

Abstract
Purpose
Coronin-1A deficiency is a recently recognized autosomal recessive primary immunodeficiency caused by mutations in CORO1A (OMIM 605000) that results in T-cell lymphopenia and is classified as T-B+NK+severe combined immunodeficiency (SCID). Only two other CORO1A-kindred are known to date, thus the defining characteristics are not well delineated. We identified a unique CORO1A-kindred.
Methods
We captured a 10-year analysis of the immuneclinical phenotypes in two affected siblings from disease debut of age 7 years. Target-specific genetic studies were pursued but unrevealing. Telomere lengths were also assessed. Whole exome sequencing (WES) uncovered the molecular diagnosis and Western blot validated findings.
Results
We found the compound heterozygous CORO1A variants: c.248_249delCT (p.P83RfsX10) and a novel mutation c.1077delC (p.Q360RfsX44) (NM_007074.3) in two affected non-consanguineous siblings that manifested as absent CD4CD45RA+ (naïve) T and memory B cells, low NK cells and abnormally increased doublenegative (DN) ϒδ T-cells. Distinguishing characteristics were late clinical debut with an unusual mucocutaneous syndrome of epidermodysplasia verruciformis-human papilloma virus (EV-HPV), molluscum contagiosum and oral-cutaneous herpetic ulcers; the older female sibling also had a disfiguring granulomatous tuberculoid leprosy. Both had bilateral bronchiectasis and the female died of EBV+ lymphomas at age 16 years. The younger surviving male, without malignancy, had reproducibly very short telomere lengths, not before appreciated in CORO1A mutations.
Conclusion
We reveal the third CORO1A-mutated kindred, with the immune phenotype of abnormal naïve CD4 and DN T-cells and newfound characteristics of a late/hypomorphiclike SCID of an EV-HPV mucocutaneous syndrome with also B and NK defects and shortened telomeres. Our findings contribute to the elucidation of the CORO1A-SCID-CID spectrum.
Keywords: CORO1A (Coronin-1A deficiency), SCID-CID, HPV-epidermodysplasia verruciformis, mucocutaneous, molluscum, leprosy, telomere, WES (whole exome sequencing)
Introduction
Severe combined immunodeficiency (SCID) represents a diverse group of primary immunodeficiency diseases that have different genetic etiologies, some with very distinguishing clinical characteristics associated with their specific mutations. All SCID types have in common the lack of autologous T-cells with risk for lifethreatening infections and early death in the absence of effective stem cell transplantation [1–8]. Since the discovery of the gene encoding the gamma chain (ϒc) of the lymphocyte interleukin-2 receptor (IL-2R) as the cause for X-linked SCID, marked as two decades from this publication, more than a dozen other SCID types have been defined by their genotype and associated immune and clinical characteristics, including their distinguishing impairments in T, B and NK-cells with pathogenesis. [9–15]. The IL2RG SCID remains the most common and the only X-linked type of SCID, with all other modes of genetic transmission for SCID defined as autosomal recessive. Despite the development of and access to advancements in genetic diagnostic and immunologic screening assays and the heightened public health outreach about primary immunodeficiency diseases, including the recent implementation of newborn screening for SCID, selected SCID types, including hypomorphic forms, may still be delayed in diagnosis or missed [16–22]. Moreover, many patients have not even had the early opportunity for newborn SCID screening because of geographic location and timing of birth.
Coronin-1A deficiency, one of the most newly discovered forms of SCID to date, has been described in only two kindred before this publication [23–25]. The first was a female who had vaccine-related disseminated varicella at 15 months of age, a profound T-cell lymphopenia and was defined as having T-B+NK+ SCID [23, 24]. She had a CORO1A null mutation, c.248_249delCT (p.P38Rfs10), on one allele that coexisted with a ∼600 kb deletion within chromosome 16p11.2, encompassing the whole CORO1A gene, on the other allele. The de novo occurring copy number variant corresponded to the recurrent deletion reported in the 16p11.2 microdeletion syndrome, known to be associated with learning difficulties, intellectual disability and autism spectrum disorders [26–29]. Intriguingly, the second CORO1A kindred, reported by Moshous et. al., was identified by whole exome sequencing (WES) as a homozygous missense mutation, c.717C>A (p.V134M), in three Moroccan siblings with consanguineous parents and the immunophenotype was described as a combined immunodeficiency (CID) [25]. All three siblings presented with EBV-lymphomas by 15 months of age, that were fatal for two of them, and had lymphopenia of naïve T-cells, CD3+CD4+ T-cells and invariant natural killer T-cells (iNKT). Our kindred is the first described to have manifested as a pervasive mucocutaneous-immunodeficiency syndrome of EV-HPV, molluscum, HSV-1 and leprosy, with such chronic, late-onset of disease presentation (not until 7 years of age) and slightly different immunophenotypic characteristics compared to the previously reported CORO1A mutated kindred. We also found immunological abnormalities in the biological parents, both carriers and assumed healthy and, in the affected, the unexpected of very short telomeres.
Coronins are a conserved family of actin-binding proteins that function in actin-dependent processes such as cytokinesis, cell motility, phagocytosis and vesicular trafficking [30–37]. Coronin-1A is an important biological regulator of the actin cytoskeleton through countering F-actin polymerization and of calcium ion mobilization through calcium-calcineurin signaling processes, particularly in T-cells [36] In addition it has also been described as a regulator of TGF-beta/SMAD3 signaling pathways [34], implicating it in the control of cellular growth and development. In mammals, Coronin-1A is predominantly expressed by hematopoietic cells and can be found in spleen, lymph nodes, and thymus, and is also expressed in brain and weakly in lung, but not in heart, kidney or muscle [32–34]. A peculiar immunophysiologic role of coronin 1 involves facilitating survival specific to mycobacterial organisms (particularly M. tuberculosis and M. leprae) in macrophages through coronin 1 recruitment and localization to mycobacteria-containing phagosomes, and this remains with unclear explanations to host advantage [35, 36]. In Coronin-1A-deficient mice, T-cell activation and calcium regulation as well as the vital process of T-cell migration from the thymus (thymic egress) were impaired [32–34, 36]. However, with the CORO1A knock-in model used, (the E26K variant), it represented a gain-of-function mutant, in contrast to the null mutations present in the initial patients described by Shiow et. al. and Moshous et. al. In human Coronin-1A deficiency, the thymus has been reported as present, despite thymic-related defects of profound T-cell lymphopenia and abnormal naïve T-cell subpopulations [23–25]. Although a T-B+NK+ immune phenotype has been described in CORO1A-deficient mice and humans, the defective outcomes to discrete lymphocyte subpopulations, have not yet been comprehensively characterized in association with the clinical phenotypes of the CORO1A mutations.
Methods
CORO1A Kindred Studies—Genetic Analyses
DNA was extracted from the peripheral blood leukocytes of the affected siblings (P1 and P2), and of the mother and father (P3 and P4), respectively. A collaborative approach to the molecular diagnostics allowed us analyses of specimens at two different centers: on P1 (the affected index case) through Necker/Rockefeller and P2 (the affected younger sibling) and P3 and P4 (the biological mother and father, respectively) through Baylor. The exome data from P1 was rerun through the identical bioinformatics pipeline that was used for P2 and P3. Specific methods by patient and center are described with addition of the pipeline and variant evaluations.
For P1, Whole Exome Sequencing (WES) was performed through The Laboratory of Human Genetics of Infectious Diseases, Necker Branch, University Paris Descartes and Inserm, Imagine Foundation, Paris, France, EU and the St. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York USA. For P2 and P3, WES were performed at The Center for Mendelian Genomics (HGSC) at Baylor College of Medicine (BCM) (BCM-HGSC), Houston, TX USA. For P4 (biological father), only Sanger sequencing was performed.
For the WES analyses, 1 to 3 micrograms (ug) of DNA from each sample was sheared into fragments of approximately 300–400 base pairs in a Covaris S2 (Necker/Rockefeller) or E210 (BCM-HGSC) system according to manufacturer instructions (Covaris Inc, Woburn MA). An adaptor-ligated library was prepared with the TruSeq DNA Sample Preparation Kit (Illumina) and exome capture was performed with 50 Mb Agilent Sure Select (Agilent).
Genomic DNA samples were constructed into Illumina paired-end pre-capture libraries according to manufacturer's protocol (Illumina Mulitplexing_SamplePrep_Guide_1005361_D) with modifications as described in the BCM-HGSC Illumina Barcoded Paired-End-Capture Library Preparation protocol. Libraries were prepared using Beckman robotic workstations (Biomek NXp and FXp models).. The complete protocol and oligonucleotide sequences are accessible from the BCM-HGSC website (https://hgsc.bcm.edu/sites/default/files/documents/Illumina_Barcoded_Paired-End_Capture_Library_Preparation.pdf). Four pre-capture libraries were pooled together (approximately 500 ng/sample, 2 ug/pool) and hybridized in solution onto the BCM-HGSC CORE design [38] (52 Mb, NimbleGen) according to the manufacturer's protocol NimbleGen SeqCap EZ Exome Library SR User's Guide (Version 2.2) with minor revisions. The captured DNA fragments were massive parallel sequenced using paired-end mode on an Illumina HiSeq 2000 platform (TruSeq SBS Kits, Part no. FC-401-3001) which produced 9–10 Gb per sample and achieved an average of 90 % of the targeted exome bases covered to a depth of 20X or greater (Necker/Rockefeller and BCM-HGSC).
At Necker/Rockefeller, for sequence alignment, variant calling and annotation, the sequences were aligned to the human genome reference sequence (GRCh37 build) using the Burrows-Wheeler Aligner (BWA) (http://biol-bwa.sourceforge.net/) [39]. Downstream processing was done with the Genome Analysis Toolkit (GATK), SAMTools and Picard (http://picard.sourceforge.net) [40]. Variant calls were made with a GATK Unified Genotyper. All calls with a read coverage of≤2X and a Phred-scaled SNP quality of≤20 were removed from consideration. WES-generated variants were annotated using an annotation software system that was developed in-house (Necker/Rockefeller).
At BCM-HGSC, Illumina sequence analysis was performed using HGSC Mercury analysis pipeline (http://www.tinyurl.com/HGSC-Mercury/) that addressed the aspects of data processing and analyses from the initial sequence generation on the instrument to annotated variant calls. This pipeline was able to generate base-call confidence values (qualities) using Illumina primary analysis software (CASAVA v1.8.0). Reads and qualities were mapped to GRCh37 Human reference genome (http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/) using the Burrows-Wheeler aligner (BWA, http://bio-bwa.sourceforge.net/) [34] producing a BAM (binary alignment/map) file [40]. BAM post-processing, with insertion/deletion (in/del) realignment and quality recalibration, was done using appropriate tools, including SAMTools and Genome Analysis Toolkit (GATK) (v 0.5.9) 93 [40, 41]. Variants were determined using the Atlas2 suite (Atlas-SNP and Atlas-in/del) [42] to call variants and produce a variant call file (VCF) [43]. Finally annotation data is added to the VCF using a suite of annotation tools “Cassandra” [44]. To compare the WES data from P1, P2 and P3 across the family members, the WES results from P1 generated from Necker/Rockefeller were processed through the Cassandra pipeline at BCM-HGSC.
Frequencies in dbSNP, HapMap data, NHLBI GO Exome Sequencing Project (ESP; http://evs.gs.washington.edu/EVS/), The Human Gene Mutation Database (HGMD®) and in-house variant databases were used for filtering, and Integrative Genomics Viewer (http//www.broadinstitute.org/igv/) for visualization. OMIM descriptions, conservation status and prediction programs (SIFT, PolyPhen, Alamut) were used for evaluation of the potential pathological relevance. The WES-identified variants were independently confirmed by an orthologous sequencing technique (Sanger), and checked for family segregation with the phenotype.
Prior to WES analyses, gene testing through clinical commercial laboratories included: CXCR4 and DOCK8 (Gene DX), Dyskeratosis Congenita (DKC) mutational panel, including DKC1, NHP2, NOP10, TERC, TERT and TINF2, (Ambry Genetics), IFNGR1/2 (Correlagen Diagnostics), RAG1/RAG2 (Gene DX) and STAT1 (Laboratory of Human Genetics of Infectious Diseases, Necker Branch, University Paris and Inserm, Paris, France) and STAT3 (Gene DX). Chromosomal microarray (CMA) analysis on genomic DNA from both P1 and P2 was performed at The Medical Genetics Laboratories BCM, Houston, TX. The BCM CMA was custom-designed and targeted intra-genetic exonic copy number variants [45]. The version 9 BCM CMA used for testing P2's sample contained 60,000 SNP probes and 400,000 oligoprobes with exon coverage of 4,900 genes including 200 genes reported in primary immunodeficiencies [15, 46].
