

Abstract
Retinoids (RA) have been used as therapeutic agents for numerous skin diseases, from psoriasis to acne and wrinkles. While RA is known to inhibit keratinocyte differentiation, the molecular effects of RA in epidermis have not been comprehensively defined. To identify the transcriptional targets of RA in primary human epidermal keratinocytes, we compared the transcriptional profiles of cells grown in the presence or absence of all-trans retinoic acid for 1, 4, 24, 48 and 72 hours, using large DNA microarrays. As expected, RA suppresses the protein markers of cornification; however the genes responsible for biosynthesis of epidermal lipids, long-chain fatty acids, cholesterol, and sphingolipids, are also suppressed. Importantly, the pathways of RA synthesis, esterification and metabolism are activated by RA; therefore, RA regulates its own bioavailability. Unexpectedly, RA regulates many genes associated with the cell cycle and programmed cell death. This led us to reveal novel effects of RA on keratinocyte proliferation and apoptosis. The response to RA is very fast: 315 genes were regulated already after 1 h. More than one-third of RA-regulated genes function in signal transduction and regulation of transcription. Using in silico analysis, we identified a set of over-represented transcription factor binding sites in the RA-regulated genes. Many psoriasis-related genes are regulated by RA, some induced, others suppressed. These results comprehensively document the transcriptional changes caused by RA in keratinocytes, add new insights into the molecular mechanism influenced by RA in the epidermis and demonstrate the hypothesis-generating power of DNA microarray analysis.
Keywords: retinoic acid, epidermis, keratinocytes, microarray
Introduction
Retinoids (RA), a group of natural and synthetic vitamin A analogues, play a major role in regulating growth, differentiation and apoptosis of many cell types, both in vivo and in vitro. Epidermis is one of the major targets for the retinoic acid signaling in adult organisms. An important contribution in the understanding of the mechanism of retinoic acid action was the discovery of its nuclear receptors (53). The retinoid signal is transduced by two families of ligand-activated nuclear hormone receptors, the retinoic acid receptors (RAR), and retinoid X receptors (RXR), each with three isotypes (α, β, γ) and multiple isoforms (26). These receptors form RXR-RAR heterodimers or RXR-RXR homodimers after activation by selective retinoids. The dimers bind consensus DNA sequence called retinoic acid response elements (RAREs) and regulate target genes (10). RARs can be activated by all-trans retinoic acid (atRA) and its stereoisomer, 9-cis retinoic acid (9-cis-RA), whereas RXRs are only activated by 9-cis-RA (11). The predominant retinoid receptors found in skin are RARγ and RXRα (21). Retinoic acid exerts its effects in epidermis through various mechanisms that may involve either direct receptor binding to epidermal genes (37, 38, 57, 69) or interference with signaling of other transcription factors (43, 70).
The efficacy of topical use of retinoids for the treatment of acne vulgaris has been recognized for 40 years (39). Retinoids have significant potential in the treatment of both preneoplastic lesions and cancers (59). Their therapeutic benefits are somewhat reduced due to the many side effects that include scaling, erythema, dryness and desquamation of skin. These changes are primarily due to specific inhibitory effects of retinoids on keratinocyte differentiation, cornification and lipid production (78). In vitro studies with cultured keratinocytes have shown that RA treatment inhibits differentiation (24, 36). In vivo studies on mice fed high doses of vitamin A have shown an increased proliferation rate in basal cells of the epidermis resulting in hyperplasia of the skin. Similarly, RA treatment of human skin results in epidermal thickening (11). RA can also induce apoptosis in cultured keratinocytes grown under conditions suitable for differentiation (35). The RA-induced apoptosis may have important implications for the understanding of how retinoids inhibit tumor growth and exert their beneficial effect in skin diseases.
The extensive therapeutic use of retinoids in dermatology makes understanding their cellular mechanisms of action and identification of their transcriptional targets important for taking full advantage of their therapeutic benefits, while avoiding their harmful side effects (78). With this in mind, we decided to define comprehensively the transcriptional targets regulated by RA in human epidermal keratinocytes, the primary cell type affected by topical retinoid treatment.
Many genes were individually shown to be regulated by RA; a list of 532 genes as regulatory targets of RA was compiled by evaluating 1,191 published papers (2). Using an array containing 475 skin-related genes, the effects of RA on the gene expression in reconstituted human epidermis at the 24 h time point were identified (5). Transcriptional profiling studies in response to RA have been performed with other cell types (8, 46), but there have been no comprehensive investigations defining the targets of RA in epidermal keratinocytes.
To understand better the complex function and the tissue-specific transcriptional effects of RA in epidermis, we used global transcriptional profiling with large oligonucleotide microarrays. We treated human epidermal keratinocytes with RA and isolated RNA from the RA treated and matched control cells after 1, 4, 24, 48 and 72 h of treatment. We hybridized the labeled RNAs to Affymetrix HGU133A 2.0 microarrays, which measure the expression of approximately 22,200 genes. We found that RA regulated multiple functional groups of genes, including those involved in differentiation, lipid metabolism, signal transduction, apoptosis and cell cycle. Furthermore, we noticed that a great proportion of genes reported in the literature as being psoriasis-related were also regulated by RA, many of them in the opposite direction.
Materials and Methods
Human Keratinocyte Culture
Keratinocytes were maintained as previously published (28), in serum-free keratinocyte medium with epidermal growth factor, bovine pituitary extract and antibiotic-antimycotic (Gibco) both in the presence and absence of 1 μM RA (Sigma). A paired set of treated and untreated cells was harvested for each time point, 1, 4, 24, 48, and 72 h, using 0.4% trypsin (Gibco) and stored in RNAlater (Ambion).
Total RNA Isolation
Total RNA was isolated using RNeasy (Qiagen). 5 μg of total RNA was reverse-transcribed, amplified, and labeled as described (83).
GeneChip Hybridization and Array Data Analysis
Labeled cRNA was hybridized to HGU133A 2.0 arrays (Affymetrix), the arrays were washed, stained and then scanned using the Agilent GeneArray Scanner system (Hewlett-Packard). Microarray Suite 5.0 (Affymetrix) was used for data extraction. With GeneChip Operating Software (GCOS) v.1.2 (Affymetrix), we checked individually the absolute expression levels and p values at all time points. We included in the analysis only those genes scored as present in at least one sample, determined by the Affymetrix algorithm (p < 0.05). Differential expressions of transcripts at each time point were then determined by using RMAExpress v.0.5 (7). This consists of three steps: a background adjustment, quintile normalization and finally summarization. Genes were considered regulated if the expression levels differed more than 1.62-fold relative to untreated control at any specific time point, i.e., 1, 4, 24, 48 and 72 h. Duplicate genes and those not regulated at any time point were eliminated.
Functional annotation of regulated genes was performed as before (28). The clustering analysis of functional annotations was performed using Database for Annotation, Visualization and Integrated Discovery (DAVID 2007), with each time point treated as a discrete experiment, and separate analyses of induced and suppressed genes (15). Gene ontology clusters with a p-value less than 10−4 were retained for evaluation.
We also compared two lists of genes, one induced and the other suppressed by RA, with the results of the expression profiling of human keratinocytes responding to a variety of cytokines and agents from our previously published microarray data (3, 4, 19, 27, 29, 66, 84), by using LOLA set of programs (List Of Lists Annotated; http://www.lola.gwu.edu). Lists of the genes regulated in common by RA and each agent were obtained.
In addition, using oPOSSUM v.2.0 program (34), we determined the over-represented transcription factor binding sites in the RA-regulated genes. We first calibrated the parameters of the oPOSSUM program to obtain the optimal statistical p-values in the one-tailed Fisher exact probability analysis. We then used 30% and 20% as “level of conservation”, and 85% and 75% as “matrix match threshold”, for the induced and suppressed genes, respectively. Those with a p- value less than 10−5 were considered statistically significant. We analyzed 2,000 bp of upstream and downstream sequences relative to the transcription start site. The complete data are submitted to Gene Expression Omnibus repository (in process).
Quantitative RT-PCR
Total RNA isolated from keratinocytes treated or untreated with RA for 1, 4, 24, 48 and 72 h was reverse transcribed using an Omniscript Reverse Transcription kit (Qiagen, Valencia, CA). Real time PCR was performed in triplicates using Maxima SYBR Green qPCR Master Mix (2x) (Fermentas, Glen Burnie, MD). The samples were amplified and quantified using Opticon 2 thermal cycler (Biorad, Hercules, CA) under the following conditions: 10 minutes at 95C; and 35 cycles 30 seconds at 95C; 30 seconds at 60C followed by 30 seconds at 60C. Relative expression was normalized for levels of Eukaryotic translation elongation factor 1 alpha 1 (EEF1A1). The primer sequences used were: EEF1A1 fw (5’-TTGTCGTCATTGGACACGTAG-3’) and rev primer (5’- TGCCACCGCATTTATAGATCAG-3’), KRT1 fw (5’-TCAATCTCGGTTGGATTCGGA-3’) and rev primer (5’-CCACCTTGGTCATATAAGCACC-3’), DSG1 (5’-GGAACCCAATCGCCAAAATTC-3’) and rev primer (5’- ACCTCTCGATCAACTATGGATGT-3’) and IL1B fw (5’-GGCTTATTACAGTGGCAATGAGGA-3’) and rev primer (5’-TCCATGGCCACAACAACTGA-3’), CAECAM6 fw (5’- TCCCCCTCAAAGGCCAATTAC-3’) and rev primer (5’- TGGAACGTCCCATTGATAAACC -3’), SERPINB1 fw (5’- TTCCTGGCGTTGAGTGAGAAC -3’) and rev primer (5’- TGTGCTGCCGTGTTACCTC- 3’), NEDD9 fw (5’- GCTCTATCAAGTGCCAAACCC -3’) and rev primer (5’- GGTTCCCCCAATGCTTCTCT - 3’) , GAS6 fw (5’- GACAAAGATGTCAACGAATGCAG -3’) and rev primer (5’- CCTGGGAGCTGTACGCAAA- 3’) and RGC32 fw (5’- GAGCGCCACTTCCACTACG-3’) and rev primer (5’- AGTGAATCTGCACTCTCCGAG- 3’). The data were analyzed and samples quantified using the software Opticon Monitor 3.
