

Abstract
Objective
The NLRP3 (NALP3, cryopyrin) inflammasome, a key component of the innate immune system, facilitates caspase-1 and interleukin (IL)–1β processing, which amplifies the inflammatory response. Here, we investigated whether NLRP3 knockdown decreases neutrophil infiltration, reduces brain edema, and improves neurological function in an intracerebral hemorrhage (ICH) mouse model. We also determined whether mitochondrial reactive oxygen species (ROS) governed by mitochondrial permeability transition pores (mPTPs) would trigger NLRP3 inflammasome activation following ICH.
Methods
ICH was induced by injecting autologous arterial blood (30μl) into a mouse brain. NLRP3 small interfering RNAs were administered 24 hours before ICH. A mPTP inhibitor (TRO-19622) or a specific mitochondria ROS scavenger (Mito-TEMPO) was coinjected with the blood. In naive animals, rotenone, which is a respiration chain complex I inhibitor, was applied to induce mitochondrial ROS production, and followed by TRO-19622 or Mito-TEMPO treatment. Neurological deficits, brain edema, enzyme-linked immunosorbent assay, Western blot, in vivo chemical cross-linking, ROS assay, and immunofluorescence were evaluated.
Results
ICH activated the NLRP3 inflammasome. NLRP3 knockdown reduced brain edema and decreased myeloper-oxidase (MPO) levels at 24 hours, and improved neurological functions from 24 to 72 hours following ICH. TRO-19622 or Mito-TEMPO reduced ROS, NLRP3 inflammasome components, and MPO levels following ICH. In naive animals, rotenone administration induced mPTP formation, ROS generation, and NLRP3 inflammasome activation, which were then reduced by TRO-19622 or Mito-TEMPO.
Interpretation
The NLRP3 inflammasome amplified the inflammatory response by releasing IL-1β and promoting neutrophil infiltration following ICH. Mitochondria ROS may be a major trigger of NLRP3 inflammasome activation. The results of our study suggest that the inhibition of the NLRP3 inflammasome may effectively reduce the inflammatory response following ICH.
Spontaneous intracerebral hemorrhage (ICH) is a devastating disease, accounting for 15 to 20% of all stroke types.1 There is currently no effective treatment for ICH.2,3 Increasing evidence indicates that inflammation mechanisms play a critical role in the pathophysiology of ICH.4 After ICH, blood components, including blood-borne leukocytes, enter into the brain parenchyma and activate resident immune cells, such as microglia. Leukocyte infiltration and activation of microglia enhance the production of proinflammatory cytokines. Among all the cytokines, interleukin (IL)–1β is regarded as a pivotal therapeutic target in ICH,5 as the observations demonstrated that the overexpression of the IL-1β antagonist via an adenovirus vector reduced brain edema and thrombin-induced inflammation in the ICH rat model.6 Our previous work has shown that the inhibition of caspase-1, the converting enzyme of active IL-1β, reduced ICH induced brain injury.7 However, how IL-1β is processed following ICH remains unclear.
Recently, several lines of evidence have suggested that inflammasome, one of the components of the innate immune system, is involved in the pathogenesis of sterile inflammatory response by processing caspase-1 and IL-1β to an active stage following human central nervous system (CNS) disorders (such as spinal cord injury,8 traumatic brain injury,9 and ischemic stroke10). Among 20 members of the human NLR protein family that have been reported, the NLRP3 (NALP3, cryopyrin) inflammasome gains notable attention, as it can sense multiple stimulus, including tissue injury and microbial invasion.11 The NLRP3 inflammasome contains NLR family, pyrin domain containing 3 (NLRP3), which is associated with the apoptosis-associated specklike protein containing a caspase recruitment domain (ASC), which recruits and activates caspase-1, therefore releasing cleaved IL-1β.12,13
In the present study, we investigated the role of the NLRP3 inflammasome in ICH-induced inflammation, especially the mechanism of its activation. We hypothesized that the NLRP3 inflammasome may respond to ICH injury, and enhance inflammatory response by facilitating caspase-1 activation and IL-1β processing. NLRP3 activation after ICH may be triggered by mitochondrial reactive oxygen species ROS (mROS), which are regulated by mitochondrial permeability transition pores (mPTPs; Supplementary Fig 2). To test this hypothesis, we first investigated the expression profiles of the NLRP3 inflammasome components, including NLRP3, caspase-1, and IL-1β, after ICH. Next, we applied small interfering RNAs (siRNAs) to knock down NLRP3 in vivo, and measured the NLRP3 component alterations as well as the functional outcomes. Additionally, specific mitochondrial ROS scavenger (Mito-TEMPO), or mPTP inhibitor (TRO-19622) was administered following ICH. In naive animals, a respiration chain complex I inhibitor, rotenone, was introduced into the brain followed by a mROS scavenger or mPTP inhibitor to determine the role of mPTP on the regulation of ROS in NLRP3 inflammasome activation.
Materials and Methods
Animals
All procedures for this study were approved by the institutional animal care and use committee at Loma Linda University. Eight-week-old male CD1 mice (weight = ∼30g, Charles River, Wilmington, MA) were housed in a 12-hour light/dark cycle at a controlled temperature and humidity with free access to food and water.
ICH Mouse Model
The general procedures for inducing ICH in the mouse using autologous arterial blood injection into the basal ganglia were performed as described in previous publications.14,15 The details are also described in the Supplementary Text and Supplementary Figure 1A.
Experimental Design
Experiments 1 and 2 were conducted in autologous arterial blood injection models, and experiment 3 was conducted in naive animals.
Experiment 1
The NLRP3 siRNAs mixture was administered intraventricularly (100pmol in 2μl) 24 hours before ICH. Vehicle animals were given the same volume injection, but with scramble siRNA (siCtrl). Samples for Western blot, enzyme-linked immunosorbent assay (ELISA), and immunostaining were collected 12 hours after ICH. Neurological deficits and brain edema were measured at 24 and 72 hours.
