitatory activity in prelimbic cortical pyramidal neurons. An in vitro study. Neuroscience. 1999;93:107–115. doi: 10.1016/s0306-4522(99)00123-2. [DOI] [PubMed] [Google Scholar]
- Centonze D, Bracci E, Pisani A, Gubellini P, Bernardi G, Calabresi P. Activation of dopamine D1-like receptors excites LTS interneurons of the striatum. Eur J Neurosci. 2002;15:2049–2052. doi: 10.1046/j.1460-9568.2002.02052.x. [DOI] [PubMed] [Google Scholar]
- Centonze D, Grande C, Usiello A, Gubellini P, Erbs E, Martin AB, Pisani A, Tognazzi N, Bernardi G, Moratalla R, et al. Receptor subtypes involved in the presynaptic and postsynaptic actions of dopamine on striatal interneurons. J Neurosci. 2003;23:6245–6254. doi: 10.1523/JNEUROSCI.23-15-06245.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Chandler SH, Shumate LW, Levine MS. Persistent Na+ conductance in medium-sized neostriatal neurons: characterization using infrared videomicroscopy and whole cell patch-clamp recordings. J Neurophysiol. 1995;74:1343–1348. doi: 10.1152/jn.1995.74.3.1343. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Colwell CS, Itri JN, Chandler SH, Levine MS. Dopaminergic modulation of NMDA-induced whole cell currents in neostriatal neurons in slices: contribution of calcium conductances. J Neurophysiol. 1998;79:82–94. doi: 10.1152/jn.1998.79.1.82. [DOI] [PubMed] [Google Scholar]
- Chalifoux JR, Carter AG. GABAB receptors modulate NMDA receptor calcium signals in dendritic spines. Neuron. 2010;66:101–113. doi: 10.1016/j.neuron.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Regehr WG. The mechanism of cAMP-mediated enhancement at a cerebellar synapse. J Neurosci. 1997;17:8687–8694. doi: 10.1523/JNEUROSCI.17-22-08687.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Greengard P, Yan Z. Potentiation of NMDA receptor currents by dopamine D1 receptors in prefrontal cortex. Proc Natl Acad Sci U S A. 2004;101:2596–2600. doi: 10.1073/pnas.0308618100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Bohanick JD, Nishihara M, Seamans JK, Yang CR. Dopamine D1/5 receptor-mediated long-term potentiation of intrinsic excitability in rat prefrontal cortical neurons: Ca2+-dependent intracellular signaling. J Neurophysiol. 2007;97:2448–2464. doi: 10.1152/jn.00317.2006. [DOI] [PubMed] [Google Scholar]
- Chen Y, Yu FH, Surmeier DJ, Scheuer T, Catterall WA. Neuromodulation of Na+ channel slow inactivation via cAMP-dependent protein kinase and protein kinase C. Neuron. 2006;49:409–420. doi: 10.1016/j.neuron.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Chiu CQ, Puente N, Grandes P, Castillo PE. Dopaminergic modulation of endocannabinoid-mediated plasticity at GABAergic synapses in the prefrontal cortex. J Neurosci. 2010;30:7236–7248. doi: 10.1523/JNEUROSCI.0736-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciliax BJ, Heilman C, Demchyshyn LL, Pristupa ZB, Ince E, Hersch SM, Niznik HB, Levey AI. The dopamine transporter: immunochemical characterization and localization in brain. J Neurosci. 1995;15:1714–1723. doi: 10.1523/JNEUROSCI.15-03-01714.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg SJ. Meaningful silences: how dopamine listens to the ACh pause. Trends Neurosci. 2006;29:125–131. doi: 10.1016/j.tins.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Day M, Wang Z, Ding J, An X, Ingham CA, Shering AF, Wokosin D, Ilijic E, Sun Z, Sampson AR, et al. Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat Neurosci. 2006;9:251–259. doi: 10.1038/nn1632. [DOI] [PubMed] [Google Scholar]
- Day M, Wokosin D, Plotkin JL, Tian X, Surmeier DJ. Differential excitability and modulation of striatal medium spiny neuron dendrites. J Neurosci. 2008;28:11603–11614. doi: 10.1523/JNEUROSCI.1840-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado A, Sierra A, Querejeta E, Valdiosera RF, Aceves J. Inhibitory control of the GABAergic transmission in the rat neostriatum by D2 dopamine receptors. Neuroscience. 2000;95:1043–1048. doi: 10.1016/s0306-4522(99)00495-9. [DOI] [PubMed] [Google Scholar]
- Deng P, Zhang Y, Xu ZC. Involvement of I(h) in dopamine modulation of tonic firing in striatal cholinergic interneurons. J Neurosci. 2007;27:3148–3156. doi: 10.1523/JNEUROSCI.5535-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Guzman JN, Tkatch T, Chen S, Goldberg JA, Ebert PJ, Levitt P, Wilson CJ, Hamm HE, Surmeier DJ. RGS4-dependent attenuation of M4 autoreceptor function in striatal cholinergic interneurons following dopamine depletion. Nat Neurosci. 2006;9:832–842. doi: 10.1038/nn1700. [DOI] [PubMed] [Google Scholar]
- Dong Y, White FJ. Dopamine D1-class receptors selectively modulate a slowly inactivating potassium current in rat medial prefrontal cortex pyramidal neurons. J Neurosci. 2003;23:2686–2695. doi: 10.1523/JNEUROSCI.23-07-02686.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Cooper D, Nasif F, Hu XT, White FJ. Dopamine modulates inwardly rectifying potassium currents in medial prefrontal cortex pyramidal neurons. J Neurosci. 2004;24:3077–3085. doi: 10.1523/JNEUROSCI.4715-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Nasif FJ, Tsui JJ, Ju WY, Cooper DC, Hu XT, Malenka RC, White FJ. Cocaine-induced plasticity of intrinsic membrane properties in prefrontal cortex pyramidal neurons: adaptations in potassium currents. J Neurosci. 2005;25:936–940. doi: 10.1523/JNEUROSCI.4715-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumartin B, Doudnikoff E, Gonon F, Bloch B. Differences in ultrastructural localization of dopaminergic D1 receptors between dorsal striatum and nucleus accumbens in the rat. Neurosci Lett. 2007;419:273–277. doi: 10.1016/j.neulet.2007.04.034. [DOI] [PubMed] [Google Scholar]
