

Abstract
Lecithin:retinol acyltransferase (LRAT) is a major enzyme involved in vitamin A/retinol metabolism, which regulates various physiological processes like cell proliferation and differentiation. LRAT expression is reduced in numerous cancers, yet the underlying mechanisms have remained undefined. We hypothesized that methylation silencing may contribute to decreased LRAT gene expression in colorectal cancer (CRC). LRAT hypermethylation status was analyzed in five CRC cell lines, 167 colorectal tumors, and 69 adjacent normal colonic mucosae, using a quantitative bisulfite/PCR/LDR/Universal Array assay. LRAT transcription levels were determined by real-time RT-PCR in a subset of tumors and matched normal tissues and in CRC cell lines that were treated with a demethylating agent, 5-aza-2′-deoxycytidine. The incidence of LRAT hypermethylation was significantly higher in colorectal tumors than in adjacent normal mucosae (p = 0.0025). Aberrant methylation occurred in 51 % of microsatellite-stable CRCs, in 84 % of microsatellite-unstable CRCs, and in 12 out of 13 colonic polyps. The number of hypermethylated LRAT events was inversely correlated with CRC stage (p < 0.0001). Importantly, LRAT hypermethylation was associated with decreased mRNA level in CRC clinical specimens, and demethylation treatment resulted in LRAT transcriptional reactivation. Our data support the idea that LRAT promoter hypermethylation associates with LRAT gene expression in CRC. The higher frequency of LRAT hypermethylation in colonic polyps and early-stage CRCs indicates that it may occur early in malignant progression.
Keywords: LRAT, CpG methylation, Colorectal cancer, Microsatellite instability, Thermostable ligase, Universal microarray
Introduction
The control of normal cell growth, cell differentiation, and apoptosis is the primary physiological function of retinoids, i.e., vitamin A (retinol) and its derivatives [1]. The physiological actions of vitamin A are either oxidized into its active metabolite, retinoic acid, or stored in the form of retinyl esters. Inside the cell, the necessary oxidation steps for retinoic acid synthesis initially involve a family of retinol dehydrogenases (RDHs) that convert all-trans retinol and 9-cis retinol into the corresponding all-trans and 9-cis retinaldehydes. Followed by a second oxidation, all retinaldehydes are converted to retinoic acid that is either degraded via hydroxylation or transferred into the nucleus, where it may bind to its nuclear receptors (RAR α, β, γ) to stimulate the transcription of target genes [2]. On the other hand, lecithin:retinol acyltransferase (LRAT) converts excess retinol into retinyl esters, which are then transported into the blood and stored in the liver [3]. An improper balance between retinol and its metabolites, collectively called retinoids, has been linked to the occurrence of various carcinomas [4–6].
Colorectal cancer (CRC) is one of the leading causes of cancer-related deaths in the Western world and in the USA [7]. The well-recognized sequential, multistep CRC model—from hyperplasia, adenoma, and eventually carcinoma—makes it an attractive study system. Several previous investigations have demonstrated a connection between retinoids and colonocyte function, and the development of colon neoplasms. In a mouse model study, vitamin A deficiency was linked to abnormal colonic epithelia, including a reduction in mucus-secreting cells and healing defects in colonic anastomosis [8]. In CRC patients, decreased serum retinol levels were associated with the neoplastic process [9, 10]. Additionally, reduced expression of RDHs (RDH5 and RDHL) in colonic adenomas and carcinomas, as compared to normal colon [11, 12], was thought to lead to decreased mucus production, expansion of proliferation zones within crypts, and ion flux alterations in colon epithelial tissues [11]. These studies have shown a relationship between tumor development and the alteration of genes involved in retinoid metabolism.
Methylation silencing has been implicated as another mechanism that may compromise retinoic acid metabolism in cancer. For instance, aberrant promoter hypermethylation of cellular retinoic acid-binding protein 1 (CRABP1), a protein that regulates retinoic acid nuclear transportation following retinaldehyde oxidization, was observed in CRC cell lines and resulted in increased cellular sensitivity to retinoic acid [13]. Cellular retinol-binding protein 1 (CRBP1), a retinol chaperone that interacts with various retinoid-metabolizing enzymes, was found hypermethylated in many cancers including 57 % of CRCs, and hypermethylation was correlated with a decreased transcription [14, 15]. CRBP1 deficiency is thought to diminish retinol transport and block the formation of retinyl esters. Altogether, these observations indicate that impairment of retinoid metabolism in tumor malignancies may be explained at least in part, by aberrant DNA hypermethylation of genes involved in retinol metabolic pathway.