Protein Isolation and Western Blot
Keratinocytes were growing and treated with RA (Sigma-Aldrich, MO) as described above. Proteins from keratinocytes were extracted using RIPA buffer with addition of Protease inhibitor Cocktail (Sigma-Aldrich, MO) and Phosphatase inhibitor Cocktail Set III (Calbiochem, Ca) according to commercial protocol. The soluble supernatants were normalized for total protein concentration using the Bradford protein assay, and the samples were stored at −20 C. Cells protein extracts were boiled for 5 min in 2x Lemlli sample buffer and were separated on 7.5% polyacrylamide gel (BioRad, CA) and transferred to nitrocellulose membrane (VWR, IL) on 100 V for 1 hrs in Tris/glycine transfer buffer. The membrane was blocked in 5% bovine serum albumin in Tris-buffered saline at room temperature for 1 hr and incubated in blocking solution with primary antibody at 4 °C overnight using antibodies specific for EGFR (Santa-Cruz, CA) and beta- tubulin (Santa Cruz, CA). Horseradish protein (HRP) conjugated anti-rabbit secondary antibody (Cell Signaling, CA) was used and immune complexes was visualized using Super Signal West Pico Chemiluminescent substrate (Pierce, IL). Immunoblot was exposed on X-ray film (HyBlot CL, Denville, NJ) according to the manufacturer's instruction.
Keratinocyte Treatment and TUNEL Assay
Cells were grown in keratinocyte growing medium and pretreated with either 1 μM RA or 1 μM Dexamethasone (Sigma) or left untreated for 24 h. After the treatment cells were washed three times with keratinocyte basal medium and irradiated with 8 mJ/cm2 of UVB (Stratagene 2000 Illuminator specially equipped with UVB light sources). We have shown that this UVB dosage induces low levels of apoptosis in keratinocytes (44). Cells were then incubated for another 24 h in the basal keratinocyte medium and fixed for the TUNEL assay. The TUNEL assay was performed following a commercial protocol for the in situ cell death detection kit (TMR red, Roche Applied Science). Cells were then mounted on slides using Dako fluorescent mounting medium (DAKO Corp.) Staining was analyzed using Nikon Eclipse E800 microscope and digital images were collected using SPOT- Camera Advanced program. All experiments were performed in triplicates. Three laboratory members blinded to the experiment counted apoptotic cells in 20 high power acquired images per condition, and median and SD values were calculated. Statistical significance between conditions was determined using Student's t-test.
Proliferation Assay
Keratinocytes were seeded at a density of 10,000 cells per 6-well dish (Falcon) and grown in Keratinocytes Serum Free Medium (Gibco, Invitrogen, Carlsbad, CA). When 80% confluence was reached, the cells were washed three times with sterile 1x Phosphate Buffered Saline (Fisher, Pittsburgh, PA), switched to Basal Keratinocyte Medium (Gibco, Invitrogen, Carlsbad, CA) and treated with 1 μM RA (Sigma, St. Louis, MO) or left untreated as controls. After 24 and 48 hours of incubation in the presence or absence of RA, the keratinocytes were trypsinized and counted. The experiment was independently performed three times in triplicates and keratinocytes were counted four times in each well using a hemocytometer (Hausser Scientific, Horsham, PA). Statistical significance was determined using a standard t-test.
Results
Initial Evaluation of the Microarray Data
We compared the transcriptional profiles of RA-treated and untreated keratinocytes using Affymetrix HGU133A 2.0 oligonucleotide microarrays. The microarrays simultaneously measure the levels of 22,200 mRNAs; of these, approximately 12,900 genes were found expressed in keratinocytes. Among them, 3,346 were regulated by RA, according to our criteria.
To confirm independently the microarray results obtained, we performed quantitative RT-PCR analysis of 8 representative genes (Fig. 1). We chose genes significantly regulated at several time points. All genes tested showed parallel direction of changes in regulation patterns, after treatment with RA while for KRT1 (keratin 1) slight variation was found only at one of the time point tested.
Figure 1. atRA promotes keratinocyte proliferation.
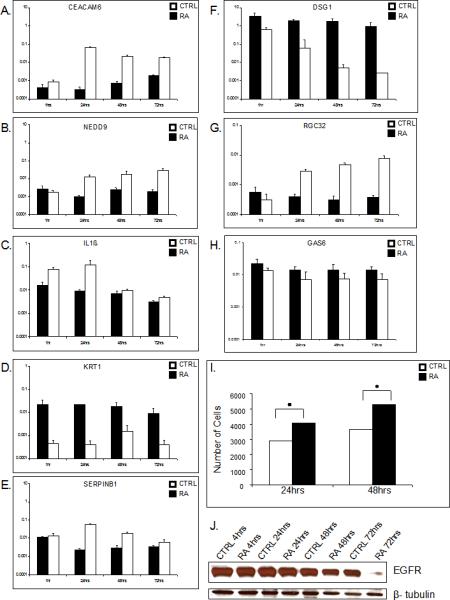
A bar graph showing the compatible regulation of cell-cycle related genes among other, obtained by using quantitative real time PCR. Mean values of expression levels were represented after normalization to expression level of EEF1A1 (A-H). Bar graph shows number of cells of triplicate experiments utilizing proliferation assay. A statistically significant increase in number of keratinocytes was found after 24 (p= 0.001) and 48 h (p=0.049) of atRA treatment when compared to untreated cells (I). Western blot analyzes confirms down regulation of EGFR (G).
Keratinocytes responded to RA very fast: 134 genes were induced and 181 suppressed already at 1 h. The maximum number of induced genes reached 768 at 48 h, and decreased to 542 at 72 h. Number of suppressed genes increased with time and reached peak of 979 at 72 h. Overall, the number of induced genes, 1,817, was slightly greater than that of suppressed, 1,782. Interestingly, 253 genes were differentially regulated at specific time points, i.e. induced at some, but suppressed at other time points. For example, the expression of CRABP2 (cellular retinoic acid binding protein II) was induced at the 4 h time point, but suppressed after 24 h.
Functional Characterization of the RA-regulated Genes
When we summarized the functional categories of the genes regulated by RA, we found that more than 75% of them fall into one of the following ontological categories: transcription factor, regulator, cytoskeletal, proteolysis, receptor, translation, transporter, adhesion, cell cycle, DNA repair/ synthesis, membrane protein, protein kinase, RNA metabolism and secreted (supplemental Table 1). In addition, apoptosis-related genes and metabolic enzymes genes are regulated by RA. To understand the biological meaning behind such a large list of genes, functional annotation clustering analysis was performed using DAVID, with each time point treated as a separate experiment (supplemental Table 2). Five clusters with the smallest p-value for each time point are listed in Figure 2.
Figure 2. Functional annotation clusters.

Five clusters, if any, with the most significant p-value for each time point are listed. For a complete list of all the clusters with a p-value less than 10−4, see supplemental Table 2. #, the number of genes in each category.
Differentiation
One of the best known effects of RA in the epidermis is to inhibit keratinocyte differentiation (24, 36). While complete differentiation does not occur in culture, our microarray data unambiguously demonstrate the suppressing transcriptional effect of RA. Specifically, the enzyme TGM1 (transglutaminase 1), which catalyzes the assembly of cornified envelope, was suppressed by RA at 24, 48, and 72 h. Epidermal differentiation markers FLG (filaggrin), a cornified envelope precursor, and LCE2B (a late cornified envelope marker) were also suppressed by RA at 48 and 72 h after treatment. CDSN (corneodesmosin) was suppressed as early as 4 h after treatment. Another group of differentiation markers, small proline-rich protein 1A (SPRR1A), 1B (SPRR1B), 2B (SPRR2B), 2C (SPRR2C) and 3 (SPRR3), were all suppressed by RA. SULT2B1 (sulfotransferase type 2 isoform B1), an important regulator of terminal keratinocyte differentiation and a mediator of barrier homeostasis, was suppressed at 24, 48, and 72 h.
In addition, the expression of KLF4 (Kruppel-like factor 4), which regulates terminal differentiation and barrier formation was suppressed (62). At 72 h, loricrin (LOR) was suppressed more than 16 fold. We also found Jagged (JAG1), a Notch ligand that promotes early differentiation, highly suppressed at 1 h. The suppression of all these genes confirmed that RA inhibits both early and late stages of keratinocyte differentiation. Unexpectedly, gene chip analysis revealed that RA induced IVL (involucrin) expression at 4 and 24 h and S100A10, a cornified envelope precursor, at 24 h. These markers may be regulated by more complex retinoid-dependent pathways.
Regulation of Keratin Gene Expression
Seventeen genes in the keratin family were regulated by RA. KRT1, KRT3, KRT6B, KRT9, KRT10, KRT16, KRT18, KRT23, KRTHA1 (keratin, hair, acidic, 1), KRTHA4, and KRTHA3A were suppressed. KRT7, KRT13, KRT15 and KRT19 were induced. KRT2A and KRT4 were induced at an early time point and suppressed later.