Experiment 2
TRO-19622 (mPTP inhibitor, 2μg/2μl per mouse)16,17 or Mito-TEMPO (mitochondrial specific ROS scavenger, 20μg/2μl per mouse)16 was simultaneously injected with autologous blood into the right basal ganglia. Vehicle groups received the same volume of phosphate-buffered saline injection. Some animals were injected with disuccinimidyl suberate (DSS)16 at a concentration of 5mM by intracerebroventricular (ICV) injection in the contralateral hemisphere 11 hours following ICH (for details see in vivo chemical cross-linking assay). Samples for Western blot, ELISA, ROS assay, and immunostaining were collected 12 hours after ICH.
Experiment 3
Rotenone (mitochondrial complex I inhibitor, 0.4μg/2μl per mouse)18 was administered into the right basal ganglia of naive animals. In some animals, TRO-19622 (mPTP inhibitor, 2μg/2μl per mouse) or Mito-TEMPO (mROS scavenger, 20μg/2μl per mouse) was simultaneously injected with rotenone into the right basal ganglia. In vivo DSS chemical cross-linking experiments were also conducted. Samples for Western blot, ELISA, and ROS assay were collected at 1 hour or 4 hours following injection.
NLRP3 siRNA Injection
Two different formats of NLRP3 siRNA were applied, according to the manufacturer's instructions, to enhance the silencing effect. For more details see Supplementary Text.
Hematoma Volume
Hematoma volume was analyzed by hemoglobin assay 24 hours after ICH operation. For more details see Supplementary Text.
Neurobehavioral Function Test
Neurobehavioral functions were evaluated by the modified Garcia test for the sensorimotor deficits as previously described.19 Four items, including side stroke, vibrissae touch, limb symmetry, and lateral turning, were tested with a maximum neurological score of 12, which represented healthy animals. The neurobehavioral function test was conducted at different time intervals following ICH induction by an investigator blinded to the study.
Western Blotting
The primary antibodies included anti-NLRP3 (1:1,000; Adipo-Gen, San Diego, CA), anti–caspase-1 p20 (1:1,000; Millipore, Billerica, MA), anti-MPO (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA), anti-VDAC (1:1,000; Millipore), and anti-CypD (1:1,000; Millipore). For more details see Supplementary Text.
ELISA Assay
Brain samples were collected at the indicated times and assayed with a Mouse IL-1β/IL-1F2 Quantikine ELISA Kit (R&D Systems, Minneapolis, MN).
ROS Assay
Brain samples were collected at the indicated times. Total ROS in brains were measured with the Oxiselect in vitro ROS/RNS assay kit (Cell Biolabs, San Diego, CA), following the manufacturer's instructions. Fluorescence of dichlorofluorescein was measured using a microplate reader and normalized to sham animals.
Immunofluorescence
The details are described in the Supplementary Text.
In Vivo Chemical Cross-Linking Assay
The formation of mPTP was detected by chemical cross-linking in vivo as described in previous studies.16 DSS (Pierce, Rock-ford, IL) at a concentration of 5mM was administered using ICV injection in the contralateral hemisphere 11 hours following ICH. Mice remained alive for 1 hour after DSS injection and were then sacrificed by decapitation. The brain from each mouse was removed quickly from the cranium and placed in an ice-cold Hanks buffer. Samples were rinsed briefly in ice-cold 50mM Tris-glycine buffer, then lysed, homogenized, and evaluated for voltage-dependent anion channel (VDAC) oligomerization by Western blotting.
Statistics
Data were expressed as the mean ± standard error of the mean. Analysis was performed using GraphPad (La Jolla, CA) Prism software. The details are described in the Supplementary Text.
Results
NLRP3, Cleaved Caspase-1, and IL-1β Were Upregulated following ICH Injury
We first investigated whether the NLRP3 inflammasome components would respond to brain injury following blood injection. Western blot results showed that NLRP3 levels were significantly increased at 3 hours, and reached a peak at around 12 hours after ICH, whereas NLRP3 levels were about 3× those of the sham group (p< 0.05; Fig 1). Accordingly, cleaved caspase-1 and IL-1β, the products of NLRP3 inflammasome activation, were significantly increased at 3 hours (p < 0.05) after ICH and peaked at 12 hours. Following this peak, NLRP3, cleaved caspase-1, and IL-1β levels declined, but still remained at high levels at 72 hours when compared with sham animals. Double immunostaining showed that NLRP3 is mainly expressed in microglia cells and not in other cell types, such as astrocytes (data not shown).
Figure 1.
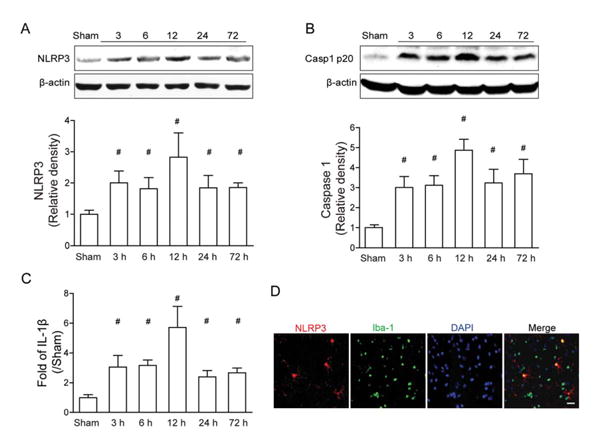
Expression profile of the NLRP3 inflammasome components after autologous arterial blood–induced intracerebral hemorrhage (ICH). (A–C) Western blot assay for the expression profiles of NLRP3 (A) and caspase-1 p20 subunit (B), and enzyme-linked immunosorbent assay assay for the expression profiles of interleukin (IL)–1β (C) in the ipsilateral hemisphere in sham and ICH mice at 3, 6, 12, 24, and 72 hours following operation; n=6 mice per group and per time point. Error bars represent mean±standard error of the mean. #p<0.05 vs sham. (D) Representative photographs of immunofluorescence staining for NLRP3 (green) expression in microglia (Iba-1, red) in the perihematomal area 12 hours following ICH. Scale bar=50μm.