Subsequent to the genomic analysis that identified the compound heterozygous CORO1A mutations in our kindred, we performed Western blot for Coronin-1A protein analyses. For Western blot analysis, frozen cell pellets from lymphoid cell lines from P2 (affected) and P3 and P4 (parental carriers) were lysed in 1 % NP-40 lysis buffer supplemented with HALT protease inhibitor as described previously [47]. Proteins were separated by 4-12 % NuPAGE gel (Invitroen) and transferred to PVDF membrane. Membranes were blocked with 3 % skim milk and incubated with primary antibody to Coronin 1A (Novus) or Myosin IIA (Sigma). Membranes were washed and incubated with secondary antibody (Rockland). Membranes were scanned on the Odyssey (Licor) and band intensities were quantified using ImageJ (NIH).
CORO1A Kindred Studies—Immune Analyses
Immunophenotyping (IP) and lymphocyte proliferation assays (LPAs) were performed by the Clinical Immunology Laboratory of the Section of Immunology, Allergy and Rheumatology of Texas Children's Hospital of the Department of Pediatrics at Baylor College of Medicine, Houston, Texas, using flow cytometry (Cytomics FC 500; Coulter Corp, Hialeah, Fla). Pediatric and adult age-matched normal values for IP were obtained from established published values [48] and laboratory controls. Markers for memory CD19+CD27+ Bcells were performed after 2008. For naïve T-cell subpopulations, although selected T-cell markers specific to these T-cell emigrants, such as CCR7 and CD62 expression, may further distinguish this proliferation-competent subset, we utilized CD4CD45RA+ as a surrogate indicator of physiologic naïve CD4+ T-cell subpopulation. Naïve CD8+ T-cell analyses were not a function of our analyses at the time of our 10-year study of this CORO1A kindred.
LPAs were performed, using tritiated thymidine proliferation assays, to mitogens: phytohemagglutinin (PHA) and pokeweed mitogen (PWM) at 10 μg/mL and concanavalin A (ConA) at 50 μg/mL and to antigens: tetanus (0.08 flocculation units/mL) and Candida albicans (10 μg/mL). Lymphocyte cultures were pulsed with mitogens (after 72 h and harvested after 96 h) and with antigens (after 5 days and harvested on day 6), and were performed in triplicate. A normal cellular specific immune response to antigens was defined as a stimulation index (SI) of greater than 2 and from counts per million (cpm) values of greater than 2,000. Normal control ranges were determined for mitogen-specific responses.
Serum immunoglobulin levels were assayed by Texas Children's Hospital Department of Pathology, Houston Texas and specific antibody titres to tetanus toxoid (Tt) and Streptococcus pneumonia (Spn) were performed by Specialty Laboratories, Santa Monica, California. Low immunoglobulin G, A, and M levels were defined as less than 2 SDs below the laboratory reported mean for age. IgE level was considered elevated at greater than 90 IU/mL. Protective antibody titres were defined as greater than 0.10 IU/mL to Tt and greater than 1.0 μg/mL to Spn. serotypes 1, 3, 4, 6B, 7F, 8, 9N, 12F, 14, 18C, 19F and 23F, as reported by the laboratory reference range. The IP, LPAs and immune analyses were repeated at several time points, with increasing ages for P1 and P2, from onset and through childhood (and for P1 through early teenage years) and compared for both affected siblings. Adenosine deaminase levels and neutrophil oxidative burst assays of nitroblue tetrozolium dye test and dihydrorhodamide flowcytometry had been performed in samples from both siblings. Interferon gamma levels and function were tested in P1 and P2 (Laboratory of Human Genetics of Infectious Diseases, Necker Branch, University Paris and Inserm, Paris, France). Telomere length assays, utilizing multicolor flow-FISH from peripheral blood, were performed on P2 at different time points through Repeat Diagnostics, Vancouver, Canada (www.repeatdiagnostics.com) where the leukocyte subsets were defined using flow cytometric histograms for telomere fluorescence on each subset of cells as identified by the monoclonal antibodies used and reported as: “lymphocytes; granulocytes; CD45RA pos (naïve T cells); CD45RA (memory T cells); CD20 pos (B cells); and CD57 pos (NK cells)”. From the antibody and gating strategies used for this telomere report, partial impure classifications of the subpopulations of the naïve and memory T cells and NK cells may have been possible. However, this telomere study did capture consistent representation of shortened telomere lengths within the lymphocytes of P2, confirmed on repeated assays.
CORO1A Kindred Studies—Clinical Analysis
Informed consent for research studies for the reported siblings and parents were obtained under approved institutional review board protocols from the following collaborating centers: Allergy and Immunology Service of the Section of Immunology, Allergy and Rheumatology of Texas Children's Hospital and the Department of Pediatrics at Baylor College of Medicine, Houston TX, and The Center for Mendelian Genomics, Baylor College of Medicine, Houston, Texas; Rockefeller University, New York City, New York; and Hospital Necker-Enfants Malades University of Paris, France.
Results
CORO1A Genotype—Compound Heterozygous Mutations, Exon 4 and 11
Compound heterozygous CORO1A mutations were discovered in P1 and P2 by WES and Sanger sequencing (Fig. 1): c.248_249delCT (p.P83RfsX10) and c.1077delC (p.Q360RfsX44) located in exon 4 and 11, respectively, according to the CORO1A transcript variant 1 (NM_001193333), and were determined in correlation with the immune and clinical phenotypes found in our CORO1A kindred as described below. Prior to WES-Sanger analyses, the genetic-specific tests performed on P1 and P2 were nondiagnostic. From the previous two CORO1A patient reports, transcript variant 2 (NM_007074.3) was used for exon number designation. In our CORO1A kindred, we detected the exact same variant in exon 4, of Louisiana origin, that was previously reported and found in exon 3 [23, 24]. The CORO1A findings segregated with the phenotype and were the causal explanation to our siblings' immunodeficiency syndrome. The exon 4 variant was detected in 13 out of 27 total reads for P1, and in 18/43 reads for P2, while the exon 11 variant was detected in 25/58 reads for P1, 10/21 reads for P2, and 12/43 reads for P3. Both CORO1A variants were confirmed by Sanger sequencing in both affected subjects. In accordance with Mendelian expectations, the biological parents were proven to be carriers of one variant each, with the father heterozygous for the c.248_249delCT Louisiana variant, and the mother heterozygous for the c.1077delC variant that is the novel CORO1A mutation we found (Fig. 1).
Fig. 1.
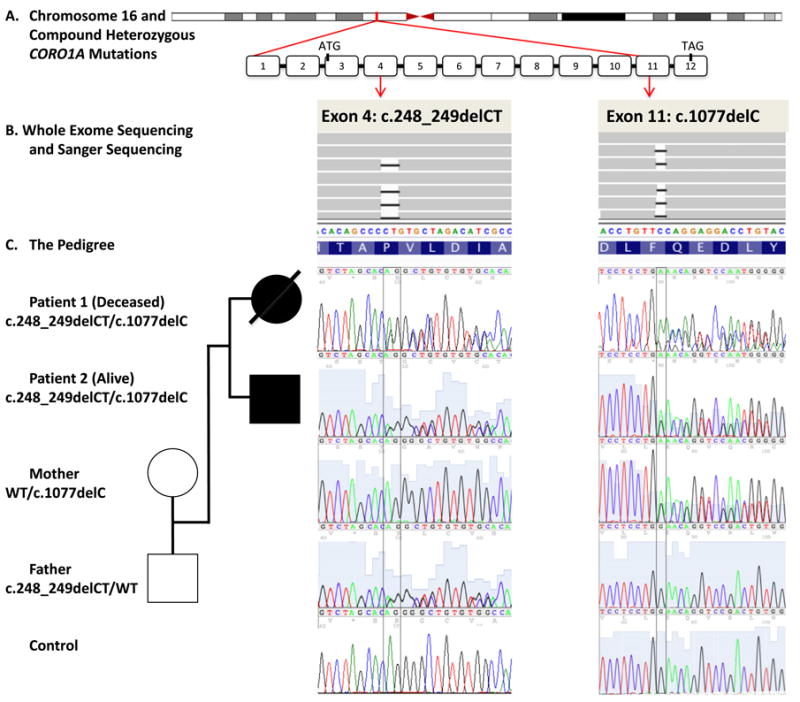
CORO1A Genotype in our Kindred: Mutations in the CORO1A gene identified by Whole Exome Sequencing (WES) in Two Affected Siblings with absent naïve T-cells and memory B cells and increased double negative T-cells manifested as a late-onset Mucocutaneous Syndrome. A The CORO1A locus at chromosome 16p11.2, in position g. 30194731–30200397 (hg19), and the genomic structure of the CORO1A gene with exons 1–12 (NM_001193333), with attention to exons 4 and 11, are illustrated to show the intragenic locations of the two mutations. B Arrows from exon 4 and 11 mutation loci define the columns matched to the results from WES and Sanger sequencing. WES results are visualized in IGV and shown in forwards strands, while the Sanger sequencing results are shown on the reverse strands. Black bars indicate the deleted nucleotides. Control sequence shown at the bottom. C The pedigree with familial segregation shown is positioned vertically to diagrammatically match family member to their mutations in row of their resultant mutations: deletion of 2 nucleotides in position g.30197968_30197969; c.248_249delCT in exon 4 (both siblings and father) and a single nucleotide deletion g.30199693; c.1077delC in exon 11 (both siblings and mother). WT, wild type.
Shared and Rare Genetic Variants in this CORO1A Kindred
There were in total 660 shared variants between the affected siblings when the FASTQ files were run through the same bioinformatic pipeline and variant frequency was limited to<1 % for the In-house database of WES data from 6,260 persons sequenced at HGSC BCM, frequency in Thousand Genome Project (TGP)<5 %, and NHLBI GO Exome Sequencing Project (ESP)<5 %. Only the rare variants in conserved sites, (ESP<1.5 %; 13 variants in total), were predicted to be deleterious and located in known and potential genes for primary immunodeficiencies [15, 46] and were shared by both siblings (Table 1). Further functional testing was performed for one maternally-inherited, shared rare variant in STAT5A. The detected variant was predicted to interfere with STAT5 tetramerization, and secondary STAT5 transcription functions, but the EBV transformed cell lines from P2 and P3 showed normal IL-2-mediated induction of the genes that are known to be dependent on STAT5 tetramers (Warren Leonard, NIH).
Table 1. CORO1A Genotype: Intersection from WES data with shared, rare variants in PID genes for affected siblings (P1 and P2) and parental carriers (P3 and P4).
