

Abstract
Objective:
We examined a cohort of Australian patients with statin exposure who developed a necrotizing autoimmune myopathy (NAM) associated with a novel autoantibody against 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR) and describe the clinical and therapeutic challenges of managing these patients and an optimal therapeutic strategy.
Methods:
Clinical, laboratory, EMG, and histopathologic results and response to immunomodulation are reported in 6 Australian patients with previous statin exposure and antibodies targeting HMGCR.
Results:
All patients presented with painless proximal weakness following statin therapy, which persisted after statin cessation. Serum creatine kinase (CK) levels ranged from 2,700 to 16,200 IU/L. EMG was consistent with a myopathic picture. Muscle biopsies revealed a pauci-immune necrotizing myopathy. Detailed graphical representation of the clinical course of these patients showed a close association with rising CK and an increase in clinical weakness signifying relapses, particularly upon weaning or ceasing steroids. All 6 patients were responsive to initial steroid therapy, with 5 relapsing upon attempts to wean steroids. Both CK and clinical strength improved with the reinstitution of immunotherapy, in particular steroids and IV immunoglobulin (IVIg). All patients required treatment with varying multiagent immunosuppressive regimens to achieve clinical remission, including prednisone (n = 6), IVIg (n = 5), plasmapheresis (n = 2), and additional therapy including methotrexate (n = 6), cyclophosphamide (n = 2), rituximab (n = 2), azathioprine (n = 1), and cyclosporine (n = 1).
Conclusions:
Recognition of HMGCR antibody–associated NAM is important because these patients are responsive to immunosuppression, and early multiagent therapy and a slow and cautious approach to withdrawing steroids may improve outcomes.
Statins are widely prescribed for the treatment of dyslipidemia and for risk reduction in cardiovascular disease.1 3-Hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR) is a key enzyme in the cholesterol biosynthesis pathway.1,2 Statins are structural analogues and competitively inhibit HMGCR.1,2 There is a wide spectrum of muscular adverse effects associated with statins, from asymptomatic elevations of creatine kinase (CK), myalgia, and exercise intolerance to toxic necrotizing myopathy and rhabdomyolysis.1 These effects may vary with the agent, dose, concurrent cholesterol pathway inhibitors, and genetic polymorphisms and usually reverse upon drug cessation.1,3,4
The idiopathic inflammatory myopathies include polymyositis (PM), dermatomyositis (DM), and necrotizing autoimmune myopathy (NAM).5,6 NAM presents with subacute proximal weakness and is defined by a distinct histologic profile with marked necrotic, degenerating, or regenerating muscle fibers undergoing myophagocytosis in the absence of an inflammatory infiltrate, with macrophages being the prominent effector cells.5,7 The absence or relative paucity of an inflammatory lymphocytic infiltrate is described as a pauci-immune necrotizing myopathy and distinguishes NAM from the characteristic histologic findings of PM or DM, which include CD8+ or CD4+ T lymphocytes and B cells, respectively.5,6
An underrecognized adverse effect of statin use is NAM that does not resolve upon statin cessation, associated with anti-HMGCR antibodies.5,8–10 We present 6 such Australian patients and detail their challenging management. Recognition of this entity is essential, as patients may respond to immunosuppression and relapse with steroid cessation. We propose that patients may have improved outcomes with the early institution of multiagent immunotherapy and describe optimal therapeutic strategies.
METHODS
We identified patients who presented to 2 tertiary referral hospitals in Australia from 2008 to 2013 in whom we suspected statin-associated NAM. Six patients fulfilled inclusion criteria, including statin exposure, clinical presentation with subacute painless proximal weakness, clinical and neurophysiologic evidence of a myopathy, and histopathologic features consistent with NAM. Exclusion criteria included a family history of muscle disease and histopathologic findings consistent with other inflammatory myopathies such as PM or DM. All patients and 6 age-matched laboratory controls had coded randomly ordered serum samples tested by a blinded investigator (A.L.M. and C.B. laboratories) for anti-HMGCR antibodies, as well as a full vasculitic and myositis-specific autoantibody screen (including anti-Ro, anti-La, anti-Sm, Scl-70, anti-Rnp, anti-Jo1, anti-Mi2, and anti-SRP antibodies). Serum was tested for HMGCR antibodies from samples collected at time of acute presentation (n = 2) or from samples taken during follow-up clinical assessment (n = 4), as 2 patients (cases 2 and 6, table) were diagnosed and treated prospectively, whereas 4 patients received a confirmatory diagnosis retrospectively. Data including serial CK levels, muscle strength as recorded using the Medical Research Council scale, and response to therapy were obtained from all patients. The 6 patients were followed up for a mean of 4.5 years (range 1.5–11 years).
