

Abstract
The development of rapid, low-cost and reliable diagnostic methods is crucial for the identification and treatment of many diseases. Screen-printed gold electrodes (Au/SPEs), coated with a ternary monolayer interface, involving hexanedithiol (HDT), a specific thiolated capture probe (SHCP), and 6-mercapto-1 hexanol (MCH) (SHCP/HDT/MCH) are shown here to offer direct and sensitive detection of nucleic acid hybridization events in untreated raw biological samples (serum, urine and crude bacterial lysate solutions). The composition of the ternary monolayer was modified and tailored to the surface of the Au/SPE. The resulting SHCP/HDT/MCH monolayer has demonstrated to be extremely useful for enhancing the performance of disposable nucleic acid sensors based on screen-printed electrodes. Compared to common SHCP/MCH binary interfaces, the new ternary self-assembled monolayer (SAM) resulted in a 10-fold improvement in the signal (S)-to-noise (N) ratio (S/N) for 1 nM target DNA. The SHCP/HDT/MCH-modified Au/SPEs allowed the direct quantification of the target DNA down to 25 pM (0.25 fmol) and 100 pM (1 fmol) in undiluted/untreated serum and urine samples, respectively, and of 16S rRNA Escherichia coli (E. coli) corresponding to 3000 CFU μL−1 in raw cell lysate samples. The new SAM-coated screen-printed electrodes also displayed favorable non-fouling properties after a 24 h exposure to raw human serum and urine samples, offering great promise as cost-effective nucleic acid sensors for a wide range of decentralized genetic tests.
Keywords: Screen-printed gold electrodes, Dithiols, Self-assembled monolayer, DNA hybridization, Raw samples
1. Introduction
Nucleic acid detection is of considerable importance to clinical diagnosis, because DNA/RNA tests can reveal genetic disorders or pathogen infection [1]. The development of inexpensive, easy-to-use, and rapid analytical measurement devices has thus been the focus of intensive research efforts. While rapid clinical diagnosis would benefit from the direct quantitation of nucleic acid in complex biological fluids, problems due to high background currents typically hinder the direct nucleic acid assays of untreated clinical samples. Electrochemical DNA hybridization biosensors have thus emerged recently as an attractive alternative to classical nucleic-acid detection technologies, which are time-consuming, labor intensive, and require centralized laboratories [2–9]. Such devices offer elegant routes for interfacing DNA recognition and signal transduction elements, and are uniquely qualified for meeting the low-cost, low-volume, simplicity and portability requirements of decentralized DNA diagnostics [2,10].
Modern microfabrication techniques have led to miniaturized devices that address the need for rapid decentralized testing [11]. In particular, the distinct advantages of screen-printed electrodes (SPEs), including their low-cost mass production along with their minimal sample volume and cross contamination, make them extremely attractive for clinical applications and potential commercialization [12,13]. In particular, screen-printed gold electrodes (Au/SPEs) have received considerable attention in the development of DNA hybridization biosensors [1,3,5,6,14–19]. Such Au/SPEs address also certain drawbacks of the conventional gold disk electrode transducers, including the requirement for surface regeneration after each measurement, high costs and large sample volumes. While the utility of Au/SPEs for detecting DNA hybridization has been demonstrated by several groups [20–22], further improvement of their performance is needed to approach the sensitivity obtained with conventional gold electrodes. The capabilities of these mass-produced disposable sensors can be improved by modifying their surfaces with different nanomaterials (metal nanoparticles, CNTs) [1,5,14,19].
Little attention has been given to the applicability of these screen-printed DNA biosensors for direct detection in raw biological samples, due to their limited sensitivity in these complex media. Only Yang et al. [23] have carried out micromolar detection of target DNA in undiluted blood serum using a folding-based electrochemical DNA (E-DNA) sensor based on a gold-plated screen-printed carbon electrode. Direct detection of target DNA in undiluted biological matrices has not been reported using these disposable electrodes.
Herein, we report for the first time on the utility of disposable Au/SPEs coated with ternary SAMs for direct nucleic acid hybridization detection in raw biological fluids. Although binary SAMs containing a SHCP and a spacer thiol, mainly MCH [24], have been widely used for modifying conventional gold disk and screen-printed electrodes improving their reproducibility and hybridization efficiency [3,5,21,25–27], such biosensors suffer from background currents and irreproducibility problems resulting from incomplete backfilling and surface defects [26–28]. Very recently, the introduction of a third thiol component [8,9,29,30] has been demonstrated to significantly improve the capabilities of conventional modified gold electrodes compared to common binary SAM, allowing ultrasensitive detection of nucleic acid target molecules in hybridization buffer [8] and direct measurement of low levels of target DNA in undiluted and untreated physiological samples [9].
In our previous study, we demonstrated picomolar detection of DNA sequences in undiluted serum and untreated urine samples using a ternary SAM of HDT, SHCP and MCH on photolithographically prepared 16-sensor Au electrode arrays [9]. This attractive performance of the new ternary SAM has prompted their evaluation for DNA hybridization detection on standard, commercially available screen-printed gold electrodes.
