

Abstract
Background
The objective of this study was to develop a novel polydatin (PLD)-loaded liposome system using the thin film hydration technique.
Methods
The delivery system was characterized in terms of morphology, size, zeta potential, encapsulation efficiency, and in vitro release. In addition, a pharmacokinetic study was carried out in rats after oral administration of PLD-loaded liposomes in vivo.
Results
Transmission electron microscopy revealed that the PLD-loaded liposomes had a homogeneous size and spherical shape. Dynamic light scattering showed that the PLD-loaded liposomes had a smaller size with a mean value of 80.2±3.7 nm and a polydispersity index of 0.12±0.06. The encapsulation efficiency of the prepared liposomes was 88.4%±3.7%. During the release process, liposome showed two distinct phases. The first was characterized by rapid release during the first 2 hours, which could be related to the release of the drug adsorbed on the surface of liposomes. In the second phase, the release rate slowed down, demonstrating a typical sustained and prolonged drug-release behavior. The release kinetic model for the PLD-loaded liposomes fitted well with the Weibull distribution equation. In vivo, relative oral bioavailability of the encapsulated PLD was 282.9%, ie, significantly enhanced (P<0.05) compared with the free drug. No histological changes occurred in the organs after administration of PLD-loaded liposomes.
Conclusion
PLD-loaded liposomes could significantly prolong the drug circulation time in vivo and increase the oral bioavailability of the drug.
Keywords: polydatin, liposome, in vitro release, oral bioavailability, histological change
Introduction
Cardiovascular disease is a serious threat to human health, and particularly so in the elderly aged over 50 years. Even with the most advanced and comprehensive treatment, still more than 50% of cerebral vascular accident survivors cannot care for themselves adequately. Up to 30 million people worldwide die each year of cardiovascular and cerebrovascular diseases, which are the most common cause of death.1 Currently, there are more than 270 million cardiovascular patients in the People’s Republic of China, inflicting huge economic losses and increasing the burden on society.
Polydatin (PLD, 3,4′,5-trihydroxystilbene-3-β-D-glucoside, Figure 1) is a monocrystalline compound originally isolated from the root and rhizome of Polygonum cuspidatum Sieb. et Zucc. (Polygonaceae), a traditional Chinese medicine that has long been used in the People’s Republic of China. Previous studies have demonstrated that PLD has many biomedical properties, such as antiplatelet aggregation, antioxidative action of low-density lipoprotein, and anti-inflammatory and immune-regulating functions.1
Figure 1.
Chemical structure of polydatin (CAS:65914-17-2).
Meanwhile, PLD has an obvious effect on the cardiovascular system, with antithrombotic, antiatherosclerotic, anti-ischemic, anti-hypercholesterolemic, and anti-inflammatory actions. It inhibits platelet aggregation, improves the microcirculation, protects the endothelium and the nerve system, alleviates cough and asthma, and can be used to treat shock.2,3
PLD can protect myocardial cells against injury caused by oxygen and glucose deprivation and chlorpromazine,4 increase calcium in myocardial cells with enhancement of the extent of myocardial cell contraction.5,6 We observed the effect of PLD on the adriamycin-injured myocardial ultrastructure in rats and discovered that PLD significantly reduced the toxicity of adriamycin on cardiomyocytes, showing an evident protective action. However, although PLD has been widely used in a number of therapeutic areas, its poor solubility (the maximum solubility is reported to be 30 μg/mL in water at 25°C) and low oral bioavailability (the half-life is only 8–14 minutes) severely limit its clinical application.7 In order to overcome the shortcomings of PLD, the development of novel dosages and formulations of PLD has attracted increasing attention in the pharmaceutical field in recent years. The currently marketed preparations, including PLD capsules and tablets, have to be administered at a large dose to obtain effective drug plasma concentrations due to low oral bioavailability. Therefore, a novel drug delivery system is needed to improve the oral bioavailability of PLD.