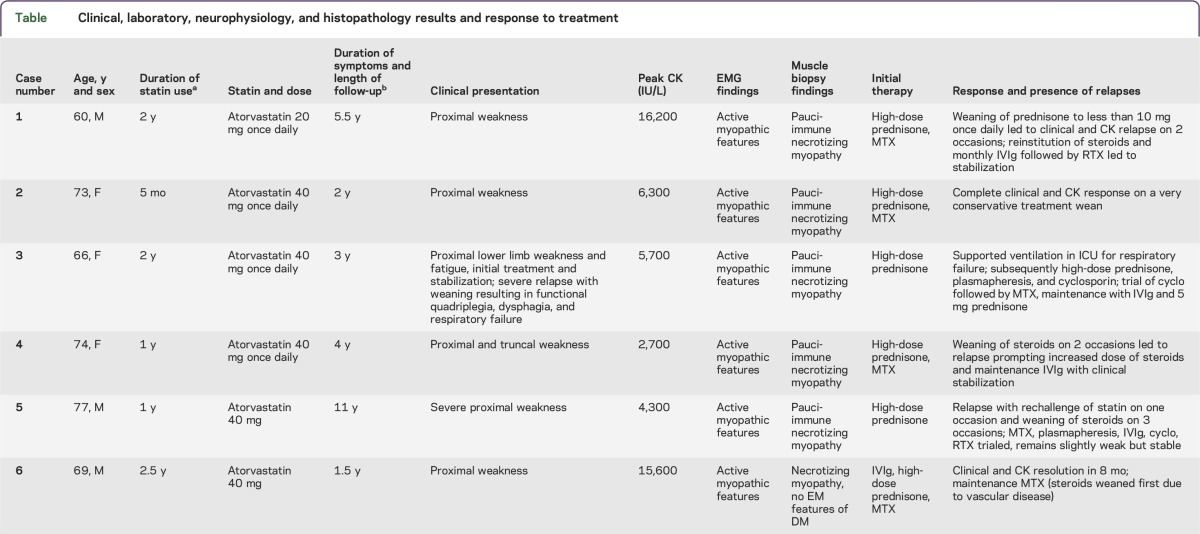
Table.
Clinical, laboratory, neurophysiology, and histopathology results and response to treatment
Standard protocol approvals, registrations, and patient consents.
Informed consent was obtained from all patients and physicians.
RESULTS
We will outline the features of this condition by means of 2 illustrative cases (cases 1 and 2, table). The clinical and investigation findings and response to therapy for all patients in this case series are summarized in the table.
Illustrative case 1.
A 60-year-old man was started on 20 mg daily of atorvastatin for the treatment of hypercholesterolemia. He first noticed the subacute onset of painless proximal upper and lower limb weakness 2 years after starting statin therapy. Examination revealed 3/5 power of neck flexion, shoulder abduction, and hip flexion. Initial CK levels were above 16,000 IU/L, prompting immediate cessation of his statin. His CK levels declined to the mid-6,000s IU/L and were monitored by his local physician. His ongoing weakness prompted referral to a specialist immunologist 14 months after symptom onset. Muscle biopsies of the deltoid muscle prior to commencement of treatment demonstrated necrotizing myopathy with multifocal polyphasic segmental muscle fiber necrosis and regeneration (figure 1, A–D). There was minimal associated inflammation, and no inclusion bodies or vascular abnormalities were noted. Macrophage infiltration was noted, with a relative paucity of lymphocytic infiltration. Major histocompatibility complex (MHC) class I expression was not upregulated at the time of this biopsy.
Figure 1. Muscle biopsy images demonstrating a pauci-immune necrotizing autoimmune myopathy in illustrative cases 1 and 2.