In the present work, the ternary layer is assembled on the surface of these disposable electrode strips by co-immobilizing the linear dithiol component (HDT) with the SHCP, followed by sequential confinement of MCH. The resulting ternary SAM-coated Au/SPEs display greatly improved S/N characteristics compared with Au/SPEs based on conventional binary interfaces, and allow for direct and rapid measurement of pM target DNA concentrations in these undiluted biological fluids. We believe that these studies will facilitate low-cost decentralized genetic testing on untreated body fluid samples.
2. Materials and methods
2.1. Apparatus and electrodes
Chronoamperometric measurements were carried out with a μ-AUTOLAB type III potentiostat using the GPEs 4.9006 software (Eco Chemie, The Netherlands). A XBAS-NS-Au gold disk electrode (φ∼3mm), Au/SPEs (DropSens-220BT and DropSens-220AT), screen-printed carbon electrodes (SPCEs) (DropSens-110) and commercial gold nanoparticles (AuNPs)-modified SPCEs (DropSens-110GNP), purchased from DropSens Inc., Oviedo-Austrias, Spain were used as the working electrodes. The design and inks were the same in the two different types of screen-printed gold electrodes, BT and AT-Au/SPEs; the only difference between them was in the printing process. AT were cured at high temperature (800–900 °C), while BT-Au/SPEs were obtained after a low temperature curing (90–100 °C). The average roughness (Ra) values of the AT and BT-Au/SPEs are 0.695 and 2.10 μm, respectively (per information provided by Dropsens S.L). The layout of the disposable planar screen-printed gold electrodes (φ∼4mm) includes a gold disk-shaped (12.6 mm2) working electrode, a Ag pseudo-reference electrode and a gold counter electrode, all of them screen-printed on a ceramic substrate (3.4 cm × 1.0 cm). An insulating layer was printed over the electrode system, leaving the electric contacts and a working area uncovered, of which the latter constitutes the reservoir of the electrochemical cell, with an actual volume of 50 μL. The format of the SPCEs and commercial AuNPs-SPCEs is similar but includes a carbon counter electrode. A specific cable connector (ref. DRP-CAC from DropSens, S.L, Oviedo-Austrias, Spain) acts as interface between the SPEs and the potentiostat.
2.2. Reagents and solutions
1,6-Hexanedithiol (HDT, 96%), DL-dithiothreitol (DTT), 6-mercapto-1-hexanol (MCH, 97%), Trizma hydrochloride (Tris–HCl), ethylenediaminetetraacetic acid (EDTA), KCl, NaCl, Na2HPO4, KH2PO4, K2HPO4·3H2O, NaH2PO4, human serum (from human male AB plasma), bovine serum albumin (BSA) and hydrogen tetrachloroaurate (III) hydrate (HAuCl4·3H2O) were purchased from Sigma–Aldrich (St. Louis, MO) and used without further purification. K3Fe(CN)6 and K4Fe(CN)6·3H2O were purchased from Fisher Scientific Company. The enzyme substrate 3,3′,5,5′-tetramethylbenzidine (TMB, Neogen K-blue enhanced activity substrate, containing H2O2) was obtained from Neogen (Lexington, KY). The conjugated anti-fluorescein-horseradish peroxidase (anti-FITC-HRP, Fab fragments) was purchased from Roche Diagnostics (Mannheim, Germany). The blocking agent casein was obtained from Pierce (Rockford, IL).
2M KOH (Panreac), prepared in deionised water, was used for the pretreatment of the gold disk electrode. A 0.5 M H2SO4 solution containing or not 10 mM KCl prepared in deionised water was used for the pretreatment of the Au/SPEs. An AuCl4− 1 mM solution, prepared in 0.1 M HCl, was used for the nanostructuration of the SPEs.
A 0.1 M KCl solution containing 5 mM of K3Fe(CN)6 and 5mM of K4Fe(CN)6 was used for the CV and EIS electrochemical characterization of the modified electrodes.
The buffers used in this study were as follows: the immobilization buffer (IB) contained 10 mM Tris–HCl, 1 mM EDTA, and 0.3 M NaCl (pH 8.0), the hybridization buffer (HB) was a 1.0 M phosphate solution containing 2.5% BSA (pH 7.2), and the binding buffer (BB), for the incorporation of the conjugated anti-FITC-HRP, was PBS (1×) containing 0.5% casein (pH 7.2).
The synthetic oligonucleotides used in the study, purchased from Integrated DNA Technologies, Inc. (CA, USA), and designed for detecting a characteristic region of E. coli 16S rRNA, are listed in Table S-1 of the Supporting Information.
Bacterial strains of E. coli NEB 5-α (New England Biolabs) and Klebsiella pneumoniae (KP210) were obtained from the Clinical Microbiology Laboratory, University of California, Los Angeles (UCLA), with approval from the UCLA and Veterans Affairs institutional review boards and appropriate Health Insurance Portability and Accountability Act exemptions. The strains were received in centrifuge tubes and were stored at −80°C until use. Overnight bacterial cultures were freshly inoculated into Luria broth (LB) and grown to logarithmic phase as measured by the optical density at 600 nm. Bacterial concentrations in the logarithmic-phase specimens were determined by serial plating.