Liposomes, a versatile and effective nanometer-scale drug delivery system, seem to be one of the most promising candidate agents, and are nontoxic, biocompatible, and biodegradable.8–10 Liposomes can carry hydrophilic and lipophilic as well as amphoteric drug molecules entrapped either in the core or in the liposome bilayer. It has been demonstrated that liposomes can increase drug solubility and stability as well as provide good drug loading for both hydrophilic and lipophilic drugs.11–14 Thus, a number of studies have shown that a liposome carrier can improve the oral bioavailability of poorly bioavailable drugs and change the in vivo distribution of entrapped drugs.15–17 For example, Jain et al developed novel polyelectrolyte-stabilized liposomes for doxorubicin, and reported a 5.94-fold increase in the oral bioavailability of doxorubicin as compared with the free drug.18
The objectives of this study was to develop a novel PLD-loaded liposome system using the thin film hydration technique to improve the oral bioavailability of PLD, and to characterize PLD-loaded liposomes in terms of morphology, size, zeta potential, encapsulation efficiency (EE), and in vitro release. A pharmacokinetic study was carried out in rats after oral administration of the PLD-loaded liposome system and a hydroxypropyl methyl cellulose (HPMC) suspension containing PLD as the reference preparation.
Materials and methods
Materials
Polydatin standard and 2,3,5,4′-tetrahydroxychrysophenine-β-D-glucoside (internal standard) were purchased from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, People’s Republic of China). Polydatin (purity >99.7%) was obtained from Xinhe Pharmaceutical Co Ltd (Hubei, People’s Republic of China). Cholesterol, sodium cholate, ethanol, and isopropyl myristate were obtained from Sinopharm Chemical Reagent Co Ltd (Shanghai, People’s Republic of China). Soybean lecithin was supplied by Phospholipid Tech Ltd (Shanghai, People’s Republic of China). Double-distilled water was produced by a Millipore water purification system (Millipore Corporation, Billerica, MA, USA). All other chemicals used in the study were of analytical grade and obtained commercially.
Preparation of PLD-loaded liposomes
Liposomes were formulated by the established thin film hydration method described in other studies.19 In brief, PLD (20 mg), soybean lecithin (100 mg), and cholesterol (50 mg) were dissolved in 20 mL of ethanol in a single-neck flask and subject to ultrasonication until the solution was clear and transparent. The solution was then evaporated using a rotary evaporator (Yarong Co, Shanghai, People’s Republic of China) to remove ethanol before sodium cholate (60 mg) and isopropyl myristate (45 mg) were added and further dissolved in 20 mL of ethanol. The evaporation process was repeated to remove the residual solvent, leaving film-like complexes at the bottom of the flask. The dried lipid film was hydrated with double-distilled water to yield the final solution. The formulation was stored at 4°C until further investigations on EE and a stability test.
Particle size and zeta potential
A 1 mg/mL PLD-loaded liposome system was diluted with double-distilled water before analysis to achieve concentrations of 0.1–0.5 mg/mL. All determinations were recorded at room temperature (25°C) using a nanoparticle size analyzer (Mastersizer 3000, Malvern Instruments, Malvern, UK). The average particle size was determined by dynamic light scattering using the software provided by Malvern. The zeta potential values were also provided directly by the same instrument.
Morphology
A transmission electron microscope (JEM-2100, JEOL, Tokyo, Japan) was used to characterize the morphology of the PLD-loaded liposomes (1 mg/mL). A drop of the diluted drug-loaded liposomes was placed on a copper grid and stained with phosphotungstic acid (2%) for observation. Upon drying at room temperature, a thin film was formed and observed by transmission electron microscopy.
Encapsulation efficiency
The percentage of PLD encapsulated in the liposome was measured using a method described in a previous report.20 First, 5.0 mL of drug-loaded liposomes (1 mg/mL) was poured onto a 0.22 μm cellulose nitrate membrane to remove the nonencapsulated PLD, after which 0.1 mL of the filtrate was collected and diluted with pure methanol to obtain a volume of 10 mL. The drug content of the formulation was analyzed by high-performance liquid chromatography (HPLC). The EE (%) was calculated according to the following equation:
where P1 represents the drug content in the liposome after filtration and P2, is the total content of drug added into the formulation.
Stability
The stability of the formulated PLD was evaluated at 4°C, 25°C, and 40°C, with a storage time of 1 month. The appearance of the liposomal formulation after centrifugation at 10,000× g for 10 minutes was inspected visually. In addition, drug content, EE, and mean particle size were monitored at selected time intervals (days 1, 14, and 30) to comprehensively evaluate the stability of the PLD-loaded liposome.