| Gene | Exon | Reference sequence |
Variant cDNA | Protein Effect | g.position_variant | P1; vR/tR | P2; vR/tR |
P3 | P4 | Inhouse 6260 |
ESP 5400 |
dbSNP & HGMD |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CORO1A | exon 4 | NM_001193333 | c.248_249delCT | p.83_83del | Chr16:30197967_CCT>C | Het; 13/27 | Het; 18/43 | WT | Het | 0,0000 | 0,0000 | 18836449 |
| CORO1A | exon 11 | NM_001193333 | c.1077delC | p.F359fs | Chr16:30199692_TC>T | Het; 25/58 | Het; 10/21 | Het | WT | 0,0000 | 0,0000 | |
| FANCD2 | exon 15 | NM_033084 | c.1275C>T | splice variant | Chr3:10088409_TAAG>T | Het; 13/88 | Het; 16/121 | WT | ND | 0,0000 | 0,0000 | |
| LIG4 | exon 2 | NM_002312 | c.1798G>A | p.E600K | Chr13:108861819_C>T | Het; 42/91 | Het; 47/116 | WT | ND | 0,0000 | 0,0068 | rs61731910 |
| RBBP8 | exon 11 | NM_203291 | c.1766G>A | p.R589H | Chr18:20573556_G>A | Het; 36/86 | Het; 18/32 | WT | ND | 0,0000 | 0,0089 | rs111445733 |
| NCF2 | exon 8 | NM_001190794 | c.755T>C | p.V252A | Chr1:183536089_A>G | Het; 30/51 | Het; 28/52 | WT | ND | 0,0000 | 0,0136 | |
| AP3B1 | exon 21 | NM_003664 | c.2409_2411delTTC | p.803_804del | Chr5:77396835_TTTC>T | Het; 23/46 | Het; 26/54 | WT | ND | 0,0118 | 0,0000 | |
| CFHR5 | exon 7 | NM_030787 | c.1135G>C | p.V379L | Chr1:196967422_G>C | Het; 26/61 | Het; 19/50 | Het | ND | 0,0110 | 0,0044 | rs111327589 & 20513133 |
| STAT5A | exon 3 | NM_003152 | c.53T>C | p.M18T | Chr17:40441482_T>C | Het; 13/25 | Het; 67/123 | Het | WT | 0,0000 | 0,0000 | |
| ASXL2 | exon 11 | NM_018263 | c.1663C>G | p.L555V | Chr2:25972762_G>C | Het; 68/136 | Het; 63/125 | WT | ND | 0,0000 | 0,0002 | |
| STAT2 | exon 24 | NM_198332 | c.2466G>T | p.Q822H | Chr12:56737251_C>A | Het; 33/62 | Het; 105/105 | WT | ND | 0,0000 | 0,0089 | |
| CLPTM1 | exon 8 | NM_001294 | c.886G> | p.E296K | Chr19:45490529_G>A | Het; 44/96 | Het; 29/63 | WT | ND | 0,0005 | 0,0003 | |
| NLRP7 | exon 1 | NM_139176.3 | c.-9C>T | p.R26W | Chr19:55453088_G>A | Het; 20/48 | Het; 29/61 | Het | ND | 0,0013 | 0,0019 | rs184816368 |
The table shows the shared, rare variants in the known genes for Primary Immunodeficiencies (PID) (Al-Herz W, Front Immunol. 2011; 2: 54.). Rare was defined as in-house database frequency < 1 % and ESP < 1.5 %. Also rare variants in potential genes for Primary Immunodeficiencies (Keerthikumar, S., RAPID Nucleic Acids Research. 37, D863-D867) are included here as long as the variant was located in a conserved site and predicted to be deleterious in 2 or more of 4 prediction programs (Phylopred, Polyphen, SIFT, LRT, Mutation Taster). The results from WES in P1, P2, and P3 are included as well as results from Sanger segregation studies for P3. The presented table is sorted according to variant frequencies within the HGSC in-house database and NHLBI GO Exome Sequencing Project (ESP)
Chr chromosome; Het heterozygote; HGMD human genome mutation database; Hom homozygote; ND not done; tR total reads; vR variant reads; WT wild type
CORO1A Immune Phenotype –Naïve T-cell & Memory B-cell Lymphopenia with Increased ϒδ T- cells
We conducted serial immunologic analyses of our Coronin-1A deficiency kindred throughout the course of their illnesses before the diagnostic CORO1A mutations were known. For P1, the immune evaluations began at age 7 years when the unique mucocutaneous syndrome of tuberculoid leprosy initially surfaced, and then immune studies were reanalyzed at increasing age time points as EV-HPV, HSV and molluscum manifested throughout her life until her death from lymphomas at age 16 years. For P2, the immune evaluations also began at age 7 years, coinciding with the death of his sister, and with the presentation of his atypical mucocutaneous disease of EV-HPV and molluscum contagiosum. Both P1 and P2 had immune and clinical phenotypes remarkable for features common with one another and for distinguishing clinical findings as compared to previous reports (Tables 2 and 3 and Figs. 2 and 3). First, they each presented significantly later than the previously reported children with CORO1A mutations and both had an unusual mucocutaneous syndrome. Because of these findings, comprehensive longitudinal immune analyses were performed (Fig. 2) and an abnormal immune phenotype was found common to both P1 and P2: (1) moderate lymphopenia of total absolute lymphocyte counts; (2) mildly low CD3+CD4+ T-cell absolute counts; (3) nearly absent “naïve” CD4CD45RA+ T cells ; (4) a preponderance of double-negative (DN) CD3+CD4-CD8- T-cells, verified as ϒδ T-cell subpopulation in P2 and expressed as 100 % of the DN T-cells; (5) undetectable memory B cells; (6) low NK cell absolute counts; (7) modestly suppressed mitogen-induced lymphoproliferation, particularly to ConA and PHA; and (8) an elevated serum IgE (peak levels at onset of presentation of their clinical diseases). For both affected siblings, despite the naive T-cell lymphopenia and abnormal TCR, percentages of total CD3+ T-cells were in the normal ranges for age and thymus was present. Moreover, the T-cell receptor representation was skewed by the increased DN T-cell subpopulation and Con A- and PHA-specific T-cellular mitogen-induced lymphoproliferative responses were persistently diminished. Strikingly, however, antigen-induced lymphoproliferative responses from stimulation with tetanus and candida were robustly present at onset of clinical disease presentation and without change (through end-of-life at 16 years of age for P1 and through 9.5 years of age for P2, alive with this publication). Both siblings had normal levels of serum IgG.
Table 2. CORO1A Immune Phenotype: Comparisons of immunological data for P1 and P2 at age of presentation (onset) and subsequently (mid and late childhood).
| Immune Data | Patient 1 | Patient 2 | Control | |||
|---|---|---|---|---|---|---|
|
|
|
|||||
| Disease periods | Onset | Mid | Late | Onset | Mid | Reference (36) |
| Age, years | 7 | 9-10 | 14-16 | 7 | 9-10 | 6-12 |
| WBC | 2800 | 3420 | 9050 | 5410 | 5000 | 5.0-14.5 × 10^3/UL |
| ALC | 812 | 581 | 1339 | 1568 | 1300 | 1.9-9.5 × 10^3/UL |
| Lymphocytes % (cells/mm3) | ||||||
| CD3+ | 63 (515) | 62 (358) | 76 (1015) | 76 (1226) | 81 (1053) | 60-76 (1200–2600) |
| CD3+CD4+ | 29 (243) | 30 (174) | 22 (293) | 29 (461) | 29 (374) | 31-48 (650–1500) |
| CD3+CD8+ | 27 (222) | 23 (136) | 40 (540) | 30 (483) | 33 (429) | 18-35 (370–1100) |
| CD3+CD4−CD8− | 7 (56) | 9 (52) | 14 (187) | 17 (257) | 19 (247) | 0-5 |
| CD4+CD45RA+ | 0.5 (5) | 0.5 (2) | 0.6 (7) | 1.6 (32) | 0 (0) | 3.3-32 (134–969) |
| CD4+CD45RO+ | 26 (266) | 31 (181) | 22 (299) | 29 (470) | 29 (377) | 15-42 (301–919) |
| CD4:CD8 ratio | 1.1 | 1.28 | 0.54 | 0.95 | 0.88 | ≥1.2 |
| CD19+ | 16.5 (166) | 25 (146) | 12 (166) | 15 (241) | 12 (156) | 11-28 (270–860) |
| CD20+ | 13 (129) | 27 (156) | 7 (99) | 15 (240) | 13 (169) | 15-42 (59–457) |
| CD19+CD27+ | ND | ND | 0.2 (3) | 0.6 (9) | 0.6 (8) | 1-5 (19–131) |
| CD3‒CD56+CD16+ | 9 (90) | 10 (60) | 31 (411) | 7.2 (116) | 5 (65) | 5-20 (128–474) |
| LPA Mitogens, cpm | ||||||
| PHA 10 ug/ml | 127, 354 | 128,483 | 86,453 | 84,852 | 132,461 | 163,500-415,000 |
| ConA 50 ug/ml | 64,584 | 40,911 | 25,105 | 40,507 | 25,026 | 80,718-286,866 |
| PWM 100 ng/ml | 88,760 | 114,721 | 36,052 | 66,374 | 91,606 | 37,000-157,955 |
| Antigens, cpm/In | ||||||
| Tetanus 80 | 86,420/18 | 14,373/7.1 | 32,054/10 | 140,569/310 | 141,942/620 | >2,000 and >2.0 |
| Candida 5 | 32,943/7.6 | 4,196/2.8 | 8,649/3.5 | 10,898/24 | 1,865/9 | >2,000 and >2.0 |
| IgG, mg/dL | 815 | 697 | 1080* | 720 | 803 | 641-1353 mg/dl |
| IgA, mg/dL | 45 | 36 | 42 | 106 | 95 | 66-295 mg/dl |
| IgM, mg/dL | 20 | 22 | 29 | 90 | 58 | 40-180 mg/dl |
| IgE, IU/ml | 2228 | 485 | 989 | 1160 | 815 | 0-90 IU/ml |
| Antibody Levels | ||||||
| Tt, μg/mL | 1.34 | 0.57 | ND | 4.1 | 4.2 | >0.10 |
| Spn, μg/mL | ND | |||||
| Type 1 | 7.2 | 0.26 | 0.65 | 0.44 | >1.0 | |
| Type 3 | 2.0 | 0.04 | 0.5 | 0.21 | >1.0 | |
| Type 4 | 2.0 | 0.09 | 11.97 | 5.9 | >1.0 | |
| Type 5 | ND | 0.56 | 5.05 | 14.5 | >1.0 | |
| Type 6B | 12.1 | 0.80 | 0.64 | 0.26 | >1.0 | |
| Type 7F | 4.8 | 0.15 | 1.42 | 0.54 | >1.0 | |
| Type 8 | 0.5 | 0.09 | 0.29 | 0.24 | >1.0 | |
| Type 9N | 0.6 | 0.08 | 1.77 | 3.50 | >1.0 | |
| Type 9V | ND | 0.10 | 35.53 | 21.05 | >1.0 | |
| Type 12F | 3.4 | 0.42 | 4.41 | 1.71 | >1.0 | |
| Type 14 | 3.7 | 0.06 | 2.0 | 0.83 | >1.0 | |
| Type 18C | 1.6 | 0.06 | 7.98 | 6.60 | >1.0 | |
| Type 19F | 14.2 | 0.13 | 16.52 | 11.11 | >1.0 | |
| Type 23F | 4.9 | 0.11 | 2.41 | 1.73 | >1.0 | |
Bolded font indicates abnormal values
ALC absolute lymphocyte count; LPA lymphoproliferation; ND not done; Spn Streptococcus pneumoniae (by serotypes); Tt tetanus toxoid; WBC white blood cell count
represents patient IgG plus exogenous intravenous immunoglobulin G (IVIG) replacement that was added due to poor specific antibody although total IgG was normal pre-IVIG
All presented data were collected before stem cell transplantation. Data on memory CD19+CD27+B-cells were not reported by the lab before year 2008
Table 3. Summary of Genotype with Immune and Clinical Phenotypes in our CORO1A Kindred.
| CORO1A Kindred | Patient 1 Female (Index) | Patient 2 Male (Younger Sibling) | Patient 3, Mother | Patient 4, Father |
|---|---|---|---|---|
| Thymus | Present | Present | ND | ND |
| Live Viral Vaccines | Tolerated | Tolerated | NA | NA |
| Immune Phenotype | ||||
| T-cells | Absent Naïve T-cells | Absent Naïve T-cells | Normal | Normal |
| High DN (ϒδ) T-cells, | High DN (ϒδ) T-cells | Normal | Normal | |
| Low CD4 T-cells | Low CD4 T-cells | Normal | Low CD4 T-cells | |
| Low Mitogen LPA | Low Mitogen LPA | Normal | Normal | |
| B-cells | Absent memory B-cells | Absent memory B-cells | Normal | Normal |
| High IgE | High IgE | ND | ND | |
| Normal IgG | Normal IgG, M, A | ND | ND | |
| Low IgM, IgA | ||||
| Specific Antibody Loss (Table 2) | Specific Antibody intact | ND | ||
| NK-cells (nadir) | Low (9%) | Low (5%) | Low (2%) | Normal |
| Clinical Phenotype | ||||
| Onset (7 years) | Cutaneous Leprosy | Cutaneous EV-HPV* | No disease | No disease |
| Mucocutaneous HSV-1 | Cutaneous Molluscum* | |||
| Rhinosinusitis* | Rhinosinusitis* | |||
| Asthma* | Asthma* | |||
| Bronchiectasis* | Bronchiectasis* | |||
| Staphylococcal cellulitis | Parvovirus B19 Sepsis | |||
| Mid (9–10 years) | Cutaneous EV-HPV*, types 5 & 17 | Mucosal HSV | No disease | No disease |
| Cutaneous Molluscum* | ||||
| Staphylococcal cellulitis | ||||
| Late (14–16 years) | Lymphoma | NA | No disease | No disease |
|
||||
| Malignancy | Yes | No | No | No |
| Survival | Death, 16 years of age | Alive | Alive | Alive |
| Other | ||||
| Autism/ADHD Disorders | No | No | No | No |
| Dysmorphic features | No | Subtle | No | No |
Abnormalities in the immunologic and clinical presentations are shown. Immune abnormalities were persistent from the onset of findings stated. The clinical findings are arranged by periods in year of presentation. All phenotypic manifestations presented prior to stem cell transplantations (SCT). SCT for P1, haploidentical maternal donor at 15 years of age before CORO1A diagnosis was known and for P2, in preparation upon confirmation of this CORO1A finding
Persistent diseases
Abbreviations: ADHD, attention deficit hyperactivity disorder, NA, not applicable; ND, not done
Fig. 2.

CORO1A Immune Phenotype: Longitudinal Lymphocyte Subsets for P1 and P2. The relative percentages of lymphocyte subsets are illustrated, at onset and at different age points, and the predominant clinical phenotype is marked in association with the timing of presentation for Patients 1 and 2. Despite normal to elevated percentages of CD3+ T-cells, the distribution of CD3+CD4+ T-cells was low with an inverted CD4:CD8 ratio and an increased double-negative T-cell population was identified as ϒδ T-cells. CD4+CD45RA+ naïve T-cells and CD19+CD27+ memory B cells were both consistently near-absent. CD19+ (not shown) and CD20+ were equivalent
Fig. 3.