NLRP3 siRNA Injection Knocked Down NLRP3 Level as Well as Cleaved Caspase-1 and IL-1β following ICH Injury
To investigate the potential role of the NLRP3 inflammasome in ICH-induced brain injury, 2 siRNAs were mixed and administered by ICV injection 24 hours before ICH surgery. After 12 hours, Western blot results showed that NLRP3 and cleaved caspase-1 levels were significantly reduced by NLRP3 siRNA injection when compared to ICH without treatment and scramble siRNA (ICH+siCtrl) injection animals (p<0.05; Fig 2). Accordingly, IL-1β levels detected by ELISA were also markedly reduced compared to ICH without treatment and siCtrl animals (p< 0.05). At 24 hours, hematoma volume was detected by hemoglobin assay, and there were no differences between the ICH group and the NLRP3 siRNA injection group (see Supplementary Fig 1B).
Figure 2.
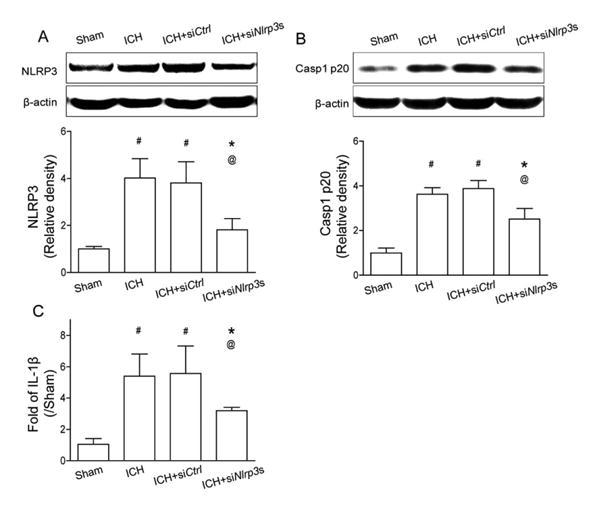
NLRP3 small interfering RNA mixture knocked down the NLRP3 inflammasome components at 12 hours following intracerebral hemorrhage (ICH). (A) Western blot assay for the expression of NLRP3 and (B) caspase-1 p20 subunit, and (C) enzyme-linked immunosorbent assay for the expression of interleukin (IL)–1β in the ipsilateral hemisphere in sham, ICH, scrambled siRNA (ICH+siCtrl), and NLRP3 siRNA mixture (ICH+siNlrp3s) groups at 12 hours following operation; n=6 mice per group. Error bars represent mean±standard error of the mean. #p<0.05 vs sham, *p<0.05 vs ICH, @p<0.05 vs ICH+ siCtrl.
NLRP3 Knockdown Improved Neurobehavioral Functions, Reduced Brain Edema, and Neutrophil Infiltration
The results at 24 and 72 hours showed that ICH animals demonstrated severe deficits compared to sham animals in the modified Garcia test (p <0.05; Fig 3). Following NLRP3 siRNA mixture administration, there was a significant improvement in the modified Garcia test at both 24 and 72 hours compared to ICH and siCtrl mice (p < 0.05). Consistent with neurological improvement, the brain edema in the ipsilateral basal ganglia (ipsi-BG) after NLRP3 siRNA injection was also significantly reduced at both 24 (ipsi-BG: 80.88±0.29% vs ICH, 82.38±0.33%, p < 0.05; vs siCtrl, 82.68±0.35%, p < 0.05) and 72 hours (ipsi-BG: 81.64±0.30% vs ICH, 83.17±0.24%, p < 0.05; vs siCtrl, 82.90±0.43%, p < 0.05) compared to ICH and siCtrl. With regard to the brain edema in the ipsilateral cortex (ipsi-CX), it was only significantly increased at 24 hours (ipsi-CX: sham, 79.12 ± 0.21% vs ICH, 80.22±0.26%, p < 0.05), but not at 72 hours (ipsi-CX: sham, 79.59±0.20% vs ICH, 80.27±0.13%, p > 0.05), and the siRNA treatment failed to reduce its level at either 24 hours (ipsi-CX: 80. 02 ± 0.20% vs ICH, 80.22±0.26%, p > 0.05; vs siCtrl, 79.62±0.82%, p > 0.05) or 72 hours (ipsi-CX: 79.52 ± 0.09% vs ICH, 80.27±0.13%, p > 0.05; vs siCtrl, 79.92±0.32%, p > 0.05).
Figure 3.
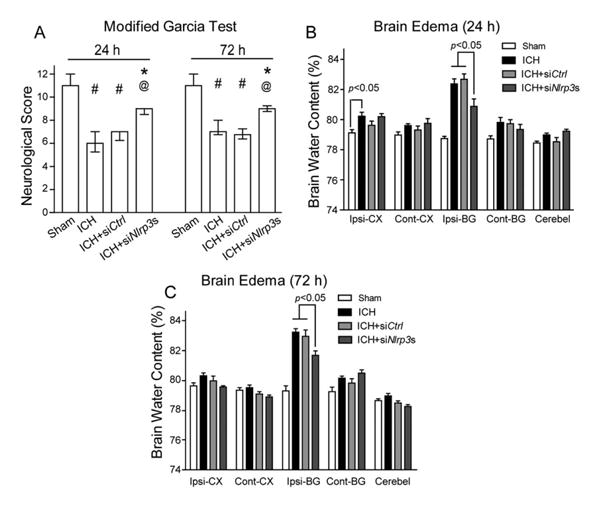
NLRP3 knockdown improved neurological functions and reduced brain edema at 24 and 72 hours following intracerebral hemorrhage (ICH). (A) Modified Garcia test at 24 and 72 hours following operation in sham, ICH, scrambled small interfering RNA (siRNA; ICH+siCtrl), and NLRP3 siRNA mixture (ICH+siNlrp3s) groups at 12 hours following operation. (B, C) Brain edema at 24 hours (B) and 72 hours (C) following operation in sham, ICH, scrambled siRNA (ICH+siCtrl), and NLRP3 siRNA mixture (ICH+siNlrp3s) groups at 12 hours following operation. Brain sections (4mm) were divided into 4 parts: ipsilateral basal ganglia (Ipsi-BG), ipsilateral cortex (Ipsi-CX), contralateral basal ganglia (Cont-BG), and contralateral cortex (Cont-CX). Cerebellum (Cerebel) is the internal control; n=6–13 mice per group. Error bars represent median±25th to 75th percen-tiles (A) or mean±standard error of the mean (B, C). #p<0.05 vs sham, *p<0.05 vs ICH, @p<0.01 vs ICH+siCtrl.