- English DF, Ibanez-Sandoval O, Stark E, Tecuapetla F, Buzsaki G, Deisseroth K, Tepper JM, Koos T. GABAergic circuits mediate the reinforcement-related signals of striatal cholinergic interneurons. Nat Neurosci. 2011;15:123–130. doi: 10.1038/nn.2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck B, Hillarp NA, Thieme G, Torp A. Fluorescence of catechol amines and related compounds condensed with formaldehyde. J Histochem Cytochem. 1962;10:348–354. doi: 10.1016/0361-9230(82)90113-7. [DOI] [PubMed] [Google Scholar]
- Fisone G. Dopamine Handbook. New York: Oxford University Press; 2010. Intracellular dopamine signaling; pp. 100–117. [Google Scholar]
- Flores-Hernandez J, Hernandez S, Snyder GL, Yan Z, Fienberg AA, Moss SJ, Greengard P, Surmeier DJ. D(1) dopamine receptor activation reduces GABA(A) receptor currents in neostriatal neurons through a PKA/DARPP-32/PP1 signaling cascade. J Neurophysiol. 2000;83:2996–3004. doi: 10.1152/jn.2000.83.5.2996. [DOI] [PubMed] [Google Scholar]
- Flores-Hernandez J, Cepeda C, Hernandez-Echeagaray E, Calvert CR, Jokel ES, Fienberg AA, Greengard P, Levine MS. Dopamine enhancement of NMDA currents in dissociated medium-sized striatal neurons: role of D1 receptors and DARPP-32. J Neurophysiol. 2002;88:3010–3020. doi: 10.1152/jn.00361.2002. [DOI] [PubMed] [Google Scholar]
- Franceschetti S, Taverna S, Sancini G, Panzica F, Lombardi R, Avanzini G. Protein kinase C-dependent modulation of Na+ currents increases the excitability of rat neocortical pyramidal neurones. J Physiol 528 Pt. 2000;2:291–304. doi: 10.1111/j.1469-7793.2000.00291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C, Wolf ME. Dopamine receptors regulate NMDA receptor surface expression in prefrontal cortex neurons. J Neurochem. 2008;106:2489–2501. doi: 10.1111/j.1471-4159.2008.05597.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao WJ, Krimer LS, Goldman-Rakic PS. Presynaptic regulation of recurrent excitation by D1 receptors in prefrontal circuits. Proc Natl Acad Sci U S A. 2001;98:295–300. doi: 10.1073/pnas.011524298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao WJ, Goldman-Rakic PS. Selective modulation of excitatory and inhibitory microcircuits by dopamine. Proc Natl Acad Sci U S A. 2003;100:2836–2841. doi: 10.1073/pnas.262796399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao WJ, Wang Y, Goldman-Rakic PS. Dopamine modulation of perisomatic and peridendritic inhibition in prefrontal cortex. J Neurosci. 2003;23:1622–1630. doi: 10.1523/JNEUROSCI.23-05-01622.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar P, Bloch B, Le Moine C. D1 and D2 receptor gene expression in the rat frontal cortex: cellular localization in different classes of efferent neurons. Eur J Neurosci. 1995;7:1050–1063. doi: 10.1111/j.1460-9568.1995.tb01092.x. [DOI] [PubMed] [Google Scholar]
- Gee S, Ellwood I, Patel T, Luongo F, Deisseroth K, Sohal VS. Synaptic activity unmasks dopamine D2 receptor modulation of a specific class of layer V pyramidal neurons in prefrontal cortex. J Neurosci. 2012;32:4959–4971. doi: 10.1523/JNEUROSCI.5835-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geijo-Barrientos E, Pastore C. The effects of dopamine on the subthreshold electrophysiological responses of rat prefrontal cortex neurons in vitro. Eur J Neurosci. 1995;7:358–366. doi: 10.1111/j.1460-9568.1995.tb00331.x. [DOI] [PubMed] [Google Scholar]
- Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization in the basal ganglia. Annu Rev Neurosci. 1992;15:285–320. doi: 10.1146/annurev.ne.15.030192.001441. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–466. doi: 10.1146/annurev-neuro-061010-113641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German DC, Manaye KF. Midbrain dopaminergic neurons (nuclei A8, A9, and A10): three-dimensional reconstruction in the rat. J Comp Neurol. 1993;331:297–309. doi: 10.1002/cne.903310302. [DOI] [PubMed] [Google Scholar]
- Gertler TS, Chan CS, Surmeier DJ. Dichotomous anatomical properties of adult striatal medium spiny neurons. J Neurosci. 2008;28:10814–10824. doi: 10.1523/JNEUROSCI.2660-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittis AH, Leventhal DK, Fensterheim BA, Pettibone JR, Berke JD, Kreitzer AC. Selective inhibition of striatal fast-spiking interneurons causes dyskinesias. J Neurosci. 2011;31:15727–15731. doi: 10.1523/JNEUROSCI.3875-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladding CM, Raymond LA. Mechanisms underlying NMDA receptor synaptic/extrasynaptic distribution and function. Mol Cell Neurosci. 2011;48:308–320. doi: 10.1016/j.mcn.2011.05.001. [DOI] [PubMed] [Google Scholar]
- Glausier JR, Khan ZU, Muly EC. Dopamine D1 and D5 receptors are localized to discrete populations of interneurons in primate prefrontal cortex. Cereb Cortex. 2009;19:1820–1834. doi: 10.1093/cercor/bhn212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JA, Wilson CJ. Control of spontaneous firing patterns by the selective coupling of calcium currents to calcium-activated potassium currents in striatal cholinergic interneurons. J Neurosci. 2005;25:10230–10238. doi: 10.1523/JNEUROSCI.2734-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JA, Reynolds JN. Spontaneous firing and evoked pauses in the tonically active cholinergic interneurons of the striatum. Neuroscience. 2011;198:27–43. doi: 10.1016/j.neuroscience.2011.08.067. [DOI] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, Heintz N. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- Gong S, Doughty M, Harbaugh CR, Cummins A, Hatten ME, Heintz N, Gerfen CR. Targeting Cre recombinase to specific neuron populations with bacterial artificial chromosome constructs. J Neurosci. 2007;27:9817–9823. doi: 10.1523/JNEUROSCI.2707-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Islas C, Hablitz JJ. Dopamine inhibition of evoked IPSCs in rat prefrontal cortex. J Neurophysiol. 2001;86:2911–2918. doi: 10.1152/jn.2001.86.6.2911. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Islas C, Hablitz JJ. Dopamine enhances EPSCs in layer II-III pyramidal neurons in rat prefrontal cortex. J Neurosci. 2003;23:867–875. doi: 10.1523/JNEUROSCI.23-03-00867.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelova N, Seamans JK, Yang CR. Mechanisms of dopamine activation of fast-spiking interneurons that exert inhibition in rat prefrontal cortex. J Neurophysiol. 2002;88:3150–3166. doi: 10.1152/jn.00335.2002. [DOI] [PubMed] [Google Scholar]