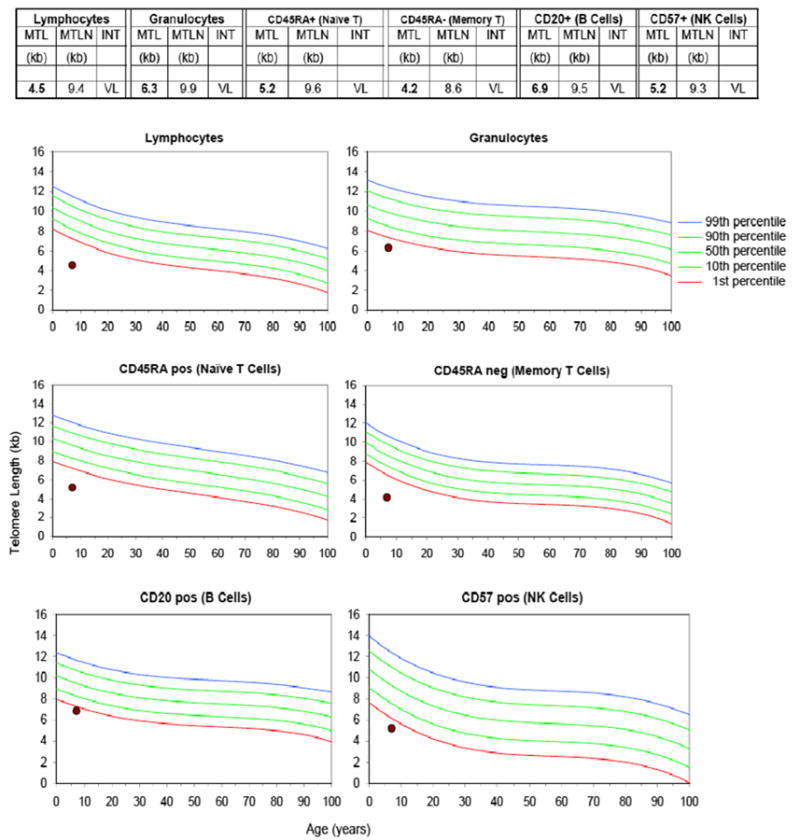
Telomere length assay results on P2. Telomere lengths were very low (<1st percentile) for all cell populations assayed by Repeat Diagnostics, Vancouver, BC Canada. MTL = Median Telomere Length; MTLN =Normal MTL at age (50th percentile); INT = Telomere Length Interpretation; VL = Very Low (< 1st percentile)
Few differences in the immune phenotype were noted between P1 and P2. Unlike P2 (the younger brother), P1 (the female index) had PWM-specific lymphoproliferative responses that became progressively abnormal at 14 years of age when then also, specific antibody diminished followed by unresponsiveness to pneumococcal polysaccharide and conjugate vaccines. By contrast, P2 had sustained normal PWM-specific T-cell responses, but had not yet reached the same age as his older sister when she had developed loss of PWM-induced mitogenic responses. P1 also had persistently low IgM and IgA from onset whereas P2 had normal IgM and IgA levels for age. For P2, very short telomere lengths were detected in lymphocytes, granulocytes, naïve and memory T-cells (telomere data no P1 not performed) (Fig. 3.) The parents had no known clinical disease, however, unexpectedly, the mother (P3) was found to have a low percentage and absolute number of NK cells, confirmed by repeat on separate blood sample (2 % with absolute number of 20, normal adult range 3–22 %/89–472). Also, the father (P4) was found to have low CD4+ T-cells (28 %, normal range 30–63 %) with an inverted CD4:C8 ratio of 0.5 (normal ratio ≥1.2).
CORO1A Clinical Phenotype—The Atypical Mucocutaneous-Immunodeficiency Syndrome
Patient 1, Female Index—Deceased (P1)
P1 was female and the first-born child of an uncomplicated pregnancy from non-consanguineous parents with normal physical and cognitive development who presented at 7 years of age with tuberculoid leprosy that manifested as a severely disfiguring ulcerative, granulomatous dermatosis over most of the body, sparing genitalia. The dermatosis was painless despite recurrent epidermal-dermal lesions (Fig. 4a and b). Multiple skin biopsies revealed caseating granulomas with dermal lymphocytic perineural infiltrates (Fig. 4c). Rare acid-fast bacilli (AFB) were uncovered (Fig. 4d). Confirmatory pathological review of the skin biopsy specimens by the National Hansen's Disease Programs in Baton Rouge, Louisiana verified that findings were consistent with Hansen's Disease due to Mycobacterium leprae, and this was further supported by the clinical presentation and family's home location of Louisiana, a geographical area endemic for leprosy [49]. Extensive evaluations for potential causes of granulomatous diseases were performed, including assays for chronic granulomatous disease (CGD) and interferon-gamma/interleukin-12 functional analyses and for human immunodeficiency virus (HIV) by serology and direct DNA testing; all tests were normal. Long-term anti-mycobacterial therapy of isoniazid, rifampin, azithromycin and dapsone resulted in resolution of active lesions by 12 years of age, but left the patient with significant cutaneous scarring. During the treatment period for the tuberculoid leprosy, at age 9–10 years, disseminated cutaneous EV-HPV (pre-malignant types 5 and 17) developed in P1. The EV-HPV was generalized mostly at extremities, chest, neck and hairline. Concurrently, she had oral mucosal and cutaneous HSV-1 (culture-confirmed) lesions that were evanescent and responsive to oral acyclovir when they occurred. At 11–12 years, a non-resistant and localized staphylococcal cellulitis developed under the site of one of the granulomatous lesions at the thigh, which resolved with intravenous nafcillin. She had a persistently low IgA and IgM from debut. However, despite an IgG that remained in normal range for age, monthly intravenous immunoglobulin (IVIG) was started. She had bronchiectasis (Fig. 5c) and was treated for persistent asthma with moderate-dosed inhaled corticosteroid, airway clearance and as-needed bronchodilator. Intravenous pentamidine or oral trimethoprim-sulfamethoxazole was also given for pneumocystis prophylaxis. No organisms were uncovered from bronchoalveolar lavage or lung biopsies. At 15 years of age, P1 developed an acute, severe leukopenia associated with fatigue, which prompted screening for malignancy. She was found to have worsening of her chronic lung disease with new cervical and mediastinal lymphadenopathy. Stage IV Hodgkin's lymphoma (HL) was confirmed by cervical lymph node biopsy (positive for Reed-Sternberg cells as CD20+ and CD4- and abnormal cells were positive for EBV by staining and encoded RNA in-situ hybridization) and comprehensive staging evaluation. While receiving immunosuppressive chemotherapy for HL, she developed a diffuse large B-cell CD20-CD38+ lymphoma (DLBCL) with plasmacytic differentiation that manifested as a conjunctival mass and was also found in the liver as two abnormal populations of kapparestricted CD20+(dim) and CD38+(bright) plasmacytic cells by flow cytometry and were also positive for EBV by in-situ analyses. Aggressive interventions, including salvage haploidentical stem cell transplantation, with her biological mother as the donor, failed to result in immunoreconstitution and the malignant processes progressed to death. It was not until just before her death at 16 years of age and after the DLBCL occurred, that peripheral blood EBV titers acutely rose within a 2-month period from 300 copies/μg to >262,144 copies/μg (the level that exceeded the linearity of the assay, Center for Cell & Gene Therapy, Baylor College of Medicine). Autopsy was not performed.
Fig. 4.
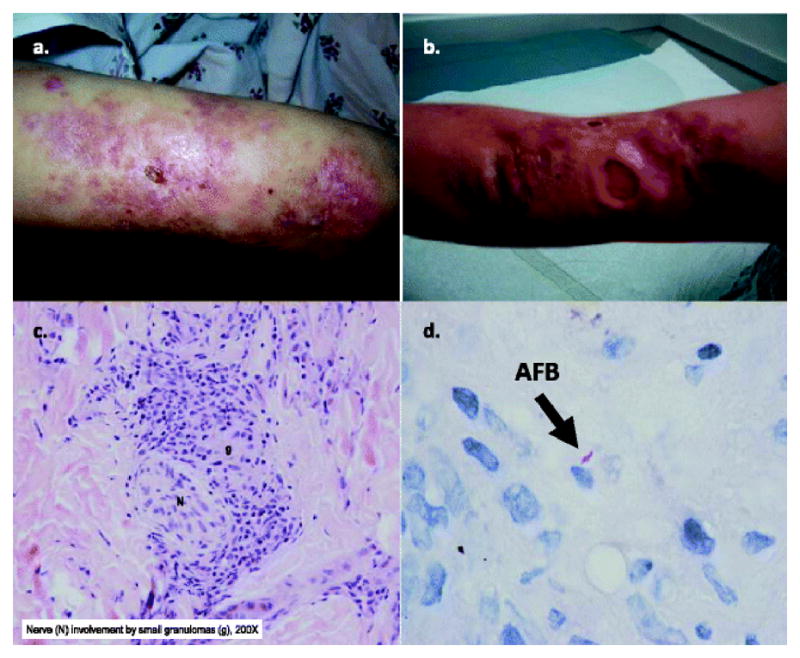
Unique CORO1A clinical phenotype in P1: cutaneous ulcerations and histopathology of tuberculoid leprae with caseating granulomas from skin biopsies from CORO1A P1, index case. Skin lesions, shown from upper extremities (Panel A. and B., respectively), were insensitive to pain despite disseminated granulomas and ulcerations. Histopathology (Panel C.) revealed subcutaneous necrotizing caseating granulomas with perineural involvement. Rare acid fast bacilli (AFB) were identified (Panel D.). Perineural granulomas by histopathology with rare AFB and skin lesions insensitive to pain were consistent with tuberculoid leprae due to Mycobacterium leprae. (Department of Pathology, Texas Children's Hospital and the National Hansen's Disease Programs, Baton Rouge, Louisiana)
Fig. 5.
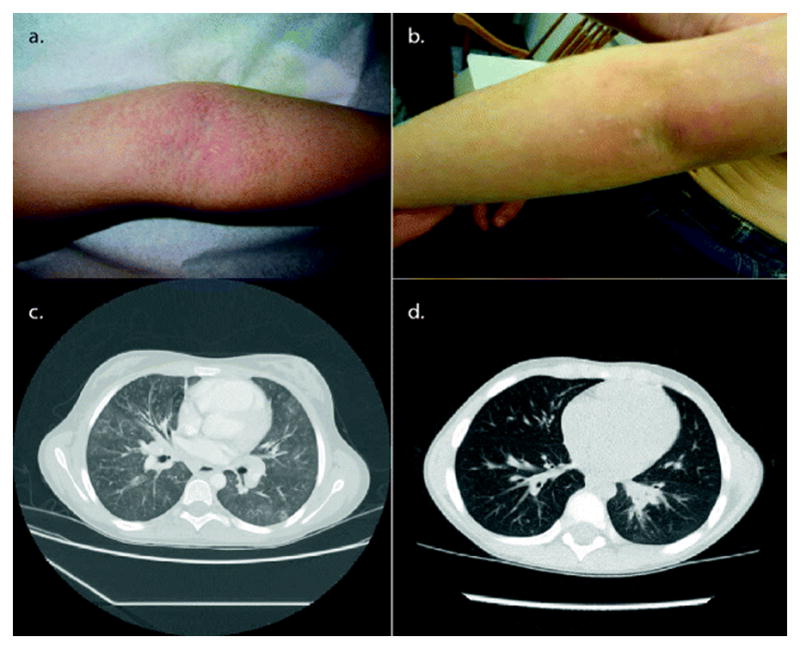
Similarities in the CORO1A Clinical Phenotype in P1 and P2: Cutaneous EV-HPV and Molluscum Contagiosum, and Bronchiectasis in the affected siblings, P1 (a and c) and P2 (b and d). The cutaneous lesions of epidermodysplasia verruciformis and molluscum are shown on extremities for P1 when she was 8 years of age (a) and Patient 2 when he was 7 years of age (b). The confluent sheaths of fine, shiny erythematous and hyperpigmented, fleshy warts were identified as epidermodysplasia verruciformis and the scattered umbillicated lesions were molluscum. Histopathology for P1 confirmed human papilloma virus types 5 and 17. Bronchiectasis was also found in both siblings (Chest CT scans are shown for P1 (c) at 9 years of age and Patient 2 (d) at 8 years of age
Patient 2, Younger Male—Alive (P2)
P2 is the younger male sibling of P1 and the product of an uneventful pregnancy, with the same parents as P1; he had normal developmental milestones. Within the year of initial presentation of the mucocutaneous disease of EV-HPV and molluscum with recurrent oral ulcers, P2 developed a severe anemia (hemoglobin 3.8 g/dL) and febrile seizures due to Parvovirus B19 infection, evident by 8.3×107 copies B19 DNA/ml plasma and erythroid maturation arrest at pronormoblast stage, including giant pronormoblasts with nuclear viral inclusions on bone marrow biopsy. Prior to deriving Parvovirus B19 as the cause to the red cell suppression (also before the CORO1A mutations were known), telomere length testing was performed. The telomere lengths were abnormal, significantly below the 1st percentile for P2 in total lymphocyte and granulocyte populations and for the subsets identified by Repeat Diagnostics, resulted as: CD45RA T cells; CD20+ B cells; and CD57+ NK cells. Recognizably, impurities to the subsets identified by Repeat Diagnostics were possible. Nonetheless, all telomere length results for P2 repeatedly resulted as “very low” (VL) within all lymphocytes tested, indicating that telomere lengths were all less than the 1st percentile for age. Within 1 month from the parvovirus B19 infection, the parvovirus DNA load reduced by 103-fold and clinical disease resolved. However, after resolution of Parvovirus B19 disease, repeated telomere studies revealed continued persistence of very low telomere lengths across all subsets (Fig. 3). He also had recurrent sinopulmonary infections, a moderate persistent asthma component confirmed by pulmonary function tests, and bilateral bronchiectasis verified on computed tomography (CT) scan (Fig. 5d). P2 also received IVIG antibody replacement monthly (at 500 mg/kg every 4 weeks), despite presence of specific antibody and normal IgG levels. Pneumocystis prophylaxis was also given with trimethoprim-sulfamethoxazole or intravenous pentamidine. At time of this publication, P2 had active EV-HPV and molluscum, asthma and bronchiectasis. He had not had any mycobacterial disease, malignancy or elevation in EBV and was in preparation for stem cell transplantation. Notably, physical exam also revealed mildly low-set posteriorly rotated ears with normal pinnae and folds, but no other abnormalities, such as intellectual disabilities or problems with behaviors or social interactions. Neither of the affected siblings had fungal or pneumocystis infections.