LRAT is essential for maintaining normal retinoic acid concentrations in the cell. Previous analyses detected a decreased LRAT activity in various cancer cell lines and cancer tissues, including prostate, breast, oral cavity, skin, bladder, and kidney [3, 16–18], suggesting that LRAT deficiency resulting in a perturbed retinoid metabolism in carcinogenesis. A very recent global methylation marker study has implied that LRAT hypermethylation linked to reduced gene expression in hepatocellular carcinoma [19]; however, the mechanisms underlying the decline in LRAT expression in CRCs are not well documented. We hypothesized that methylation silencing may play an important role in the modulation of LRAT function in retinoid metabolism, which could have a significant impact on CRC development and progression.
Materials and methods
Study population
Details of study population have been described previously [20]. Briefly, the population consisted of 167 patients who presented at Memorial Sloan-Kettering Cancer Center with a sporadic colorectal neoplasm between 1992 and 2004. Primary tumors were located in the proximal, the distal colon, or the rectum. The study cohort included White Non-Hispanic, White Hispanic, Black Non-Hispanic, Asian, and American Indian patients. Age ranged from 17 to 86 years with a median age of 66. All specimens were thoroughly examined by pathologists and microdissected. Tissue procurement and sample usage were done under IRB-approved protocols.
Quantitative real-time RT-PCR
Total RNA from frozen tissues was extracted using TRIzol reagent (Invitrogen) and purified by the RNeasy mini columns (QIAgen). DNase-treated RNA concentration was measured using NanoDrop 1000 spectrophotometer (Thermo Scientific), and the quality was analyzed with Bioanalyzer 2100 (Agilent). cDNA was synthesized from 1 μg total RNA using multiscribe reverse transcriptase. LRAT RNA levels were measured using predeveloped LRAT TaqMan® assay (Hs00428109_m1, Applied Biosystems). The human GAPDH (Hs99999905_m1) gene was used for normalization. Quantitative PCR was performed with the Stratagene Mx3005p real-time thermal cycler (Agilent Technologies, Santa Clara, CA). Each sample was measured in triplicates, and the average Ct (threshold cycle) was used for calculating mRNA levels.
Determination of promoter hypermethylation status
Details of the methylation study using bisulfite/PCR/LDR/Universal Array were described previously [20–22]. Standard curves were established for each interrogated cytosine to calibrate the methylation ratio (Supplementary Fig. 1). Generally, methylation levels at six CpG sites located in LRAT intronic region were averaged to determine the overall promoter methylation value in a sample. Hypermethylation was considered when a sample’s overall methylation value was greater than one standard deviation of the mean of normal tissues’ overall promoter methylation values. This cutoff scored samples with 15 % or more methylation in a promoter region.
Cell cultures and 5-aza-2′-deoxycytidine treatment
All CRC cell lines were originally obtained from ATCC, and their genetic origin has been confirmed by DNA fingerprinting (STR profiling). Cell cultures subjected to a demethylating treatment were incubated with 2uM 5-aza-2′-deoxycytidine (5-aza-dC) (Sigma, MO) for a period of 3 days. The drug and media were replaced every 24 h. Cells were harvested at the end of the third day, and promoter methylation status was determined using the bisulfite/PCR/LDR/Universal Array assay. Mock-treated cells were prepared in parallel as controls, using the same volumes of PBS and/or ethanol.
Statistical analysis
Statistical comparisons were performed by either Student’s t test or Wilcoxon signed-rank test. The correlation studies were computed using the chi-square or Fisher’s exact (twosided) tests of the n × m contingency tables. p values of <0.05 were considered significant. Multiple-testing correction was performed using the Benjamini–Hochberg method to find those tests with a false discovery rate (FDR) of at most 5 %.
Results
CRC cell lines exhibit various degrees of LRAT promoter hypermethylation
A CpG island was observed in the LRAT promoter starting near the 5′ untranslated region (UTR), continuing across the first intron and the translation start site, and then extending into the second exon. LRAT methylation status was initially assessed in three CRC cell lines using methylation-specific PCR (MSP) (Fig. 1a). LRAT hypermethylation was detected in all cell lines; however, DLD1 and HT29 cells appeared to have higher proportion of LRAT hypermethylation than the event in SW620. The data suggested a common occurrence of aberrant LRAT promoter hypermethylation in CRC cell lines. To accurately quantify methylation levels in the LRAT promoter, a bisulfite/PCR/LDR/Universal Array assay was employed to interrogate six cytosines interspersed among the first exon and the first intron (Fig. 1b, c). Using this approach, DLD1 cells exhibited the highest methylation level than HT29 and other CRC cell lines (Fig. 1d). These differences in hypermethylation levels were consistent with the observations using MSP, thereby corroborating that a variation in the extent of aberrant LRAT methylation among CRC cell lines.
Fig. 1.