2.3. Procedures
2.3.1. Pretreatment of the working electrodes
Before carrying out the assembly of the ternary recognition interface, the Au/SPEs were pretreated by placing a 50 μL drop of a 0.5 M H2SO4 solution either containing (AT electrodes) or not containing (BT electrodes) 10 mM KCl onto and covering the surface of the three electrodes. 10 cyclic voltammograms from 0.00 to 1.25 V were recorded at a scan rate of 100 mV s−1, and the electrodes were washed with deionised water and dried with nitrogen.
A conventional three-electrode setup with a gold disk (AuE) as working electrode, a BAS MF-2052 Ag/AgCl/KCl (3 M) reference electrode and a Pt wire counter electrode were employed for the comparison of the performance of the new ternary monolayers assembled on Au/SPEs vs. AuE. The AuE was pretreated as described previously [31].
2.3.2. Modification of the SPEs with gold nanoparticles
The home-made AuNPs-SPCEs and AuNPs-BT-Au/SPEs were prepared by electrodeposition from a HAuCl4 solution. Gold nanostructuring of these surfaces was achieved placing a 50 μL drop of 1 mM AuCl4− acidic solution on the electrode surfaces, and applying a constant potential of −200 mV vs. the pseudo-reference electrodes for 2min. The modified electrodes were rinsed with water and dried under a nitrogen flow.
2.3.3. Assembling of the sensing interfaces on the working electrodes
A mixture of the SHCP with or without the freshly prepared dithiol compound to be examined (HDT or DTT), at the appropriate concentration, was prepared in IB buffer solution and allowed to stand for 10 min. Then, this mixture (10 μL) was cast on the working electrode under study (AT or BT-Au/SPEs, commercial GNP-SPCEs, home-made AuNPs-SPCEs, home-made AuNPs-BT-Au/SPEs or AuE). The chemisorption process was allowed to proceed overnight in a humidified chamber. After washing with water and drying with nitrogen, the modified sensors were subsequently treated with 10 μL of 1 mM MCH aqueous solution (in IB buffer) for 50 min to obtain the final SAM interfaces. Finally, the sensors were thoroughly rinsed with water and dried under nitrogen.
Unless otherwise specified, all experiments were done using 5 μM of SHCP, 600 μM of HDT and 400 μM of DTT.
2.3.4. Electrochemical characterization by CV and EIS
Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were performed with a CHI 660D Electrochemical Analyzer (CH Instruments, Austin, TX, USA). The measurements were carried out by placing a 50 μL drop of a 0.1 M KCl solution containing 5 mM of K3Fe(CN)6 and 5 mM of K4Fe(CN)6 onto the electrode surfaces. Electrochemical impedance spectra were obtained at +0.25 V under an AC amplitude of 0.01 V and a frequency range from 0.01 to 10,000 Hz. The impedance data were analyzed by non-linear least squares using the EQUIVCTR.PAS (EQU) program by Boukamp.
2.3.5. DNA hybridization procedure in HB and raw biofluid samples
The electrochemical response of the prepared working electrodes was evaluated using a sandwich-type hybridization assay. In these experiments, fluorescein (FITC) and anti-FITC-HRP were used as the tracer and the reporter molecules, respectively. TMB substrate was used for the electrochemical measurement of the activity of the HRP reporter. Firstly, target DNA at different concentrations were mixed with FITC-DP (0.25 mM) in HB and left for 15 min to obtain homogeneous hybridization. Then, 10 μL of this solution was cast on the SAM-modified working electrode. After 15 min of incubation, the electrode was sssssgen and 10 μL of a 0.5UmL−1 anti-FITC-HRP solution (prepared in BB) was applied to the working electrode and incubated for an additional 15 min. Subsequently, the prepared electrode was washed and dried under nitrogen. For electrochemical detection, a 50 μL of the TMB–H2O2 K-Blue reagent solution was placed on the modified Au/SPEs, covering the three electrodes area or by immersion of the AuE in a glass electrochemical cell containing 1.5 mL of the enzymatic substrate. In all cases, chronoampero-metric measurements were performed after 30 s; at −200 mV vs. Ag pseudo-reference (SPEs) or Ag/AgCl reference electrodes (AuE) and the current was sampled for 60 s. In order to minimize the variability of the reference potential and to ensure the reproducibility between different measurements when working with an Ag pseudo-reference electrode (SPEs) we optimized the detection potential. Since no significant differences in sensitivity of the HRP-TMB–H2O2 reaction product were observed over the wide 0 and −300 mV range, a working potential of −200 mV was selected to minimize possible potential variations.
For hybridization studies in raw clinical samples, HB was substituted for the clinical sample under study; both target DNA and FITC-DP (0.25 μM) were prepared directly either in undiluted commercial human serum or fresh untreated urine. The homogeneous hybridization between different target concentrations and the detection probe was carried out in the untreated samples and then 10 μL of the hybrid solution was applied to each modified sensor and incubated for 15 min. The following steps: capture of anti-FITC-HRP and electrochemical detection were carried out using the same protocol described above for the determination of target DNA in HB. All procedures were carried out at ambient temperature (22–25 °C).