CORO1A Clinical Phenotype—Deficiency of Coronin-1A Protein
To validate that our genetic and clinical findings could be explained by a deficiency of Coronin-1A, we also performed Western blot analyses on the surviving members of this CORO1A kindred. Specimens available for protein analyses were on P2 (the surviving younger male sibling), P3 (biological mother) and P4 (biological father). Results are shown in Fig. 6 that illustrate absence of Coronin-1A protein and diminished Myosin IIA protein in the affected P2. Presence of Coronin-1A and Myosin IIA proteins were identified in both parents, P3 and P4. Western blot studies were not able to be formed on the deceased index, P1. Normal controls were applied for Coronin-1A and Myosin IIA assays (Fig. 6).
Fig. 6.
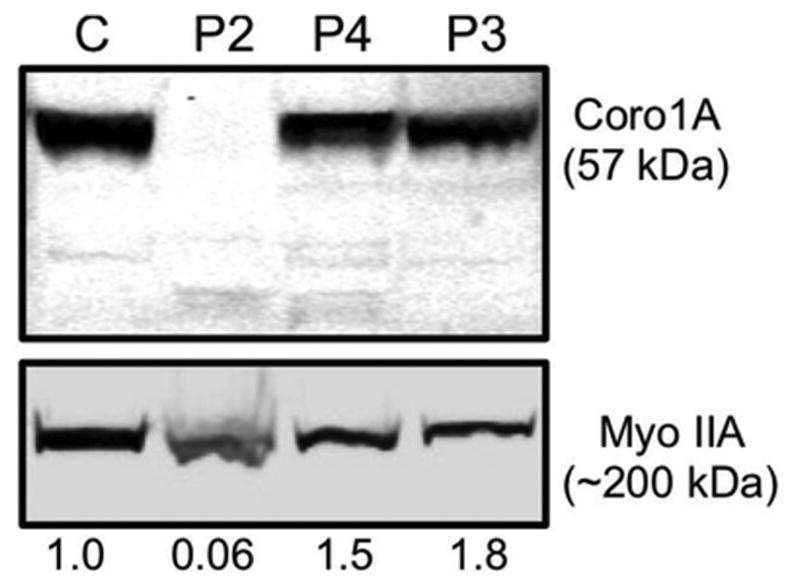
Coronin 1A novel compound heterozygous mutation results in loss of protein expression. Lymphoblastoid cell lines from a healthy donor control (C) Patient 2 (P2), his father (P4), and mother (P3) were lysed and evaluated by Western blot analysis for Coronin 1A and Myosin IIA, which was used as a loading control. Equal protein amounts were loaded into each lane as measured by quantitative colormetric protein assay. The density of Coronin 1A protein expression was quantified relative to the Myosin IIA loading control and expressed below each lane as normalized value relative to the healthy donor control
Discussion
We uncovered the rare Coronin-1A deficiency from WES genomic studies and Sanger confirmation that documented the trans localization and compound heterozygous CORO1A mutations in our two affected siblings, with a novel exon 11 variant identified, inherited from the mother. The paternal exon 4 Louisiana variant was previously found to represent a null allele that manifested as SCID with intolerance to varicella vaccine [23, 24]. The two nucleotide deletions in genomic position g.30197968-30197969 (hg19) caused a frameshift at codon 83 (proline substituted with arginine) that, after 10 codons, ended in a premature stop, and the shortened, aberrant mRNA transcript likely would be quickly eliminated by nonsense-mediated mRNA decay (NMD). By contrast, the nucleotide deletion of cytidine in position g.30199693 (hg19) in exon 11 (NM_001193333) caused a frameshift from codon 360 and the further 44 codons were aberrantly transcribed before the premature stop codon at the junction between the C-terminal extension and the coiled coil (cc) domain. Interestingly, the exon 11 mutation is located within a loop between the propeller and the Unique region of the C-terminal extension (CE) (Fig. 7a). This loop is highly conserved across species (Fig. 7b), and is responsible for stabilization of Coronin-1A protein's 3-dimensional-propeller structure. [39, 50]. The novel mutation's localization is illustrated in the murine model of Coronin-1A (Fig. 7c). The available murine protein mode 2AQ5 did not include the cc domain. A functional cc domain is required for the Coronin-1A homotrimerization [51, 52], which is indispensable for the Coronin-1A's downstream activity since the cc domain contains the binding site for F-actin. Thus, in the event of a NMD escape with transient expression of a truncated Coronin-1A protein (when of a destabilized propeller structure and lacking the cc domain), Coronin-1A would be predicted to occur as a monomer only, rendering F-actin defective for binding and its downstream action.
Fig. 7.
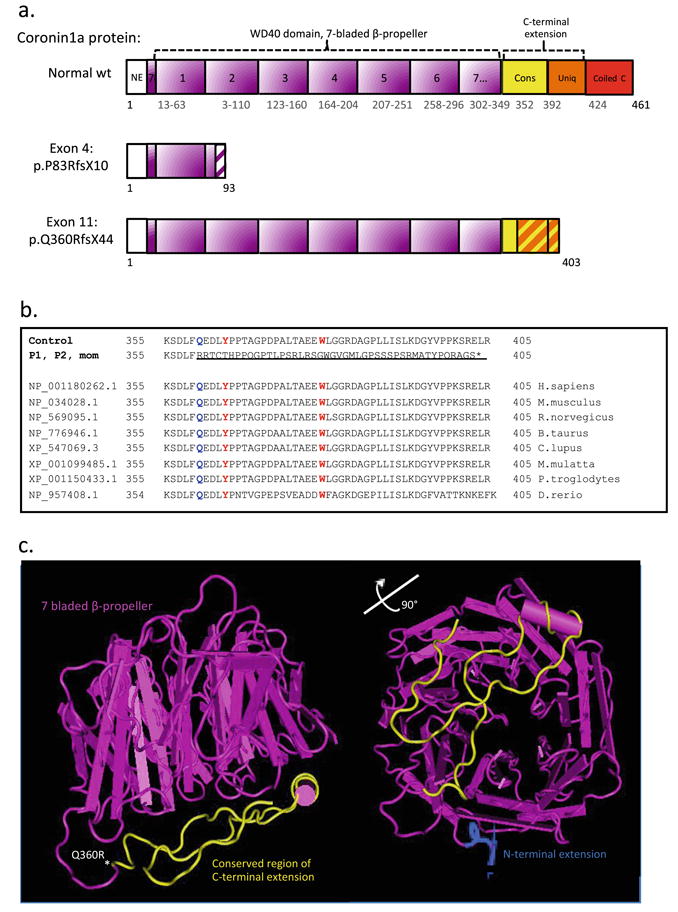
The predicted protein product compared to the normal structure and amino acid sequence of wild type (wt) Coronin-1A protein. a Predicted schematic protein structure of mutant versus wt Coronin-1A. Abbreviations: Coiled c, Coiled coil domain; Cons, conserved region of the C-terminus extension; NE, N-terminal extension; Uniq, Unique region of the C-terminal extension. b Multiple sequence alignment showing the conservation across species of CORO1A's C-terminal extension domain. The aberrant transcript caused by the exon 11 mutation is shown and underlined. The site for the first amino acid substitution (Q360R) is marked in blue, the two sites (Y366 and W379) important for stabilization of the propeller structure, marked in yellow, and * shows location of the stop codon. c Murine Model Coronin-1A, 2AQ5, visualized in Cn3D, version 4.32. This 3-dimensional murine 2AQ5 model includes parts (amino acids 8–10) of N-terminal (blue), the whole WD40 domain with the 7-bladed beta propeller (pink) and main parts of the C-terminal extension, but not the coiled coil domain. The exon 11 mutation is marked * at the first frame shifted amino acid (Q360R) and the whole frame shifted region (amino acids 360–403) in the C-terminal extension is marked in yellow. The open access wt murine 3D model is available at http://www.ncbi.nlm.nih.gov/Structure/CN3D/cn3d.shtml
The first reported CORO1A patient with two null mutations was by Shiow et. al. and the child presented with a severe SCID clinical phenotype. In the second CORO1A kindred, reported by Moshous et. al, three siblings were homozygous for the missense mutation and had features consistent with both CID and SCID. Our CORO1A patients shared selected immune and clinical findings with these reports, yet also had novel phenotypic properties not previously reported such as the even later, insidious presentations with the unusual mucocutaneous-immunodeficiency syndrome and shortened telomeres not before associated with Coronin-1A deficiency. Moreover, we found abnormalities in also B and NK cells in the face of normal percentages of total CD3+ T-cells, revealed as a skewed TCR with absence of the naive T-cell subpopulation in the affected siblings; the parents had subclinical mild immunodeficiencies isolated to NK cells for the mother and T-cells for the father, of undetermined significance. Additionally we confirmed absence of Coronin-1A protein expression in the affected (P2) and found presence in both parents (P3 and P4). Since our patients were found to be compound heterozygous for one null mutation, it may have been possible that the novel exon 11 variant could potentially confer unique disease-modifying factors responsible for the immune and clinical phenotypes we observed in this CORO1A kindred.
As found in our kindred and in the two existing reports, the Coronin-1A deficiency immune signature appears to be a profoundly low to absent CD4+CD45RA+ naïve T-cell subpopulation with a preponderance of DN T-cells defined as ϒδ T-cells, a subpopulation described as limited in diversity as compared to the a αβ-heterodimeric TCR. In addition, CD19+CD27+ memory B cells can be very low or absent and NK cells can be abnormally low. Yet, normal levels may be found for total IgG, specific humoral memory, percentages of total CD3+ T-cells and antigen-induced T-cell lymphoproliferation.. An elevated serum IgE, also seen, may represent an integral part of the Coronin-1A deficiency syndromes or may be an atopy overlap.
Remarkably, live viral vaccines were tolerated (varicella and measles-mumps-rubella) in both of our CORO1A-affected siblings. Classically in all SCID types, the result of exposure to live viral vaccines is life-threatening in severe T-cell defects [53]. However, tolerance to inadvertent live-vaccine exposure has been seen in patients with deletion 22q11.2 as partial DiGeorge Syndrome, who did not have absence of naïve T-cells but who did have mild T-cell impairments [54]. Despite the severe naïve T-cell and memory B-cell lymphopenias found in our affected siblings with Coronin-1A deficiency their clinical presentations were not typical for SCID and more so consistent with a CID or a hypomorphic SCID given their clinical onset as pre-teenagers and manifestations of the chronic bronchiectasis and lymphoma susceptibility. In the CORO1A mutation cases published to date, when lymphomas did develop as EBV-associated, they were severely lifeshortening whether the presentation of Coronin-1A deficiency was as SCID and CID. EV-HPV, particularly with types 5 and 17, plays a role in malignant transformation risks [55-59], but HPV-driven cancers were not seen in our patients. However, with one of the shared variants in STAT2 or STAT5A identified in our affected siblings, a malignancy risk was considered since somatic occurring variants in these genes are linked to apoptosis resistance and reported by others in lymphomas [60–62]. But, functional testing of the consequences of the particular STAT5A variant in our CORO1A kindred did not reveal any secondary transcriptional changes compared to normal controls (Warren Leonard NIH/NHLBI). Thus, malignant susceptibility and the EBV-induced lymphomas observed in Coronin-1A deficient patients may be partially explained by the impaired lymphocyte integrity associated with abnormal or absent Coronin-1A.