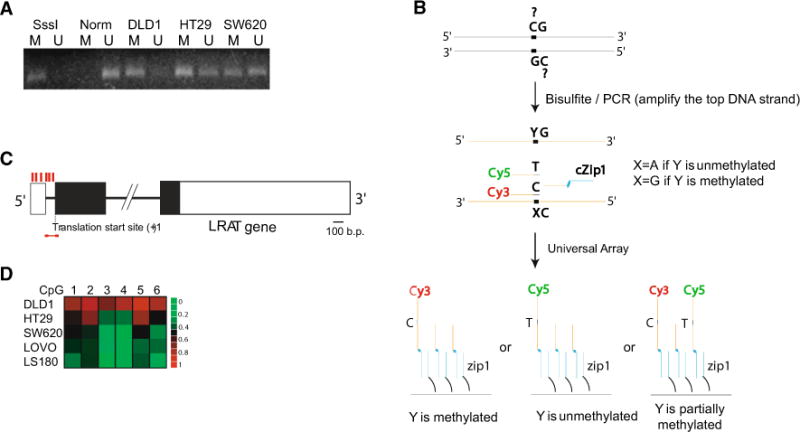
Evaluation of LRAT proximal promoter methylation in CRC cell lines. a LRAT methylation status determined by methylation-specific PCR (MSP). MSP products were resolved on a 3 % agarose gel. SssI: in vitro methylated normal human lymphocyte DNA. Norm: normal human lymphocyte DNA. DLD1, HT29, and SW620: genomic DNAs extracted from the corresponding CRC cell lines. M methylation-specific PCR product, U unmethylation-specific PCR product. b Schematic diagram of the bisulfite/PCR/LDR/Universal Array assay. Upstream LDR primers with distinguishable nucleotides (cytosine or thymine) are labeled with either Cy3 (detection of methylated cytosine) or Cy5 (detection of unmethylated cytosine), respectively. Downstream LDR primer contains a zip-code complementary sequence that may direct the ligation product to a proper address on the Universal Array. A thermostable ligase seals the upstream and downstream LDR primers, provided they form a perfect match to the DNA template. Ligation products with varied methylation levels are captured on the Universal Array. c Schematic diagram of LRAT gene structure. Positions of the six CpG sites that were analyzed are symbolized by red vertical bars. A red horizontal line indicates the position of MSP primers. Black rectangles represent coding regions of the LRAT gene. d LRAT methylation status determined using the bisulfite/PCR/LDR/Universal Array assay. DNA methylation levels are rendered into a heat map diagram: Names of CRC cell lines are shown vertically, and analyzed CpG sites (1–6) appear horizontally. Sequences of all PCR and LDR primers are provided in Supplementary Table 1
High incidence of LRAT hypermethylation in primary colorectal tumors
The frequency of aberrant LRAT methylation was subsequently analyzed in CRC tissues using the bisulfite/PCR/LDR/Universal Array assay. Analysis of genomic DNA extracted from microsatellite instability (MSI) tumors was performed initially, as those tumors tend to possess many hypermethylated genomic loci [20]. Indeed, LRAT hypermethylation was found in 26 of 31 (84 %) MSI tumors (Table 1 and Supplementary Fig. 2A). Because MSI accounts for only 10–15 % of sporadic CRCs, we next sought to determine hypermethylation incidence in the majority of CRCs, by examining another 205 clinical specimens comprised of 13 colonic polyps, 123 microsatellite-stable (MSS) tumors, and 69 adjacent normal mucosae (Fig. 2a). Compared to normal colorectal mucosae, nearly half of the tumors showed medium- to highlevel and tumor-specific (p = 0.0025) cytosine methylation at sites 1–6 (Fig. 2b, and Supplementary Fig. 2B). The differential methylation pattern of the cytosines was validated in a subset of 10 MSS tumors using bisulfite sequencing (Supplementary Fig. 3A). The concordance of the results obtained with two independent methylation assays confirmed the common occurrence of aberrant LRAT hypermethylation in CRC tissues (Supplementary Fig. 3B). The LRAT methylation profile also revealed an inverse relationship between CRC stages and the number of hypermethylated LRAT events (p < 0.0001, Table 1). Namely, instances of hypermethylation were more frequent in earlier tumor stages (I/II) than in later stages (III/IV). Because CRC patients with MSI or early-stage MSS are known for better prognosis than patients with advanced MSS tumors [23], a higher incidence of LRAT hypermethylation in MSI and early-stage CRC may correlate with a better survival or more favorable clinical outcome. Moreover, LRAT hypermethylation was found in 12 of the 13 examined colorectal polyps (Table 1), indicating that this epigenetic phenomenon occurred early during CRC development.
Table 1.
Detection of aberrant LRAT methylation in colorectal tumor and adjacent normal tissues, according to MSI status and tumor stages
| CRC types | Stages | LRAT hypermethylation (%) |
|---|---|---|
| MSI | Various (n = 31) | 83.9 |
| MSS | Polyps (n = 13) | 92.3 |
| Stage I (n = 20) | 60.0 | |
| Stage II (n = 25) | 68.0 | |
| Stage III (n = 33) | 45.5 | |
| Stage IV (n = 45) | 33.3 | |
| Normal | Adjacent mucosa (n = 69) | 21.7 |
The methylation difference among MSS samples was calculated by Mantel–Haenszel chi-square analysis (p<0.0001)
Fig. 2.