2.3.6. Non-fouling properties of the modified surfaces
For the investigation of the non-fouling properties of the SHCP/HDT/MCH and SHCP/MCH-modified BT-Au/SPEs, a 10 μL droplet of the untreated biological samples was applied to a SAM-modified gold working electrode and left overnight at 4°C in a humidified chamber. The electrode was washed with deionised water and dried with nitrogen and the same protocol described above was used for the determination of target DNA in HB. For comparison purposes, a control experiment was also performed by applying 10 μL of HB on SAM-modified gold working electrode and left overnight at 4°C in a humidified chamber.
2.3.7. Hybridization with bacterial rRNA in raw lysate solutions
The bacteria were lysed by resuspension of the appropriate pellet containing ∼107 CFU bacteria in 10 μL of 1 M NaOH and incubation for 5 min [8,32]. A 50 μL aliquot of FITC-DP (0.25 μM) in HB was added to this 10 μL bacterial lysate, yielding a concentration of nucleic acids corresponding to ∼107 CFU per 60 μL. This solution was serially diluted in the FITC-DP (0.25 μM) to provide different concentrations of bacterial nucleic acids (including 16S rRNA). Aliquots (10 μL) of this raw bacterial lysate target solution were applied to each capture probe-modified sensor and incubated for 15 min, followed by the same volume of anti-FITC-HRP and the electrochemical detection steps described earlier for the synthetic target DNA. All procedures were carried out at room temperature.
3. Results and discussion
The present detection strategy involves a “sandwich” hybridization strategy of target DNA to specific capture and detector probes. Capture probe is anchored to the surface of the Au/SPEs and the detector probe is FITC-linked to allow binding of a HRP labeled anti-FITC antibody. Application of HRP substrate (H2O2) and its cofactor and of a measurement potential allow chronoamperometric detection of probe-target-enzyme complexes on the working electrode surface. Schematic illustration of the Au/SPEs, optimal surface chemistry and detection strategy are shown in Scheme 1.
Scheme. 1.
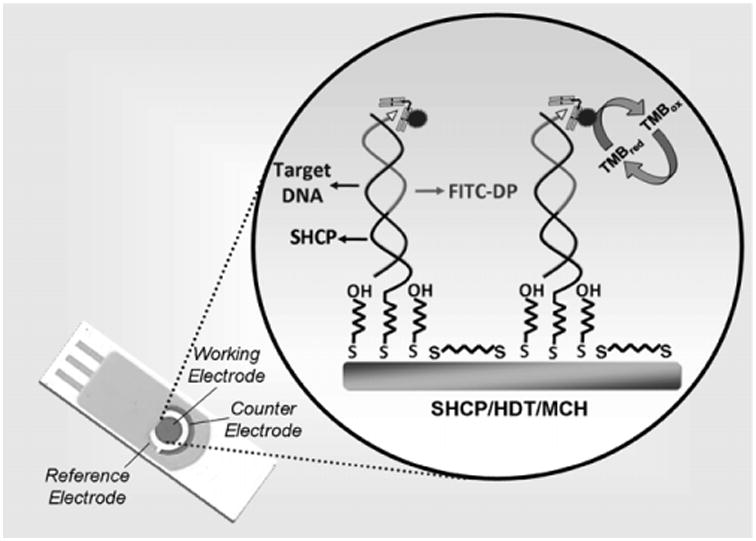
Schematic representations of the Au/SPE, the SHCP/HDT/MCH interface, and the sandwich detection strategy.
3.1. Comparison of different gold surfaces and recognition interfaces
Initially, we compared the performance of the conventional SHCP/MCH and the new SHCP/HDT/MCH SAMs assembled on different gold substrates (rough BT-Au/SPEs, smooth AT-Au/SPEs and a conventional gold disk electrode). As illustrated in Fig. 1 and from the data of Table S-2, considerably higher S/N ratio was observed using the SHCP/HDT/MCH ternary layer applied to the 3 electrode substrates. While keeping the current response of 1nM target DNA at the level of the binary layer or increased slightly, the coimmobilization of SHCP with HDT causes a significant decrease of the background currents. These lower nonspecific background contributions, along with similar or slightly better signals, lead to a substantially higher S/N ratio (up to 10-fold improvement with the BT-Au/SPEs). As will be discussed below the new interface offers convenient detection of picomolar levels of target DNA in raw clinical samples as compared to the conventional binary layer assembly where, due to the large background current, such picomolar detection, even in HB, is impossible.
Fig. 1.

Signal (S) and noise (N) values obtained for 1 nM (fill columns) and 0 nM (empty columns) target DNA and resulting S/N ratios obtained with different electrode substrates modified with the binary SHCP/MCH and ternary SHCP/HDT/MCH monolayers in HB. Chronoamperometric responses obtained for 1 nM (solid lines) and 0 nM of target DNA (dashed lines) using BT-Au/SPEs modified with the SHCP/MCH and SHCP/HDT/MCH SAMs. Error bars were estimated from 3 parallel experiments.
Comparing the different gold surfaces, BT-Au/SPEs provide higher signals than the AT-Au/SPEs and the conventional gold disk electrode. This improvement in sensitivity can be attributed to the high surface area of the rough BT-Au/SPEs and hence to a higher number of capture-probe sites and display of SHCP that enhances the probe accessibility and lead to a faster and more efficient target hybridization [33].