- Gorelova NA, Yang CR. Dopamine D1/D5 receptor activation modulates a persistent sodium current in rat prefrontal cortical neurons in vitro. J Neurophysiol. 2000;84:75–87. doi: 10.1152/jn.2000.84.1.75. [DOI] [PubMed] [Google Scholar]
- Graziane NM, Yuen EY, Yan Z. Dopamine D4 Receptors Regulate GABAA Receptor Trafficking via an Actin/Cofilin/Myosin-dependent Mechanism. J Biol Chem. 2009;284:8329–8336. doi: 10.1074/jbc.M807387200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greif GJ, Lin YJ, Liu JC, Freedman JE. Dopamine-modulated potassium channels on rat striatal neurons: specific activation and cellular expression. J Neurosci. 1995;15:4533–4544. doi: 10.1523/JNEUROSCI.15-06-04533.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge AT, Jaffe DB. Dopamine decreases the excitability of layer V pyramidal cells in the rat prefrontal cortex. J Neurosci. 1998;18:9139–9151. doi: 10.1523/JNEUROSCI.18-21-09139.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge AT, Jaffe DB. Multiple effects of dopamine on layer V pyramidal cell excitability in rat prefrontal cortex. J Neurophysiol. 2001;86:586–595. doi: 10.1152/jn.2001.86.2.586. [DOI] [PubMed] [Google Scholar]
- Gulledge AT, Stuart GJ. Action potential initiation and propagation in layer 5 pyramidal neurons of the rat prefrontal cortex: absence of dopamine modulation. J Neurosci. 2003;23:11363–11372. doi: 10.1523/JNEUROSCI.23-36-11363.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman JN, Hernandez A, Galarraga E, Tapia D, Laville A, Vergara R, Aceves J, Bargas J. Dopaminergic modulation of axon collaterals interconnecting spiny neurons of the rat striatum. J Neurosci. 2003;23:8931–8940. doi: 10.1523/JNEUROSCI.23-26-08931.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakansson K, Galdi S, Hendrick J, Snyder G, Greengard P, Fisone G. Regulation of phosphorylation of the GluR1 AMPA receptor by dopamine D2 receptors. J Neurochem. 2006;96:482–488. doi: 10.1111/j.1471-4159.2005.03558.x. [DOI] [PubMed] [Google Scholar]
- Hallett PJ, Spoelgen R, Hyman BT, Standaert DG, Dunah AW. Dopamine D1 activation potentiates striatal NMDA receptors by tyrosine phosphorylation-dependent subunit trafficking. J Neurosci. 2006;26:4690–4700. doi: 10.1523/JNEUROSCI.0792-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey J, Lacey MG. A postsynaptic interaction between dopamine D1 and NMDA receptors promotes presynaptic inhibition in the rat nucleus accumbens via adenosine release. J Neurosci. 1997;17:5271–5280. doi: 10.1523/JNEUROSCI.17-14-05271.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suarez-Farinas M, Schwarz C, Stephan DA, Surmeier DJ, et al. A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008;135:738–748. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henze DA, Gonzalez-Burgos GR, Urban NN, Lewis DA, Barrionuevo G. Dopamine increases excitability of pyramidal neurons in primate prefrontal cortex. J Neurophysiol. 2000;84:2799–2809. doi: 10.1152/jn.2000.84.6.2799. [DOI] [PubMed] [Google Scholar]
- Hernandez-Echeagaray E, Starling AJ, Cepeda C, Levine MS. Modulation of AMPA currents by D2 dopamine receptors in striatal medium-sized spiny neurons: are dendrites necessary? Eur J Neurosci. 2004;19:2455–2463. doi: 10.1111/j.0953-816X.2004.03344.x. [DOI] [PubMed] [Google Scholar]
- Hernandez-Lopez S, Bargas J, Surmeier DJ, Reyes A, Galarraga E. D1 receptor activation enhances evoked discharge in neostriatal medium spiny neurons by modulating an L-type Ca2+ conductance. J Neurosci. 1997;17:3334–3342. doi: 10.1523/JNEUROSCI.17-09-03334.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Lopez S, Tkatch T, Perez-Garci E, Galarraga E, Bargas J, Hamm H, Surmeier DJ. D2 dopamine receptors in striatal medium spiny neurons reduce L-type Ca2+ currents and excitability via a novel PLC[beta]1-IP3-calcineurin-signaling cascade. J Neurosci. 2000;20:8987–8995. doi: 10.1523/JNEUROSCI.20-24-08987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley MJ, Sabatini BL. Competitive regulation of synaptic Ca(2+) influx by D2 dopamine and A2A adenosine receptors. Nat Neurosci. 2010 doi: 10.1038/nn.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnasko TS, Chuhma N, Zhang H, Goh GY, Sulzer D, Palmiter RD, Rayport S, Edwards RH. Vesicular glutamate transport promotes dopamine storage and glutamate corelease in vivo. Neuron. 2010;65:643–656. doi: 10.1016/j.neuron.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover BR, Marshall JF. Molecular, chemical, and anatomical characterization of globus pallidus dopamine D2 receptor mRNA-containing neurons. Synapse. 2004;52:100–113. doi: 10.1002/syn.20007. [DOI] [PubMed] [Google Scholar]
- Hopf FW, Seif T, Mohamedi ML, Chen BT, Bonci A. The small-conductance calcium-activated potassium channel is a key modulator of firing and long-term depression in the dorsal striatum. Eur J Neurosci. 2010;31:1946–1959. doi: 10.1111/j.1460-9568.2010.07231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu XT, Dong Y, Zhang XF, White FJ. Dopamine D2 receptor-activated Ca2+ signaling modulates voltage-sensitive sodium currents in rat nucleus accumbens neurons. J Neurophysiol. 2005;93:1406–1417. doi: 10.1152/jn.00771.2004. [DOI] [PubMed] [Google Scholar]