In vitro drug release studies
The in vitro release of the PLD-loaded liposomes was measured in phosphate-buffered saline (pH 7.4) or 0.1 N hydrochloric acid solution (pH 1.2) to simulate gastric fluid at a temperature of 37°C±0.5°C by the dialysis method. PLD-loaded liposome samples (PLD ~5 mg) were suspended in 100 mL of release medium and stirred at 50× g using the USP paddle method. At predetermined times of 0.25, 0.5, 1, 2, 4, 6, 8, 10, 16, and 24 hours, samples (2 mL) were withdrawn with a syringe filter (0.45 μm pore size) from the release medium and replaced with an equal volume of the corresponding fresh medium to maintain a constant volume. The test solution was analyzed by HPLC. Triplicates were conducted and the results averaged.
Pharmacokinetics study
In vivo experiments were carried out in 12 male Sprague-Dawley rats (aged 6–8 weeks, weight 220–250 g) maintained on a half-light cycle in an animal facility with unlimited access to food and water (approved by the Hospital Animal Ethical Committee and the Guide for Care and Use of Laboratory Animals, Shanghai, People’s Republic of China).
Before oral administration of the study formulations, all rats were fasted for 12 hours, but allowed free access to water. One group of rats was given a 10 mg/kg dose of free PLD suspension (1 mg/mL, suspended in 0.5 % [w/v] HPMC aqueous solution) and the other group was given the same dose of PLD-loaded liposomes (10 mg/kg). Blood samples were taken at 5 and 15 minutes, and at 0.5, 1, 2, 4, 6, 8, 12, and 24 hours after oral administration. The plasma was then obtained by centrifugation at 4,000× g for 10 minutes for further assay.
The pharmacokinetic parameters of the drug, namely the peak concentration in plasma (Cmax), the time taken to attain peak concentration (Tmax), mean residence time, area under the concentration-time curve from time 0 to infinity (AUC0–∞), and half-life of the terminal phase (t1/2), were investigated. A linear-up/log-down method of estimation was used to calculate AUC0–∞ and was obtained by adding Clast/Ke to AUC0–t. The terminal elimination rate constant (Ke) was determined according to the slope of the terminal exponential phase of the logarithmic plasma AUC. The elimination half-life was calculated using 0.693/Ke.
Histopathological studies
Twenty-four hours after administration of the formulation, the study animals (including the control saline group) were euthanized by excess anesthesia, and the hearts, livers, spleens, lungs, and kidneys were dissected and washed with cold saline. The organs were pressed between filter pads and weighed. Heart, liver, spleen, lung, and kidney tissues were fixed in 10% neutral formalin and embedded in paraffin. The paraffin sections were cut on glass slides and stained with eosin for histopathological examination. The sections were examined under a light microscope with 500× magnification to detect damage to the tissue.
HPLC analysis
The amount of PLD in each sample was determined by HPLC (LC-10A, Shimadzu Co Ltd, Kyoto, Japan). Chromatographic separation was achieved using a Dikma Diamonsil™ C18 column (Dikma Co Ltd, Beijing, People’s Republic of China, 5 μm, 250 mm ×4.6 mm) and a precolumn (Nova-Pak, 10 μm, C18, Waters, Milford, MA, USA) at 40°C. The mobile phase was a mixture of methanol and water (35:65, v/v) at a flow rate of 1 mL per minute. The detection wavelength was set at 310 nm.
Next, 100 μL of acetonitrile containing the internal standard (1 mg/mL, 10 μL) was added to a 100 μL aliquot of rat plasma. The mixture was shaken for 30 seconds and 200 μL of the mobile phase was added. After vortex-mixing, the samples were centrifuged at 4,000× g for 10 minutes and a 20 μL aliquot of the supernatant was injected directly into the HPLC system.
Statistical analysis
All experimental data are expressed as the mean ± standard deviation, and the statistical differences between the different groups were evaluated using the Student’s t-test. The data were analyzed using Statistical Package for the Social Sciences version 15.0 software (SPSS Inc, Chicago, IL, USA). Differences between two groups were considered to be statistically significant at P<0.05.