The performance of the ternary layer prepared with a cyclic dithiol (SHCP/DTT/MCH SAM) instead of a linear one (SHCP/HDT/MCH SAM) was also evaluated. The results obtained (Table S-2 of the Supporting Information) showed that while the presence of DTT keeps the noise at the level of the HDT-based ternary layer, DTT also decreases the resulting signal as compared to SHCP/MCH and SHCP/HDT/MCH, probably due to the lower amount of thiolated capture probe on the DTT interface. In the view of these results over the 3 electrode substrates the performance of the different SAMs studied follows the order: SHCP/MCH < SHCP/DTT/MCH < SHCP/HDT/MCH.
BT-Au/SPEs modified with the binary and the new dithiol-based ternary layers were characterized using CV and EIS. To the best of our knowledge the modification and hence the characterization of this rough Au/SPEs surfaces with ternary layers has not yet been attempted. The results are illustrated in Fig. 2.
Fig. 2.

Nyquist plots (−Z″ vs. Z′) for the faradaic impedance measurements and the corresponding cyclic voltammograms (inset) obtained with a bare BT-Au/SPE surface and with BT-Au/SPEs modified with SHCP/MCH, SHCP/DTT/MCH, and SHCP/HDT/MCH SAMs. Parameters: EIS, 5 mM [Fe(CN)6]3−/4− (1:1) in 0.1 M KCl, 0.01–10,000 Hz frequency range with a 0.01 V rms signal at +0.25 V (vs. Ag/AgCl); CV, ν = 100 m Vs−1.
The surface coverage values were determined by CV and EIS. As expected based on previous studies [34], lower fractional coverages of the monolayers are estimated from CV compared to those obtained from EIS. According to the charge-transfer resistance values (see Table 1, obtained after modeling these experimental EIS data using the Randles equivalent circuit, the SAM-modified BT-Au/SPEs coverage values were estimated to be 67% for the binary layer and 77% for both ternary layers [35]. These results suggest that the presence of the dithiol (DTT or HDT) third component leads to the assembly of more dense monolayers with fewer pinholes conferring the modified printed surfaces with lower background currents and attractive anti-fouling properties, as we will present below. The lower coverage values obtained for all the tested SAMs on BT-Au/SPEs, compared with those obtained on conventional gold disk electrodes, confirm the influence of the surface roughness upon the fractional surface coverage of the monolayers. Yet, despite the rough surface of these disposable Au/SPEs, the SAM coverage was generally high, around 70–80%.
Table 1.
Electrochemical impedance data for different SAMs. Data were calculated using Randles equivalent circuit.
| Surface | AuE | BT-Au/SPEs | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||||
| iap (μF) | θCVi | Q (μF) | Ret (Ω) | θISR | iap (μF) | θCVi | Q (μF) | Ret (Ω) | θISR | |
| Bare | 39.83 | – | 4.96 | 1029.6 | – | 120.3 | – | 1.95 | 1153.8 | – |
| SHCP/MCH | 21.71 | 0.4549 | 0.398 | 5562.8 | 0.8149 | 59.13 | 0.5085 | 0.375 | 3500.2 | 0.6704 |
| SHCP/DTT/MCH | 8.757 | 0.7801 | 0.637 | 32,595.9 | 0.9684 | 48.68 | 0.5953 | 0.664 | 5103.7 | 0.7739 |
| SHCP/HDT/MCH | 21.12 | 0.4697 | 0.335 | 5914.6 | 0.8259 | 39.82 | 0.6690 | 0.382 | 5019.3 | 0.7701 |
The performance of the SHCP/HDT/MCH layer on BT-Au/SPEs was also compared with the attributes of different AuNPs-modified SPEs: commercial AuNPs-modified SPCEs (GNP-SPCEs), SPCEs modified with home-made AuNPs (AuNPs-SPCEs), and BT-Au/SPEs modified with home-made AuNPs (AuNPs-BT-Au/SPE) for 1 nM target DNA in HB. All these AuNPs-modified SPEs yielded a lower signal and a higher noise, leading to inferior S/N ratios compared to those observed with the unmodified BT-Au/SPEs (Table S-3 of the Supporting Information). This difference can be attributed to less Au active surface area and roughness (commercial GNP-SPCEs and home-made AuNPs-SPCEs or to the blocking effect of the AuNPs on the electrode surface (home-made AuNPs-BT-Au/SPEs) in comparison with the results achieved with the unmodified BT-Au/SPEs.
The S/N ratio characteristics obtained for the different SAM interfaces and electrode substrates clearly demonstrate the influence of the substrate morphology on the accessibility of the SHCP [33]. The S/N ratio findings also show that the presence of a 3rd SAM component, including its structure and adopted configuration during the assembly, are key contributors to the attractive non-fouling properties and hybridization efficiency achieved with the new layers on both conventional gold disk and disposable electrodes. Subsequent work thus focused on the optimization and performance evaluation of the SHCP/HDT/MCH interface on BT-Au/SPEs.