- Jocoy EL, Andre VM, Cummings DM, Rao SP, Wu N, Ramsey AJ, Caron MG, Cepeda C, Levine MS. Dissecting the contribution of individual receptor subunits to the enhancement of N-methyl-d-aspartate currents by dopamine D1 receptor activation in striatum. Front Syst Neurosci. 2011;5:28. doi: 10.3389/fnsys.2011.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan ZU, Gutierrez A, Martin R, Penafiel A, Rivera A, De La Calle A. Differential regional and cellular distribution of dopamine D2-like receptors: an immunocytochemical study of subtype-specific antibodies in rat and human brain. J Comp Neurol. 1998;402:353–371. doi: 10.1002/(sici)1096-9861(19981221)402:3<353::aid-cne5>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Kisilevsky AE, Mulligan SJ, Altier C, Iftinca MC, Varela D, Tai C, Chen L, Hameed S, Hamid J, Macvicar BA, Zamponi GW. D1 receptors physically interact with N-type calcium channels to regulate channel distribution and dendritic calcium entry. Neuron. 2008;58:557–570. doi: 10.1016/j.neuron.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Kitai ST, Surmeier DJ. Cholinergic and dopaminergic modulation of potassium conductances in neostriatal neurons. Adv Neurol. 1993;60:40–52. [PubMed] [Google Scholar]
- Kittler JT, Moss SJ. Modulation of GABAA receptor activity by phosphorylation and receptor trafficking: implications for the efficacy of synaptic inhibition. Curr Opin Neurobiol. 2003;13:341–347. doi: 10.1016/s0959-4388(03)00064-3. [DOI] [PubMed] [Google Scholar]
- Kohnomi S, Koshikawa N, Kobayashi M. D(2)-like dopamine receptors differentially regulate unitary IPSCs depending on presynaptic GABAergic neuron subtypes in rat nucleus accumbens shell. J Neurophysiol. 2011;107:692–703. doi: 10.1152/jn.00281.2011. [DOI] [PubMed] [Google Scholar]
- Kotecha SA, Oak JN, Jackson MF, Perez Y, Orser BA, Van Tol HH, MacDonald JF. A D2 class dopamine receptor transactivates a receptor tyrosine kinase to inhibit NMDA receptor transmission. Neuron. 2002;35:1111–1122. doi: 10.1016/s0896-6273(02)00859-0. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature. 2007;445:643–647. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- Kroener S, Chandler LJ, Phillips PE, Seamans JK. Dopamine modulates persistent synaptic activity and enhances the signal-to-noise ratio in the prefrontal cortex. PLoS One. 2009;4:e6507. doi: 10.1371/journal.pone.0006507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroner S, Krimer LS, Lewis DA, Barrionuevo G. Dopamine increases inhibition in the monkey dorsolateral prefrontal cortex through cell type-specific modulation of interneurons. Cereb Cortex. 2007;17:1020–1032. doi: 10.1093/cercor/bhl012. [DOI] [PubMed] [Google Scholar]
- Lammel S, Ion DI, Roeper J, Malenka RC. Projection-specific modulation of dopamine neuron synapses by aversive and rewarding stimuli. Neuron. 2011;70:855–862. doi: 10.1016/j.neuron.2011.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin A, Grace AA. Stimulation of D1-type dopamine receptors enhances excitability in prefrontal cortical pyramidal neurons in a state-dependent manner. Neuroscience. 2001;104:335–346. doi: 10.1016/s0306-4522(01)00096-3. [DOI] [PubMed] [Google Scholar]
- Le Moine C, Gaspar P. Subpopulations of cortical GABAergic interneurons differ by their expression of D1 and D2 dopamine receptor subtypes. Brain Res Mol Brain Res. 1998;58:231–236. doi: 10.1016/s0169-328x(98)00118-1. [DOI] [PubMed] [Google Scholar]
- Lee FJ, Xue S, Pei L, Vukusic B, Chery N, Wang Y, Wang YT, Niznik HB, Yu XM, Liu F. Dual regulation of NMDA receptor functions by direct protein-protein interactions with the dopamine D1 receptor. Cell. 2002;111:219–230. doi: 10.1016/s0092-8674(02)00962-5. [DOI] [PubMed] [Google Scholar]
- Lee SP, So CH, Rashid AJ, Varghese G, Cheng R, Lanca AJ, O’Dowd BF, George SR. Dopamine D1 and D2 receptor Co-activation generates a novel phospholipase C-mediated calcium signal. J Biol Chem. 2004;279:35671–35678. doi: 10.1074/jbc.M401923200. [DOI] [PubMed] [Google Scholar]
- Lester J, Fink S, Aronin N, DiFiglia M. Colocalization of D1 and D2 dopamine receptor mRNAs in striatal neurons. Brain Res. 1993;621:106–110. doi: 10.1016/0006-8993(93)90303-5. [DOI] [PubMed] [Google Scholar]
- Levine MS, Altemus KL, Cepeda C, Cromwell HC, Crawford C, Ariano MA, Drago J, Sibley DR, Westphal H. Modulatory actions of dopamine on NMDA receptor-mediated responses are reduced in D1A-deficient mutant mice. J Neurosci. 1996a;16:5870–5882. doi: 10.1523/JNEUROSCI.16-18-05870.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine MS, Li Z, Cepeda C, Cromwell HC, Altemus KL. Neuromodulatory actions of dopamine on synaptically-evoked neostriatal responses in slices. Synapse. 1996b;24:65–78. doi: 10.1002/syn.890240102. [DOI] [PubMed] [Google Scholar]
- Li YC, Xi D, Roman J, Huang YQ, Gao WJ. Activation of glycogen synthase kinase-3 beta is required for hyperdopamine and D2 receptor-mediated inhibition of synaptic NMDA receptor function in the rat prefrontal cortex. J Neurosci. 2009;29:15551–15563. doi: 10.1523/JNEUROSCI.3336-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YC, Liu G, Hu JL, Gao WJ, Huang YQ. Dopamine D(1) receptor-mediated enhancement of NMDA receptor trafficking requires rapid PKC-dependent synaptic insertion in the prefrontal neurons. J Neurochem. 2010;114:62–73. doi: 10.1111/j.1471-4159.2010.06720.x. [DOI] [PubMed] [Google Scholar]
- Lin JY, Dubey R, Funk GD, Lipski J. Receptor subtype-specific modulation by dopamine of glutamatergic responses in striatal medium spiny neurons. Brain Res. 2003;959:251–262. doi: 10.1016/s0006-8993(02)03757-5. [DOI] [PubMed] [Google Scholar]