(A–D) Case 1 deltoid muscle. (A) Frozen section showing scattered necrotic fibers with associated macrophage infiltration but no discernible lymphocytic infiltration (arrow) (hematoxylin & eosin [H&E] stain, magnification 100×). (B) Immunohistochemistry for major histocompatibility complex class I highlights macrophages associated with muscle fiber necrosis and background capillaries, but there is no generalized upregulation on uninvolved muscle fibers in this biopsy sample (arrow) (magnification 100×). (C) Immunohistochemistry for complement membrane attack complex highlights foci of muscle fiber necrosis, but there is no microvascular deposition (arrow) (magnification 100×). (D) Immunohistochemistry for CD3 reveals a paucity of T cells in areas of fiber degeneration. (E, F) Case 2 deltoid muscle. (E) H&E stain (magnification 40×). (F) H&E stain (magnification 100×). There are necrotic fibers with inflammatory cells (arrow) involved in the process of myophagocytosis and a few regenerating fibers, consistent with a pauci-immune necrotizing myopathy.
Oral prednisone was started at 50 mg daily with a plan for slow weaning, in conjunction with azathioprine. Improvement in CK was noted; however, intolerance to azathioprine prompted its replacement with methotrexate 20 mg weekly as a steroid-sparing agent. This patient had brittle disease with his turbulent clinical course, depicted in figure 2A. Ongoing treatment with weekly methotrexate and slow weaning of prednisone initially resulted in stable disease with normalization of power and decline in CK level to 1,500 IU/L. However, reduction of prednisone to less than 10 mg daily coincided with a rise in CK and a recurrence of weakness. IV immunoglobulin (IVIg) as acute treatment as well as monthly maintenance doses, an increase in the dose of prednisone, and continuation of methotrexate permitted disease stabilization for more than a year and a half, with normalization of power and decline in CK. A second attempt to wean prednisone again prompted a relapse, requiring rescue therapy with rituximab and pulsed IV steroids. At this stage, IVIg was ceased due to patient preference. Over the next 6 months, the CK rose to >11,000 IU/L, despite ongoing B-cell depletion, and correlated with clinical deterioration. Recommencement of maintenance IVIg resulted in improvement. Testing of serum confirmed the presence of antibodies to HMGCR. The patient has remained stable with maintenance therapy consisting of methotrexate, prednisone, and monthly IVIg. His power is currently normal, and his CK is stable albeit slightly elevated at 400 IU/L more than 5 and a half years after symptom onset.
Figure 2. Clinical course and response to therapy in 6 Australian patients with HMGCR antibodies.

The clinical course, creatine kinase (CK) levels, Medical Research Council (MRC) scale for power grading of shoulder abduction strength (appendix e-1 at Neurology.org/nn), and therapeutic regimens in the 6 patients in this cohort are graphically represented. Timing of statin commencement and cessation are indicated. Horizontal arrows indicate the duration of use of various immunosuppressive therapies. All patients were asymptomatic with no reported weakness at the time of statin commencement based on assessment by their general practitioner. Most patients experienced relapses with attempts to wean steroids, with a rise in CK often preceding clinical weakness. (A) Illustrative case 1 (case 1, table). The CK levels of this 60-year-old man are graphed from the time of statin commencement through 93 months of follow-up. Acute relapses characterized by worsening proximal weakness and coinciding with a rise in CK accompanied attempts to wean the dose of oral prednisone below 10 mg daily. Reinstitution of steroids and the use of IV immunoglobulin (IVIg) had a good correlation with clinical improvement and a reduction in CK. (B) Illustrative case 2 (case 2, table). The CK levels of this 73-year-old woman are shown for 30 months of follow-up. Early recognition of this diagnosis and initiation of high-dose prednisone and methotrexate (MTX) resulted in a rapid improvement in CK and clinical resolution of weakness. A slow and cautious approach to weaning steroids was associated with clinical remission without any relapses, and maintenance immunosuppression with prednisone 5 mg once daily and MTX 20 mg weekly has resulted in clinical stabilization. (C) Case 3, table. This 66-year-old woman was on a statin for 24 months prior to symptom onset. Early treatment with statin cessation and high-dose steroids resulted in an immediate improvement, but rapid steroid tapering prompted a severe relapse necessitating respiratory support and multiple immunosuppressive agents to allow for clinical stabilization. (D) Case 4, table. This 74-year-old woman responded well to steroid therapy, but attempts to wean steroids prompted relapses associated with both a CK rise and clinical weakness. Maintenance therapy with prednisone and IVIg has allowed for clinical remission. (E) Case 5, table. This 77-year-old man was on a statin for 1 year prior to symptom onset. Initial therapy with steroids, IVIg, and MTX allowed for an early recovery. However, a rechallenge with statin therapy prompted a further relapse. This patient required multiple immunosuppressive agents to maintain remission, with attempts to wean prednisone below 15 mg prompting 2 further relapses and ongoing disease activity almost 11 years after statin cessation. Maintenance therapy with prednisone and IVIg has permitted clinical remission. (F) Case 6, table. This 69-year-old man was on a statin for 2.5 years prior to symptom onset. He responded well to treatment with prednisone, IVIg, and MTX. Steroids were weaned due to his significant vascular comorbidities, and he was maintained on MTX. Recent results herald a possible early relapse, with CK levels trending up and mild clinical weakness, suggesting reinstitution of steroid therapy may be required. HMGCR = 3-hydroxy-3-methylglutaryl-coenzyme A reductase; IV MP = IV methylprednisolone; PE = plasmapheresis.