3.2. Optimization of the parameters that influence the performance of the SHCP/HDT/MCH SAM
In order to optimize the composition of the SHCP/HDT/MCH SAM assembled on the surface of BT-Au/SPEs, we examined the effect of the concentration of the SHCP and the HDT components upon the S/N ratio obtained for 1 nM target DNA. Fig. 3a shows the dependence of the S/N ratio on the concentration of the SHCP of the ternary layer (prepared with a fixed HDT concentration of 300 μM). These data illustrate that the S/N ratio increased with the SHCP concentration up to 5 μM, decreased sharply until 10 μM of SHCP and leveled off thereafter. This profile indicates a lower hybridization efficiency at higher surface coverages and that the accessibility and configuration of the probes were more important factors in promoting efficient target capture than any benefits of high probe density [33]. Thus, 5 μM SHCP was chosen as optimum concentration for further experiments.
Fig. 3.

Dependence of the S/N ratio obtained for 1 nM target DNA with the concentration of SHCP (a) and HDT (b) on the ternary SHCP/HDT/MCH surface layer on BT-Au/SPEs. Error bars were estimated from 3 parallel experiments.
Next, we investigated the dependence of the S/N ratio on the concentration of the dithiol component of the ternary SAMs (Fig. 3b). The concentration of the dithiol component can affect the surface coverage and the spacing of the co-assembled SHCP molecules. As illustrated in Fig. 3b, low concentrations of HDT still led to high nonspecific adsorptions and thus to lower S/N values. On the other hand, relatively high concentrations of the HDT (>600 μM) could displace some of the SHCP molecules, decreasing the hybridization efficiency, and hence the resulting S/N. Optimal behavior was thus observed at a HDT concentration of 600 μM in connection with 5 μM SHCP, along with 1 mM MCH. Apparently, these concentrations provide the most favorable trade-off between resistance to nonspecific adsorption (induced by HDT) and hybridization efficiency (determined by the SHCP coverage).
3.3. Electrochemical detection of DNA hybridization in raw biological samples
Serum and urine are complex biological samples containing multiple components that can be adsorbed nonspecifically onto the sensing interface, interfering with the binding of the target DNA and/or increasing the background signal. We observed that the new ternary surface layers allowed direct detection of trace target DNA in undiluted human serum and urine. To the best of our knowledge, this represents the first report using disposable SPEs for detection of picomolar target DNA concentrations in raw biological samples. As a first step toward this goal, we compared the chronoamperometric response at 1 nM target DNA concentration in 100% human serum or urine using the commonly employed SHCP/MCH binary interface with those observed using the new HDT-based ternary layer (Table S-4 in the Supporting Information). These data indicate that the HDT-based ternary SAM layer offered 3-fold and 2.5-fold improvements in the S/N, over the binary layers, in undiluted human serum and untreated urine, respectively.
The performance of the SHCP/HDT/MCH-modified BT-Au/SPEs was tested with different concentrations of target DNA in 100% human serum (Fig. 4 left) and 100% urine (Fig. 4 right). As shown in Fig. 4, the chronoamperometric signal increased linearly with the target DNA concentration up to 1 nM, with a detection limit of 25 pM (0.25 fmol) in human serum and 100 pM (1 fmol) in urine. It is worth noting that the attractive attributes of these ternary-SAM modified Au/SPEs allowed the detection of target DNA in 100% of human serum and urine with a 52.9 and 79.5%, respectively of the sensitivity achieved in pure HB (slope values in μA pM−1 of 2.37, 1.26 and 1.88 × 10−3 in 100% HB, serum and urine, respectively). The detection limits obtained with the HDT-ternary monolayer in undiluted clinical samples are substantially lower than those reported previously for SAM-based electrochemical DNA sensors in diluted samples. For example, the detection limit of the new HDT-based monolayer is 40-fold lower than that reported recently in 20% serum by Patterson et al. [36]; it is also 4.5-fold lower than that reported by Pei et al. [37] in 50% serum using conventional gold electrodes.
Fig.4.

Chronoamperometric responses obtained after hybridizationin100% of human serum or human urine with the 25 and 250 pM target DNA concentrations, respectively, along with the response without the target (dashed lines). Calibration plots for different target DNA concentrations obtained after hybridization in these untreated and undiluted biological samples (and background subtraction). Error bars were estimated from 3 parallel experiments.
This attractive performance in undiluted clinical samples was coupled with highly reproducible surface modification properties. A series of 6 measurements of 1nM of target DNA in HB yielded a RSD of 9.2% (conditions, as in right hand side of Fig. 1).
3.4. Non-fouling properties of the SHCP/HDT/MCH and SHCP/MCH monolayers
The resistance to biofouling of screen-printed electrodes modified with ternary and binary monolayers was examined by exposing them to undiluted human serum and untreated urine. These complex physiological samples have components that can be non-specifically absorbed onto the interface, thus interfering with the binding of the target DNA and/or increasing the background signal. Comparison of the S and N values obtained after 24 h of incubation in HB, 100% human serum and 100% urine obtained after carrying out the hybridization with 1 nM and 0 nM target DNA over BT-Au/SPEs modified with SHCP/MCH and SHCP/HDT/MCH are shown on left and right hand sides, respectively, of Fig. 5, along with Table S-5.