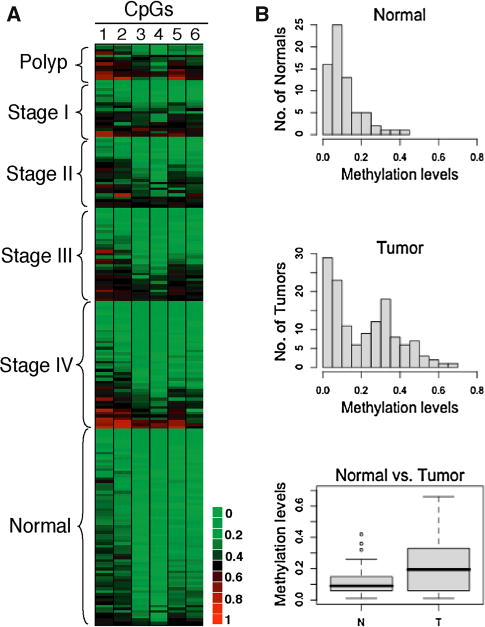
Evaluation of LRAT proximal promoter methylation in CRC tissue specimens. Methylation levels were determined using the bisulfite/PCR/LDR/Universal Array assay. a DNA methylation levels are rendered into a heat map diagram: Clinical samples, which are comprised of 136 MSS colorectal tumors and 69 adjacent normal mucosae, are shown vertically and classified by disease stage; average methylation levels of six CpG sites (1–6) appear horizontally. b Histograms and box plots of the LRAT methylation levels shown in (a)
LRAT hypermethylation correlates with reduced transcription in matched primary tumors
To determine whether LRAT promoter hypermethylation correlated with decreased transcription in CRC, the methylation and RNA levels of the LRAT gene were examined in ten paired (tumor and matched normal) colorectal specimens (Fig. 3a). Each of the ten tumor samples was previously identified as hypermethylated in the LRAT gene, contrary to their adjacent normal tissue counterparts. We observed that colorectal tumors with LRAT hypermethylation display at least an eightfold or greater decrease (ΔΔCt ≥ 3) in LRAT mRNAs, as compared to the transcript levels in the corresponding adjacent normal mucosae. The association between LRAT hypermethylation and transcription levels was further scrutinized in an additional 20 CRC samples (Fig. 3b). Overall, samples with higher LRAT methylation levels were associated with greater real-time PCR cycle threshold (Ct) values, i.e., lower LRAT transcript levels, indicating that hypermethylation at these CpG sites correlated with decreased LRAT transcription.
Fig. 3.
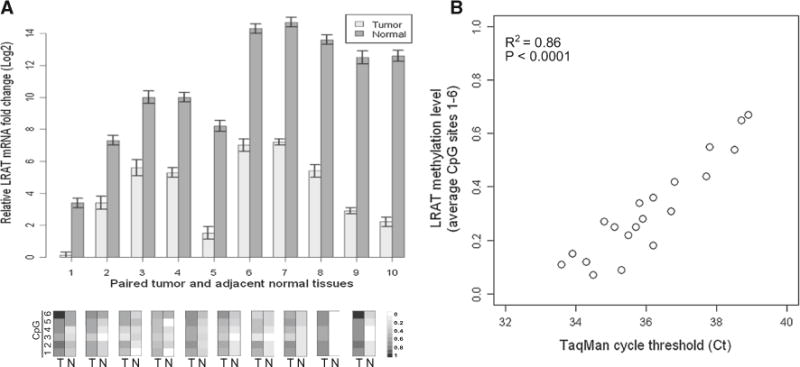
Correlation between LRAT promoter hypermethylation and mRNA expression levels in CRC samples. a LRAT mRNA levels in 10 paired colorectal tissues (adjacent normal vs. tumor) were determined by real-time RT-PCR and quantified using the delta Ct method (ΔCt). ΔCt was defined as Ct(LRAT, normal)–Ct(GAPDH, normal) or Ct (LRAT, tumor)–Ct(GAPDH, tumor). Average methylation levels of six CpG sites (1–6) for each tissue sample are shown below their corresponding mRNA measurement. T tumor, N adjacent normal tissue. b Correlation plot of LRAT mRNA and promoter methylation levels in a total of 20 CRC specimens. The extent of LRAT promoter methylation was determined by averaging the methylation levels of six CpG sites
Reactivation of LRAT gene expression by 5-aza-2′-deoxycytidine in cancer cell lines
To demonstrate that hypermethylation at the measured CpG dinucleotides was functionally relevant in the modulation of LRAT expression, we examined the effect of a demethylating agent, 5-aza-dC, on LRAT mRNA levels in CRC cell lines showing LRAT hypermethylation. A 72-h demethylating treatment resulted in reactivation of LRAT mRNAs in DLD1 (~1000-fold, ΔΔCt ~ 10), HT29 (~5.7-fold, ΔΔCt ~ 2.5), and SW620 (~4-fold, ΔΔCt ~ 2) cells (Fig. 4). Overall, our data linked demethylation of the LRAT promoter with reactivation of its transcription, implying that aberrant LRAT promoter hypermethylation impaired its gene function.