Illustrative case 2.
A 73-year-old woman with hypertension, hypercholesterolemia, and TIAs was started on atorvastatin 40 mg daily. Five months later, she developed painless progressive shoulder weakness and difficulty climbing stairs, suggestive of proximal myopathy. There were no features of systemic illness. Despite cessation of the statin, her weakness progressed such that her mobility was impaired within 2 months of onset, with examination demonstrating shoulder abduction and adduction weakness and hip flexion and extension weakness (graded 3/5). CK levels were above 6,000 IU/L. An EMG was supportive of irritable muscle with active myopathic changes and abnormal spontaneous activity represented by positive sharp waves, fibrillation potentials, and small motor unit action potentials with an increased pattern of recruitment. A deltoid muscle biopsy (performed on the contralateral side to the EMG) demonstrated features of NAM (figure 1, E and F). A serum sample was positive for antibodies to HMGCR.
She received immunosuppression with high-dose oral steroids (prednisone 60 mg daily, weaned over subsequent months) and methotrexate 20 mg weekly, with CK and clinical resolution after 11 months at a prednisone dose of 5 mg (figure 2B). She has continued prednisone 5 mg and methotrexate 20 mg weekly for the past 14 months without relapse.
Clinical presentation.
Clinical data from 3 female and 3 male patients are summarized in the table. The patients had an average age at symptom onset of 70 years (range 60–77 years). All patients were on a statin for varying lengths of time (ranging from 5 months to 2 and a half years) before the onset of symptoms. Illustrative cases 2 and 6 were the 2 most recently identified patients, with the diagnosis being confirmed relatively early in their disease course. All patients presented with painless proximal weakness of varying severity.
Investigations.
Investigation findings demonstrated elevated CK in all patients. These results ranged from 2,700 to 16,200 IU/L at presentation. A complete vasculitic screen and myositis-specific autoantibody screen were negative in all patients except one. One patient (case 6, table) had anti-Mi2 antibodies but no other clinical or histopathologic features consistent with DM. All patients had antibodies targeting HMGCR, as detected by ELISA. All age-matched laboratory controls were negative for HMGCR antibodies. Needle EMG demonstrated features consistent with an active myopathic process. Muscle biopsies showed a pauci-immune necrotizing myositis in all patients. None of the patients were found to have an associated malignancy on initial presentation or on follow-up.
Immunomodulatory therapy trialed and patient outcomes.
All patients were initially treated with high-dose steroids and subsequently with varying regimens of other immunosuppressive agents, including IVIg (n = 5), plasmapheresis (n = 2), and additional therapy including methotrexate (n = 6), cyclophosphamide (n = 2), rituximab (n = 2), azathioprine (n = 1), and cyclosporine (n = 1). Despite therapy, 5/6 patients had a relapse, with between 0 and 3 relapses per patient after a mean of 4.5 years of follow-up (range 1.5–11 years). There was a total of 10 relapses in the 6 patients, with 9/10 relapses being associated with steroid tapering or cessation. All 6 patients were steroid-responsive, with 5 relapsing upon weaning of steroids. The relapse in case 3 was so severe that diaphragmatic weakness ensued and supported ventilation was required. Cessation of statin therapy alone did not result in improvement, with most patients demonstrating myopathic features for periods ranging from 6 months to 11 years after cessation despite the institution of immunosuppressive therapy.