Results and discussion
HPLC assay validation
The analytical performance of the HPLC system depended on the analytes used for the estimate. In this study, the in vivo assay was linear, ranging from 20 to 500 ng/mL. The standard curve of PLD gave Y =36.7C +6.54 (n=5, r=0.9993). The intraday and interday precision coefficients of variation in vivo ranged from 4.5% to 7.6% at the three different concentrations (20, 100, and 250 ng/mL). The average recovery of the analytes was greater than 96.9% in vivo. The detection limits and quantitation limits of HPLC analysis were respectively determined to be 20 and 50 ng/mL for in vivo plasma/tissue samples. No interfering peaks was observed in any of the chromatograms. It is noteworthy that the HPLC technique, although simple, was an effective method for analyzing PLD in the liposomal system.
Characterization of PLD-loaded liposome
Transmission electron microscopy revealed that the drug-loaded liposomes had a homogeneous size and a spherical shape (Figure 2). For further study, the size of the liposome was determined by dynamic light scattering. In dynamic light scattering, the PLD-loaded liposome showed a smaller size with a mean value of 80.2±3.7 nm and a polydispersity index of 0.12±0.06. The zeta potential of the PLD-loaded liposomal system was −32.2±1.28 mV. The EE of the prepared liposomes was 88.4%±3.7% (n=3). It is generally accepted that a higher value of the absolute zeta potential (above 30 mV) will confer stability on the examined nanosystem. A negative zeta potential value may be caused by the composition used when preparing liposomes. Many researchers have reported that if phospholipid, cholesterol, and anionic surfactants are used to prepare anionic liposomes, the negatively charged anionic surfactant may contribute to a negative zeta potential.21 In our study, sodium cholate was incorporated into the liposomal layers as an anionic surfactant, and thus the prepared liposomes had a negative zeta potential. These results indicate that the liposome approached a monodispersed stable system and could deliver the drug effectively.22 Moreover, the relatively small size of the liposomes may have the potential to facilitate oral absorption of PLD. The liposome was expected to have good physical stability due to its high absolute zeta potential values that reduce the probability of coalescence, thereby maintaining a homogeneous particle size.23
Figure 2.
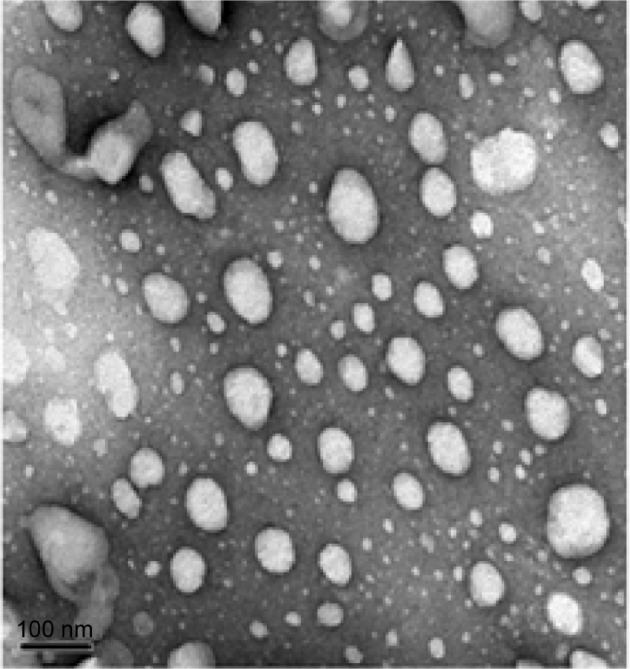
Transmission electron micrograph of polydatin-loaded liposome formulation. 2,500×.
Stability
Stability test data showed that the PLD-loaded liposome system was stable, although there were slight changes in particle diameter and percent EE (Table 1). During the observation period, the liposomes maintained a good round shape and the polydispersity index drifted between 0.12±0.06 and 0.13±0.07. The particle size remained substantially unchanged at 80.2 nm and 83.4 nm (at 4°C and 25°C, respectively). With prolonged storage, the data showed that the percent EE decreased by 3.6% and 6.7% at 4°C and 25°C. The accelerated stability data showed that the diameter increased by 8.2 nm and percent EE decreased by 7.4% (time 30 days, at 40°C). These results indicate that the liposome is an ideal carrier for poorly water-soluble drugs.