Fig. 4.
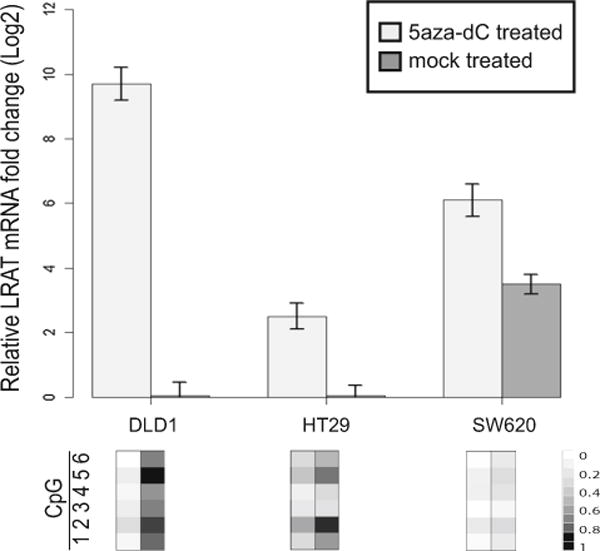
Reactivation of LRAT gene expression and function by demethylating agent 5-aza-dC. a Restoration of LRAT transcript levels in CRC cell lines in the presence of 5-aza-dC in CRC cell lines. mRNA fold changes were measured by real-time RT-PCR and calculated using the delta Ct method (ΔCt). ΔCt was defined as Ct(LRAT, mock treated)–Ct(GAPDH, mock treated) or Ct (LRAT, treated)–Ct(GAPDH, treated). Average methylation levels of six CpG sites (1–6) in the presence or absence of 5-aza-dC are shown below their corresponding mRNA fold changes. b Restoration of LRAT enzymatic activity in the DLD1 cells in the presence of 5-aza-dC. HPLC analytical profiles are displayed. RE retinyl esters, ROH retinols
Discussion
It has been proposed that LRAT, in conjunction with cytochrome P450 hydroxylases, plays essential roles in regulating the availability of retinol as a retinoic acid precursor [24]. Different mechanisms were reported to modulate LRAT gene expression and therefore may have a crucial impact on retinol metabolism. Firstly, vitamin A and retinoic acid regulate LRAT gene expression in a tissue-specific manner [25]. Study in murine has shown that liver LRAT mRNA level and its activity responded dose dependently to vitamin A, while LRAT expression remained constitutively active in other tissues such as small intestine and testis. Secondly, it was also reported that the level of CRBP may regulate LRAT gene expression [24, 26]. The ratio of free CRBP to holo-CRBP (i.e., retinol bound) directly reflects retinol concentration and thus influences retinol’s ability to regulate LRAT activity. In the present work, we propose LRAT promoter hypermethylation as another mechanism in the modulation of LRAT gene expression and function. Methylation silencing may abolish the ability of retinol and retinoic acid positively regulating LRAT gene expression and disrupt retinoid homeostasis, namely the dynamic balance between retinoic acid and retinol concentrations. Such an imbalance may interfere with regular cell growth, differentiation, and apoptosis.
The determination of LRAT methylation status was based on the averaging cytosine methylation levels at multiple CpG sites. This strategy not only allowed a representational mapping of cytosine methylation levels over a relatively large window, it also minimized scoring of false hypermethylation events that were resulted from sporadic methylation, such as one or two methylated CpG sites in the analyzed promoter region. Our study, which focused on the proximal CpG island of LRAT promoter, provided evidence for a correlation between LRAT methylation and expression levels in clinical specimens. Moreover, our methylation assay was validated by bisulfite, sequencing in ten clinical CRC samples. Although we cannot completely rule out the contribution of another CpG island in the vicinity of an alternative transcription start site located further upstream, our results strongly argue in favor of a direct effect of LRAT proximal promoter methylation on its gene function.
In the present work, LRAT hypermethylation was the most frequent in MSI and early-stage MSS CRCs, which are typically associated with a better prognosis. One interpretation is that LRAT hypermethylation may be attributed to a CpG island methylator phenotype (CIMP), characteristic of a subset of CRCs that have multiple hypermethylated genes and significantly correlate with MSI [20, 21]. Hence, LRAT hypermethylation was detected in 9 out of the 12 CIMP-positive cases identified in a subset of 58 CRC samples that were previously analyzed for CIMP (Supplementary Fig. 4). However, in the same sample cohort, aberrant LRAT hypermethylation was equally found in CIMP-negative tumors, thus ruling out the possibility that frequent LRAT hypermethylation was part of the CIMP-dependent features.