DISCUSSION
We present a series of patients with anti-HMGCR antibody–associated NAM with the aim of increasing awareness of this diagnosis. NAM has been associated with malignancies, HIV, antibodies to signal recognition particle, connective tissue diseases, and certain medications, but it can also occur in isolation.5,7 This underrecognized condition was recently described in association with statin exposure.8,9 Christopher-Stine et al.9 reviewed a cohort of patients with necrotizing myopathy on muscle biopsy and demonstrated that 16/26 patients' sera immunoprecipitated a pair of proteins with approximate sizes of 200 and 100 kDa. Sixty-three percent of these patients had had statin exposure at some point prior to symptom onset. This antigen was later characterized as HMGCR, the pharmacologic target of statins, with immunoprecipitation assays demonstrating that the antibodies had a specific binding affinity for the carboxy-terminus of this enzyme.10 Further work by this group has demonstrated that these antibodies are not present in healthy controls; in the majority of patients with DM, PM, and inclusion body myositis; or in patients with statin exposure and isolated CK, myalgia, or self-limiting statin intolerance, thus suggesting they are highly specific for statin-associated NAM.9–11 It has been shown that the human leukocyte antigen class II allele DRB1*11:01 may be a genetic risk factor for developing anti-HMGCR–associated NAM, whereas DQA1 and DQB6 may be protective, perhaps helping to explain why this condition is relatively rare despite the prevalence of statin use.12
Our series of patients highlights the characteristic clinical features of this condition that should prompt consideration of anti-HMGCR–associated NAM as a diagnosis. All patients had been exposed to statins for periods of time ranging from 5 months to 2 years. Because anti-HMGCR antibody–associated NAM may occur long after initial exposure or even after statin cessation, the triggering role of statins may not be recognized. Investigation findings supportive of this diagnosis include elevated CK levels and EMG evidence of an active myopathic process, which all 6 patients demonstrated. However, these investigation findings in isolation are not diagnostic of this condition and may be part of the clinical profile of other inflammatory myopathies. A key diagnostic finding that distinguished these patients from those with DM or PM was the presence of a pauci-immune necrotizing myositis on muscle biopsy, which was present in all 6 patients in this series.
One of the most notable features of this series of patients was the persistence of active myopathy for prolonged periods of time after statin cessation (table). Most other self-limiting adverse effects of statins, such as cramps and myalgia, cease within weeks of statin discontinuation.8 However, muscle biopsies of some of our patients done months to years after the onset of proximal weakness still demonstrated an active necrotizing myopathy. Furthermore, patients had significant clinical weakness and an active myopathic EMG up to 11 years after statin cessation, as demonstrated by case 5 (table). Mammen et al.10 demonstrated that muscle expression of HMGCR is upregulated not only with statin exposure but also in regenerating muscle cells compared to resting myocytes. Statins may increase the expression of HMGCR as the first step to initiating autoimmunity. However, the immune-mediated muscle damage initiated in the presence of statins may be sustained long after statin cessation through persistently increased HMGCR expression in regenerating muscle fibers.10 This hypothesis of a persistent self-antigen that perpetuates the immune response may explain our patients' propensity to relapse, often dramatically, with reduction or tapering of immunosuppression. This hypothesis rests on increased antigen density as the trigger for autoimmunity, but other possibilities such as formation of a neoantigen conformation of HMGCR such as a splice variant are not excluded.