Table 1.
Stability data for PLD-loaded liposomes
| Formulation | Time (days) | Diameter (nm)
|
EE (%)
|
||||
|---|---|---|---|---|---|---|---|
| 4°C | 25°C | 40°C | 4°C | 25°C | 40°C | ||
| PLD-loaded liposome | 0 | 80.2±3.7 | 80.2±3.7 | 80.2±3.7 | 88.4±3.7 | 88.4±3.7 | 88.4±3.7 |
| 14 | 80.8±2.6 | 82.7±4.1 | 84.9±4.3 | 86.9±3.1 | 84.1±4.1 | 84.2±2.8 | |
| 30 | 81.1±2.9 | 83.4±3.9 | 88.4±5.3 | 85.2±2.8 | 82.5±2.9 | 81.9±2.3 | |
Abbreviations: EE, encapsulation efficiency; PLD, polydatin.
In vitro release
The release of PLD from the encapsulated formulation and that of the free drug was investigated using the dialysis method. The in vitro cumulative release profile for the PLD-loaded liposomes in phosphate-buffered saline (pH 7.4) and in simulated gastric fluid (pH 1.2) are shown in Figure 3. It was observed that the dissolution rate of free PLD was higher than that of the PLD-loaded liposome in both types of medium. Whereas the free drug was released almost completely at around 6 hours (85% in phosphate-buffered saline and 82% in simulated gastric fluid), the release from the liposome system was only 43% in phosphate-buffered saline and 41% in simulated gastric fluid. In the following 2 hours, the liposomes entered a slow-release phase and released up to about 69% by the end of 24 hours of observation. During the release process, the liposomes showed two distinct release phases: the first was characterized by rapid release during the first 2 hours, which could be related to release of drug adsorbed on the surface of the liposomes; in the second phase, the release rate slowed down, demonstrating typical sustained and prolonged drug-release behavior. As shown in Table 2, the in vitro drug-release kinetic model for the PLD-loaded liposomes in phosphate-buffered saline fits well with the Weibull distribution equation: lnln (1/[1−Q]) =13.298 lnt −2.726 (r=0.994). Therefore, it was speculated that the sustained-release properties of the PLD-loaded liposomal system could enhance the absorption of PLD in the gastrointestinal tract.
Figure 3.
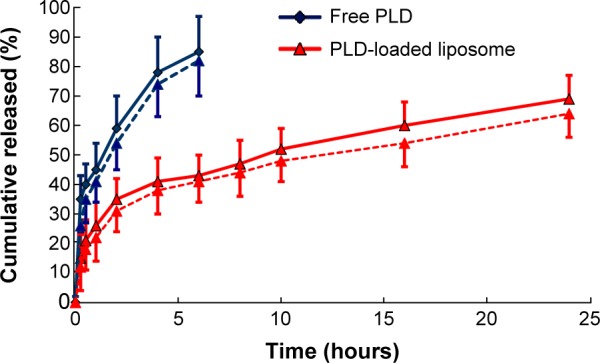
In vitro release of the polydatin-loaded liposome in phosphate-buffered saline (pH 7.4, n=3, solid line) and simulated gastric fluid (pH 1.2, n=3, dotted line).
Notes: Red line indicates polydatin (PLD) loaded liposome; blue line indicates free polydatin.
Table 2.
Dissolution kinetic parameters for PLD from PLD-loaded liposomes in phosphate-buffered saline
| PLD-loaded liposome
|
||
|---|---|---|
| Equation | Correlation coefficient (R) | |
| Zero-order equation | Q =4.261t +25.372 | 0.923 |
| First-order equation | Ln (1−Q) =−1.282t +0.214 | 0.968 |
| Higuchi | Q =19.287t1/2 +8.562 | 0.982 |
| Weibull’s equation | lnln (1/[1−Q]) =13.298 lnt −2.726 | 0.994 |
Abbreviations: PLD, polydatin; Ln (1−Q), natural logarithm.