In Wiskott-Aldrich Syndrome protein (WASp) immunodeficiency disorders, F-actin and Arp2/3 have been determined as essential to the lymphocyte cytoskeleton and functional immune synapse, yet the mechanisms regulating these physiologic functions are not exactly understood [63–66]. In Coronin-1A deficiency, F-actin and Arp2/3 may also change immune cell morphology and function with direct consequences on T as well as B and NK cellular development and survival, with potential for like mechanisms to those found in WASp, particularly related to cellular communications critical to immune function [65, 66] and may be linked to the susceptibility to mycobacterial organisms. [35, 67]. The regulatory mechanisms of lymphocyte production, maturation and migration connected to immune function during cellular development when faced with F-actin distortion in Coronin 1A-deficiency have yet to be explained [68–70] as well as the impact of these changes on micro-organism susceptibility and lymphoma risks.
Before CORO1A was a known etiology, we had pursued specific immunodeficiency diagnoses, all of which were not proven. In particular, we highly considered known mutations: in EVER1/EVER2 given the disseminated HPV-EV [55–59]; CXCR4, which results in WHIM (warts, hypogammaglobulinemia, infections and myelokathexis) syndrome and may have naïve T-cell abnormalities [71–73]; DOCK8, with viral infections and an immunodeficiency with elevated IgE [74]; inherited MST1 deficiency and RHOH defects, with increased EV-HPV susceptibility and T-cell defects[75, 76]; hypomorphic mutant forms of SCID, particularly due to defects in RAG1/RAG2 and Omenn syndrome; and immune defects that impact STAT function. [14, 21, 22, 77]. Interferon-gamma/IL-12 pathway defects, including those of low penetrance, and NFkB immune defects known for susceptibility to mycobacterial and viral diseases, were also examined. [78–80]. The shortened telomere findings drove further analyses, especially in the absence of confirming Dyskeratosis Congenita (DC). Although we were subsequently unable to determine definitively whether the very short telomeres (Fig. 3) tracked with the Coronin-1A deficiency state, inspection of P2's WES variant file did not reveal any potentially or clearly deleterious variants among genes previously implicated in telomere biology (81–83). It is accepted that telomere length <1st percentile in the lymphocyte populations in telomere length flow FISH assays is highly specific and sensitive for the telomere biology disorder DC [81–84]. However, we proved absence of Coronin-1A protein in our CORO1A affected siblings who did not have DC mutations, and our CORO1A affected siblings did not manifest the mucocutaneous triad specific to DC of oral leukoplakia, nail dystrophy and abnormal reticulated skin pigmentation or bone marrow failure, which are highly penetrant in DC. Whereas the increased cancer risk has been reported in patients with DC, neither cancer types that occurred in our P1 (HL or DLBCL) were prevalent in well-studied DC cohorts [84–86]. Furthermore, the immune defects in DC may be progressive, as in Hoyeraal-Hreidarsson Syndrome [87], but have not been defined as specific to a naïve T-cell lymphopenia [87–89]. Therefore, we suspect that CORO1A does not represent a new DC-associated gene, but rather that the abnormally short telomeres are likely related to the CORO1A mutations identified and the Coronin-1A deficient cells may have either altered telomere maintenance or accelerated shortening. Notably, a genome-wide yeast two-hybrid screen detected an interaction between Coronin-1A and Pot1 [90], a telomere-associated protein and a key regulator of telomerase, (the telomere replication enzyme [91]), raising the possibility that the Coronin-1A deficiency state may result in altered cellular localization of Pot1, which transits through the cytoplasm [92]. Alternatively, the F-actin interruptions observed in the context of a Coronin-1A deficiency may also affect filamentous chromosomal networks, and secondarily, telomere stability. The shortened telomere lengths may then represent clonal exhaustion in the setting of persistent stimulation on an impaired immune system when Coronin-1A is defective.
Although learning disorders were identified in the two previous reports, dysmorphic facial features were not mentioned as part of CORO1A-mutation immunodeficiencies as observed in P2 of our kindred, albeit mild The particular gene(s) responsible, when haploinsufficient, for the cognitive impairments, learning difficulties and autism spectrum disorders within the recurrent 16p11.2 microdeletion region, still remain to be determined. KCTD13 gene is the strongest candidate, since deletions and duplications of this gene cause macrocephaly and microcephaly in mice, resembling the human del16p11.2 and dup16p11.2 phenotype and persons with del16p11.2 with the reciprocal dup16p11.2 do have mild dysmorphic facial features [29, 93, 94]. Coronin-1A is highly expressed in microglia in the brain and may also play an instrument in the neurocognitive orchestration reflected by the dynamic properties of microglia [95]. However, no severe learning difficulties or social interaction problems were reported in our CORO1A kindred.
Conclusion
We report the first compound heterozygous CORO1A mutations with a novel exon 11 variant, identified by WES, and is now the third known Coronin-1A deficiency kindred. Patients with CORO1A mutations may present as SCID or CID clinical phenotypes and all lymphocyte lineages may be impaired, with naïve T-cell lymphopenia as the most severe immunologic hallmark, even when percentages of total CD3+ T- cells may be within or near normal range for age. Risks for bronchiectasis and lymphomas, exacerbated by EBV, are likely. A mucocutaneous-immunodeficiency syndrome, especially of EV-HPV, molluscum and HSV, and even leprosy, may be one of the primary clinical presentations in Coronin-1A deficiencies, and may not be apparent until pre-teen years in otherwise seemingly well-appearing pediatric patients who are described as atopic with recurrent sinopulmonary disease and a chronic dermatitis. Neurocognitive disorders may not be present in all Coronin-1A deficient patients, yet a range of dysmorphic and developmental disorders may be possible. Shortened telomere length in lymphocytes and granulocytes may be found not only in DC but also in Coronin-1A deficiency, however, the relationship and its interface with the described CORO1A immunodeficiency is a point for further analyses.
Newfound Coronin-1A deficiency associations will continue to surface as more kindred with CORO1A mutations are identified, especially with the application of new-generation deep sequencing technology [96, 97]. We now have validated and expanded the important distinguishing immunologic and clinical characteristics that define CORO1A mutations and reveal an association of very short telomere lengths in Coronin-1A deficiency. Further analyses of the role of Coronin 1A [98] on immune cell development and function and the immunogenetics of Coronin-1A deficiency states are necessary to further explain the impact of differing CORO1A mutations on the affected and on carriers.
Acknowledgments
This original work is in heartfelt memory of P1 and is dedicated to our CORO1A kindred. We graciously thank each of the medical and social service professionals of Texas Children's Hospital and Baylor College of Medicine who contributed to the excellence in patient care, support, counseling and well-being of this CORO1A family and who continue to give their expertise and compassion on behalf of the affected siblings and their family members. Special appreciation is given to Drs. Robert Krance and Caridad Martinez, along with their team, of the Bone Marrow Transplantation Service of Texas Children's Hospital-Baylor College of Medicine who are leading the transplantation efforts for the surviving sibling of this CORO1A kindred. With appreciation to Warren Leonard, MD, NIH Distinguished Investigator of the Laboratory of Molecular Immunology, National Institutes of Health, and to Karen Eldin, MD, Department of Pathology and Immunology of Baylor College of Medicine at Texas Children's Hospital. Collaboratively, the co-authors respectfully acknowledge all patients with primary immunodeficiencies and their families and express gratitude for the privilege of serving as their clinicians and scientists. This work was funded in part by Immunology, Allergy and Rheumatology of Texas Children's Hospital-Department of Pediatrics of Baylor College of Medicine, the David Center and the Jeffrey Modell Diagnostic and Research Center to EMM, LMN and JSO the National Human Genome Research Institute, NIH Grant 5U54HG006542 to JRL and the Cancer Prevention Research Institute of Texas Grant RP120076 to AAB.
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
Contributor Information
Asbjorg Stray-Pedersen, Allergy & Immunology, Section of Immunology, Allergy and Rheumatology, Texas Children's Hospital, Houston, TX, USA, Baylor-Hopkins Center for Mendelian Genomics of the Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA, Department of Medical Genetics, Oslo University Hospital, Oslo, Norway, Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA, Human Genome Sequencing Center of Baylor College of Medicine, Houston, TX, USA.
Emmanuelle Jouanguy, St. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY, USA, Laboratory of Human Genetics of Infectious Diseases, Necker Branch, University Paris Descartes and Inserm, Imagine Foundation, Paris, FranceEU.
Amandine Crequer, St. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY, USA, Laboratory of Human Genetics of Infectious Diseases, Necker Branch, University Paris Descartes and Inserm, Imagine Foundation, Paris, FranceEU.
Alison A. Bertuch, Hematology/Oncology, Texas Children's Hospital, Houston, TX, USA, Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA
Betty S. Brown, Allergy & Immunology, Section of Immunology, Allergy and Rheumatology, Texas Children's Hospital, Houston, TX, USA, Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA, Clinical Immunology Laboratory, Section of Immunology, Allergy and Rheumatology, Texas Children's Hospital/Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA
Shalini N. Jhangiani, Baylor-Hopkins Center for Mendelian Genomics of the Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA, Human Genome Sequencing Center of Baylor College of Medicine, Houston, TX, USA
Donna M. Muzny, Baylor-Hopkins Center for Mendelian Genomics of the Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA, Human Genome Sequencing Center of Baylor College of Medicine, Houston, TX, USA
Tomasz Gambin, Baylor-Hopkins Center for Mendelian Genomics of the Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA, Human Genome Sequencing Center of Baylor College of Medicine, Houston, TX, USA.
Hanne Sorte, Department of Medical Genetics, Oslo University Hospital, Oslo, Norway, Human Genome Sequencing Center of Baylor College of Medicine, Houston, TX, USA.
Ghadir Sasa, Hematology/Oncology, Texas Children's Hospital, Houston, TX, USA, Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA.
Denise Metry, Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA, Department of Dermatology, Texas Children's Hospital, Houston, TX, USA.
Judith Campbell, Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA, Infectious Diseases, Texas Children's Hospital, Houston, TX, USA.
Marianna M. Sockrider, Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA, Pulmonary Medicine, Texas Children's Hospital, Houston, TX, USA
Megan K. Dishop, Department of Pathology, University of Colorado, Denver, CO, USA, Department of Pathology, Texas Children's Hospital, Houston, TX
David M. Scollard, National Hansen's Disease Programs, Baton Rouge, LA, USA
Richard A. Gibbs, Baylor-Hopkins Center for Mendelian Genomics of the Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA, Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA, Human Genome Sequencing Center of Baylor College of Medicine, Houston, TX, USA
Emily M. Mace, Allergy & Immunology, Section of Immunology, Allergy and Rheumatology, Texas Children's Hospital, Houston, TX, USA, Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA, Center for Human Immunobiology, Texas Children's Hospital-Baylor College of Medicine, Houston, TX, USA
Jordan S. Orange, Allergy & Immunology, Section of Immunology, Allergy and Rheumatology, Texas Children's Hospital, Houston, TX, USA, Hematology/Oncology, Texas Children's Hospital, Houston, TX, USA, Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA, Clinical Immunology Laboratory, Section of Immunology, Allergy and Rheumatology, Texas Children's Hospital/Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA, Center for Human Immunobiology, Texas Children's Hospital-Baylor College of Medicine, Houston, TX, USA
James R. Lupski, Baylor-Hopkins Center for Mendelian Genomics of the Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA, Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA, Human Genome Sequencing Center of Baylor College of Medicine, Houston, TX, USA
Jean-Laurent Casanova, St. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY, USA, Laboratory of Human Genetics of Infectious Diseases, Necker Branch, University Paris Descartes and Inserm, Imagine Foundation, Paris, FranceEU.
Lenora M. Noroski, Email: Lnoroski@bcm.edu, Allergy & Immunology, Section of Immunology, Allergy and Rheumatology, Texas Children's Hospital, Houston, TX, USA, Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA, Clinical Immunology Laboratory, Section of Immunology, Allergy and Rheumatology, Texas Children's Hospital/Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA.