Anti-HMGCR–associated NAM is believed to have an immunologic etiology. MHC class I upregulation on the surface of non-necrotic muscle fibers in many cases supports an immune-mediated pathogeneisis.9 A humorally mediated pathogenesis is suggested by microvascular deposition of membrane attack complex in some cases.5 The relative specificity of anti-HMGCR antibodies to the NAM patient population compared to patients with DM and PM, healthy controls with statin exposure, or patients with self-limiting statin intolerance further argues for a specific autoimmune pathogenesis.9–11 The favorable and rapid response of all our patients to immunosuppression, as well as the significant clinical relapses with attempts to wean immunotherapy, is once again supportive of an immune-mediated pathogenesis. Muscle biopsy performed months after statin cessation in our patients still demonstrated features of NAM, which differentiates this clinical entity from a toxic myopathy secondary to statin use, in which cessation of the offending statin usually results in clinical and histologic improvement.8 In a subgroup of patients with statin exposure and anti-HMGCR associated–NAM, initial antibody levels were significantly correlated with CK levels and arm and leg strength.13 Long-term follow-up demonstrated that with immunosuppressive therapy, antibody levels and CK declined, with a corresponding improvement in arm and leg strength, once again demonstrating the immune responsive nature of this condition. This close correlation with CK and clinical progress was demonstrated in all our patients (figure 2).
All the patients in this case series had statin exposure prior to the onset of weakness. However, larger series have demonstrated the prevalence of statin use to be 44%–92% in patients with HMGCR antibodies.9,10,14 This suggests that in a minority of patients, HMGCR can be a primary autoimmune muscle target antigen as well. Statin-naive patients were younger, had higher CK levels, and were more likely to be African American.10,13
This study provides a detailed description and graphical representation of the clinical course and potential pitfalls in treating patients with this challenging condition. All the patients in this series had previous statin exposure and clinically responded to immunotherapy with an initial improvement in power and a reduction in CK. All patients except case 2 (table; figure 2B) experienced recurrent relapses when attempts were made to wean their maintenance steroid therapy. Figure 2 clearly shows the close association of rising CK levels and clinical weakness with steroid taper or cessation. While all patients required multiple immunosuppressive agents to achieve clinical remission, the greatest improvement in both CK levels and clinical power seemed to occur after the reinstitution of high-dose steroids or rescue therapy with IVIg (cases 1, 3, 4, and 5, table; figure 2, A, C–E) rather than other forms of maintenance immunosuppression. This case series also highlights the difficulty in achieving clinical remission in patients with this condition. Early recognition of the diagnosis, de novo use of multiple immunotherapeutic agents, and a slow and cautious approach to weaning steroid therapy as in case 2 may result in earlier remission and avoidance of relapse. As this series has a relatively small number of patients, with a retrospective diagnosis made in 4 patients, larger prospective series with long follow-up periods are needed to determine optimal induction strategies, whether remission is sometimes maintained after cessation of all therapy, and whether biomarkers such as the persistence of HMGCR antibodies or a mildly elevated CK are predictive of relapse.
This case series describes a subgroup of patients with statin exposure who experienced a rare side effect in the form of anti-HMGCR antibody–associated NAM. This condition has a distinct clinical, histologic, and therapeutic profile that distinguishes it from other inflammatory myopathies. Early recognition is essential, as this condition is responsive to immunosuppressive therapy, with a propensity to clinically significant relapses with weaning of therapy. Identification of the diagnosis, aggressive immunotherapy, and close monitoring may improve the long-term outcome of these patients.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Dr. Min Xia Wang, Dr. Roger Pamphlett, and the Neuropathology Laboratory, University of Sydney for providing us with the biopsy slides for case 2. The authors thank the following physicians for referring and providing clinical information on the patients in this series: Associate Professor Michael Prowse, Dr. Chris Needs, Dr. Noel Saines, Dr. Alan Henderson, Dr. Michael Walsh, and Dr. David Heyworth-Smith. The authors thank Professor Frank Mastaglia for leadership in neuromuscular diseases generally and of the muscle laboratory, Perth, Western Australia.