Pharmacokinetic studies
The mean plasma concentration-time profiles for PLD after oral administration as the free drug and when loaded into liposomes are presented in Figure 4. The pharmacokinetic parameters are shown in Table 3. Significant differences in the pharmacokinetic parameters of PLD were observed between PLD-HPMC and PLD-loaded liposomes. The plasma concentration of PLD increased within the first 2 hours and decreased slowly for up to 24 hours. The mean t1/2 increased from 2.78±0.89 hours to 18.36±3.54 hours. The Cmax for PLD-HPMC was 1,562.7±453.2 ng/mL, which is different from that in the PLD-loaded liposome formulation (1,898.3±376.5 ng/mL). The AUC0-t for the PLD in the form of a liposome was significantly increased and was 3.07-fold that of PLD-HPMC. The increased Cmax and AUC0-t indicate increased oral absorption. The relative oral bioavailability of the encapsulated PLD was 282.9%, and thus significantly enhanced (P<0.05) when compared with the free drug. However, compared with PLD-HPMC, the apparent V and clearance of PLD were reduced in the case of the PLD-loaded liposome, which might result in decreased liver metabolism, thus improving the oral bioavailability of PLD. In addition, the increased oral bioavailability of PLD when in the form of the PLD-loaded liposome might be attributed to the combination of several effects.24,25 First, the particle size of the PLD-loaded liposome played a key role in the nanoparticle absorption rate; in general, a decreased particle size may improve the drug dissolution rate by increasing the surface area, which would increase absorption in the gastrointestinal tract. Second, the bioadhesive nature of the PLD-loaded liposome could increase the affinity between the nanoliposomes and the gastrointestinal membrane. Therefore, the extended residence time at the site of absorption would help to increase the uptake of PLD.24,25
Figure 4.
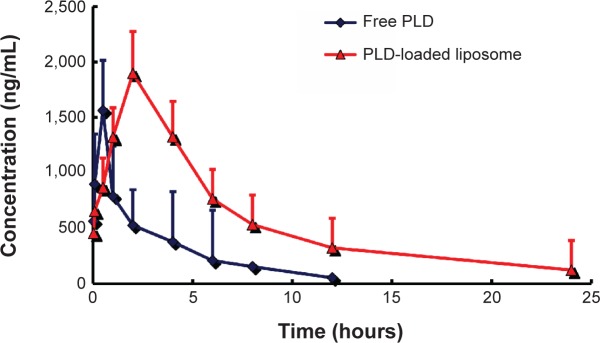
Mean plasma polydatin concentration in rats after intravenous administration of the two formulations (n=6).
Abbreviation: PLD, polydatin.
Table 3.
Pharmacokinetic parameters of the two formulations
| Parameter | Formulations
|
|
|---|---|---|
| PLD-HPMC | PLD-loaded liposome | |
| t1/2 (hours) | 2.78±0.89 | 18.36±3.54* |
| Cmax (ng/mL) | 1,562.7±453.2 | 1,898.3±376.5 |
| AUC0–t (ng·hour/mL) | 4,399.3±476.9 | 13,516.1±876.4* |
| AUC0–∞ (ng·hour/mL) | 5,387.4±527.4 | 15,241.5±945.5* |
| MRT (hours) | 6.8±2.3 | 32.2±3.4* |
| V (L/kg) | 122.5±10.1 | 28.6±2.7* |
| CL (L/hour/kg) | 15.7±2.2 | 7.9±1.7* |
Note:
P<0.05: PLD-HPMC versus PLD-loaded liposome.
Abbreviations: AUC, area under the concentration-time curve; HPMC, hydroxypropyl methyl cellulose; MRT, mean residence time; PLD, polydatin; t1/2, half-life period; Cmax, concentration maximum; V, apparent volume of distribution; CL, clearance.
Histopathological studies
In a targeted drug delivery system such as a liposome formulation, most of the drug and excipient are accumulated in specific tissues, so determining the compatibility between these tissues and the liposome formulation is necessary in order to ensure the safety of the formulation. On comparison of the liposome formulation with placebo, the cytoarchitecture of the tissue did not show any major difference (ie, degenerative changes) as shown in Figure 5, indicating no histological organ changes after administration of the PLD-loaded liposome formulation.
Figure 5.