To determine the effect of NLRP3 on neutrophil infiltration, we detected myeloperoxidase (MPO) levels in brain tissue by Western blot as well as immunostaining 24 hours following ICH. Western blot results showed that MPO levels in the ipsilateral hemisphere significantly increased following ICH but were markedly reduced after NLRP3 siRNA treatment compared to ICH or siCtrl mice (p < 0.05; Fig 4A). Consistently, results from immunostaining indicated that NLRP3 siRNA treatment significantly reduced the number of MPO-positive cells in the perihematomal area compared to ICH or siCtrl mice (p < 0.05; see Fig 4B, D).
Figure 4.
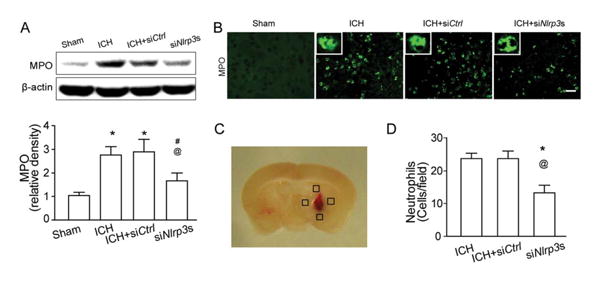
NLRP3 knockdown–attenuated neutrophils infiltration at 24 hours following intracerebral hemorrhage (ICH). (A) Western blot assay for the myeloperoxidase (MPO) level in the ipsilateral hemisphere in sham, ICH, scrambled small interfering RNA (siRNA; ICH+siCtrl), and NLRP3 siRNA mixture (ICH+siNlrp3s) groups at 12 hours following operation; n=9 mice per group. Error bars represent mean±standard error of the mean. *p<0.05 vs sham; #p<0.05 vs ICH; @p<0.05 vs ICH+siCtrl. (B) Representative photographs of immunofluorescence staining for MPO-positive cells (green) in the perihematomal area in sham, ICH, scrambled siRNA (ICH+siCtrl), and NLRP3 siRNA mixture (ICH+siNlrp3s) groups 12 hours following ICH. Scale bar=50μm. (C) The schematic diagram shows the 4 areas (black squares) for the MPO-positive cell counting in the perihemato-mal region. (D) Bar graph illustrates the quantification of MPO-positive cells in the perihematomal region in ICH, scrambled siRNA (ICH+siCtrl), and NLRP3 siRNA mixture (ICH+siNlrp3s) groups at 24 hours following operation (12 fields/brain); n=6 mice per group. Error bars represent mean±standard error of the mean. *p<0.05 vs ICH, @p<0.01 vs ICH+siCtrl. [Color figure can be viewed in the online issue, which is available at www.annalsofneurology.org.]
mPTP Formed and mROS Increased following ICH, Which Was Reversed by VDAC Inhibitor or mROS-Specific Scavenger
We tested whether the mPTP components expressed themselves in brain tissue and whether the mPTP formed following ICH. Our Western blot results showed that both VDAC and cyclophilin D (CypD) monomers were expressed in brain tissue, but their protein levels did not change in either the ipsilateral hemisphere or the contra-lateral hemisphere 12 hours after ICH (Fig 5A). With regard to mPTP formation, we examined VDAC dimer formation by using in vivo chemical cross-linking with DSS injection.16 Western blotting of cross-linked brain tissue from sham and ICH showed that 30kDa monomeric VDAC was the predominant form in both the ICH and sham samples, whereas the dimer form of approximately 60kDa20,21 was increased in the ipsilateral hemisphere in ICH animals compared to the contralateral hemisphere and sham with DSS injection (see Fig 5B).
Figure 5.
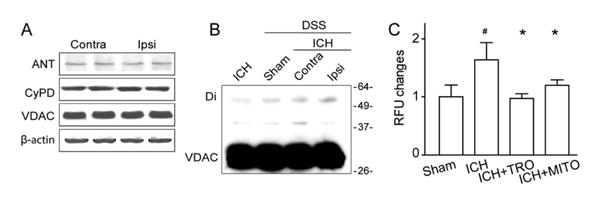
Mitochondrial reactive oxygen species ROS (mROS) production was associated with the formation of mitochondrial permeability transition pore (mPTP) protein assemblies. (A) Western blot assay for the 3 core components of the mPTP (voltage-dependent anion channel [VDAC], adenine nucleotide translocase [ANT], and cyclophilin D [CypD]) in the ipsilateral (Ipsi) versus contralateral (Contra) hemisphere at 12 hours following intracerebral hemorrhage (ICH); n=4 mice per group. (B) Western blots for VDAC in ICH without or with chemical cross-linker (disuccinimidyl suberate [DSS]) at 12 hours following operation. Monomeric VDAC is located at 30kDa. Dimeric (Di) VDAC is detected around 60kDa. (C) Quantification of ROS changes in the perihematomal region in sham, ICH, ICH+TRO-19622 (ICH+TRO), and ICH+Mito-TEMPO (ICH+MITO) groups at 12 hours following operation; n=6 mice per group. Error bars represent mean±standard error of the mean. #p<0.05 vs sham, *p<0.05 vs ICH. RFU=relative fluorescence units.
The effects of pharmacological inactivation of the mPTP on mROS production were assessed by intraventricular injection of TRO-19622, a VDAC inhibitor, and Mito-TEMPO, a mitochondrial ROS-specific scavenger. ROS assay showed that ROS was significantly increased 12 hours following ICH, and both TRO-19622 and Mito-TEMPO significantly reduced ROS levels (p < 0.05; see Fig 5C).
mPTP Suppression or mROS Scavenging Attenuated NLRP3, Cleaved Caspase-1, and Active IL-1β Levels as Well as Neutrophil Infiltration following ICH
Western blot showed that both TRO-19622 and Mito-TEMPO significantly reduced NLRP3 and cleaved caspase-1 levels (Fig 6). Similarly, ELISA showed that IL-1β was also significantly reduced compared to ICH groups (p < 0.05).
Figure 6.