- Liu XY, Chu XP, Mao LM, Wang M, Lan HX, Li MH, Zhang GC, Parelkar NK, Fibuch EE, Haines M, et al. Modulation of D2R-NR2B interactions in response to cocaine. Neuron. 2006;52:897–909. doi: 10.1016/j.neuron.2006.10.011. [DOI] [PubMed] [Google Scholar]
- Lobo MK, Covington HE, 3rd, Chaudhury D, Friedman AK, Sun H, Damez-Werno D, Dietz DM, Zaman S, Koo JW, Kennedy PJ, et al. Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science. 2010;330:385–390. doi: 10.1126/science.1188472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet N, Micklem BR, Henny P, Brown MT, Williams C, Bolam JP, Nakamura KC, Magill PJ. Dichotomous organization of the external globus pallidus. Neuron. 2012;74:1075–1086. doi: 10.1016/j.neuron.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda W, Furuta T, Nakamura KC, Hioki H, Fujiyama F, Arai R, Kaneko T. Single Nigrostriatal Dopaminergic Neurons Form Widely Spread and Highly Dense Axonal Arborizations in the Neostriatum. The Journal of Neuroscience. 2009;29:444–453. doi: 10.1523/JNEUROSCI.4029-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice N, Tkatch T, Meisler M, Sprunger LK, Surmeier DJ. D1/D5 dopamine receptor activation differentially modulates rapidly inactivating and persistent sodium currents in prefrontal cortex pyramidal neurons. J Neurosci. 2001;21:2268–2277. doi: 10.1523/JNEUROSCI.21-07-02268.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice N, Mercer J, Chan CS, Hernandez-Lopez S, Held J, Tkatch T, Surmeier DJ. D2 dopamine receptor-mediated modulation of voltage-dependent Na+ channels reduces autonomous activity in striatal cholinergic interneurons. J Neurosci. 2004;24:10289–10301. doi: 10.1523/JNEUROSCI.2155-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momiyama T, Koga E. Dopamine D(2)-like receptors selectively block N-type Ca(2+) channels to reduce GABA release onto rat striatal cholinergic interneurones. J Physiol. 2001;533:479–492. doi: 10.1111/j.1469-7793.2001.0479a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AR, Zhou WL, Potapenko ES, Kim EJ, Antic SD. Brief dopaminergic stimulations produce transient physiological changes in prefrontal pyramidal neurons. Brain Res. 2011;1370:1–15. doi: 10.1016/j.brainres.2010.10.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve KA, Seamans JK, Trantham-Davidson H. Dopamine receptor signaling. J Recept Signal Transduct Res. 2004;24:165–205. doi: 10.1081/rrs-200029981. [DOI] [PubMed] [Google Scholar]
- Nicola SM, Malenka RC. Dopamine depresses excitatory and inhibitory synaptic transmission by distinct mechanisms in the nucleus accumbens. J Neurosci. 1997;17:5697–5710. doi: 10.1523/JNEUROSCI.17-15-05697.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola SM, Malenka RC. Modulation of synaptic transmission by dopamine and norepinephrine in ventral but not dorsal striatum. J Neurophysiol. 1998;79:1768–1776. doi: 10.1152/jn.1998.79.4.1768. [DOI] [PubMed] [Google Scholar]
- Nicola SM, Surmeier J, Malenka RC. Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annu Rev Neurosci. 2000;23:185–215. doi: 10.1146/annurev.neuro.23.1.185. [DOI] [PubMed] [Google Scholar]
- Oldenburg IA, Ding JB. Cholinergic modulation of synaptic integration and dendritic excitability in the striatum. Curr Opin Neurobiol. 2011;21:425–432. doi: 10.1016/j.conb.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco-Cano MT, Bargas J, Hernandez-Lopez S, Tapia D, Galarraga E. Inhibitory action of dopamine involves a subthreshold Cs(+)-sensitive conductance in neostriatal neurons. Exp Brain Res. 1996;110:205–211. doi: 10.1007/BF00228552. [DOI] [PubMed] [Google Scholar]
- Penit-Soria J, Audinat E, Crepel F. Excitation of rat prefrontal cortical neurons by dopamine: an in vitro electrophysiological study. Brain Res. 1987;425:263–274. doi: 10.1016/0006-8993(87)90509-9. [DOI] [PubMed] [Google Scholar]
- Pennartz CM, Dolleman-Van der Weel MJ, Kitai ST, Lopes da Silva FH. Presynaptic dopamine D1 receptors attenuate excitatory and inhibitory limbic inputs to the shell region of the rat nucleus accumbens studied in vitro. J Neurophysiol. 1992;67:1325–1334. doi: 10.1152/jn.1992.67.5.1325. [DOI] [PubMed] [Google Scholar]
- Perez MF, White FJ, Hu XT. Dopamine D(2) receptor modulation of K(+) channel activity regulates excitability of nucleus accumbens neurons at different membrane potentials. J Neurophysiol. 2006;96:2217–2228. doi: 10.1152/jn.00254.2006. [DOI] [PubMed] [Google Scholar]
- Perreault ML, Hasbi A, O’Dowd BF, George SR. The dopamine d1–d2 receptor heteromer in striatal medium spiny neurons: evidence for a third distinct neuronal pathway in Basal Ganglia. Front Neuroanat. 2011;5:31. doi: 10.3389/fnana.2011.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson JD, Wolf ME, White FJ. Repeated amphetamine administration decreases D1 dopamine receptor-mediated inhibition of voltage-gated sodium currents in the prefrontal cortex. J Neurosci. 2006;26:3164–3168. doi: 10.1523/JNEUROSCI.2375-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisani A, Bonsi P, Centonze D, Calabresi P, Bernardi G. Activation of D2-like dopamine receptors reduces synaptic inputs to striatal cholinergic interneurons. J Neurosci. 2000;20:RC69. doi: 10.1523/JNEUROSCI.20-07-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisani A, Martella G, Tscherter A, Bonsi P, Sharma N, Bernardi G, Standaert DG. Altered responses to dopaminergic D2 receptor activation and N-type calcium currents in striatal cholinergic interneurons in a mouse model of DYT1 dystonia. Neurobiol Dis. 2006;24:318–325. doi: 10.1016/j.nbd.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Plotkin JL, Day M, Surmeier DJ. Synaptically driven state transitions in distal dendrites of striatal spiny neurons. Nat Neurosci. 2011;14:881–888. doi: 10.1038/nn.2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu S, Ondo WG, Zhang X, Xie WJ, Pan TH, Le WD. Projections of diencephalic dopamine neurons into the spinal cord in mice. Exp Brain Res. 2006;168:152–156. doi: 10.1007/s00221-005-0075-1. [DOI] [PubMed] [Google Scholar]