References
- 1.Notarangelo LD. Primary immunodeficiencies. J Allergy Clin Immunol. 2010;125:S182–94. doi: 10.1016/j.jaci.2009.07.053. [DOI] [PubMed] [Google Scholar]
- 2.Casanova JL, Abel L. Primary immunodeficiencies: a field in its infancy. Science. 2007;317:617–9. doi: 10.1126/science.1142963. [DOI] [PubMed] [Google Scholar]
- 3.Kalman L, Lindegren M, Kobrynski L, Vogt R, Hannon H, Howard JT, et al. Mutations in selected genes required for Tcell development: IL7R, CD45, IL2R gamma chain, JAK3, RAG1, RAG2, ARTEMIS and ADA and severe combined immunodeficiency. Genet Med. 2004;6:16–26. doi: 10.1097/01.GIM.0000105752.80592.A3. [DOI] [PubMed] [Google Scholar]
- 4.Buckley RH. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu Rev Immunol. 2004;22:625–55. doi: 10.1146/annurev.immunol.22.012703.104614. doi:10.1146/annurev.immunol.22. 012703.104614. [DOI] [PubMed] [Google Scholar]
- 5.Patel NC, Chinen J, Rosenblatt HM, Hanson IC, Krance RA, Paul ME, et al. Outcomes of patients with severe combined immunodeficiency treated with hematopoietic stem cell transplantation with and without preconditioning. J Allergy Clin Immunol. 2009;124(5):1062–9. e1–4. doi: 10.1016/j.jaci.2009.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dvorak C, Cowan MJ, Logan BR, Griffith L, Puck J, Notarangelo LD, et al. The natural history of children with severe combined immunodeficiency disease (SCID): the first fifty patients of the primary immune deficiency treatment consortium (PIDTC) prospective study 6901. Biol Blood Marrow Transplant. 2013;19(2):S161–2. doi: 10.1016/j.bbmt.2012.11.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Antoine C, Müller S, Cant A, Cavazzana-Calvo M, Veys P, Vossen J, et al. Long-term survival and transplantation of haemopoietic stem cells for immunodeficiencies: report of the European experience 1968-1999. Lancet. 2003;361(9357):553–60. doi: 10.1016/S0140-6736(03)12513-5. [DOI] [PubMed] [Google Scholar]
- 8.Myers LA, Patel DD, Puck JM, Buckley RH. Hematopoietic stem cell transplantation for severe combined immunodeficiency in the neonatal period leads to superior thymic output and improved survival. Blood. 2002;99(3):872–8. doi: 10.1182/blood.V99.3.872. [DOI] [PubMed] [Google Scholar]
- 9.Noguchi M, Yi H, Rosenblatt HM, Filipovich AH, Adelstein S, Modi WS, et al. Interleukin-2 receptor gamma chain mutation results in x-linked severe combined immunodeficiency in humans. Cell. 1993;73(1):147–57. doi: 10.1016/0092-8674(93)90167-O. [DOI] [PubMed] [Google Scholar]
- 10.Puck JM, Deschenes SM, Porter JC, Dutra AS, Brown CJ, Willard HF, et al. The interleukin-2 receptor gamma chain maps to Xq13.1 and is mutated in X-linked severe combined immunodeficiency, SCIDX1. Hum Mol Genet. 1993;2(8):1099–104. doi: 10.1093/hmg/2.8.1099. [DOI] [PubMed] [Google Scholar]
- 11.Macchi P, Villa A, Giliani S, Sacco MG, Frattini A, Porta F, et al. Mutations of JAK-3 gene in patients with autosomal severe combined immune deficiency (SCID) Nature. 1995;377:65–8. doi: 10.1038/377065a0. [DOI] [PubMed] [Google Scholar]
- 12.Dadi JK, Simon AJ, Roifman CM. Effect of CD3δ deficiency on maturation of α/β and γ/δ t-cell lineages in severe combined immunodeficiency. N Engl J Med. 2003;349(19):1821–8. doi: 10.1056/NEJMoa031178. [DOI] [PubMed] [Google Scholar]
- 13.Schwarz K, Gaus GH, Ludwig L, Pannicke U, Li Z, Linderner D, et al. RAG mutations in human B cell-negative SCID. Science. 1996;274(5284):97–9. doi: 10.1126/science.274.5284.97. [DOI] [PubMed] [Google Scholar]
- 14.Schuetz C, Huck K, Gudowius S, Megahed M, Feyen O, Hubner B, et al. An immunodeficiency disease with rag mutations and granulomas. N Engl J Med. 2008;358:2030–8. doi: 10.1056/NEJMoa073966. [DOI] [PubMed] [Google Scholar]
- 15.Al-Herz W, Bousfiha A, Casanova JL, Chapel H, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol. 2011;2(54):1–26. doi: 10.1111/j.1365-2249.2012.04561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Immunodeficiency Foundation. www.primaryimmune.org.
- 17.Puck JM. Laboratory technology for population-based screening for severe combined immunodeficiency in neonates: the winner is T-cell receptor excision circles. JACI. 2012;129(3):607–16. doi: 10.1016/j.jaci.2012.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lindegren ML, Kobrynski L, Rassmussen SA, Moore CA, Grosse SD, Vanderford ML, et al. Applying public health strategies to primary immunodeficiency diseases: a potential approach to genetic disorders. MMWR Recomm Rep Morb Mortal Wkly Rep Recomm Rep/Centers Dis Control. 2004;53(RR-1):1–29. [PubMed] [Google Scholar]
- 19.Chinen J, Shearer WT. Advances in basic and clinical immunology in 2010. J Allergy Clin Immunol. 2011;127(2):336–41. doi: 10.1016/j.jaci.2010.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Byun M, Abhyankar A, Lelarge V, Plancoulaine S, Palanduz A, Telhan L, et al. Whole-exome sequencing-based discovery of STIM1 deficiency in a child with fatal classic kaposi sarcoma. JEM. 2010;207(11):2307–12. doi: 10.1084/jem.20101597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Avila EM, Uzel G, Hsu A, Milner JD, Turner ML, Pittalupa S, et al. Highly variable clinical phenotypes of hypomorphic RAG1 mutations. Pediatrics. 2010;126:e1248. doi: 10.1542/peds.2009-3171. [DOI] [PubMed] [Google Scholar]
- 22.De Villartay JP, Lim A, Al-Mousa H, Dupont S, Dechanet-Mervill J, Coumau-Gatbois E, et al. A novel immunodeficiency associated with hypomorphic RAG1 mutations and CMV infection. J Clin Investig. 2005;115(11):3291–9. doi: 10.1172/JCI25178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shiow LR, Roadcap DW, Paris K, Watson SR, Grigorova IL, Lebet T, et al. The actin regulator coronin-1A is mutated in a thymic egress deficient mouse strain and in a T-B+NK+SCID patient. Nat Immunol. 2008;9(11):1307–15. doi: 10.1038/ni.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shiow LR, Paris K, Akan MC, Cyster JG, Sorensen RU, Puck JM. Severe combined immunodeficiency (SCID) and attention deficit hyperactivity disorder (ADHD) associated with a coronin-1A mutation and a chromosome 16p11.2 deletion. Clin Immunol. 2009;131(1):24–30. doi: 10.1016/j.clim.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moshous D, Martin E, Carpentier W, Lim A, Callebaut I, Canioni D, et al. Whole-exome sequencing identifies coronin-1A deficiency in 3 siblings with immunodeficiency and EBV-associated B-cell lymphoproliferation. J Allergy Clin Immunol. 2013;131(6):1594–603. doi: 10.1016/j.jaci.2013.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar RA, KaraMohamed S, Sudi J, Conrad DF, Brune C, Badner JA, et al. Recurrent 16p11.2 microdeletions in Autism. Hum Mol Genet. 2008;17:628–38. doi: 10.1093/hmg/ddm376. [DOI] [PubMed] [Google Scholar]
- 27.Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, et al. Structural variations of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82:477–88. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, et al. Association between microdeletion and microduplication at 16p11.2 and Autism. N Engl J Med. 2008;358(7):667–75. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 29.Shinawi M, Liu P, Kang SH, Shen J, Belmont JW, Scott DA, et al. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioral problems, dysmorphism, epilepsy and abnormal head size. J Med Genet. 2010;47(5):332–41. doi: 10.1136/jmg.2009.073015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eckert C, Hammesfahr B, Kollmar M. A holistic phylogeny of the coronin gene family reveals an ancient origin of the tandem-Coronin, defines a new subfamily and predicts protein function. BMC Evol Biol. 2011;11(268):1–17. doi: 10.1186/1471-2148-11-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Foger N, Rangell L, Dnailenko DM, Chan AC. Requirement for coronin 1 in T lymphocyte trafficking and cellular homeostasis. Science. 2006;313:839–42. doi: 10.1126/science.1130563. [DOI] [PubMed] [Google Scholar]
- 32.Mueller P, Massner J, Jayachandran R, Combaluzier B, Albrecht I, Gatfield J, et al. Regulation of T cell survival through coronin-1-mediated generation of inositol-1,4,5-trisphosphate and calcium mobilization after T cell receptor triggering. Nat Immunol. 2008;9(4):424–31. doi: 10.1038/ni1570. [DOI] [PubMed] [Google Scholar]
- 33.Coronin 1A. CORO1A Online Mendelian Inheritance in Man. http://omim.org/entry/605000.
- 34.Kaminski S, Hermann-Kleiter N, Meisel M, Thuille N, Cronin S, Hara H, et al. Coronin-1A is an essential regulator of the TGF beta receptor/SMAD3 signaling pathway in Th17 CD4+ T cells. J Autoimmun. 2011;37(3):198–208. doi: 10.1016/j.aut.2011.05.018. [DOI] [PubMed] [Google Scholar]
- 35.Ferrari G, Lange H, Naito M, Pieters J. A coat protein on phagosomes involved in the intracellular survival of mycobacteria. Cell. 1999;97:435–47. doi: 10.1016/S0092-8674(00)80754-0. [DOI] [PubMed] [Google Scholar]
- 36.Pieters J, Muller P, Jayachandran R. On guard: coronin proteins in innate and adaptive immunity. Nat Rev Immunol. 2013;13:510–18. doi: 10.1038/nri3465. [DOI] [PubMed] [Google Scholar]
- 37.Sl L, Needham KM, May JR, Nolen BJ. Mechanism of a concentration-dependent switch between activation and inhibition of Arp2/3 complex by coronin. J Biol Chem. 2011;286:17039–46. doi: 10.1074/jbc.M111.219964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bainbridge MN, Wang M, Wu Y, Newsham I, Muzny DM, Jefferies JL, et al. Targeted enrichment beyond the consensus coding DNA sequence exome reveals exons with higher variant densities. Genome Biol. 2011;12(7):R68. doi: 10.1186/gb-2011-12-7-r68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H, Durbin R. Fast and accurate short read alignment with burrowswheeler transform. Bioinformatics. 2009;25(14):1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map (SAM) format and SAM tools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DePristo M, Banks E, Poplin R, Garimella K, Maguire J, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Challis D, Yu J, Evani US, Jackson AR, Paithankar S, Coarfa C, et al. An integrative variant analysis suite for whole exome next-generation sequencing data. BMC Bioinforma. 2012;13:8. doi: 10.1186/1471-2105-13-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–8. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bainbridge MN, Wiszniewski W, Murdock DR, Friedman J, Gonzaga-Jauregui C, Newsham I, et al. Sci Transl Med. 2011;3(87):87re3. doi: 10.1126/scitranslmed.3002243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boon PM, Bacin CA, Shaw CA, Eng PA, Hixon PM, Pursley AN, et al. Detection of clinically relevant exonic copy-number changes by array CGH. Hum Mutat. 2010;31(12):1326–42. doi: 10.1002/humu.21360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keerthikumar S, Raju R, Kandasamy K, Hijikata A, Ramabadran S, Balakrishnan L, et al. RAPID: resource of Asian primary immunodeficiency diseases. Nucleic Acids Res. 2009;37(database issue):D863–7. doi: 10.1093/nar/gkn682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Orange JS, Ghanta SR, Mace EM, Maru S, Rak GD, Sanborn KB, et al. IL2 induces a WAVE2-dependent pathway for actin reorganization that enables WASp-independent human NK cell function. J Clin Invest. 2011;121(4):1535–48. doi: 10.1172/JCI44862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shearer WT, Rosenblatt HM, Gelman RS, Oyomopito R, Plaeger S, Stiehm ER, et al. Lymphocyte subsets in healthy children from birth through 18 years of Age: the pediatric AIDS clinical trials group P1009 study. J Allergy Clin Immunol. 2003;112:973–80. doi: 10.1016/j.jaci.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 49.Scollard DM, Adams LB, Gillis TP, Krahenbuhl JL, Truman RW, Williams DL. The continuing challenges of leprosy. Clin Microbiol Rev. 2006;19(2):338–81. doi: 10.1128/CMR.19.2.338-381.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McKenna A, Hanna M, Banks E, Sivachendo A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit : a mapreduce franmework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gatfield J, Albrecht I, Zanolari B, Steinmetz MO, Pieters J. Association of the leukocyte plasma membrane with the actin cytoskeleton through coiled coil-mediated trimeric coronin 1 molecules. Mol Biol Cell. 2005;16(6):2786–98. doi: 10.1092/mbc.E05-01-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Appleton BA, Wu P, Wiesmann C. The crystal structure of murine coronin-1A: a regulator of actin cytoskeletal dynamics in lymphocytes. Structure. 2006;14:87–96. doi: 10.1016/j.str.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 53.Patel NC, Hertel PM, Estes MK, de la Morena M, Petru AM, Noroski LM, et al. Vaccine-acquired rotavirus in infants with severe combined immunodeficiency. NEJM. 2010;362:314–9. doi: 10.1056/NEJMoa0904485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moylett EH, Wasan AN, Noroski LM, Shearer WT. Live Viral Vaccines in patients with Partial DiGeorge Syndrome: Clinical Experience and Cellular Immunity. Clin Immunol. 2004;112(1):106–112. doi: 10.1016/j.clim.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 55.Orth G, Favre M, Majewski S, Jablonska K, Antonsson A, Forslund O, et al. Epidermodysplasia verruciformis defines a subset of cutaneous human papillomaviruses. J Virol. 2001;75(10):4952–3. doi: 10.1128/JVI.75.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Orth G. Host defences against human papillomaviruses : lessons from epidermodysplasia verruciformis. Curr Top Microbiol Immunol. 2008;321:59–83. doi: 10.1007/978-3-540-75203-5_3. [DOI] [PubMed] [Google Scholar]