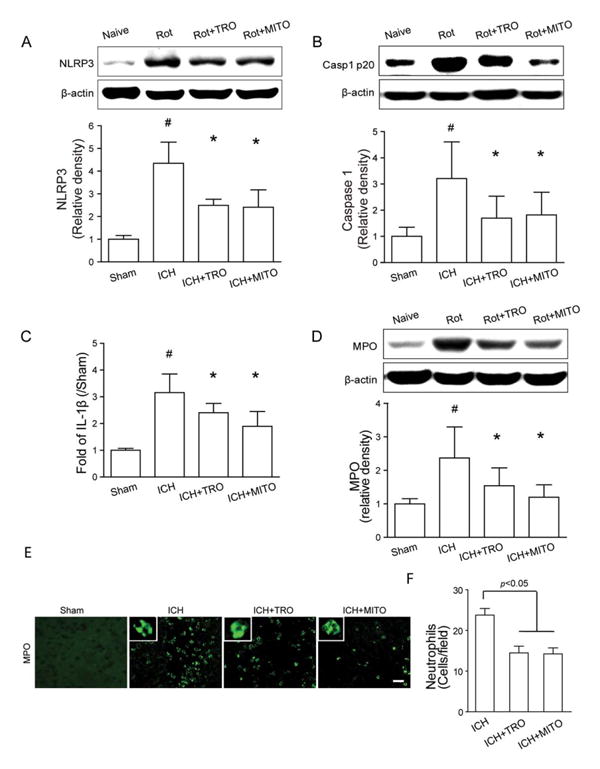
Mitochondrial reactive oxygen species inhibition reduced NLRP3 inflammasome components expression and neutrophil infiltration following intracerebral hemorrhage (ICH). (A–C) Western blot assay for the expression of NLRP3 (A) and caspase-1 p20 subunit (B), and enzyme-linked immunosorbent assay for the expression of interleukin (IL)–1β (C) in the ipsilateral hemisphere in sham, ICH, ICH+TRO-19622 (ICH+TRO), and ICH+Mito-TEMPO (ICH+MITO) groups at 12 hours following operation; n=6 mice per group. Rot=rotenone injection. Error bars represent mean±standard error of the mean. #p<0.05 vs sham, *p<0.05 vs ICH. (D) Western blot assay for the myeloperoxidase (MPO) level in the ipsilateral hemisphere in sham, ICH, ICH+TRO, and ICH+MITO groups at 12 hours following operation; n=6 mice per group. Error bars represent mean±standard error of the mean. #p<0.05 vs sham, *p<0.05 vs ICH. (E) Representative photographs of immunofluorescence staining for MPO-positive cells (green) in the perihematomal area in sham, ICH, ICH+TRO, and ICH+MITO groups at 12 hours following operation. Scale bar= 50μm. (F) Bar graph illustrating the quantification of MPO-positive cells in the perihematomal region in ICH, ICH+TRO, and ICH+MITO groups at 24 hours following operation (12 fields/brain); n=6 mice per group. Error bars represent mean±standard error of the mean. [Color figure can be viewed in the online issue, which is available at www.annalsofneurology.org.]
Similar to the NLRP3 siRNA injection, the Western blot showed that mPTP inhibition and Mito-TEMPO significantly reduced MPO levels in brain tissue 24 hours following ICH (see Fig 6D). Consistently, results from immunostaining indicated that the number of MPO-positive cells in the perihematomal area was significantly reduced compared to the number in ICH mice (p < 0.05; see Fig 6E, F).
mROS Induced by Rotenone Promoted mPTP Formation in Naive Animals, Which Was Reversed by VDAC Inhibitor or mROS-Specific Scavenger
To confirm the role of mROS in NLRP3 inflammasome activation, rotenone was intraventricularly injected to induce mROS production22 in naive mice. ROS assay revealed that mROS was significantly induced in the brain as early as 1 hour following the rotenone injection. At 4 hours after injection, mROS levels were less than those at 1 hour, but still higher than in the naive group (p < 0.05; Fig 7). The inhibition of mPTP or mROS reversed the effect of rotenone at 4 hours after injection (p < 0.05). Western blot results of in vivo chemical cross-linking16 showed that VDAC dimer was increased in the ipsilateral hemisphere after rotenone injection compared to contralateral and naive samples. In naive mice, NLRP3 and cleaved caspase-1 were significantly increased 4 hours after rotenone injection, which were then reversed by mPTP inhibition or mitochondrial ROS scavenger (p < 0.05).
Figure 7.
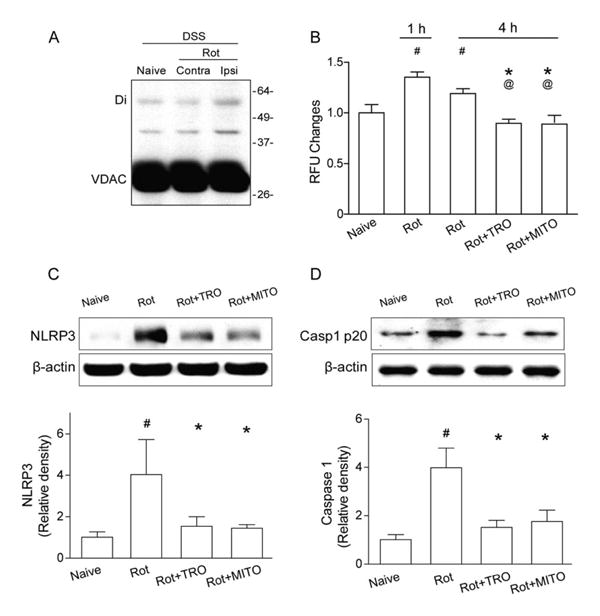
Mitochondrial reactive oxygen species (mROS) promotes mitochondrial permeability transition pore formation and NLRP3 inflammasome component expression in naive animals. (A) Western blots for voltage-dependent anion channel (VDAC) in naive and intracerebral hemorrhage (ICH) animals with chemical cross-linker (disuccinimidyl suberate [DSS]) at 1 hour following rotenone injection (Rot). Monomeric VDAC is located at 33kDa. Dimeric (Di) VDAC is detected at 45kDa. Contra=contra-lateral hemisphere; Ipsi = ipsilateral hemisphere. (B) Quantification of mROS alterations in the ipsilateral hemisphere in naive mice, 1 or 4 hours following rotenone injection (Rot), or TRO-19622 (Rot+TRO) or Mito-TEMPO treatment (Rot+MITO) 4 hours following rotenone injection; n=6 mice per group. Error bars represent mean±standard error of the mean. #p<0.05 vs naive, *p<0.05 vs Rot 1 hour; @p<0.05 vs Rot 4 hours. (C, D) Western blot assay for the expression of NLRP3 (C) and caspase-1 p20 subunit (D) in the ipsilateral hemisphere in naive, Rot, ICH+TRO, and ICH+MITO groups at 4 hours following operation; n=6 mice per group. #p<0.05 vs naive, *p<0.05 vs Rot.