Lipid Metabolism
An essential characteristic of epidermal differentiation is the formation of the extracellular lipid barrier in the stratum corneum. As keratinocytes differentiate, they synthesize lipids and package them into lamellar granules, which are extruded into the extracellular spaces, forming the lipid envelope of the stratum corneum (16, 82). The lipid barrier mainly consists of free fatty acids, cholesterol and ceramides. Because the synthesis of specific lipids is a part of keratinocyte differentiation (56), we examined whether RA also affects lipid metabolism. Lipid metabolism-related genes, such as those involved in steroid metabolism, sterol biosynthesis and cholesterol biosynthesis, were suppressed by RA from 24 h to 72 h (supplemental Table 2). Our microarray data show that the genes involved in sphingolipid metabolism, e.g. ceramide glucosyltransferase (UGCG, also called glucosylceramide synthase), acid beta-glucosidase (GBA) and acid sphingomyelinase (SMPD1), were all suppressed by RA at 72 h. UGCG transfers glucose to ceramide, forming glucosylceramide, GBA hydrolyzes glucosylceramide and SMPD1 converts sphingomyelin to ceramide. Fatty acid synthetase (FASN), catalyzing the formation of long-chain fatty acids from acetyl-CoA, malonyl-CoA and NADPH, was suppressed by RA at 72 h; in contrast, fatty acid desaturase 1 (FADS1), catalyzing the biosynthesis of highly unsaturated fatty acid, was induced.
A series of genes along the pathway of cholesterol biosynthesis were also suppressed by RA. These include 3-hydroxy-3-methylglutaryl-coA synthase 1 (HMGCS1), 3-hydroxy-3-methylglutaryl-coA reductase (HMGCR), mevalonate kinase (MVK), phosphomevalonate kinase (PMVK), diphosphomevalonate decarboxylase (MVD), isopentenyl-diphosphate delta-isomerase 1 (IDI1), farnesyl diphosphate synthase (FDPS), farnesyl-diphosphate farnesyltransferase 1 (FDFT1) and lanosterol synthase (LSS). Sterol regulatory element binding transcription factor 2 (SREBF2), a transcription factor controlling the transcription of enzymes required for cholesterol and fatty acid synthesis, was suppressed as well. Phospholipase D2 (PLD2), phospholipase D3 (PLD3), several members of phospholipase A2 group (PLA2G3, PLA2G4A, PLA2G4B, PLA2G7) and phospholipase C group (PLCB3, PLCG1, PLCL4, PLCXD1) were also suppressed by RA.
RA Receptors and the Enzymes of RA Metabolism
Importantly, RA affects the enzymes of its own metabolism in keratinocytes. For example, RARB (RARβ) was induced at 72 h while RARG (RARγ) suppressed at 48 h. RXRB (RXRβ) was also suppressed at 4 h. RARA (RARα) and RXRA (RXRα) were not regulated by RA. RXRG (RXRγ) was not expressed in keratinocytes. CRBP (cellular retinol binding protein), CRABP2, LRAT (lecithin:retinol acyltransferase), ALDH1A3 (aldehyde dehydrogenase 1 family, member A3), DHRS9 (dehydrogenase/reductase, SDR family, member 9), RDH11 (retinol dehydrogenase 11, all-trans and 9-cis) and CYP26B1 (Cytochrome P450, 26B1) all play important roles in retinoic acid metabolism and maintenance of vitamin A homeostasis (22). In our gene chips analysis, CRABP2 was induced at 4 h and then suppressed after 24 h. LRAT was 2.83 fold induced at 24 h, ALDH1A3 10.2 fold at 24 h, DHRS9 3.5 fold at 24 h, RDH11 1.88 fold at 4 h, and CYP26B1 12.81 fold at 4 h. CRBP is not represented in HGU133A 2.0 chips, while CRABP1, ALDH1A1 and ALDH1A2 were expressed below detection levels in our samples. Using DAVID analysis suite, we noted that UDP-glucuronosyltransferases were significantly suppressed as a category at 24, 48 and 72 h (Figure 2). This family of enzymes conjugate RA and glucuronic acid into retinoyl-β-glucuronide.
Proliferation
Analysis of the microarray data using DAVID revealed that genes contributing to proliferation, clustered into categories such as mitotic cell cycle, spindle and cell division, were significantly induced by RA at 48 h (supplemental Table 2). Most cell cycle inhibitors, e.g. CDKN1B (cyclin-dependent kinase inhibitor 1B), CDKN1C, CDKN2B, GAS1 (growth arrest-specific 1), GAS6, and GAS7 were suppressed. Other genes related to cell proliferation including HAS2 (hyaluronan synthase 2), HBEGF (heparin-binding EGF-like growth factor), RGC32 (response gene to complement 32) , NEDD9 (neural precursor cell expressed, developmentally down-regulated 9 ), IL1ß (inetelukin 1-beta) were induced, while EGFR/ERBB1 (epidermal growth factor receptor) and ERBB2 (v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 2) were suppressed. We confirmed suppression of EGFR on protein level (Figure 1J) .
These data led us to hypothesize that RA influences keratinocyte proliferation, a previously not recognized effect of RA. However, because both positive and negative regulators of proliferation were both induced and suppressed by RA, we wanted to establish directly the functional relevance of data obtained by microarrays. Therefore, we tested the effects of RA in a proliferation assay in which we quantified the cell number of RA-treated and untreated, control keratinocytes after 24 and 48 h. Number of keratinocytes 24 and 48 h after RA treatment was moderately, ~30%, but statistically significantly higher than number of untreated cells (Figure 1I). We concluded that RA promotes keratinocyte proliferation, thus confirming the microarray data.
Apoptosis
Interestingly, as early as 4 h, RA began to induce many genes in the functional category of “apoptosis” or “regulation of programmed cell death”. This category remains significantly induced at 24 h. By 48 h, as many as 48 apoptosis-related genes and at 72 h 38 genes were suppressed by RA. Of those genes suppressed at the later time points, 13 were induced at an earlier time point. These include e.g., BCL2L1 (B-cell CLL/lymphoma 2-like 1, Bcl-xL), CFLAR (CASP8 and FADD-like apoptosis regulator), CDK5R1 (cyclin-dependent kinase 5, regulatory subunit 1), IL1α (interleukin 1 alpha), IL18 (interleukin 18), IL24 (interleukin 24) and SERPINB2 (serpin peptidase inhibitor, clade B, member 2). A list of gene symbols in the category of “apoptosis” or “regulation of programmed cell death” at different time points is presented in supplemental Table 3. The caspases CASP-3, -6, -7, -9 were induced by RA; however, CASP-1 and -4 were suppressed. Twelve genes with anti-apoptotic effects, such as BCL2L2 (B-cell CLL/lymphoma 2-like 2), PSEN1 (presenilin 1) and VEGF (vascular endothelial growth factor), were suppressed. Most pro-apoptotic genes, such as FAS, BID (BH3 interacting domain death agonist), MCL1 (myeloid cell leukemia sequence 1), and APAF1 (apoptotic protease activating factor), were induced while there were 20 pro-apoptotic genes suppressed by RA, e.g. BAX (BCL2-associated X protein). BCL2 was not expressed and BAK (BCL2-antagonist/killer 1) was not regulated by RA in keratinocytes.
The data above suggested that RA influences apoptosis in keratinocytes. By using a TUNEL assay to quantify the apoptotic cells directly, we confirmed that the net effect of RA on regulating the apoptosis-related genes was to promote apoptosis (Figure 3). We further hypothesized that RA may accentuate the effect of other apoptosis-inducing agents, e.g., UVB irradiation. However, this clearly was not the case: RA did not enhance the UVB-caused apoptosis (Figure 3). In contrast, keratinocytes treated with glucocorticoids, a negative control, showed less apoptotic cells, with or without UV-irradiation, as reported previously (66).
Figure 3. Retinoids have a net pro-apoptotic effect in keratinocytes, but do not synergize with UVB irradiation.
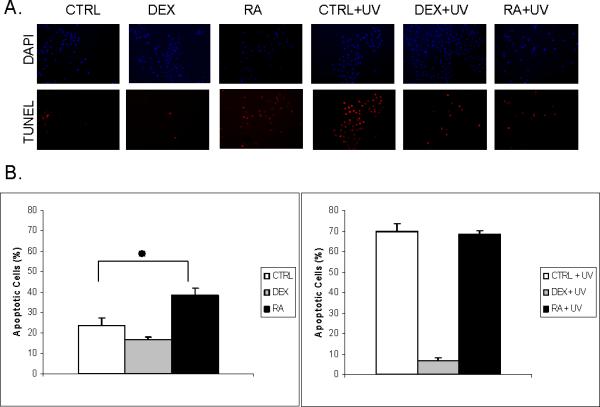
A. TUNEL staining. More apoptotic cells, showing red fluorescence, are noted in RA-treated cells than in untreated control. UVB irradiation induced massive apoptosis but without significant difference between keratinocytes treated with and without RA. Treatment with glucocorticoid (DEX), capable of inhibiting apoptosis, was used as a negative control. Blue staining with DAPI represents the nuclei. B. Quantitative analysis of apoptotic cells; *, p=0.007.
Cytoskeletal Proteins and Cell Migration
Extracellular, cell adhesion, and cytoskeletal structural proteins represent an important category of the RA regulated genes. 249 genes were induced in this category, while 288 were suppressed. For example, PKP3 (plakophilin 3), a unique binding element in desmosomes, was suppressed at all time points, and PKP1, protein with a key role in increasing desmosomal protein content and in desmosome assembly, was suppressed after 4 h, while PKP2 was induced at 4 and 24 h. DSG1 (desmoglein 1), a component of intercellular desmosome junctions, was suppressed after 4 h while DSG2 induced at 24 h.
MMP1 (matrix metalloproteinase 1), MMP3, MMP9 and MMP14 were suppressed while MMP2 and MMP7 induced. Other genes related to keratinocyte migration, including PLAT (tissue plasminogen activator), PLAUR (urokinase plasminogen activator receptor), SERPINE1 (plasminogen activator inhibitor, type I) and SERPINE2 were induced by RA. PLAU (urokinase plasminogen activator) was induced at 4 h but suppressed at 48 h and 72 h. Genes of cell motility and cell migration were among the significant clusters at 24 h (supplemental Table 2). We have previously shown that RA has a mild inhibitory effect on keratinocyte migration (43).