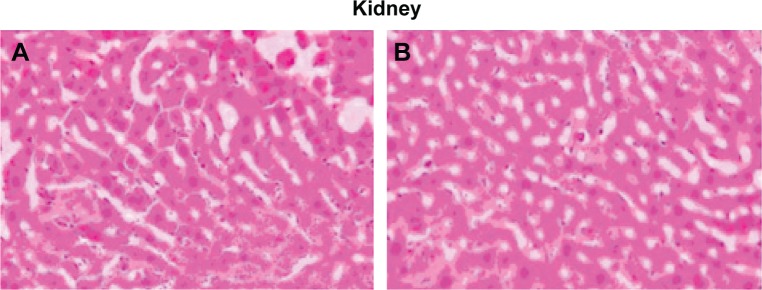
Histopathological studies of heart, liver, spleen, lung, and kidney.
Notes: (A) Saline control group. (B) polydatin-loaded liposome. 500×.
Footnotes
Disclosure
The product was codeveloped in partnership with a pharmaceutical institute responsible for designing, supervising and coordinating the study. Otherwise, the authors report no conflicts of interest in this work.
References
- 1.Du QH, Peng C, Zhang H. Polydatin: a review of pharmacology and pharmacokinetics. Pharm Biol. 2013;51(11):1347–1354. doi: 10.3109/13880209.2013.792849. [DOI] [PubMed] [Google Scholar]
- 2.Liu LT, Guo G, Wu M, Zhang WG. The progress of the research on cardio-vascular effects and acting mechanism of polydatin. Chin J Integr Med. 2012;18(9):714–719. doi: 10.1007/s11655-012-1060-8. [DOI] [PubMed] [Google Scholar]
- 3.Kimura Y. Pharmacological studies on resveratrol. Methods Find Exp Clin Pharmacol. 2003;25(4):297–310. doi: 10.1358/mf.2003.25.4.727207. [DOI] [PubMed] [Google Scholar]
- 4.Luo SF, Yu CL, Zhang PW. Influences of 3,40,5-trihydroxystibene 3-beta-mono-D-glucoside on beat rate and injury of cultured newborn rat myocardial cells. Acta Pharmacol Sin. 1990;11(2):147–150. [PubMed] [Google Scholar]
- 5.Zhao KS, Jin C, Huang X, et al. The mechanism of polydatin in shock treatment. Clin Hemorheol Microcirc. 2003;29(3–4):211–217. [PubMed] [Google Scholar]
- 6.Zhao J, Li HY, Wang ZH, et al. Effect of polydatin on ultrastructure of cardiac myocytes in rats with adriamycin-induced myocardial damage. Acta Acad Med CPAPF. 2010;19:629–634. [Google Scholar]
- 7.Das M, Das DK. Resveratrol and cardiovascular health. Mol Aspects Med. 2010;31(6):503–512. doi: 10.1016/j.mam.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 8.Dicheva BM, Koning GA. Targeted thermosensitive liposomes: an attractive novel approach for increased drug delivery to solid tumors. Expert Opin Drug Deliv. 2014;11(1):83–100. doi: 10.1517/17425247.2014.866650. [DOI] [PubMed] [Google Scholar]
- 9.Liu M, Li M, Sun S, et al. The use of antibody modified liposomes loaded with AMO-1 to deliver oligonucleotides to ischemic myocardium for arrhythmia therapy. Biomaterials. 2014;35(11):3697–3707. doi: 10.1016/j.biomaterials.2013.12.099. [DOI] [PubMed] [Google Scholar]
- 10.Dai Y, Zhou R, Liu L, Lu Y, Qi J, Wu W. Liposomes containing bile salts as novel ocular delivery systems for tacrolimus (FK506): in vitro characterization and improved corneal permeation. Int J Nanomedicine. 2013;8:1921–1933. doi: 10.2147/IJN.S44487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramos-Cabrer P, Campos F. Liposomes and nanotechnology in drug development: focus on neurological targets. Int J Nanomedicine. 2013;8:951–960. doi: 10.2147/IJN.S30721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wei Y, Xue Z, Ye Y, Huang Y, Zhao L. Paclitaxel targeting to lungs by way of liposomes prepared by the effervescent dispersion technique. Arch Pharm Res. 2014;37(6):728–737. doi: 10.1007/s12272-013-0181-8. [DOI] [PubMed] [Google Scholar]
- 13.Wei Y, Xue Z, Ye Y, Wang P, Huang Y, Zhao L. Pharmacokinetic and tissue distribution of paclitaxel in rabbits assayed by LC-UV after intravenous administration of its novel liposomal formulation. Biomed Chromatogr. 2014;28(2):204–212. doi: 10.1002/bmc.3005. [DOI] [PubMed] [Google Scholar]