Discussion
Inflammation plays a critical role in the pathophysiology of ICH-induced brain injury23; however, the mechanism by which ICH stimulates the inflammatory response remains to be elucidated. In the present study, we investigated an innate immune system component, the NLRP3 inflammasome, which may be involved in the inflammatory signal amplification following ICH. The NLRP3 inflammasome components were upregulated as early as 3 hours following ICH, and NLRP3 knockdown reduced IL-1β levels and neutrophil infiltration in the perihematoma. Our results indicated that mPTP and mROS contributed to NLRP3 inflammasome activation in both the ICH model and naive animals with rotenone injection. These observations suggested that blocking NLRP3 may prove to be a useful therapeutic intervention after ICH.
Mounting evidence indicates that the infiltration of systemic immune cells, specifically blood-borne leukocytes, is among the earliest events after ICH.23 The infiltrated leukocytes may activate resident cells in the brain, especially microglia, which are regarded as the first non-neuronal cells to respond to brain injury.23 Consequently, blood-borne leukocytes and activated glial cells release proinflammatory cytokines, such as IL-1β,6,24,25 and subsequently amplify leukocyte recruitment signals, which have been proposed to exacerbate ICH-induced brain injury in animal models.26
After establishing that IL-1β levels were enhanced after ICH, the sources of IL-1β still need to be determined. Recent evidence has alluded to the notion that a multiple-protein complex, inflammasome, is the major source of IL-1β.12 More than 20 members have been reported in the inflammasome family. Unlike other inflammasomes, the NLRP3 inflammasome exhibited its functionality in both sterile inflammatory response and antimicrobial response11; therefore, it gained more attention in the pathophysiology study of human disease.27,28 It has been reported that NLRP3 played an important role in a model of multiple sclerosis by exacerbating CNS inflammation, and this was partly mediated by caspase-1 and IL-18.28 Consistent with these studies, our study showed that a transient increase of NLRP3, activation of caspase-1, and releasing of IL-1β occurred as early as 3 hours after ICH, peaking at 12 hours and returning to the baseline by 72 hours. This resulted in a significant increase in brain edema and neurological deficits at 24 and 72 hours. Although previous observations showed that caspase-1 inhibition reduced ICH induced brain injury,7 NLRP3 inflammasome silencing with siRNAs also showed similar protective effects by reducing caspase-1 and IL-1β levels, thereby decreasing neutrophil infiltration. Overall, our data indicate that the NLRP3 inflammasome may contribute to the amplification of ICH-induced inflammation.
We have determined that the NLRP3 inflammasome contributes to ICH-induced inflammatory response; however, the molecular basis of NLRP3 inflammasome activation in ICH-induced brain injury is far from complete. Recent studies have linked ROS and potassium efflux to inflammasomes activation in response to diverse inflammatory danger signals.29 ROS production was regarded as a necessary step for NLRP3 activation, as ROS inhibitor (N-acetyl-L-cysteine) suppressed NLRP3 inflammasome activation.30 The source of ROS is currently unclear, but mitochondria are suspected to be the main site of intracellular ROS generation.30,31 Zhou and colleagues found that the NLRP3 inflammasome senses mitochondrial dysfunction, and both ROS generation and inflammasome activation are suppressed when mitochondrial activity is dysregulated.13 Intriguingly, mitochondrial dysfunction also occurred in ICH patients in the acute stage.32 One of the major causes of mitochondrial dysfunction was cellular injury; for instance, necrosis cell death happened at about 3 to 6 hours after ICH.33 Based on this evidence, we proposed that mROS production following ICH resulted in the initial inflammation response through the activation of the NLRP3 inflammasome. The released active IL-1β provides a signal to accumulate blood-borne leukocytes, such as neutrophils to the perihematomal region. We found a marked increase of ROS after ICH, and a specific mROS scavenger, Mito-TEMPO, reduced NLRP3 levels as well as casppas-1 and IL-1β levels. Accordingly, the infiltrated neutrophils numbers were also reduced, as expected, by Mito-TEMPO. To further confirm the role of mitochondrial ROS in inflammasome activation, rotenone22 (a respiration chain complex I inhibitor) was administered in naive animals to induce mROS. This led to the activation of the NLRP3 inflammasome being consistently obtained, which was then suppressed by Mito-TEMPO. Therefore, mitochondria ROS may be responsible for NLRP3 inflammasome activation following ICH.