Signal Transduction and Transcription
More than one-third of RA-regulated genes belong to this functional group (supplement, Table 1). RA induced and suppressed the expression of 192 and 151 transcription factors, respectively, and thereby triggered subsequent cellular responses. For those genes functioning as regulators in the RAS, WNT, NFκB, Myc, JNK/ERK/p38, inositol, diacylglycerol (DAG) and p53 pathways, comparable numbers were induced and suppressed, without a predominant effect of RA on these pathways.
Correlation Between the RA-regulated Genes and Genes Affected in Psoriasis
Psoriasis is an inflammatory and proliferative skin disease in which the expression and activity of many cytokines and growth factors is dysregulated; these include IL-1, TNFα, and IFNγ. The effects of IL-1, in particular, have been associated with this disease (30, 31, 75). Oral (acitretin) and topical (tazarotene) synthetic retinoids have been established as effective treatment for psoriasis (76), although their mechanisms of action have not been fully elucidated. Recently, we performed a thorough and meticulous literature search of genes reported to be induced or suppressed in psoriatic epidermis or lymphocytes and extracted a list of 186 genes shown to be associated with psoriasis, of which 146 genes are differentially expressed in psoriatic epidermal cells (84). We compared those genes with the genes regulated by RA in our microarray experiments. Of the 146 psoriasis-associated genes, one was not represented on the HGU133A 2.0 chips and 28 were not expressed in our samples. Of the remaining 117 genes, 86 were regulated by RA, 29 were induced, 42 suppressed and 15 both induced and suppressed, but at different time points. Among the 29 induced and 42 suppressed genes, 54 were specified either as induced or as suppressed in psoriasis in the literature. Thirty-three (61%) were regulated in opposite directions by RA and in psoriasis (Figure 4). For example, TGM1 is induced in psoriasis, but suppressed by RA; on the contrary, KRT15 is suppressed in psoriasis and induced by RA. In addition, we also noted that RARRES1 (retinoic acid receptor responder 1, tazarotene induced) and RARRES3 were induced by RA.
Figure 4. Genes affected in psoriasis and regulated by RA.

List of genes specified in the literature either as induced (up) or as suppressed (down) in psoriasis and also regulated by RA, either up- or down-regulated, in the present study.
Comparison of Lists of Genes Regulated by RA with Those Regulated by Proinflammatory Cytokines
By using LOLA set of programs (List Of Lists Annotated; http://www.lola.gwu.edu), we compared the list of RA-regulated genes with the results of the expression profiling of human keratinocytes from our previously published microarray data (3, 4, 19, 27, 29, 34, 66, 84), as well as our unpublished results. These include the targets of transforming growth factor-β (TGFβ), epidermal growth factor, tumor necrosis factor-α (TNFα), IL1, Oncostatin-M, interferon-γ, calcium, triiodothyronine, glucocorticoids, SP600125 (a specific JNK inhibitor), PD98059 (ERK inhibitor), SB203580 (p38-MAPK inhibitor), and tyrphostin (EGF receptor kinase inhibitor). The number of regulated genes shared by RA and listed cytokines or inhibitors were compiled in the supplemental Table 4. Surprisingly, almost all lists of regulated genes, either induced or suppressed, shared significant number of genes regulated by RA, either induced or suppressed. For example, 339 genes were induced by both RA and TGFβ (p=2.1E-185) and 256 suppressed by both (p=9.1E-136); conversely, 187 genes were induced by RA but suppressed by TGFβ (p=2.14E-70) and 347 were suppressed by RA but induced by TGFβ (p=2.4E-197). We reanalyzed specifically these four groups of genes with DAVID program. We found that the most significant category in those genes induced by RA and suppressed by TGF-β was “interferon induction” (p=2.98E-17), while in those suppressed by both, “steroid biosynthesis” was in the first rank (p=2.89E-16).
Promoter Analysis
Because RA regulates many transcription factors, we performed an analysis of transcription factor binding sites (TFBS) in the RA regulated genes. Using oPOSSUM algorithm, we determined the over-represented TFBS in the RA-regulated genes (34). A characteristic set of transcription factors were identified for genes regulated at each time point, although these sets overlap to a certain degree (Figure 5). The number of RA-induced genes (1,817) was slightly greater than that of the suppressed (1,782), but, surprisingly, the number of TFBS identified by oPOSSUM in the induced genes was much less than that in the suppressed genes.
Figure 5. Transcription factors whose binding sites are over-represented in the RA-regulated genes.
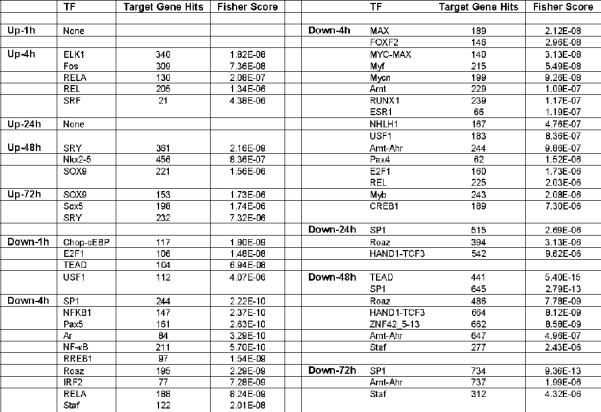
TF, names of transcription factor; target gene hits, the number of genes induced (up) or suppressed (down) at that time point showing the given transcription factor binding site; Fisher score, p-value in the one-tailed Fisher exact probability analysis. Those with a p-value less than 10−5 are listed in this table.
The SP1 binding site was over-represented in the suppressed genes at almost all time points and was the most significant one at 4, 24 and 72 h. This may reflect the known interaction between SP1 and RA receptors (63). Interestingly, RELA and REL binding sites were over-represented both in the suppressed and induced genes at 4 h, implying that these transcription factors can be both inducers and suppressors at the same time. Binding sites of two transcription factors, SRY and SOX9, both belonging to the HMG class, were over-represented in the induced genes at 48 and 72 h.
Discussion
In this study we compared the transcriptional profiles of 22,200 genes in the RA-treated and untreated keratinocytes, using Affymetrix HGU133A 2.0 oligonucleotide microarrays. Not surprisingly, for a critical regulator of many physiological functions in skin, RA regulated as many as 3,346 genes present on our gene chips. We categorized the regulated genes according to the direction of regulation (induced vs. suppressed), the time course of regulation (at 1, 4, 24, 48 and 72 h time points of RA treatment) and their biological and molecular ontological functions. We also correlated the RA-regulated genes with genes affected in psoriasis, as well as compared the list of RA-regulated genes with the lists of genes regulated by other signaling agents. In addition, we analyzed the transcription factor binding sites in the regulatory sequences associated with the RA-regulated genes.
RA metabolism
The intracellular levels of retinoic acid in tissues are tightly regulated, although the mechanisms underlying the control of retinoid metabolism at the level of specific enzymes are not completely understood. Importantly, our microarray data indicate that the pathways of retinol esterification, atRA biosynthesis and metabolism were all activated by atRA itself (Figure 6). Specifically, RA induces LRAT, DHRS9, RDH11, ALDH1A3, CRABP2 and CYP26B1 in human keratinocytes. All-trans retinol, from the circulation or applied topically, is either esterified to retinyl esters, catalyzed by LRAT, or oxidized to all-trans retinaldehyde, catalyzed by DHRS or alcohol dehydrogenase, and then oxidized to RA, which is catalyzed by ALDH (17, 20). Oxidation of all-trans retinol to all-trans retinaldehyde is reversible by the enzyme of RDH11. LRAT activity, normally low in cultured keratinocytes, increases formation of retinyl esters that function as a storage form of retinol, and reduces RA biosynthesis (20). All-trans retinoic acid, synthesized from all-trans retinol or applied externally, binds to CRABP2 and is either hydroxylated by CYP26 or enters the nucleus (20). CRABP2 is the major form of cytoplasmic binding protein in keratinocytes and skin and regulates the availability of RA for binding to nuclear receptors (1). CRBP, CRABP2 and CYP26 are inducible by retinoids in vivo (20).There are opposite reports on CRABP2 regulation by retinoic acid in reconstituted human epidermis (RHE) (60), wheras in normal human skin CRABP2 is upregulated after topical retinoid acid application (18). “UDP glucuronosyltransferase” was a significantly suppressed ontological category at 24, 48 and 72h. Conversely a significant increase in transferase specific activity was observed in the vitamin A deficient rat liver (50). This family of UDP glucuronosyltransferases conjugates RA and glucuronic acid into retinoyl-β-glucuronide. The retinoyl-β-glucuronide possesses 30-100% of the biological activity of RA (52) or greater activity than RA (64), and is considered to be of functional importance (50). Our study showed induction of both RARB and KRT19 genes by RA, as reported before for HEK, RHE and normal skin after the RA treatment (13) RARB may also mediate apoptosis in keratinocytes (49).
Figure 6. Simplified pathways of retinol esterification, atRA biosynthesis and metabolism.
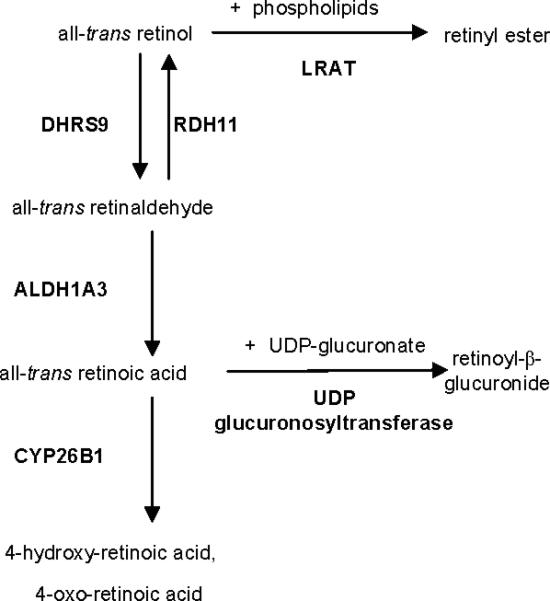
Only the genes regulated in the present study are shown.