It is noteworthy that LRAT hypermethylation occurred in nearly 22 % of adjacent normal colonic mucosae and tended to be in older CRC patients (Tables 1, 2). Methylated genomic loci with similar features are often classified as a “type A” (age-related) methylation marker [27, 28], suggesting a predisposing factor with increased CRC risk during aging [29]. The presence of methylated loci in the adjacent normal-appearing tissues may also imply a field effect in which abnormal gene expression happens not only within few centimeters of the tumor margin, but possibly extends to the entire mucosal field [30]. The potential field effect of LRAT points to its contribution to early colorectal carcinogenesis, which is consistent with the frequent LRAT hypermethylation found in colonic polyps. Additionally, LRAT hypermethylation in adjacent normal-appearing tissues seemed more frequent in female than in male patients (Table 2). Although this gender-associated result was another potential characteristic of type A markers and was in agreement with a previous work by Kawakami et al. [31], a recent study of type A markers in normal colonic mucosae reported male-associated methylation events [32]. These divergences may be related to differences in patient populations, sample sizes, as well as different genetic loci, or CpG islands selected in these analyses. In summary, our study shows that LRAT promoter hypermethylation occurs frequently in CRCs. Promoter hypermethylation resulting in gene silencing provides a plausible mechanism for the reduced LRAT expression observed in various human cancers.
Table 2.
The relationships between LRAT promoter methylation status and clinical features in CRCs
| Tumor (CpG 1–6)
|
p value | Adjacent normal (CpG 1–6)
|
p value | |||
|---|---|---|---|---|---|---|
| Hypermethylated | Unmethylated | Hypermethylated | Unmethylated | |||
| Location | ||||||
| Distal | 39 | 42 | 5 | 32 | ||
| Proximal | 20 | 22 | 0.96 | 7 | 25 | 0.36 |
| Gender | ||||||
| Male | 29 | 30 | 4 | 29 | ||
| Female | 30 | 34 | 0.8 | 11 | 25 | 0.06 |
| Age | ||||||
| <55 | 8 | 20 | 0 | 15 | ||
| 55–64 | 17 | 13 | 4 | 13 | ||
| 65–70 | 14 | 13 | 2 | 11 | ||
| 71–85 | 20 | 18 | 0.13 | 6 | 18 | 0.07 |
| AJCC stage | ||||||
| I | 12 | 8 | ||||
| II | 17 | 8 | ||||
| III | 15 | 18 | ||||
| IV | 15 | 30 | 0.03 | |||
| Differentiation | ||||||
| Well | 0 | 1 | ||||
| Moderate | 48 | 52 | ||||
| Poor | 7 | 5 | ||||
| No info | 4 | 6 | 0.73 | |||
| Mucinous histology | ||||||
| No | 37 | 47 | ||||
| Yes | 18 | 12 | ||||
| No info | 4 | 5 | 0.32 | |||
| Mutations | ||||||
| K-ras | 23 | 18 | ||||
| B-raf | 3 | 2 | ||||
| WT | 33 | 44 | 0.31 | |||
Supplementary Material
Acknowledgments
The authors thank WeiJi Shi for the advice in statistical analysis. We also thank Owen Parker for insightful discussion and critical reading of the manuscript, Ludwig Institute for Cancer Research/Conrad N. Hilton Foundation joint Hilton-Ludwig Cancer Metastasis Initiative, and the Gilbert Family Foundation.
Abbreviations
- CIMP
CpG island methylator phenotype
- MSP
Methylation-specific PCR
- LDR
Ligase detection reaction
- LRAT
Lecithin:retinol acyltransferase
Footnotes
Conflict of interest None.
Electronic supplementary material The online version of this article (doi:10.1007/s12032-014-0254-7) contains supplementary material, which is available to authorized users.
Contributor Information
Yu-Wei Cheng, Email: chengy@ccf.org, Department of Laboratory Medicine, Cleveland Clinic, Cleveland, OH 44195, USA.
Hanna Pincas, Department of Neurology and Neuroscience, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA.
Jianmin Huang, Department of Microbiology and Immunology, Weill Medical College of Cornell University, New York, NY 10021, USA.
Emmanuel Zachariah, Rutgers-Cancer Institute of New Jersey, New Brunswick, NJ 08903–0019, USA.
Zhaoshi Zeng, Department of Surgery, Colorectal Surgery Service, Memorial Sloan-Kettering Cancer Center, New York, NY 10021, USA.
Daniel A. Notterman, PennState University Hershey Medical Center, Hershey, PA 17033, USA
Philip Paty, Department of Surgery, Colorectal Surgery Service, Memorial Sloan-Kettering Cancer Center, New York, NY 10021, USA.