- 14.Wei Y, Zhao L. Passive lung-targeted drug delivery systems via intravenous administration. Pharm Dev Technol. 2014;19(2):129–136. doi: 10.3109/10837450.2012.757782. [DOI] [PubMed] [Google Scholar]
- 15.Jain S, Kumar D, Swarnakar NK, Thanki K. Polyelectrolyte stabilized multilayered liposomes for oral delivery of paclitaxel. Biomaterials. 2012;33(28):6758–6768. doi: 10.1016/j.biomaterials.2012.05.026. [DOI] [PubMed] [Google Scholar]
- 16.Niu M, Lu Y, Hovgaard L, et al. Hypoglycemic activity and oral bio-availability of insulin-loaded liposomes containing bile salts in rats: the effect of cholate type, particle size and administered dose. Eur J Pharm Biopharm. 2012;81(2):265–272. doi: 10.1016/j.ejpb.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 17.Gradauer K, Barthelmes J, Vonach C, et al. Liposomes coated with thiolated chitosan enhance oral peptide delivery to rats. J Control Release. 2013;172(3):872–878. doi: 10.1016/j.jconrel.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jain S, Patil SR, Swarnakar NK, Agrawal AK. Oral delivery of doxorubicin using novel polyelectrolyte-stabilized liposomes (layersomes) Mol Pharm. 2012;9(9):2626–2635. doi: 10.1021/mp300202c. [DOI] [PubMed] [Google Scholar]
- 19.Yi C, Fu M, Cao X, et al. Enhanced oral bioavailability and tissue distribution of a new potential anticancer agent, Flammulina velutipes sterols, through liposomal encapsulation. J Agric Food Chem. 2013;61(25):5961–5971. doi: 10.1021/jf3055278. [DOI] [PubMed] [Google Scholar]
- 20.Wu Y, Yang Y, Zhang FC, Wu C, Lu WL, Mei XG. Epirubicin-encapsulated long-circulating thermosensitive liposome improves pharmacokinetics and antitumor therapeutic efficacy in animals. J Liposome Res. 2011;21(3):221–228. doi: 10.3109/08982104.2010.520273. [DOI] [PubMed] [Google Scholar]
- 21.Fang JY, Hwang TL, Huang YL, Fang CL. Enhancement of the transdermal delivery of catechins by liposomes incorporating anionic surfactants and ethanol. Int J Pharm. 2006;310(1–2):131–138. doi: 10.1016/j.ijpharm.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 22.Bshara H, Osman R, Mansour S, El-Shamy Ael-H. Chitosan and cyclodextrin in intranasal microemulsion for improved brain buspirone hydrochloride pharmacokinetics in rats. Carbohydr Polym. 2014;99:297–305. doi: 10.1016/j.carbpol.2013.08.027. [DOI] [PubMed] [Google Scholar]
- 23.Choudhury H, Gorain B, Karmakar S, et al. Improvement of cellular uptake, in vitro antitumor activity and sustained release profile with increased bioavailability from a nanoemulsion platform. Int J Pharm. 2014;460(1–2):131–143. doi: 10.1016/j.ijpharm.2013.10.055. [DOI] [PubMed] [Google Scholar]
- 24.Hu K, Cao S, Hu F, Feng J. Enhanced oral bioavailability of docetaxel by lecithin nanoparticles: preparation, in vitro, and in vivo evaluation. Int J Nanomedicine. 2012;7:3537–3545. doi: 10.2147/IJN.S32880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Venkatesan N, Uchino K, Amagase K, Ito Y, Shibata N, Takada K. Gastro-intestinal patch system for the delivery of erythropoietin. J Control Release. 2006;111(1–2):19–26. doi: 10.1016/j.jconrel.2005.11.009. [DOI] [PubMed] [Google Scholar]