Skin expresses both lecithin:retinol acyltransferase (LRAT) and acyl CoA:retinol acyltransferase (ARAT) activities, the two enzymes known to catalyze retinyl ester synthesis (41, 71). The normally low LRAT activity of cultured keratinocytes is induced by retinoids, probably via RARs, within 12 h (41). Induction of LRAT activity by retinoids increases retinyl ester formation and reduces RA biosynthesis (41). In summary, these results show that RA tightly regulates its own bioavailability in the epidermis by regulating the expression of the enzymes involved in RA metabolism, and protein involved in RA delivery and sequestration.
Epidermal Differentiation
one of the well established effects of RA is the suppression of keratinocyte differentiation, manifested as suppression of expression of differentiation markers such as TGM1, FLG and CDSN, as well as inhibition of formation of cornified envelopes. Approximately 20 structural protein genes, including keratins, desmosomal proteins, integrins, and the terminal differentiation markers LOR, FLG and TGM1, were downregulated in keratinocytes after treatment with retinoids. Several of the modulated genes have well-characterized retinoid response elements in their regulatory sequences and all have already shown to be directly downregulated via retinoic acid nuclear receptors (14). These genes include several cytokeratins (K1, K10), desmosomal proteins (CDSN) and important cornified envelope precursors (FLG, LOR). The differentiation genes listed are downregulated by retinoids in RHE in chemically defined medium (for review see (14). All of these genes are suppressed by RA in our experiments, as expected. Interestingly, the involucrin gene transcription, another canonical epidermal differentiation marker, was actually induced by RA, confirming previously published data (55), whereas in RHE involucrin expression was not modified by retinoids (5) .
Synthesis of specific lipids is an integral part of keratinocyte differentiation and RA affects lipid metabolism (78). These lipids, which mainly comprise ceramides, free fatty acids, and cholesterol, are important for the cohesion of the stratum corneum and the maintenance of normal epidermal barrier function (16, 61). The inhibition of keratinocyte differentiation by RA includes the suppression of genes responsible for biosynthesis of epidermal lipids. Genes involved in sphingolipid metabolism, in the pathway of cholesterol biosynthesis, a gene catalyzing the formation of long-chain fatty acids, FASN, and a transcription factor, SREBF2, were suppressed. SREBFs stimulate the transcription of key genes of cholesterol metabolism, such as HMGCS1 and HMGCR, and genes of fatty acid synthesis, such as FASN (32). In RHE, strong suppression of HMGCR and FAS was reported after the treatment with retinoic acid (5). During keratinocyte differentiation, glucosylceramide is synthesized by the enzyme UGCG, which is induced during human keratinocyte differentiation (79). Glucosylceramide is stored in intracellular lamellar granules and eventually extruded into the intercellular space where it is hydrolyzed by the enzyme GBA to ceramide, the major lipid component of the epidermal permeability barrier (79). Another precursor of ceramide, sphingomyelin, is converted into ceramide by the enzyme SMPD1 (9). All three of these enzymes, UGCG, GBA and SMPD1, were suppressed in the present study.
The overall effect of these transcriptional changes is to inhibit the synthesis of the lipid components of the stratum corneum. Thus, RA suppresses not only the synthesis of the protein markers of cornification but in addition other epidermal differentiation-associated processes, such as the synthesis of the stratum corneum lipids.
Retinoids mediate changes in keratin synthesis, reflecting the different stages of keratinocyte differentiation. In our microarray experiments, 11 keratin genes were suppressed, 4 induced and 2 induced at early time points and then suppressed later. The suppression of KRT1 biosynthesis and elevation in KRT13 synthesis occurred within 2 days of RA treatment of keratinocyte raft cultures (12). In our microarray data, KRT1 was suppressed at 24 h and KRT13 induced as early as 1 h. Previous studies demonstrated a strong correlation between KRT6 and KRT16 expression and epidermal hyperproliferation, including in psoriasis, actinic keratosis and regions of epidermis adjacent to tumors (67). KRT6B and KRT16 were suppressed in the present study. In response to elevated levels of retinoids in the medium, both KRT13 and KRT19, normally found in non-cornified epithelia, are induced (24, 40). We have suggested that RA has a two-fold effect on keratin gene expression: by regulating keratinocyte differentiation it determines which keratins are expressed and by direct interaction between its receptor and keratin genes, RA determines the total amount of keratin protein within the cell (6). We have also demonstrated that the nuclear receptors for RA can suppress the promoters of some keratin genes through direct binding of these receptors to the RAREs of the keratin gene promoters in a ligand-dependent way (69).
Retinoic acid affects different tissue cell types in a diverse ways. It can act as an inhibitor or inducing factor for main cellular processes such as proliferation, differentiation and apoptosis. Different effects of retinoids on proliferation and apoptosis have been reported depending on weather fetal or adult, normal or pathological cells were used for experiments (65). This diversity can be accounted on diversity of retinoic acid receptors present in the specific cell/tissue. Treatment with retinoic acid simultaneously suppresses proliferation and significantly induced differentiation in astrocytoma cell line deriving from glioblastoma multiforme and exhibits the same effects in retinoblastoma cells. PMID: (86, 87). Pre-treatment of follicular thyroid carcinoma cell lines with RA resulted in decreased in-vitro proliferation rates and reduced tumor cell growth (72)Incubation with all-trans retinoic acid (RA) induces embryonic carcinoma cells to cease proliferation, induce differentiation and undergo apoptosis (33). In Leydig and cultured Sertoli cells RA treatment induced an increase in apoptosis (73, 85). However in purified peripheral blood eosinophils RA is a potent inhibitor of spontaneous apoptosis (74). In the recent study using microarray analysis, Mamoon et al. (2008) characterized RA-responsive genes in hepatocytes using non-transformed hepatic cell line thus providing a simple and physiologically relevant system to study RA action in hepatocytes (46). They reported that all-trans RA increased expression of proliferation and differentiation associated genes. They also reported that genes that promote apoptosis are inhibited and, conversely, genes that inhibit apoptosis are induced (46). Furthermore they expanded their work by investigating the effect of v-erbA, an oncogene known to exert dominant – negative effect on the expression of retinoic acid responsive genes in order to understand nuclear receptor mediated transcriptional regulation in murine hepatocytes in culture (77).
Keratinocyte terminal differentiation shares several cellular and molecular characteristics with apoptosis (47). While our data confirm the well-established effect of retinoids on inhibiting keratinocyte differentiation, we find that RA, in contrast, induces genes that promote apoptosis. These results advance the hypothesis that apoptosis and terminal differentiation of epidermal keratinocytes are two distinct processes (25). In most programmed cell deaths, permeabilization of the outer mitochondrial membrane is the point of no return and Bcl-2 family proteins control mitochondrial membrane permeability. Some Bcl-2 family proteins (Bcl-2, ced-9 homologue, and Bcl-xL) may block apoptosis whereas others (Bax, Bak, and Bid) promote apoptosis (58). Reports of RA affects on the expression of Bcl-2 family in vitro are controversial. Islam et al.(2005) studied the effects of RA on mouse keratinocyte cell line and found that the mRNA level of bcl-2 was unaltered or even increased, depending upon growth conditions, and that Bax mRNA was expressed at high levels under all conditions (35). Thomková et al. ( 1999) treated human keratinocytes with RA under both low and high Ca2+ conditions, without observing any change in bax, bak, bcl-2, bcl-xL, and mcl-1 protein levels (68). We demonstrated that BID and MCL1 were induced, while BAX and BCL2L2 were suppressed by RA. BCL2L1 was induced at 4 h and 24 h, and suppressed at 48 h. BCL2 was not expressed and BAK not regulated by RA in our data. In addition to the genes of Bcl-2 family, we found several other pro-apoptotic and anti-apoptotic genes regulated by RA, even at as early as 4 h. Because of these complex and contrasting findings, in order to evaluate the net effect of RA on apoptosis in cultured keratinocytes, we performed the TUNEL assay. As expected, we found that RA actually promotes apoptosis of keratinocytes. The pro-apoptotic effect of RA may play an important role in the treatment of skin diseases, e.g. non-melanoma skin cancers.
RA treatment of aged skin results in photosensitivity, which led us to analyze the combined effects of RA and UVB light on the apoptosis of keratinocytes. Contrary to our expectations, RA did not exacerbate the severity of apoptosis induced by UV-irradiation. This implies that the RA-induced photosensitivity is not directly related to apoptosis of keratinocytes.
When the dying keratinocytes differentiate and form the stratum corneum they appear to fragment their DNA in a manner characteristic for cells undergoing apoptosis (48), and activation of caspases has also been observed during this process (80). These and other observations have led to the suggestion that keratinocyte terminal differentiation may be regarded as a special form of apoptosis, sharing several cellular and molecular events with the apoptosis induced by external agents such as ultraviolet (UV) radiation, and with apoptosis in other cell types (54). These conclusions stand in contrast to results from other authors, among them Gandarillas et al. (1999) who showed that terminally differentiated keratinocytes rarely underwent apoptosis as determined by in situ end labeling (TUNEL), DNA laddering or morphological examination (25). Mitra et al. (1997) have shown that apoptosis can occur in keratinocytes without evidence of differentiation, and vice versa (51). Study by Bernard et al ( 2002) revealed strong induction of THYB10 transcript, the gene suspected to play a role in control of apoptosis in skin after the topical treatment with retinoic acid (5).In this respect, the TGMs are of special interest in view of a potential effector role in the formation of apoptotic bodies and DNA fragmentation. Retinoids have been reported to induce TGM activity linked to tissue type TGM (TGM2) in actively proliferating cultured keratinocytes, while suppressing the expression of TGM1, normally induced when cells undergo differentiation (23, 45), further indicating that these enzymes fulfill their roles in different cellular compartments. Importantly, both TGM1 and TGM2 were suppressed by RA in our chips. These data support the hypothesis that epidermal differentiation and apoptosis are separate and independent processes.