- Rankin ML, Hazelwood LA, Free RB, Namkung Y, Rex EB, Roof RA, Sibley DR. Dopamine Handbook. In: Iversen LL, Dunnett SB, Iversen SD, Bjorklund A, editors. Molecular pharmacology of the dopamine receptors. New York: Oxford University Press; 2010. pp. 63–87. [Google Scholar]
- Rivera A, Alberti I, Martin AB, Narvaez JA, de la Calle A, Moratalla R. Molecular phenotype of rat striatal neurons expressing the dopamine D5 receptor subtype. Eur J Neurosci. 2002;16:2049–2058. doi: 10.1046/j.1460-9568.2002.02280.x. [DOI] [PubMed] [Google Scholar]
- Rotaru DC, Lewis DA, Gonzalez-Burgos G. Dopamine D1 receptor activation regulates sodium channel-dependent EPSP amplification in rat prefrontal cortex pyramidal neurons. J Physiol. 2007;581:981–1000. doi: 10.1113/jphysiol.2007.130864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu A, Tyeryar KR, Vongtau HO, Sibley DR, Undieh AS. D5 dopamine receptors are required for dopaminergic activation of phospholipase C. Mol Pharmacol. 2009;75:447–453. doi: 10.1124/mol.108.053017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado H, Tecuapetla F, Perez-Rosello T, Perez-Burgos A, Perez-Garci E, Galarraga E, Bargas J. A reconfiguration of CaV2 Ca2+ channel current and its dopaminergic D2 modulation in developing neostriatal neurons. J Neurophysiol. 2005;94:3771–3787. doi: 10.1152/jn.00455.2005. [DOI] [PubMed] [Google Scholar]
- Santana N, Mengod G, Artigas F. Quantitative analysis of the expression of dopamine D1 and D2 receptors in pyramidal and GABAergic neurons of the rat prefrontal cortex. Cereb Cortex. 2009;19:849–860. doi: 10.1093/cercor/bhn134. [DOI] [PubMed] [Google Scholar]
- Schiffmann SN, Lledo PM, Vincent JD. Dopamine D1 receptor modulates the voltage-gated sodium current in rat striatal neurones through a protein kinase A. J Physiol. 1995;483(Pt 1):95–107. doi: 10.1113/jphysiol.1995.sp020570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seamans JK, Durstewitz D, Christie BR, Stevens CF, Sejnowski TJ. Dopamine D1/D5 receptor modulation of excitatory synaptic inputs to layer V prefrontal cortex neurons. Proc Natl Acad Sci U S A. 2001a;98:301–306. doi: 10.1073/pnas.011518798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seamans JK, Gorelova N, Durstewitz D, Yang CR. Bidirectional dopamine modulation of GABAergic inhibition in prefrontal cortical pyramidal neurons. J Neurosci. 2001b;21:3628–3638. doi: 10.1523/JNEUROSCI.21-10-03628.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seamans JK, Yang CR. The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol. 2004;74:1–58. doi: 10.1016/j.pneurobio.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Seong HJ, Carter AG. D1 receptor modulation of action potential firing in a subpopulation of layer 5 pyramidal neurons in the prefrontal cortex. J Neurosci. 2012;32:10516–10521. doi: 10.1523/JNEUROSCI.1367-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesack SR, Aoki C, Pickel VM. Ultrastructural localization of D2 receptor-like immunoreactivity in midbrain dopamine neurons and their striatal targets. J Neurosci. 1994;14:88–106. doi: 10.1523/JNEUROSCI.14-01-00088.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- Shi WX, Zheng P, Liang XF, Bunney BS. Characterization of dopamine-induced depolarization of prefrontal cortical neurons. Synapse. 1997;26:415–422. doi: 10.1002/(SICI)1098-2396(199708)26:4<415::AID-SYN9>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Skagerberg G, Lindvall O. Organization of diencephalic dopamine neurones projecting to the spinal cord in the rat. Brain Res. 1985;342:340–351. doi: 10.1016/0006-8993(85)91134-5. [DOI] [PubMed] [Google Scholar]
- Skeberdis VA, Chevaleyre V, Lau CG, Goldberg JH, Pettit DL, Suadicani SO, Lin Y, Bennett MV, Yuste R, Castillo PE, Zukin RS. Protein kinase A regulates calcium permeability of NMDA receptors. Nat Neurosci. 2006;9:501–510. doi: 10.1038/nn1664. [DOI] [PubMed] [Google Scholar]
- Snyder GL, Allen PB, Fienberg AA, Valle CG, Huganir RL, Nairn AC, Greengard P. Regulation of phosphorylation of the GluR1 AMPA receptor in the neostriatum by dopamine and psychostimulants in vivo. J Neurosci. 2000;20:4480–4488. doi: 10.1523/JNEUROSCI.20-12-04480.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song WJ, Surmeier DJ. Voltage-dependent facilitation of calcium channels in rat neostriatal neurons. J Neurophysiol. 1996;76:2290–2306. doi: 10.1152/jn.1996.76.4.2290. [DOI] [PubMed] [Google Scholar]
- Stuber GD, Hnasko TS, Britt JP, Edwards RH, Bonci A. Dopaminergic Terminals in the Nucleus Accumbens But Not the Dorsal Striatum Corelease Glutamate. The Journal of Neuroscience. 2010;30:8229–8233. doi: 10.1523/JNEUROSCI.1754-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Zhao Y, Wolf ME. Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J Neurosci. 2005;25:7342–7351. doi: 10.1523/JNEUROSCI.4603-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Milovanovic M, Zhao Y, Wolf ME. Acute and chronic dopamine receptor stimulation modulates AMPA receptor trafficking in nucleus accumbens neurons cocultured with prefrontal cortex neurons. J Neurosci. 2008;28:4216–4230. doi: 10.1523/JNEUROSCI.0258-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun XD, Lee EW, Wong EH, Lee KS. ATP-sensitive potassium channels in freshly dissociated adult rat striatal neurons: activation by metabolic inhibitors and the dopaminergic receptor agonist quinpirole. Pflugers Arch. 2000;440:530–547. doi: 10.1007/s004240000322. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Eberwine J, Wilson CJ, Cao Y, Stefani A, Kitai ST. Dopamine receptor subtypes colocalize in rat striatonigral neurons. Proc Natl Acad Sci U S A. 1992;89:10178–10182. doi: 10.1073/pnas.89.21.10178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Kitai ST. D1 and D2 dopamine receptor modulation of sodium and potassium currents in rat neostriatal neurons. Prog Brain Res. 1993;99:309–324. doi: 10.1016/s0079-6123(08)61354-0. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Bargas J, Hemmings HC, Jr, Nairn AC, Greengard P. Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron. 1995;14:385–397. doi: 10.1016/0896-6273(95)90294-5. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Song WJ, Yan Z. Coordinated expression of dopamine receptors in neostriatal medium spiny neurons. J Neurosci. 1996;16:6579–6591. doi: 10.1523/JNEUROSCI.16-20-06579.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna S, Canciani B, Pennartz CM. Dopamine D1-receptors modulate lateral inhibition between principal cells of the nucleus accumbens. J Neurophysiol. 2005;93:1816–1819. doi: 10.1152/jn.00672.2004. [DOI] [PubMed] [Google Scholar]
- Tecuapetla F, Koos T, Tepper JM, Kabbani N, Yeckel MF. Differential dopaminergic modulation of neostriatal synaptic connections of striatopallidal axon collaterals. J Neurosci. 2009;29:8977–8990. doi: 10.1523/JNEUROSCI.6145-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tecuapetla F, Patel JC, Xenias H, English D, Tadros I, Shah F, Berlin J, Deisseroth K, Rice ME, Tepper JM, Koos T. Glutamatergic signaling by mesolimbic dopamine neurons in the nucleus accumbens. J Neurosci. 2010;30:7105–7110. doi: 10.1523/JNEUROSCI.0265-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepper JM, Tecuapetla F, Koos T, Ibanez-Sandoval O. Heterogeneity and diversity of striatal GABAergic interneurons. Front Neuroanat. 2011;4:150. doi: 10.3389/fnana.2010.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threlfell S, Lalic T, Platt NJ, Jennings KA, Deisseroth K, Cragg SJ. Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron. 2012;75:58–64. doi: 10.1016/j.neuron.2012.04.038. [DOI] [PubMed] [Google Scholar]
- Tong H, Gibb AJ. Dopamine D1 receptor inhibition of NMDA receptor currents mediated by tyrosine kinase-dependent receptor trafficking in neonatal rat striatum. J Physiol. 2008;586:4693–4707. doi: 10.1113/jphysiol.2008.158931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towers SK, Hestrin S. D1-like dopamine receptor activation modulates GABAergic inhibition but not electrical coupling between neocortical fast-spiking interneurons. J Neurosci. 2008;28:2633–2641. doi: 10.1523/JNEUROSCI.5079-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tozzi A, de Iure A, Di Filippo M, Tantucci M, Costa C, Borsini F, Ghiglieri V, Giampa C, Fusco FR, Picconi B, Calabresi P. The distinct role of medium spiny neurons and cholinergic interneurons in the D(2)/A(2)A receptor interaction in the striatum: implications for Parkinson’s disease. J Neurosci. 2011;31:1850–1862. doi: 10.1523/JNEUROSCI.4082-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trantham-Davidson H, Neely LC, Lavin A, Seamans JK. Mechanisms underlying differential D1 versus D2 dopamine receptor regulation of inhibition in prefrontal cortex. J Neurosci. 2004;24:10652–10659. doi: 10.1523/JNEUROSCI.3179-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trantham-Davidson H, Kroner S, Seamans JK. Dopamine modulation of prefrontal cortex interneurons occurs independently of DARPP-32. Cereb Cortex. 2008;18:951–958. doi: 10.1093/cercor/bhm133. [DOI] [PubMed] [Google Scholar]
- Tritsch NX, Ding JB, Sabatini BL. Dopaminergic neurons inhibit striatal output via non-canonical release of GABA. Nature. 2012 doi: 10.1038/nature11466. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudeau LE, Emery DG, Haydon PG. Direct modulation of the secretory machinery underlies PKA-dependent synaptic facilitation in hippocampal neurons. Neuron. 1996;17:789–797. doi: 10.1016/s0896-6273(00)80210-x. [DOI] [PubMed] [Google Scholar]
- Tseng KY, O’Donnell P. Dopamine-glutamate interactions controlling prefrontal cortical pyramidal cell excitability involve multiple signaling mechanisms. J Neurosci. 2004;24:5131–5139. doi: 10.1523/JNEUROSCI.1021-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchimura N, North RA. Actions of cocaine on rat nucleus accumbens neurones in vitro. Br J Pharmacol. 1990;99:736–740. doi: 10.1111/j.1476-5381.1990.tb12999.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Bertran-Gonzalez J, Herve D, Fisone G, Girault JA. Looking BAC at striatal signaling: cell-specific analysis in new transgenic mice. Trends Neurosci. 2009;32:538–547. doi: 10.1016/j.tins.2009.06.005. [DOI] [PubMed] [Google Scholar]
- Vergara R, Rick C, Hernandez-Lopez S, Laville JA, Guzman JN, Galarraga E, Surmeier DJ, Bargas J. Spontaneous voltage oscillations in striatal projection neurons in a rat corticostriatal slice. J Physiol. 2003;553:169–182. doi: 10.1113/jphysiol.2003.050799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilchis C, Bargas J, Ayala GX, Galvan E, Galarraga E. Ca2+ channels that activate Ca2+-dependent K+ currents in neostriatal neurons. Neuroscience. 2000;95:745–752. doi: 10.1016/s0306-4522(99)00493-5. [DOI] [PubMed] [Google Scholar]
- Wang H, Pickel VM. Dopamine D2 receptors are present in prefrontal cortical afferents and their targets in patches of the rat caudate-putamen nucleus. J Comp Neurol. 2002;442:392–404. doi: 10.1002/cne.10086. [DOI] [PubMed] [Google Scholar]