GLOSSARY
- CK
creatine kinase
- DM
dermatomyositis
- HMGCR
3-hydroxy-3-methylglutaryl-coenzyme A reductase
- IVIg
IV immunoglobulin
- MHC
major histocompatibility complex
- NAM
necrotizing autoimmune myopathy
- PM
polymyositis
Footnotes
Supplemental data at Neurology.org/nn
AUTHOR CONTRIBUTIONS
Study conception: S.R., D.L., S.W.R. Study design: all authors. Sample acquisition: D.L., S.W.R. Acquisition of laboratory data: C.B., A.R.-U., T.R., A.L.M. Acquisition of clinical data: S.R., D.L., S.W.R. Study supervision, analysis and interpretation of data: S.R., D.L., S.W.R. Writing of first draft: S.R., S.W.R. Editing and revising final draft for intellectual content: all authors.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
S. Ramanathan has received research support from NHMRC (Australia). D. Langguth served on the scientific advisory board for University of Queensland, received speaker fees from Roche, is employed by Sullivan Nicolaides Pathology, and received research support from NHMRC. T.A. Hardy has been an associate editor for Advances in Clinical Neurosciences. N. Garg's fellowship is partially funded from Allergan. C. Bundell and A. Rojana-Udomsart report no disclosures. R.C. Dale has served on the research advisory committee for Queensland Children's Medical Institute; received honoraria from Biogen Idec; is an Editorial board member for Multiple Sclerosis and Related Disorders, Neurology: Neuroimmunology & Neuroinflammation, and European Journal of Paediatric Neurology; received publishing royalties from Biogen Idec, and received research support from NHMRC. T. Robertson reports no disclosures. A.L. Mammen has served on the advisory boards for aTYR Pharmaceuticals and Biogen, is on the editorial board for Experimental Neurology and Arthritis and Rheumatism, holds a patent for and has received royalty payments from test for anti-HMGCR antibodies, and received research support from NIH. S.W. Reddel has served on the scientific advisory boards for CSL, Baxter, Genzyme, Biogen, and National Immunoglobulin Governmental Advisory Council, Australia; has received travel funding and/or speaker honoraria from CSL, Genzyme, and Biogen; is employed by NSW Health; has received research support from Genzyme and Beeren Foundation; and holds stock or has received board of directors compensation from Sydney Neurology and Medical Safety Systems. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Radcliffe KA, Campbell WW. Statin myopathy. Curr Neurol Neurosci Rep 2008;8:66–72. [DOI] [PubMed] [Google Scholar]
- 2.van der Most PJ, Dolga AM, Nijholt IM, Luiten PGM, Eisel ULM. Statins: mechanisms of neuroprotection. Prog Neurobiol 2009;88:64–75. [DOI] [PubMed] [Google Scholar]
- 3.Sirtori CR, Mombelli G, Triolo M, Laaksonen R. Clinical response to statins: mechanism(s) of variable activity and adverse effects. Ann Med 2012;44:419–432. [DOI] [PubMed] [Google Scholar]
- 4.Bellosta S, Corsini A. Statin drug interactions and related adverse reactions. Expert Opin Drug Saf 2012;11:933–946. [DOI] [PubMed] [Google Scholar]
- 5.Dimachkie MM. Idiopathic inflammatory myopathies. J Neuroimmunol 2011;231:32–42. [DOI] [PubMed] [Google Scholar]
- 6.Mammen AL. Autoimmune myopathies: autoantibodies, phenotypes and pathogenesis. Nat Rev Neurol 2011;7:343–354. [DOI] [PubMed] [Google Scholar]
- 7.Liang C, Needham M. Necrotizing autoimmune myopathy. Curr Opin Rheumatol 2011;23:612–619. [DOI] [PubMed] [Google Scholar]
- 8.Grable-Esposito P, Katzberg HD, Greenberg SA, Srinivasan J, Katz J, Amato AA. Immune-mediated necrotizing myopathy associated with statins. Muscle Nerve 2010;41:185–190. [DOI] [PubMed] [Google Scholar]
- 9.Christopher-Stine L, Casciola-Rosen LA, Hong G, Chung T, Corse AM, Mammen AL. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum 2010;62:2757–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mammen AL, Chung T, Christopher-Stine L, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum 2011;63:713–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mammen AL, Pak K, Williams EK, et al. Rarity of anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase antibodies in statin users, including those with self-limited musculoskeletal side effects. Arthritis Care Res (Hoboken) 2012;64:269–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mammen AL, Gaudet D, Brisson D, et al. Increased frequency of DRB1*11:01 in anti-hydroxymethylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Care Res (Hoboken) 2012;64:1233–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Werner JL, Christopher-Stine L, Ghazarian SR, et al. Antibody levels correlate with creatine kinase levels and strength in anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Rheum 2012;64:4087–4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Allenbach Y, Drouot L, Rigolet A, et al. Anti-HMGCR autoantibodies in European patients with autoimmune necrotizing myopathies: inconstant exposure to statin. Medicine (Baltimore) 2014;93:150–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.