Despite the demonstrated clinical success of retinoid therapy in psoriasis, its mechanism of action is not completely understood. We found that out of 117 genes affected in psoriasis and expressed in our chips, 86 genes were also regulated by RA. A large proportion of these were even regulated in opposite directions (Figure 4). It can be postulated that RA counteracts those “abnormally-regulated” genes in psoriasis. Of them, 11 genes were in the functional category of “signal transduction and transcription”, another 11 in “inflammation and immunity” and 8 in “structural and cytoskeletal” (including the function of “epidermal differentiation”). However, none of them was directly related to apoptosis, though it has been shown that physiological apoptosis in psoriatic skin lesions was decreased (42), and we have revealed that RA promotes keratinocyte apoptosis in this study. This might provide a clue to how retinoids work to alleviate psoriasis.
We found genes encoding growth factors such as IGFBP2, HBEGF and PDGFA previously reported as being down-regulated in RHEafter topical retinoic acid treatment regulated in our microarray data (5). Interestingly, the expression of VEGF was severely suppressed by RA, 7.2 fold. The reduction in VEGF protein in human keratinocyte cultures by RA was paralleled by a strong down-regulation of VEGF mRNA levels (81), which may contribute to the therapeutic effects of retinoids in diseases that are accompanied by angioproliferation, such as psoriasis.
Our comparison of genes regulated by RA with lists of genes regulated by other cytokines or agents revealed the complexity of the effects of those agents on different genes. For example, hundreds of genes were regulated by both RA and TGFβ, either in the same or in opposite directions. Specifically, we noticed in TGFβ-suppressed genes, RA also significantly suppressed those in the category of “steroid biosynthesis” and contrarily, RA induced those in the category of “interferon induction”. There is a complex interrelation between TGFβ and RA in epidermal differentiation (12). In some regards, e.g., suppression of KRT1 and FLG expression, the actions of TGFβ and RA can be synergistic and in other regards, e.g., proliferation and KRT6/KRT16 expression, the action of TGFβ can be both antagonistic and dominant over RA.
In summary, these data present a comprehensive documentation and description of the transcriptional changes caused by all-trans retinoic acid treatment of primary human keratinocytes, demonstrating the complexity of the effects that RA may have on epidermis. RA itself activates the pathways of retinol esterification, RA synthesis and metabolism. It inhibits keratinocyte differentiation and lipid metabolism associated with the formation of epidermal barrier function. Furthermore, RA regulates pro- and anti-apoptotic genes and specifically the genes reported to be affected in psoriasis.
Supplementary Material
Acknowledgements
We thank the NYU Genomic Facility for technical assistance with microarray.
Contract grant sponsor: National Institutes of Health Contract grant number: AR45974 Contract grant sponsor: National Institutes of Health Contract grant number: NR08029 Contract grant sponsor: National Institutes of Health Contract grant number: AG030673 Contract grant sponsor: DebRA-UK Foundation Contract grant number: N/A
Literature Cited
- 1.Astrom A, Tavakkol A, Pettersson U, Cromie M, Elder JT, Voorhees JJ. Molecular cloning of two human cellular retinoic acid-binding proteins (CRABP). Retinoic acid-induced expression of CRABP-II but not CRABP-I in adult human skin in vivo and in skin fibroblasts in vitro. Journal of Biological Chemistry. 1991;266:17662–17666. [PubMed] [Google Scholar]
- 2.Balmer JE, Blomhoff R. Gene expression regulation by retinoic acid. J Lipid Res. 2002;43:1773–1808. doi: 10.1194/jlr.r100015-jlr200. [DOI] [PubMed] [Google Scholar]
- 3.Banno T, Adachi M, Mukkamala L, Blumenberg M. Unique keratinocyte-specific effects of interferon-gamma that protect skin from viruses, identified using transcriptional profiling. Antivir Ther. 2003;8:541–554. [PubMed] [Google Scholar]
- 4.Banno T, Gazel A, Blumenberg M. Effects of tumor necrosis factor-alpha (TNF alpha) in epidermal keratinocytes revealed using global transcriptional profiling. J Biol Chem. 2004;279:32633–32642. doi: 10.1074/jbc.M400642200. [DOI] [PubMed] [Google Scholar]
- 5.Bernard FX, Pedretti N, Rosdy M, Deguercy A. Comparison of gene expression profiles in human keratinocyte mono-layer cultures, reconstituted epidermis and normal human skin; transcriptional effects of retinoid treatments in reconstituted human epidermis. Exp Dermatol. 2002;11:59–74. doi: 10.1034/j.1600-0625.2002.110107.x. [DOI] [PubMed] [Google Scholar]
- 6.Blumenberg M, Connolly DM, Freedberg IM. Regulation of keratin gene expression: the role of the nuclear receptors for retinoic acid, thyroid hormone and vitamin D3. Journal of Investigative Dermatology. 1992;98:42S–49S. doi: 10.1111/1523-1747.ep12462194. [DOI] [PubMed] [Google Scholar]
- 7.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 8.Boucherat O, Franco-Montoya ML, Thibault C, Incitti R, Chailley-Heu B, Delacourt C, Bourbon JR. Gene expression profiling in lung fibroblasts reveals new players in alveolarization. Physiol Genomics. 2007;32:128–141. doi: 10.1152/physiolgenomics.00108.2007. [DOI] [PubMed] [Google Scholar]
- 9.Breiden B, Gallala H, Doering T, Sandhoff K. Optimization of submerged keratinocyte cultures for the synthesis of barrier ceramides. Eur J Cell Biol. 2007;86:657–673. doi: 10.1016/j.ejcb.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 10.Chambon P. A decade of molecular biology of retinoic acid receptors. Faseb J. 1996;10:940–954. [PubMed] [Google Scholar]
- 11.Cheepala SB, Syed Z, Trutschl M, Cvek U, Clifford JL. Retinoids and skin: microarrays shed new light on chemopreventive action of all-trans retinoic acid. Mol Carcinog. 2007;46:634–639. doi: 10.1002/mc.20346. [DOI] [PubMed] [Google Scholar]
- 12.Choi Y, Fuchs E. TGF-B and retinoic acid: regulators of growth and modifiers of differentiation in human epidermal cells. Cell Regulation. 1990;1:791–809. doi: 10.1091/mbc.1.11.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crowe DL, Hu L, Gudas LJ, Rheinwald JG. Variable expression of retinoic acid receptor (RAR beta) mRNA in human oral and epidermal keratinocytes; relation to keratin 19 expression and keratinization potential. Differentiation. 1991;48:199–208. doi: 10.1111/j.1432-0436.1991.tb00258.x. [DOI] [PubMed] [Google Scholar]
- 14.Darmon M, Blumenberg M. Retinoic acid in epithelial and epidermal differentiation. In: Darmon M, Blumenberg M, editors. Molecular Biology of the Skin: The Keratinocyte. New York: 1993. Academic Press, Inc.. pp. 181–198. [Google Scholar]
- 15.Dennis G, Jr., Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. Epub 2003 Apr 2003. [PubMed] [Google Scholar]
- 16.Downing DT. Lipid and protein structures in the permeability barrier of mammalian epidermis. J Lipid Res. 1992;33:301–313. [PubMed] [Google Scholar]
- 17.Duester G. Families of retinoid dehydrogenases regulating vitamin A function: production of visual pigment and retinoic acid. Eur J Biochem. 2000;267:4315–4324. doi: 10.1046/j.1432-1327.2000.01497.x. [DOI] [PubMed] [Google Scholar]
- 18.Elder JT, Astrom A, Pettersson U, Tavakkol A, Griffiths CE, Krust A, Kastner P, Chambon P, Voorhees JJ. Differential regulation of retinoic acid receptors and binding proteins in human skin. Journal of Investigative Dermatology. 1992;98:673–679. doi: 10.1111/1523-1747.ep12499896. [DOI] [PubMed] [Google Scholar]
- 19.Finelt N, Gazel A, Gorelick S, Blumenberg M. Transcriptional responses of human epidermal keratinocytes to Oncostatin-M. Cytokine. 2005;31:305–313. doi: 10.1016/j.cyto.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 20.Fisher GJ, Datta SC, Talwar HS, Wang ZQ, Varani J, Kang S, Voorhees JJ. Molecular basis of sun-induced premature skin ageing and retinoid antagonism. Nature. 1996;379:335–339. doi: 10.1038/379335a0. [DOI] [PubMed] [Google Scholar]
- 21.Fisher GJ, Talwar HS, Xiao JH, Datta SC, Reddy AP, Gaub MP, Rochette-Egly C, Chambon P, Voorhees JJ. Immunological identification and functional quantitation of retinoic acid and retinoid X receptor proteins in human skin. Journal of Biological Chemistry. 1994;269:20629–20635. [PubMed] [Google Scholar]