- Wang W, Dever D, Lowe J, Storey GP, Bhansali A, Eck EK, Nitulescu I, Weimer J, Bamford NS. Regulation of prefrontal excitatory neurotransmission by dopamine in the nucleus accumbens core. J Physiol. 2012 doi: 10.1113/jphysiol.2012.235200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhong P, Yan Z. Dopamine D4 receptors modulate GABAergic signaling in pyramidal neurons of prefrontal cortex. J Neurosci. 2002;22:9185–9193. doi: 10.1523/JNEUROSCI.22-21-09185.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhong P, Gu Z, Yan Z. Regulation of NMDA receptors by dopamine D4 signaling in prefrontal cortex. J Neurosci. 2003;23:9852–9861. doi: 10.1523/JNEUROSCI.23-30-09852.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Goldman-Rakic PS. D2 receptor regulation of synaptic burst firing in prefrontal cortical pyramidal neurons. Proc Natl Acad Sci U S A. 2004;101:5093–5098. doi: 10.1073/pnas.0400954101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Markram H, Goodman PH, Berger TK, Ma J, Goldman-Rakic PS. Heterogeneity in the pyramidal network of the medial prefrontal cortex. Nat Neurosci. 2006;9:534–542. doi: 10.1038/nn1670. [DOI] [PubMed] [Google Scholar]
- Wickens J, Arbuthnott G. Structural and functional interactions in the striatum at the receptor level. In: Dunnett SB, Bentivoglio M, Bjorklund A, Hokfelt T, editors. Dopamine. San Diego: Elsevier; 2005. pp. 199–236. [Google Scholar]
- Wilson CJ. Basal Ganglia. In: Shepherd GM, editor. The synaptic organization of the brain. New York: Oxford University Press; 2004. pp. 361–415. [Google Scholar]
- Witkowski G, Szulczyk B, Rola R, Szulczyk P. D(1) dopaminergic control of G protein-dependent inward rectifier K(+) (GIRK)-like channel current in pyramidal neurons of the medial prefrontal cortex. Neuroscience. 2008;155:53–63. doi: 10.1016/j.neuroscience.2008.05.021. [DOI] [PubMed] [Google Scholar]
- Witten IB, Lin SC, Brodsky M, Prakash R, Diester I, Anikeeva P, Gradinaru V, Ramakrishnan C, Deisseroth K. Cholinergic interneurons control local circuit activity and cocaine conditioning. Science. 2010;330:1677–1681. doi: 10.1126/science.1193771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Hablitz JJ. Cooperative activation of D1 and D2 dopamine receptors enhances a hyperpolarization-activated inward current in layer I interneurons. J Neurosci. 2005;25:6322–6328. doi: 10.1523/JNEUROSCI.1405-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu TX, Yao WD. D1 and D2 dopamine receptors in separate circuits cooperate to drive associative long-term potentiation in the prefrontal cortex. Proc Natl Acad Sci U S A. 2010;107:16366–16371. doi: 10.1073/pnas.1004108107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Song WJ, Surmeier J. D2 dopamine receptors reduce N-type Ca2+ currents in rat neostriatal cholinergic interneurons through a membrane-delimited, protein-kinase-C-insensitive pathway. J Neurophysiol. 1997;77:1003–1015. doi: 10.1152/jn.1997.77.2.1003. [DOI] [PubMed] [Google Scholar]
- Yan Z, Surmeier DJ. D5 dopamine receptors enhance Zn2+-sensitive GABA(A) currents in striatal cholinergic interneurons through a PKA/PP1 cascade. Neuron. 1997;19:1115–1126. doi: 10.1016/s0896-6273(00)80402-x. [DOI] [PubMed] [Google Scholar]
- Yan Z, Hsieh-Wilson L, Feng J, Tomizawa K, Allen PB, Fienberg AA, Nairn AC, Greengard P. Protein phosphatase 1 modulation of neostriatal AMPA channels: regulation by DARPP-32 and spinophilin. Nat Neurosci. 1999;2:13–17. doi: 10.1038/4516. [DOI] [PubMed] [Google Scholar]
- Yang CR, Seamans JK. Dopamine D1 receptor actions in layers V-VI rat prefrontal cortex neurons in vitro: modulation of dendritic-somatic signal integration. J Neurosci. 1996;16:1922–1935. doi: 10.1523/JNEUROSCI.16-05-01922.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HH, Lovinger DM. Frequency-specific and D2 receptor-mediated inhibition of glutamate release by retrograde endocannabinoid signaling. Proc Natl Acad Sci U S A. 2006;103:8251–8256. doi: 10.1073/pnas.0510797103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young CE, Yang CR. Dopamine D1/D5 receptor modulates state-dependent switching of soma-dendritic Ca2+ potentials via differential protein kinase A and C activation in rat prefrontal cortical neurons. J Neurosci. 2004;24:8–23. doi: 10.1523/JNEUROSCI.1650-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XF, Hu XT, White FJ. Whole-cell plasticity in cocaine withdrawal: reduced sodium currents in nucleus accumbens neurons. J Neurosci. 1998;18:488–498. doi: 10.1523/JNEUROSCI.18-01-00488.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XF, Cooper DC, White FJ. Repeated cocaine treatment decreases whole-cell calcium current in rat nucleus accumbens neurons. J Pharmacol Exp Ther. 2002;301:1119–1125. doi: 10.1124/jpet.301.3.1119. [DOI] [PubMed] [Google Scholar]
- Zhang ZW, Burke MW, Calakos N, Beaulieu JM, Vaucher E. Confocal Analysis of Cholinergic and Dopaminergic Inputs onto Pyramidal Cells in the Prefrontal Cortex of Rodents. Front Neuroanat. 2010;4:21. doi: 10.3389/fnana.2010.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng P, Zhang XX, Bunney BS, Shi WX. Opposite modulation of cortical N-methyl-D-aspartate receptor-mediated responses by low and high concentrations of dopamine. Neuroscience. 1999;91:527–535. doi: 10.1016/s0306-4522(98)00604-6. [DOI] [PubMed] [Google Scholar]
- Zhou FM, Hablitz JJ. Dopamine modulation of membrane and synaptic properties of interneurons in rat cerebral cortex. J Neurophysiol. 1999;81:967–976. doi: 10.1152/jn.1999.81.3.967. [DOI] [PubMed] [Google Scholar]
- Zhou WL, Antic SD. Rapid Dopaminergic and GABA-ergic Modulation of Calcium and Voltage Transients in Dendrites of Prefrontal Cortex Pyramidal Neurons. J Physiol. 2012 doi: 10.1113/jphysiol.2011.227157. [DOI] [PMC free article] [PubMed] [Google Scholar]