How are mitochondria ROS generated during mitochondrial dysfunction? A special structure called mPTP is associated with mROS production.16,34 It has been reported that mPTP functions to enhance ROS production, and the inhibition of mPTP attenuated reactive oxygen/nitrogen species in target derived neurons in the adult mouse brain.16,35 The mPTP assembles between the inner and outer mitochondrial membranes. Generally, it consists of a dimer of VDAC in the outer mitochondrial membrane and a dimer of adenine nucleotide translocase in the inner membrane together with a regulatory protein, CypD.35–38 The role of mPTP in normal mitochondrial physiology is still unclear. However, under pathophysiological conditions, such as oxidative stress and mitochondrial dysfunction, the subunits of mPTP are stimulated to form a pore between the inner and outer membranes that allows free diffusion of metabolites across the membranes. ROS production leads to the induction of mPTP formation39 and consequently, more ROS production is promoted by the mPTP opening. The opening of the mPTP ultimately leads to mitochondrial swelling, mitochondrial Ca2+ efflux, ROS production, and the release of apoptotic proteins.35,40 VDAC is an indispensable subunit for mPTP formation and opening, which induces apoptosis41,42 and is ultimately required for ROS production.13 Previous studies found that endostatin-induced apoptosis is mediated by mPTP opening, and VDAC siRNA attenuated endostatin-induced apoptosis in endothelial cells.42 Being consistent with previous reports, our data showed that VDAC dimer was observed in the ipsilateral hemisphere, but not contralateral hemisphere in both ICH and naive mice with rotenone injection, which indicated the mPTP formation and opening. The VDAC inhibitor reduced ROS production, and NLRP3, caspase-1, and IL-1β levels. In this study, VDAC and mPTP were detected at an early stage of ICH in which ROS was mainly induced by primary injury, and less leukocyte infiltration and cell death occurred. The ROS level in brain tissue was rather low; therefore, a relatively weak VDAC dimer band was observed compared to VDAC monomer, which meant the formation and opening of mPTP.41,42 Previous studies suggested that moderate ROS production stimulated by endogenous or exogenous danger signals leads to inflammasome activation, and high ROS level causes cell death.43 Our observation about VDAC level and mPTP in ICH corresponded to the ROS level at the early stage of ICH. Another possible explanation for the weak dimer band was the use of a polyclonal antibody for VDAC detection in our immunoblot. It has been reported that the polyclonal antibody presented weaker dimer signals compared to the monoclonal antibody.20
It is important to note that IL-1β production may also be conducted through caspase-1 independent pathways, such as matrix metalloproteinases, which may also process pro–IL-1β to the active form.44 Because multiple factors are involved in the inflammatory response, other cytokines processed through inflammasome-dependent or independent pathways may also contribute to the inflammatory response following ICH. Additionally, there are no NLRP3-specific inhibitors or neutralizing antibodies, although several NLRP3 antagonists are commercially available, such as glibenclamide, a proton pump inhibitor, or Z-VAD-FMK, a caspase-1 inhibitor; however, neither of them shows NLRP3-specific inhibition.
In conclusion, our findings indicate that the NLRP3 inflammasome may contribute to ICH-induced inflammatory activation, which increases caspase-1 and IL-1β, and promotes neutrophil infiltration. Mitochondria ROS may be a major trigger of NLRP3 inflammasome activation (see Supplementary Fig 2). This study provided new information on the NLRP3 inflammasome following ICH, and the NLRP3 inflammasome may be a novel therapeutic target for intervention in ICH patients.
Supplementary Material
Acknowledgments
This study was supported by the NIH, NINDS (NS060936, J.T.; NS053407, J.H.Z.) and The National Basic Research Program of China (973), grant number: 2014CB541600 to H.F.
Footnotes
Additional Supporting Information may be found in the online version of this article.
Authorship: Q.M. and S.C. contributed equally to this work.
Potential Conflicts of Interest: Nothing to report.
References
- 1.Ribo M, Grotta JC. Latest advances in intracerebral hemorrhage. Curr Neurol Neurosci Rep. 2006;6:17–22. doi: 10.1007/s11910-996-0004-0. [DOI] [PubMed] [Google Scholar]
- 2.Fisher M, Vasilevko V, Cribbs DH. Mixed cerebrovascular disease and the future of stroke prevention. Transl Stroke Res. 2012;3(suppl 1):39–51. doi: 10.1007/s12975-012-0185-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qureshi AI, Tuhrim S, Broderick JP, et al. Spontaneous intracerebral hemorrhage. N Engl J Med. 2001;344:1450–1460. doi: 10.1056/NEJM200105103441907. [DOI] [PubMed] [Google Scholar]
- 4.Lively S, Schlichter LC. Age-related comparisons of evolution of the inflammatory response after intracerebral hemorrhage in rats. Transl Stroke Res. 2012;3(suppl 1):132–146. doi: 10.1007/s12975-012-0151-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lok J, Zhao S, Leung W, et al. Neuregulin-1 effects on endothelial and blood-brain-barrier permeability after experimental injury. Transl Stroke Res. 2012;3(suppl 1):S119–S124. doi: 10.1007/s12975-012-0157-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Masada T, Hua Y, Xi G, et al. Attenuation of intracerebral hemorrhage and thrombin-induced brain edema by overexpression of interleukin-1 receptor antagonist. J Neurosurg. 2001;95:680–686. doi: 10.3171/jns.2001.95.4.0680. [DOI] [PubMed] [Google Scholar]
- 7.Wu B, Ma Q, Khatibi N, et al. Ac-YVAD-CMK decreases blood-brain barrier degradation by inhibiting caspase-1 activation of interleukin-1beta in intracerebral hemorrhage mouse model. Transl Stroke Res. 2010;1:57–64. doi: 10.1007/s12975-009-0002-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Rivero Vaccari JP, Lotocki G, Marcillo AE, et al. A molecular platform in neurons regulates inflammation after spinal cord injury. J Neurosci. 2008;28:3404–3414. doi: 10.1523/JNEUROSCI.0157-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tomura S, de Rivero Vaccari JP, Keane RW, et al. Effects of therapeutic hypothermia on inflammasome signaling after traumatic brain injury. J Cereb Blood Flow Metab. 2012;32:1939–1947. doi: 10.1038/jcbfm.2012.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abulafia DP, de Rivero Vaccari JP, Lozano JD, et al. Inhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in mice. J Cereb Blood Flow Metab. 2009;29:534–544. doi: 10.1038/jcbfm.2008.143. [DOI] [PubMed] [Google Scholar]