- 22.Fisher GJ, Voorhees JJ. Molecular mechanisms of retinoid actions in skin. Faseb Journal. 1996;10:1002–1013. doi: 10.1096/fasebj.10.9.8801161. [DOI] [PubMed] [Google Scholar]
- 23.Floyd EE, Jetten AM. Regulation of type I (epidermal) transglutaminase mRNA levels during squamous differentiation: down regulation by retinoids. Molecular & Cellular Biology. 1989;9:4846–4851. doi: 10.1128/mcb.9.11.4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fuchs E, Green H. Regulation of terminal differentiation of cultured human keratinocytes by vitamin A. Cell. 1981;25:617–625. doi: 10.1016/0092-8674(81)90169-0. [DOI] [PubMed] [Google Scholar]
- 25.Gandarillas A, Goldsmith LA, Gschmeissner S, Leigh IM, Watt FM. Evidence that apoptosis and terminal differentiation of epidermal keratinocytes are distinct processes. Exp Dermatol. 1999;8:71–79. doi: 10.1111/j.1600-0625.1999.tb00350.x. [DOI] [PubMed] [Google Scholar]
- 26.Gann AA, Gates PB, Stark D, Brockes JP. Receptor isoform specificity in a cellular response to retinoic acid. Proc Biol Sci. 1996;263:729–734. doi: 10.1098/rspb.1996.0109. [DOI] [PubMed] [Google Scholar]
- 27.Gazel A, Nijhawan RI, Walsh R, Blumenberg M. Transcriptional profiling defines the roles of ERK and p38 kinases in epidermal keratinocytes. J Cell Physiol. 2008;215:292–308. doi: 10.1002/jcp.21394. [DOI] [PubMed] [Google Scholar]
- 28.Gazel A, Ramphal P, Rosdy M, De Wever B, Tornier C, Hosein N, Lee B, Tomic-Canic M, Blumenberg M. Transcriptional profiling of epidermal keratinocytes: comparison of genes expressed in skin, cultured keratinocytes, and reconstituted epidermis, using large DNA microarrays. J Invest Dermatol. 2003;121:1459–1468. doi: 10.1111/j.1523-1747.2003.12611.x. [DOI] [PubMed] [Google Scholar]
- 29.Gazel A, Rosdy M, Bertino B, Tornier C, Sahuc F, Blumenberg MA. Characteristic Subset of Psoriasis-Associated Genes Is Induced by Oncostatin-M in Reconstituted Epidermis. J Invest Dermatol. 2006;17:17. doi: 10.1038/sj.jid.5700461. [DOI] [PubMed] [Google Scholar]
- 30.Gottlieb AB, Chamian F, Masud S, Cardinale I, Abello MV, Lowes MA, Chen F, Magliocco M, Krueger JG. TNF inhibition rapidly down-regulates multiple proinflammatory pathways in psoriasis plaques. J Immunol. 2005;175:2721–2729. doi: 10.4049/jimmunol.175.4.2721. [DOI] [PubMed] [Google Scholar]
- 31.Groves RW, Sherman L, Mizutani H, Dower SK, Kupper TS. Detection of interleukin-1 receptors in human epidermis. Induction of the type II receptor after organ culture and in psoriasis. American Journal of Pathology. 1994;145:1048–1056. [PMC free article] [PubMed] [Google Scholar]
- 32.Harris IR, Farrell AM, Holleran WM, Jackson S, Grunfeld C, Elias PM, Feingold KR. Parallel regulation of sterol regulatory element binding protein-2 and the enzymes of cholesterol and fatty acid synthesis but not ceramide synthesis in cultured human keratinocytes and murine epidermis. J Lipid Res. 1998;39:412–422. [PubMed] [Google Scholar]
- 33.Herget T, Specht H, Esdar C, Oehrlein SA, Maelicke A. Retinoic acid induces apoptosis-associated neural differentiation of a murine teratocarcinoma cell line. J Neurochem. 1998;70:47–58. doi: 10.1046/j.1471-4159.1998.70010047.x. [DOI] [PubMed] [Google Scholar]
- 34.Ho Sui SJ, Mortimer JR, Arenillas DJ, Brumm J, Walsh CJ, Kennedy BP, Wasserman WW. oPOSSUM: identification of over-represented transcription factor binding sites in co-expressed genes. Nucleic Acids Res. 2005;33:3154–3164. doi: 10.1093/nar/gki624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Islam TC, Skarin T, Sumitran S, Toftgard R. Retinoids induce apoptosis in cultured keratinocytes. Br J Dermatol. 2000;143:709–719. doi: 10.1046/j.1365-2133.2000.03823.x. [DOI] [PubMed] [Google Scholar]
- 36.Jetten AM. Multi-stage program of differentiation in human epidermal keratinocytes: regulation by retinoids. J Invest Dermatol. 1990;95:44S–46S. doi: 10.1111/1523-1747.ep12505757. [DOI] [PubMed] [Google Scholar]
- 37.Jho SH, Radoja N, Im MJ, Tomic-Canic M. Negative response elements in keratin genes mediate transcriptional repression and the cross-talk among nuclear receptors. J Biol Chem. 2001;276:45914–45920. doi: 10.1074/jbc.M103144200. Epub 42001 Oct 45918. [DOI] [PubMed] [Google Scholar]
- 38.Jho SH, Vouthounis C, Lee B, Stojadinovic O, Im MJ, Brem H, Merchant A, Chau K, Tomic-Canic M. The Book of Opposites: The Role of the Nuclear Receptor Co-regulators in the Suppression of Epidermal Genes by Retinoic Acid and Thyroid Hormone Receptors. J Invest Dermatol. 2005;124:1034–1043. doi: 10.1111/j.0022-202X.2005.23691.x. [DOI] [PubMed] [Google Scholar]
- 39.Kligman AM, Fulton JE, Jr., Plewig G. Topical vitamin A acid in acne vulgaris. Arch Dermatol. 1969;99:469–476. [PubMed] [Google Scholar]
- 40.Kopan R, Traska G, Fuchs E. Retinoids as important regulators of terminal differentiation: examining keratin expression in individual epidermal cells at various stages of keratinization. J Cell Biol. 1987;105:427–440. doi: 10.1083/jcb.105.1.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kurlandsky SB, Duell EA, Kang S, Voorhees JJ, Fisher GJ. Auto-regulation of retinoic acid biosynthesis through regulation of retinol esterification in human keratinocytes. Journal of Biological Chemistry. 1996;271:15346–15352. doi: 10.1074/jbc.271.26.15346. [DOI] [PubMed] [Google Scholar]
- 42.Laporte M, Galand P, Fokan D, de Graef C, Heenen M. Apoptosis in established and healing psoriasis. Dermatology. 2000;200:314–316. doi: 10.1159/000018394. [DOI] [PubMed] [Google Scholar]
- 43.Lee B, Vouthounis C, Stojadinovic O, Brem H, Im M, Tomic-Canic M. From an enhanceosome to a repressosome: molecular antagonism between glucocorticoids and EGF leads to inhibition of wound healing. J Mol Biol. 2005;345:1083–1097. doi: 10.1016/j.jmb.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 44.Li D, Turi TG, Schuck A, Freedberg IM, Khitrov G, Blumenberg M. Rays and arrays: the transcriptional program in the response of human epidermal keratinocytes to UVB illumination. Faseb J. 2001;15:2533–2535. doi: 10.1096/fj.01-0172fje. [DOI] [PubMed] [Google Scholar]
- 45.Lichti U, Ben T, Yuspa SH. Retinoic acid-induced transglutaminase in mouse epidermal cells is distinct from epidermal transglutaminase. Journal of Biological Chemistry. 1985;260:1422–1426. [PubMed] [Google Scholar]
- 46.Mamoon A, Ventura-Holman T, Maher JF, Subauste JS. Retinoic acid responsive genes in the murine hepatocyte cell line AML 12. Gene. 2008;408:95–103. doi: 10.1016/j.gene.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 47.Maruoka Y, Harada H, Mitsuyasu T, Seta Y, Kurokawa H, Kajiyama M, Toyoshima K. Keratinocytes become terminally differentiated in a process involving programmed cell death. Biochem Biophys Res Commun. 1997;238:886–890. doi: 10.1006/bbrc.1997.7405. [DOI] [PubMed] [Google Scholar]
- 48.McCall CA, Cohen JJ. Programmed cell death in terminally differentiating keratinocytes: role of endogenous endonuclease. J Invest Dermatol. 1991;97:111–114. doi: 10.1111/1523-1747.ep12478519. [DOI] [PubMed] [Google Scholar]
- 49.Mendelsohn C, Ruberte E, LeMeur M, Morriss-Kay G, Chambon P. Developmental analysis of the retinoic acid-inducible RAR-beta 2 promoter in transgenic animals. Development. 1991;113:723–734. doi: 10.1242/dev.113.3.723. [DOI] [PubMed] [Google Scholar]
- 50.Miller DA, DeLuca HF. Biosynthesis of retinoyl-beta-glucuronide, a biologically active metabolite of all-trans-retinoic acid. Arch Biochem Biophys. 1986;244:179–186. doi: 10.1016/0003-9861(86)90107-4. [DOI] [PubMed] [Google Scholar]
- 51.Mitra RS, Wrone-Smith T, Simonian P, Foreman KE, Nunez G, Nickoloff BJ. Apoptosis in keratinocytes is not dependent on induction of differentiation. Laboratory Investigation. 1997;76:99–107. [PubMed] [Google Scholar]
- 52.Nath K, Olson JA. Natural occurrence and biological activity of vitamin A derivatives in rat bile. J Nutr. 1967;93:461–469. doi: 10.1093/jn/93.4.461. [DOI] [PubMed] [Google Scholar]
- 53.Petkovich M, Brand NJ, Krust A, Chambon P. A human retinoic acid receptor which belongs to the family of nuclear receptors. [Review]. Nature. 1987;330:444–450. doi: 10.1038/330444a0. [DOI] [PubMed] [Google Scholar]
- 54.Polakowska RR, Piacentini M, Bartlett R, Goldsmith LA, Haake AR. Apoptosis in human skin development: morphogenesis, periderm, and stem cells. Dev Dyn. 1994;199:176–188. doi: 10.1002/aja.1001990303. [DOI] [PubMed] [Google Scholar]