Fig. 5.

Non-fouling properties of the SHCP/MCH and SHCP/HDT/MCH monolayers on BT-Au/SPEs after dipping 24 h in HB and 100% of biological samples. S and N values obtained after hybridization with 1 nM (solid columns) and 0 nM (empty columns) of target DNA in HB. Error bars were estimated from 3 parallel experiments.
The screen-printed electrodes modified with the binary SHCP/MCH layer lost between 50 and 70% of its S and N after incubation for 24 h in serum and urine, respectively, indicating considerable surface passivation by the non-specific adsorption of components of these complex matrices. In contrast, SPEs modified with the SHCP/HDT/MCH retained 80 or 85% of their original signal while maintaining similarly low background currents. These results confirm that only the SHCP/HDT/MCH bioplatform displays favorable non-fouling properties after 24 h immersion of the strip biosensor in the selected clinical samples, a result with high relevance to real-world applications.
At this point it should be noted that the slight diminution observed mainly in the S and hence in the S/N after incubation of a SHCP/HDT/MCH-modified screen-printed electrode in HB in comparison with a freshly prepared one (comparison of the data in HB with SHCP/HDT/MCH-modified Au/SPEs in Tables S-2 and S-5) could be attributed to the rapid displacement of SHCP by MCH [38–40].
3.5. Application to the detection of E. coli 16S rRNA in raw bacterial lysate solutions
We also investigated the practical utility of the SHCP/HDT/MCH-modified disposable electrodes for the direct detection of E. coli pathogenic 16S rRNA in a raw bacterial lysate solution without isolation or purification steps. As can be seen in Fig. 6 the disposable modified electrodes were able to clearly distinguish the signals obtained for a bacterial lysate solution corresponding to 3 × 104 CFU per sensor from those obtained without the bacterial rRNA target. Fig. 6a (inset) displays the calibration plot obtained for bacterial lysate solutions corresponding to different E. coli cell concentrations, indicating a nonlinear logarithmic dependence between the chronoamperometric signals and the level of E. coli between 3 × 104 and 3 × 106 CFU per sensor. Considering the 10 μL sample volume, such a detection limit corresponds to 3000 CFU μL−1. Taking into account that E. coli contains approximately 2 × 104 copies of 16S rRNA per cell [41], the present detection limit of 3000 CFU μL−1 can be translated to the detection of 250 pM ribosome target copies in these raw bacterial lysate solutions.
Fig.6.
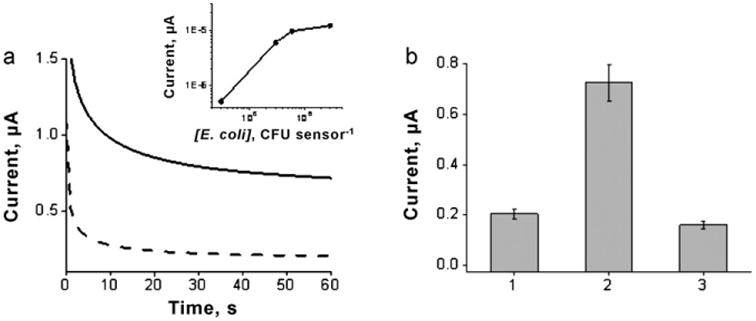
(a) Chronoamperometric signals obtained for 16S rRNA of 3 × 104 CFU per sensor along with the corresponding blank (0 CFU). Inset: calibration plot for E. coli 16S rRNA corresponding to different pathogen bacterial concentrations after background subtraction. (b) Column bar corresponding to the chronoamperometric responses obtained with 0 nM target DNA (1), 16S rRNA corresponding to 3 × 104 CFU per sensor E. coli (2), and 16S rRNA corresponding to 7.3 × 105 CFU per sensor K. pneumoniae (3). Error bars were estimated from 3 parallel experiments. BT-Au/SPEs modified with a SHCP/DTT/MCH SAM.
The specificity of the disposable Au/SPE biosensor was also tested using control K. pneumoniae, another Gram-negative pathogenic member of Enterobacteriaceae, as the no-target biological control [42]. In contrast to the chronoamperometric signal obtained for E. coli 16S rRNA, the response observed in the presence of a 24-fold excess of K. pneumoniae 16S rRNA was similar to that observed for the negative control without target DNA (Fig. 6b), reflecting the high specificity and negligible nonspecific adsorption of the detection probe onto the SHCP/HDT/MCH-modified disposable electrodes.
4. Conclusions
We report for the first time the application of disposable Au/SPEs modified with ternary SAM interfaces to the direct measurement of target DNA in undiluted and untreated human serum and urine samples. Despite the rough surface of BT-Au/SPEs, such ternary SAMs form highly packed monolayers with minimal defects that impart significantly higher resistance to nonspecific adsorption. The improved capabilities of the modified disposable electrodes allowed direct, sensitive and rapid (30 min) detection of picomolar levels of target nucleic acids in untreated and undiluted microliter clinical samples or bacterial lysate samples solutions. In addition, this new strip platform displayed excellent antifouling properties during prolonged exposure to raw body fluids. These results demonstrate that a rational design of the surface chemistry of screen-printed electrodes can offer significant analytical improvements and can facilitate direct assays of unprocessed body fluids. As a result, the new SAM-coated Au/SPEs offer excellent prospects for a wide range of applications in diverse environments, including single-use genetic testing in resource-poor settings.