- 57.Orth G. Genetics of epidermodysplasia verruciformis : insights into host defense against papillomaviruses. Semin Immunol. 2006;18(6):362–74. doi: 10.1016/j.smim.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 58.Kremsdorf D, Favre M, Jablonska S, Obalek S, Rueda LA, Lutzner MA, et al. Molecular cloning and characterization of the genomes of nine newly recognized human papillomavirus types associated with epidermodysplasia verruciformis. J Virol. 1984;52(3):1013–8. doi: 10.1128/jvi.52.3.1013-1018.1984. http://jvi.asm.org/content/52/3/1013.short. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ramoz N, Rueda LA, Bouadjar B, Montoya LS, Orth G, Favre M. Mutations in two adjacent novel genes are associated with epidermodysplasia verruciformis. Nat Genet. 2002;32:579–81. doi: 10.1038/ng1044. [DOI] [PubMed] [Google Scholar]
- 60.Butterbach K, Beckmann L, de Sanjose S, Benavente Y, Becker N, Foretova L, et al. Association of JAK-STAT pathway related genes with lymphoma risk: results of a european case–control study (EpiLymph) Br J Haematol. 2011;153(3):318–33. doi: 10.1111/j.1365-2141.2011.0832.x. [DOI] [PubMed] [Google Scholar]
- 61.Abraham N, Ma MC, Snow JW, Miners MJ, Herndier BG, Goldsmith MA. Haploinsufficiency identifies STAT5 as a modifier of IL-7-induced lymphomas. Oncogene. 2005;24(33):5252–7. doi: 10.1038/sj.onc.1208726. [DOI] [PubMed] [Google Scholar]
- 62.Romero-Weaver AL, Wang HW, Steen HC, Scarzello AJ, Hall VL, Sheikh R, et al. Resistance to IFN-alpha-induced apoptosis is linked to a loss of STAT2. Mol Cancer Res. 2010;8(1):80–92. doi: 10.1158/1541-7786.MCR-08-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Veltman DM, Insall RH. WASP family proteins: their evolution and its physiological implications. Mol Biol Cell. 2010;21:2880–93. doi: 10.1091/mbc.E10-04-0372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Millard TH, Sharp SJ, Machesky LM. Millard TH, Sharp SJ, Machesky LM. Signaling to actin assembly via the WASP-family proteins and the Arp2/3 complex. Biochem J. 2004;380:1–17. doi: 10.1042/BJ20040176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chereau D, Kerff F, Graceffa P, Grabarek Z, Langsetmo K, Dominguez F. Actin-bound structures of Wiskott-aldrich syndrome protein (WASP)-homology domain 2 and the implications for filament assembly. PNAS. 2005;102(46):166644–9. doi: 10.1073/pnas.0507021102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Orange J. Formation and function of the lytic NK-cell immunological Synapse. Nat Rev Immunol. 2008:713–725. doi: 10.1038/nri2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ghosh A, Touski S, Bhatttacharya D, Samuchiwal SK, Bhalla K, Tharad M, et al. Expression of the ARPC4 subunit of human Arp2/3 severely affects mycobacterium tuberculosis growth and suppresses immunogenic response in murine macrophages. PLoS One. 2013;8(e69949):1–12. doi: 10.1372/journal.pone.0069949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mugnier B, Nal B, Verthuy C, Boyer C, Lam D, Chasson L. Coronin-1A links cytoskeleton dynamics to TCR alpha beta-induced cell signaling. PLoS One. 2008;3:e3467. doi: 10.1371/journal.pone.0003467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Weinreich MA, Hogquist KA. Thymic emigration: when and how T cells leave home. J Immunol. 2008;181:2265–70. doi: 10.4049/jimmunol.181.4.2265. www.jimmunol.org/content/181/4/2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weill JC, Weller S, Reynaud CA. Human marginal zone B cells. Annu Rev Immunol. 2009;27:267–85. doi: 10.1146/annurev.immunol.021908.132607. [DOI] [PubMed] [Google Scholar]
- 71.Kawai T, Malech HL. WHIM syndrome: congenital immune deficiency disease. Curr Opin Hematol. 2009;16(1):20–6. doi: 10.1097/MOH.0b013e32821ac557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tarzi MD, Jenner M, Hattotuwa K, Faruqi AZ, Diaz GA, Longhurst HJ. Sporadic case of warts, hypogammaglobulinemia, immunodeficiency and myelokathexis syndrome. J All Clin Immunol. 2005;116(5):1101–5. doi: 10.1016/j.jaci.2005.08.040. [DOI] [PubMed] [Google Scholar]
- 73.Gulino AV, Moratto D, Sozzani S, Cavadini P, Otero K, Tassone L, et al. Altered leukocyte response to CXCL12 in patients with warts, hypogammaglobulinemia, infections myelokathexis (WHIM) syndrome. Blood. 2004;104:444–52. doi: 10.1182/blood-2003-10-3532. [DOI] [PubMed] [Google Scholar]
- 74.Qian Z, Davis JC, Lamborn IT, Freeman AF, Jing H, Favreau AJ, et al. Combined immunodeficiency associated with DOCK8 mutations. New Engl J Med. 2009;361:2046–55. doi: 10.1056/NEJMoa0905506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Crequer A, Troeger A, Patin E, Ma CS, Picard C, Pedergnana V, et al. Human RHOH deficiency causes T cell defects and susceptibility to EV-HPV infections. J Clin Invest. 2012;122(9):3239–47. doi: 10.1172/JCI162949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Crequer A, Picard C, Patin E, D'Amico A, Abhyankar A, Munzer M, Debre M, Zhang SY, de Saint-Basile G, Fischer A, Abel L, Orth G, Casanova JL, Jouanguy E. Inherited MST1 deficiency underlies susceptibility to EV-HPV infections. PLOS ONE. 2012;(7):1–7. doi: 10.1371/journal/pone.0044010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Casanova JL, Holland SM, Notarangelo LD. Inborn errors of human JAKs and STATs. Immunity. 2012;36:515–28. doi: 10.1016/j.immuni.2012.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fieschi C, Dupuis S, Catherinot E, Feinberg J, Bustamonte J, Breiman A, et al. Low penetrance, broad resistance, and favorable outcome of inerleukin 12 receptor β1 deficiency: medical and immunological implications. J Exp Med. 2003;197(4):527–35. doi: 10.1084/jem.20021769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Feinberg J, Fieschi C, Doffinger R, Feinberg M, Beclerc T, Boisson-Dupuis S, et al. Bacillus Calmette Guerin triggers the IL-12/IFNgamma axis by an IRAK4 and NEMO-dependent Non-cognate interaction between monocytes, NK and T lymphocytes. Eur J Immunolo. 2004;34:3276–84. doi: 10.1002/eji.200425221. [DOI] [PubMed] [Google Scholar]
- 80.Casanova JL, Abel L. Genetic dissection of immunity to mycobacteria: the human model. Annu Rev Immunol. 2002;20:581–620. doi: 10.1146/annurev.immunol.20.081501.125851. [DOI] [PubMed] [Google Scholar]
- 81.Alter BP, Baerlocher GM, Savage SA, Chanock SJ, Weksler BB, Willner JP, et al. Very short telomere length by flow fluorescence in situ hybridization identifies patients with dyskeratosis congenita. Blood. 2007;110:1439–47. doi: 10.1182/blood-2007-02-075598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Baerlocher GM, Vulto I, de Jong G, Lansdorp PM. Flow cytometry and FISH to measure the average length of telomeres (flow FISH) Nat Protoc. 2006;1:2365–76. doi: 10.1038/nprot.2006.263. [DOI] [PubMed] [Google Scholar]
- 83.Savage SA, Bertuch AA. The genetics and clinical manifestations of telomere biology disorders. Genet Med. 2010;12(12):753–64. doi: 10.1097/GIM.0b013e3181f415b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in dyskeratosis congenita. Blood. 2009;113:6549–6557. doi: 10.1182/blood-2008-12-192880. 2710915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yamada O, Ozaki K, Akiyama M, et al. JAK-STAT and JAK-PI3K-mTORC1 pathways regulate telomerase transcriptionally and posttranslationally in ATL cells. Mol CancerTher. 2012;11:1112–21. doi: 10.1158/1535-7163. [DOI] [PubMed] [Google Scholar]
- 86.Baykal C, Buyukbabani N, Kavak A. Dyskeratosis congenita associated with Hodgkin's disease. Eur J Dermatol. 1998;8:385–7. http://www.jle.com/en/revues/medecine/ejd/e-docs/00/01/88/6A/article.phtml. [PubMed] [Google Scholar]
- 87.Jyonouchi S, Forbes L, Ruchelli E, Sullivan KE. Dyskeratosis congenita: a combined immunodeficiency with broad clinical spectrum-a single center pediatric experience. Pediatr Allergy Immunol. 2011;22:313–9. doi: 10.1111/j.1399-3038.2010.01136.x. [DOI] [PubMed] [Google Scholar]
- 88.Berthet F, Tuchshmid P, Boltshauser E, Seger RA. The hoyeraalhreidarsson syndrome: don't forget the associated immunodeficiency. Eur J Pediatr. 1995;154:998. doi: 10.1007/BF01958649. [DOI] [PubMed] [Google Scholar]
- 89.Knudson M, Kulkarni S, Ballas ZK, Bessler M, Goldman F. Association of immune abnormalities with telomere shortening in autosomal-dominant dyskeratosis congenita. Blood. 2005;105:682–8. doi: 10.1182/blood-2004-04-1673. [DOI] [PubMed] [Google Scholar]
- 90.Wang J, Huo K, Ma L, Tang L, Li D, Huang X, et al. Toward an understanding of the protein interaction network of the human liver. Mol Syst Biol. 2011;7(536):1–10. doi: 10.1038/msb.2011.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nandakumar J, Cech TR. Finding the end: recruitment of telomerase to telomeres. Nat Rev Mol Cell Biol. 2013;14:69–82. doi: 10.1038/nrm3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chen LY, Liu D, Songyang Z. Telomere maintenance through spatial control of telomeric proteins. Mol Cell Biol. 2007;27(16):5898–909. doi: 10.1128/MCB.00603-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Angkustsiri K, Krakowiak P, Moghaddam B, Wardinsky T, Gardner J, Kalamkarian N, et al. Autism. 2011;15(6):746–60. doi: 10.1177/1362361310397620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Golzio C, Willer J, Talkowski ME, Oh EC, Taniguchi Y, Jacquemont S, et al. KCTD13 is a major driver of the mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature. 2012;485(7398):363–7. doi: 10.1038/nature11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ahmen Z, Shaw G, Sharma VP, Yang C, McGowan E, Dickeson DW. Actin-binding proteins coronin-1A are effective microglial markers for immunohistochemistry. J Histochem Cytochem. 2007;55(7):687–700. doi: 10.1369/jhc.6A7156.2007. [DOI] [PubMed] [Google Scholar]
- 96.Alcais A, Quintana-Murci L, Thaler DS, Schurr E, Abel L, Casanova JL. Life-threatening infectious diseases of childhood: single-gene inborn errors of immunity? Ann NY Acad Sci. 2010;1214:18–33. doi: 10.1111/j.1749-6621.2010.05834.x. [DOI] [PubMed] [Google Scholar]
- 97.Casanova JL, Abel L. The genetic theory of infectious diseases: a brief history and selected illustrations. Annu Rev Genom Hum Genet. 2013 doi: 10.1146/annurev-genom-091212-153448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Morgan RO, Fernandez MP. Molecular phylogeny and evolution of the coronin gene family. In: Clemen ChristophS, Eichinger Ludwig, Rybakin Vasily., editors. The Coronin family of proteins. Lands Bioscience and Springer Science+Business Media; 2008. Chapter 4. [DOI] [Google Scholar]