- 55.Poumay Y, Herphelin F, Smits P, De Potter IY, Pittelkow MR. High-cell-density phorbol ester and retinoic acid upregulate involucrin and downregulate suprabasal keratin 10 in autocrine cultures of human epidermal keratinocytes. Mol Cell Biol Res Commun. 1999;2:138–144. doi: 10.1006/mcbr.1999.0165. [DOI] [PubMed] [Google Scholar]
- 56.Proksch E, Holleran WM, Menon GK, Elias PM, Feingold KR. Barrier function regulates epidermal lipid and DNA synthesis. Br J Dermatol. 1993;128:473–482. doi: 10.1111/j.1365-2133.1993.tb00222.x. [DOI] [PubMed] [Google Scholar]
- 57.Radoja N, Diaz DV, Minars TJ, Freedberg IM, Blumenberg M, Tomic-Canic M. Specific organization of the negative response elements for retinoic acid and thyroid hormone receptors in keratin gene family. J Invest Dermatol. 1997;109:566–572. doi: 10.1111/1523-1747.ep12337483. [DOI] [PubMed] [Google Scholar]
- 58.Raj D, Brash DE, Grossman D. Keratinocyte apoptosis in epidermal development and disease. J Invest Dermatol. 2006;126:243–257. doi: 10.1038/sj.jid.5700008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rook AH, Jaworsky C, Nguyen T, Grossman RA, Wolfe JT, Witmer WK, Kligman AM. Beneficial effect of low-dose systemic retinoid in combination with topical tretinoin for the treatment and prophylaxis of premalignant and malignant skin lesions in renal transplant recipients. Transplantation. 1995;59:714–719. doi: 10.1097/00007890-199503150-00013. [DOI] [PubMed] [Google Scholar]
- 60.Sanquer S, Eller MS, Gilchrest BA. Retinoids and state of differentiation modulate CRABPII gene expressioninaskin equivalent. J Invest Dermatol. 1993;100:148–153. doi: 10.1111/1523-1747.ep12462785. [DOI] [PubMed] [Google Scholar]
- 61.Schurer NY, Elias PM. The biochemistry and function of stratum corneum lipids. Adv Lipid Res. 1991;24:27–56. doi: 10.1016/b978-0-12-024924-4.50006-7. [DOI] [PubMed] [Google Scholar]
- 62.Segre JA, Bauer C, Fuchs E. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat Genet. 1999;22:356–360. doi: 10.1038/11926. [DOI] [PubMed] [Google Scholar]
- 63.Shimada J, Suzuki Y, Kim SJ, Wang PC, Matsumura M, Kojima S. Transactivation via RAR/RXR-Sp1 interaction: characterization of binding between Sp1 and GC box motif. Mol Endocrinol. 2001;15:1677–1692. doi: 10.1210/mend.15.10.0707. [DOI] [PubMed] [Google Scholar]
- 64.Sietsema WK, DeLuca HF. A new vaginal smear assay for vitamin A in rats. J Nutr. 1982;112:1481–1489. doi: 10.1093/jn/112.8.1481. [DOI] [PubMed] [Google Scholar]
- 65.Simoni D, Tolomeo M. Retinoids, apoptosis and cancer. Curr Pharm Des. 2001;7:1823–1837. doi: 10.2174/1381612013397168. [DOI] [PubMed] [Google Scholar]
- 66.Stojadinovic O, Lee B, Vouthounis C, Vukelic S, Pastar I, Blumenberg M, Brem H, Tomic-Canic M. Novel genomic effects of glucocorticoids in epidermal keratinocytes: inhibition of apoptosis, interferon-gamma pathway, and wound healing along with promotion of terminal differentiation. J Biol Chem. 2007;282:4021–4034. doi: 10.1074/jbc.M606262200. [DOI] [PubMed] [Google Scholar]
- 67.Stoler A, Kopan R, Duvic M, Fuchs E. Use of monospecific antisera and cRNA probes to localize the major changes in keratin expression during normal and abnormal epidermal differentiation. J Cell Biol. 1988;107:427–446. doi: 10.1083/jcb.107.2.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thomkova H, Fujimoto W, Arata J. Expression of the bcl-2 family of genes in the course of keratinocyte differentiation. Eur J Dermatol. 1999;9:191–196. [PubMed] [Google Scholar]
- 69.Tomic-Canic M, Day D, Samuels HH, Freedberg IM, Blumenberg M. Novel regulation of keratin gene expression by thyroid hormone and retinoid receptors. Journal of Biological Chemistry. 1996;271:1416–1423. doi: 10.1074/jbc.271.3.1416. [DOI] [PubMed] [Google Scholar]
- 70.Tomic-Canic M, Freedberg IM, Blumenberg M. Codominant regulation of keratin gene expression by cell surface receptors and nuclear receptors. Experimental Cell Research. 1987;224:96–102. doi: 10.1006/excr.1996.0115. 1996. [DOI] [PubMed] [Google Scholar]
- 71.Torma H, Vahlquist A. Retinol esterification by mouse epidermal microsomes: evidence for acyl-CoA:retinol acyltransferase activity. J Invest Dermatol. 88:398–402. doi: 10.1111/1523-1747.ep12469726. [DOI] [PubMed] [Google Scholar]
- 72.Trojanowicz B, Winkler A, Hammje K, Chen Z, Sekulla C, Glanz D, Schmutzler C, Mentrup B, Hombach-Klonisch S, Klonisch T, Finke R, Kohrle J, Dralle H, Hoang-Vu C. Retinoic acid-mediated down-regulation of ENO1/MBP-1 gene products caused decreased invasiveness of the follicular thyroid carcinoma cell lines. J Mol Endocrinol. 2009;42:249–260. doi: 10.1677/JME-08-0118. [DOI] [PubMed] [Google Scholar]
- 73.Tucci P, Cione E, Perri M, Genchi G. All-trans-retinoic acid induces apoptosis in Leydig cells via activation of the mitochondrial death pathway and antioxidant enzyme regulation. J Bioenerg Biomembr. 2008;40:315–323. doi: 10.1007/s10863-008-9156-8. [DOI] [PubMed] [Google Scholar]
- 74.Ueki S, Mahemuti G, Oyamada H, Kato H, Kihara J, Tanabe M, Ito W, Chiba T, Takeda M, Kayaba H, Chihara J. Retinoic acids are potent inhibitors of spontaneous human eosinophil apoptosis. J Immunol. 2008;181:7689–7698. doi: 10.4049/jimmunol.181.11.7689. [DOI] [PubMed] [Google Scholar]
- 75.Uyemura K, Yamamura M, Fivenson DF, Modlin RL, Nickoloff BJ. The cytokine network in lesional and lesion-free psoriatic skin is characterized by a T-helper type 1 cell-mediated response. J Invest Dermatol. 1993;101:701–705. doi: 10.1111/1523-1747.ep12371679. [DOI] [PubMed] [Google Scholar]
- 76.van de Kerkhof PC. Update on retinoid therapy of psoriasis in: an update on the use of retinoids in dermatology. Dermatol Ther. 2006;19:252–263. doi: 10.1111/j.1529-8019.2006.00082.x. [DOI] [PubMed] [Google Scholar]
- 77.Ventura-Holman T, Mamoon A, Subauste JS. Modulation of expression of RA-regulated genes by the oncoprotein v-erbA. Gene. 2008;425:23–27. doi: 10.1016/j.gene.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 78.Voorhees JJ. Clinical effects of long-term therapy with topical tretinoin and cellular mode of action. J Int Med Res 18 Suppl. 1990;3:26C–28C. [PubMed] [Google Scholar]
- 79.Watanabe R, Wu K, Paul P, Marks DL, Kobayashi T, Pittelkow MR, Pagano RE. Up-regulation of glucosylceramide synthase expression and activity during human keratinocyte differentiation. J Biol Chem. 1998;273:9651–9655. doi: 10.1074/jbc.273.16.9651. [DOI] [PubMed] [Google Scholar]
- 80.Weil M, Raff MC, Braga VM. Caspase activation in the terminal differentiation of human epidermal keratinocytes. Curr Biol. 1999;9:361–364. doi: 10.1016/s0960-9822(99)80162-6. [DOI] [PubMed] [Google Scholar]
- 81.Weninger W, Rendl M, Mildner M, Tschachler E. Retinoids downregulate vascular endothelial growth factor/vascular permeability factor production by normal human keratinocytes. J Invest Dermatol. 1998;111:907–911. doi: 10.1046/j.1523-1747.1998.00393.x. [DOI] [PubMed] [Google Scholar]
- 82.Wertz PW, Swartzendruber DC, Abraham W, Madison KC, Downing DT. Essential fatty acids and epidermal integrity. Archives of Dermatology. 1987;123:1381–1384. [PubMed] [Google Scholar]
- 83.Wodicka L, Dong H, Mittmann M, Ho MH, Lockhart DJ. Genome-wide expression monitoring in Saccharomyces cerevisiae. Nat Biotechnol. 1997;15:1359–1367. doi: 10.1038/nbt1297-1359. [DOI] [PubMed] [Google Scholar]
- 84.Yano S, Banno T, Walsh R, Blumenberg M. Transcriptional responses of human epidermal keratinocytes to cytokine interleukin-1. J Cell Physiol. 2008;214:1–13. doi: 10.1002/jcp.21300. [DOI] [PubMed] [Google Scholar]
- 85.Zanotto-Filho A, Cammarota M, Gelain DP, Oliveira RB, Delgado-Canedo A, Dalmolin RJ, Pasquali MA, Moreira JC. Retinoic acid induces apoptosis by a non-classical mechanism of ERK1/2 activation. Toxicol In Vitro. 2008;22:1205–1212. doi: 10.1016/j.tiv.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 86.Zeng Y, Yang Z, Xu JG, Yang MS, Zeng ZX, You C. Differentially expressed genes from the glioblastoma cell line SHG-44 treated with all-trans retinoic acid in vitro. J Clin Neurosci. 2009;16:285–294. doi: 10.1016/j.jocn.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 87.Zhang H, Yan JH, Li YP, Cai Y. Growth inhibition and differentiation of retinoblastoma cells induced by all trans retinoic acid. Zhonghua Yan Ke Za Zhi. 2008;44:549–553. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.