Francis Barany, Department of Microbiology and Immunology, Weill Medical College of Cornell University, New York, NY 10021, USA.
References
- 1.Das BC, Thapa P, Karki R, Das S, Mahapatra S, Liu TC, et al. Retinoic acid signaling pathways in development and diseases. Bioorg Med Chem. 2014;22(2):673–83. doi: 10.1016/j.bmc.2013.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mark M, Ghyselinck NB, Chambon P. Function of retinoid nuclear receptors: lessons from genetic and pharmacological dissections of the retinoic acid signaling pathway during mouse embryogenesis. Annu Rev Pharmacol Toxicol. 2006;46:451–80. doi: 10.1146/annurev.pharmtox.46.120604.141156. [DOI] [PubMed] [Google Scholar]
- 3.Guo X, Ruiz A, Rando RR, Bok D, Gudas LJ. Esterification of all-trans-retinol in normal human epithelial cell strains and carcinoma lines from oral cavity, skin and breast: reduced expression of lecithin:retinol acyltransferase in carcinoma lines. Carcinogenesis. 2000;21(11):1925–33. doi: 10.1093/carcin/21.11.1925. [DOI] [PubMed] [Google Scholar]
- 4.Mahmoud LA, Robinson WA. Vitamin A levels in human bladder cancer. Int J Cancer. 1982;30(2):143–5. doi: 10.1002/ijc.2910300203. [DOI] [PubMed] [Google Scholar]
- 5.Hayden LJ, Satre MA. Alterations in cellular retinol metabolism contribute to differential retinoid responsiveness in normal human mammary epithelial cells versus breast cancer cells. Breast Cancer Res Treat. 2002;72(2):95–105. doi: 10.1023/a:1014815112078. [DOI] [PubMed] [Google Scholar]
- 6.Crowe DL, Kim R, Chandraratna RA. Retinoic acid differentially regulates cancer cell proliferation via dose-dependent modulation of the mitogen-activated protein kinase pathway. Mol Cancer Res. 2003;1(7):532–40. [PubMed] [Google Scholar]
- 7.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57(1):43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 8.Okada M, Bothin C, Blomhoff R, Kanazawa K, Midtvedt T. Vitamin A deficiency impairs colonic healing but not adhesion formation in germ-free and conventional rats. J Invest Surg. 1999;12(6):319–25. doi: 10.1080/089419399272304. [DOI] [PubMed] [Google Scholar]
- 9.Ostrowski J, Janik P, Nowacki M, Janczewska I, Przybyszewska M, Szaniawska B, et al. Serum retinol level in patients with colorectal premalignant and malignant lesions. Br J Cancer. 1987;55(2):203–5. doi: 10.1038/bjc.1987.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sawicki J, Ostrowski J, Swietochowska B, Janik P, Sikora J, Słowik-Gabryelska A, et al. Vitamin A (retinol) level in colon and lung cancer patient sera. Neoplasma. 1985;32(2):225–7. [PubMed] [Google Scholar]
- 11.Jette C, Peterson PW, Sandoval IT, Manos EJ, Hadley E, Ireland CM, et al. The tumor suppressor adenomatous polyposis coli and caudal related homeodomain protein regulate expression of retinol dehydrogenase L. J Biol Chem. 2004;279(33):34397–405. doi: 10.1074/jbc.M314021200. [DOI] [PubMed] [Google Scholar]
- 12.Nadauld LD, Sandoval IT, Chidester S, Yost HJ, Jones DA. Adenomatous polyposis coli control of retinoic acid biosynthesis is critical for zebrafish intestinal development and differentiation. J Biol Chem. 2004;279(49):51581–9. doi: 10.1074/jbc.M408830200. [DOI] [PubMed] [Google Scholar]
- 13.Huang Y, dela Chapelle A, Pellegata NS. Hypermethylation, but not LOH, is associated with the low expression of MT1G and CRABP1 in papillary thyroid carcinoma. Int J Cancer. 2003;104(6):735–44. doi: 10.1002/ijc.11006. [DOI] [PubMed] [Google Scholar]
- 14.Napoli JL. A gene knockout corroborates the integral function of cellular retinol-binding protein in retinoid metabolism. Nutr Rev. 2000;58(8):230–6. doi: 10.1111/j.1753-4887.2000.tb01870.x. [DOI] [PubMed] [Google Scholar]
- 15.Esteller M. CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene. 2002;21(35):5427–40. doi: 10.1038/sj.onc.1205600. [DOI] [PubMed] [Google Scholar]
- 16.Guo X, Knudsen BS, Peehl DM, Ruiz A, Bok D, Rando RR, et al. Retinol metabolism and lecithin:retinol acyltransferase levels are reduced in cultured human prostate cancer cells and tissue specimens. Cancer Res. 2002;62(6):1654–61. [PubMed] [Google Scholar]