- 11.Stutz A, Golenbock DT, Latz E. Inflammasomes: too big to miss. J Clin Invest. 2009;119:3502–3511. doi: 10.1172/JCI40599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tschopp J, Schroder K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10:210–215. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- 13.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 14.Rynkowski MA, Kim GH, Komotar RJ, et al. A mouse model of intracerebral hemorrhage using autologous blood infusion. Nat Protoc. 2008;3:122–128. doi: 10.1038/nprot.2007.513. [DOI] [PubMed] [Google Scholar]
- 15.Ma Q, Huang B, Khatibi N, et al. PDGFR-alpha inhibition preserves blood-brain barrier after intracerebral hemorrhage. Ann Neurol. 2011;70:920–931. doi: 10.1002/ana.22549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin LJ, Adams NA, Pan Y, et al. The mitochondrial permeability transition pore regulates nitric oxide-mediated apoptosis of neurons induced by target deprivation. J Neurosci. 2011;31:359–370. doi: 10.1523/JNEUROSCI.2225-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bordet T, Buisson B, Michaud M, et al. Identification and characterization of cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J Pharmacol Exp Ther. 2007;322:709–720. doi: 10.1124/jpet.107.123000. [DOI] [PubMed] [Google Scholar]
- 18.Thiffault C, Langston JW, Di Monte DA. Increased striatal dopamine turnover following acute administration of rotenone to mice. Brain Res. 2000;885:283–288. doi: 10.1016/s0006-8993(00)02960-7. [DOI] [PubMed] [Google Scholar]
- 19.Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation Stroke. 1995;26:627–634. doi: 10.1161/01.str.26.4.627. discussion 635. [DOI] [PubMed] [Google Scholar]
- 20.Abu-Hamad S, Arbel N, Calo D, et al. The VDAC1 N-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. J Cell Sci. 2009;122(pt 11):1906–1916. doi: 10.1242/jcs.040188. [DOI] [PubMed] [Google Scholar]
- 21.Keinan N, Tyomkin D, Shoshan-Barmatz V. Oligomerization of the mitochondrial protein voltage-dependent anion channel is coupled to the induction of apoptosis. Mol Cell Biol. 2010;30:5698–5709. doi: 10.1128/MCB.00165-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li N, Ragheb K, Lawler G, et al. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem. 2003;278:8516–8525. doi: 10.1074/jbc.M210432200. [DOI] [PubMed] [Google Scholar]
- 23.Wang J, Dore S. Inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2007;27:894–908. doi: 10.1038/sj.jcbfm.9600403. [DOI] [PubMed] [Google Scholar]
- 24.Barone FC, Feuerstein GZ. Inflammatory mediators and stroke: new opportunities for novel therapeutics. J Cereb Blood Flow Metab. 1999;19:819–834. doi: 10.1097/00004647-199908000-00001. [DOI] [PubMed] [Google Scholar]
- 25.Lu A, Tang Y, Ran R, et al. Brain genomics of intracerebral hemorrhage. J Cereb Blood Flow Metab. 2006;26:230–252. doi: 10.1038/sj.jcbfm.9600183. [DOI] [PubMed] [Google Scholar]
- 26.Aronowski J, Hall CE. New horizons for primary intracerebral hemorrhage treatment: experience from preclinical studies. Neurol Res. 2005;27:268–279. doi: 10.1179/016164105X25225. [DOI] [PubMed] [Google Scholar]
- 27.Chen S, Ma Q, Krafft PR, et al. P2X7R/cryopyrin inflammasome axis inhibition reduces neuroinflammation after SAH. Neurobiol Dis. 2013;58:296–307. doi: 10.1016/j.nbd.2013.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jha S, Srivastava SY, Brickey WJ, et al. The inflammasome sensor, NLRP3, regulates CNS inflammation and demyelination via caspase-1 and interleukin-18. J Neurosci. 2010;30:15811–15820. doi: 10.1523/JNEUROSCI.4088-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou R, Tardivel A, Thorens B, et al. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 30.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao X, Aronowski J. Nrf2 to pre-condition the brain against injury caused by products of hemolysis after ICH. Transl Stroke Res. 2013;4:71–75. doi: 10.1007/s12975-012-0245-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim-Han JS, Kopp SJ, Dugan LL, Diringer MN. Perihematomal mitochondrial dysfunction after intracerebral hemorrhage. Stroke. 2006;37:2457–2462. doi: 10.1161/01.STR.0000240674.99945.4e. [DOI] [PubMed] [Google Scholar]
- 33.Zhu X, Tao L, Tejima-Mandeville E, et al. Plasmalemma permeability and necrotic cell death phenotypes after intracerebral hemorrhage in mice. Stroke. 2012;43:524–531. doi: 10.1161/STROKEAHA.111.635672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion—a target for cardioprotection. Cardiovasc Res. 2004;61:372–385. doi: 10.1016/S0008-6363(03)00533-9. [DOI] [PubMed] [Google Scholar]
- 35.Abou-Sleiman PM, Muqit MM, Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nat Rev Neurosci. 2006;7:207–219. doi: 10.1038/nrn1868. [DOI] [PubMed] [Google Scholar]
- 36.Miura T, Tanno M. The mPTP and its regulatory proteins: final common targets of signalling pathways for protection against necrosis. Cardiovasc Res. 2012;94:181–189. doi: 10.1093/cvr/cvr302. [DOI] [PubMed] [Google Scholar]
- 37.McEnery MW, Snowman AM, Trifiletti RR, Snyder SH. Isolation of the mitochondrial benzodiazepine receptor: association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc Natl Acad Sci U S A. 1992;89:3170–3174. doi: 10.1073/pnas.89.8.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rasola A, Bernardi P. The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis. 2007;12:815–833. doi: 10.1007/s10495-007-0723-y. [DOI] [PubMed] [Google Scholar]
- 39.Javadov S, Karmazyn M, Escobales N. Mitochondrial permeability transition pore opening as a promising therapeutic target in cardiac diseases. J Pharmacol Exp Ther. 2009;330:670–678. doi: 10.1124/jpet.109.153213. [DOI] [PubMed] [Google Scholar]
- 40.Brustovetsky N, Brustovetsky T, Purl KJ, et al. Increased susceptibility of striatal mitochondria to calcium-induced permeability transition. J Neurosci. 2003;23:4858–4867. doi: 10.1523/JNEUROSCI.23-12-04858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tajeddine N, Galluzzi L, Kepp O, et al. Hierarchical involvement of Bak, VDAC1 and Bax in cisplatin-induced cell death. Oncogene. 2008;27:4221–4232. doi: 10.1038/onc.2008.63. [DOI] [PubMed] [Google Scholar]
- 42.Yuan S, Fu Y, Wang X, et al. Voltage-dependent anion channel 1 is involved in endostatin-induced endothelial cell apoptosis. FASEB J. 2008;22:2809–2820. doi: 10.1096/fj.08-107417. [DOI] [PubMed] [Google Scholar]
- 43.Finkel T. Signal transduction by mitochondrial oxidants. J Biol Chem. 2012;287:4434–4440. doi: 10.1074/jbc.R111.271999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schonbeck U, Mach F, Libby P. Generation of biologically active IL-1 beta by matrix metalloproteinases: a novel caspase-1-independent pathway of IL-1 beta processing. J Immunol. 1998;161:3340–3346. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.