- 17.Boorjian S, Tickoo SK, Mongan NP, Yu H, Bok D, Rando RR, et al. Reduced lecithin:retinol acyltransferase expression correlates with increased pathologic tumor stage in bladder cancer. Clin Cancer Res. 2004;10(10):3429–37. doi: 10.1158/1078-0432.CCR-03-0756. [DOI] [PubMed] [Google Scholar]
- 18.Guo X, Nanus DM, Ruiz A, Rando RR, Bok D, Gudas LJ. Reduced levels of retinyl esters and vitamin A in human renal cancers. Cancer Res. 2001;61(6):2774–81. [PubMed] [Google Scholar]
- 19.Revill K, Wang T, Lachenmayer A, Kojima K, Harrington A, Li J, et al. Genome-wide methylation analysis and epigenetic unmasking identify tumor suppressor genes in hepatocellular carcinoma. Gastroenterology. 2013;145(6):1424–35. doi: 10.1053/j.gastro.2013.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng YW, Pincas H, Bacolod MD, Schemmann G, Giardina SF, Huang J, et al. CpG island methylator phenotype associates with low-degree chromosomal abnormalities in colorectal cancer. Clin Cancer Res. 2008;14(19):6005–13. doi: 10.1158/1078-0432.CCR-08-0216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng YW, Shawber C, Notterman D, Paty P, Barany F. Multiplexed profiling of candidate genes for CpG island methylation status using a flexible PCR/LDR/Universal Array assay. Genome Res. 2006;16(2):282–9. doi: 10.1101/gr.4181406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng YW, Idrees K, Shattock R, Khan SA, Zeng Z, Brennan CW, et al. Loss of imprinting and marked gene elevation are 2 forms of aberrant IGF2 expression in colorectal cancer. Int J Cancer. 2010;127(3):568–77. doi: 10.1002/ijc.25086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nash GM, Gimbel M, Shia J, Culliford AT, Nathanson DR, Ndubuisi M, et al. Automated, multiplex assay for high-frequency microsatellite instability in colorectal cancer. J Clin Oncol. 2003;21(16):3105–12. doi: 10.1200/JCO.2003.11.133. [DOI] [PubMed] [Google Scholar]
- 24.Ross AC, Zolfaghari R. Regulation of hepatic retinol metabolism: perspectives from studies on vitamin A status. J Nutr. 2004;134(1):269S–75S. doi: 10.1093/jn/134.1.269S. [DOI] [PubMed] [Google Scholar]
- 25.Zolfaghari R, Ross AC. Lecithin:retinol acyltransferase from mouse and rat liver. CDNA cloning and liver-specific regulation by dietary vitamin a and retinoic acid. J Lipid Res. 2000;41(12):2024–34. [PubMed] [Google Scholar]
- 26.Napoli JL. Interactions of retinoid binding proteins and enzymes in retinoid metabolism. Biochim Biophys Acta. 1999;1440(2–3):139–62. doi: 10.1016/s1388-1981(99)00117-1. [DOI] [PubMed] [Google Scholar]
- 27.Issa JP, Ottaviano YL, Celano P, Hamilton SR, Davidson NE, Baylin SB. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat Genet. 1994;7(4):536–40. doi: 10.1038/ng0894-536. [DOI] [PubMed] [Google Scholar]
- 28.Worthley DL, Whitehall VL, Buttenshaw RL, Irahara N, Greco SA, Ramsnes I, et al. DNA methylation within the normal colorectal mucosa is associated with pathway-specific predisposition to cancer. Oncogene. 2010;29(11):1653–62. doi: 10.1038/onc.2009.449. [DOI] [PubMed] [Google Scholar]
- 29.Issa JP. Aging, DNA methylation and cancer. Crit Rev Oncol Hematol. 1999;32(1):31–43. doi: 10.1016/s1040-8428(99)00019-0. [DOI] [PubMed] [Google Scholar]
- 30.Polley AC, Mulholland F, Pin C, Williams EA, Bradburn DM, Mills SJ, et al. Proteomic analysis reveals field-wide changes in protein expression in the morphologically normal mucosa of patients with colorectal neoplasia. Cancer Res. 2006;66(13):6553–62. doi: 10.1158/0008-5472.CAN-06-0534. [DOI] [PubMed] [Google Scholar]
- 31.Kawakami K, Ruszkiewicz A, Bennett G, Moore J, Grieu F, Watanabe G, et al. DNA hypermethylation in the normal colonic mucosa of patients with colorectal cancer. Br J Cancer. 2006;94(4):593–8. doi: 10.1038/sj.bjc.6602940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Belshaw NJ, Elliott GO, Foxall RJ, Dainty JR, Pal N, Coupe A, et al. Profiling CpG island field methylation in both morphologically normal and neoplastic human colonic mucosa. Br J Cancer. 2008;99(1):136–42. doi: 10.1038/sj.bjc.6604432. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.