Supplementary Material
Acknowledgments
Financial support from the National Institutes of Health (Award U01 AI075565) is gratefully acknowledged. FK and SC acknowledge fellowships from The Scientific and Technical Council of Turkey (TUBITAK) and Programa Becas Complutense del Amo (2010-2011), respectively. The authors would like to acknowledge S. Marx and C. Halford for their assistance.
Footnotes
Appendix A. Supplementary data: Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.talanta.2011.06.012.
References
- 1.Martínez-Paredes G, González-García MB, Costa-García A. Sens Actuators B. 2010;49:329–335. [Google Scholar]
- 2.Wang J. Anal Chim Acta. 2002;469:63–71. [Google Scholar]
- 3.Carpini G, Lucarelli F, Marrazza G, Mascini M. Biosens Bioelectron. 2004;20:167–175. doi: 10.1016/j.bios.2004.02.021. [DOI] [PubMed] [Google Scholar]
- 4.Paleček E. Electroanalysis. 2009;21:239–251. [Google Scholar]
- 5.Miranda-Castro R, Lobo-Castañón MJ, Miranda-Ordieres AJ, Tuñón-Blanco P. Electroanalysis. 2009;21:267–273. [Google Scholar]
- 6.He Y, Zeng K, Zhang X, Gurung AS, Baloda M, Xu H, Liu G. Electrochem Commun. 2010;12:985–988. [Google Scholar]
- 7.Singha R, Sumana G, Verma R, Sood S, Pandeya MK, Guptac RK, Mal-hotra BD. J Biotechnol. 2010;50:357–365. doi: 10.1016/j.jbiotec.2010.09.935. [DOI] [PubMed] [Google Scholar]
- 8.Wu J, Campuzano S, Halford C, Haake DA, Wang J. Anal Chem. 2010;82:8830–8837. doi: 10.1021/ac101474k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Campuzano S, Kuralay F, Lobo-Castañón MJ, Bartošík M, Vyavahare K, Paleček E, Haake DA, Wang J. Biosens Bioelectron. 2011;26:3577–3583. doi: 10.1016/j.bios.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang J. Analyst. 2005;130:421–426. doi: 10.1039/b414248a. [DOI] [PubMed] [Google Scholar]
- 11.Albers J, Grunwald T, Nebling E, Piechotta G, Hintsche R. Anal Bioanal Chem. 2003;377:521–527. doi: 10.1007/s00216-003-2192-7. [DOI] [PubMed] [Google Scholar]
- 12.Alonso-Lomillo MA, Dominguez-Renedo O, Arcos-Martinez MJ. Talanta. 2010;82:1629–1636. doi: 10.1016/j.talanta.2010.08.033. [DOI] [PubMed] [Google Scholar]
- 13.Metters JP, Kadara RO, Banks CE. Analyst. 2011;136:1067–1076. doi: 10.1039/c0an00894j. [DOI] [PubMed] [Google Scholar]
- 14.Martínez-Paredes G, González-García MB, Costa-García A. Electroanalysis. 2009;21:379–385. [Google Scholar]
- 15.García T, Fernández-Barrena MG, Revenga-Parra M, Núñez A, Casero E, Pariente F, Prieto J, Lorenzo E. Anal Bioanal Chem. 2010;398:1385–1393. doi: 10.1007/s00216-010-4029-5. [DOI] [PubMed] [Google Scholar]
- 16.de la Escosura-Muñiz A, Merkoci A. Chem Commun. 2010;46:9007–9009. doi: 10.1039/c0cc02683b. [DOI] [PubMed] [Google Scholar]
- 17.Zhang D, Peng Y, Qi H, Gao Q, Zhang C. Biosens Biolectron. 2010;25:1088–1094. doi: 10.1016/j.bios.2009.09.032. [DOI] [PubMed] [Google Scholar]
- 18.Tran LD, Nguyen BH, Hieu NV, Tran HV, Nguyen HL, Nguyen PX. Mater Sci Eng C. 2011;31:477–485. [Google Scholar]
- 19.Ren R, Leng C, Zhang S. Biosens Bioelectron. 2010;25:2089–2094. doi: 10.1016/j.bios.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 20.Lucarelli F, Marrazza G, Mascini M. Biosens Bioelectron. 2005;20:2001–2009. doi: 10.1016/j.bios.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 21.Loaiza O, Campuzano S, Pedrero M, García P, Pingarrón JM. Electroanalysis. 2008;20:1397–1405. [Google Scholar]
- 22.Moreno M, Rincona E, Pérez JM, González VM, Domingo A, Dominguez E. Biosens Bioelectron. 2009;25:778–783. doi: 10.1016/j.bios.2009.08.028. [DOI] [PubMed] [Google Scholar]
- 23.Yang W, Gerasimov JY, Lai RY. Chem Commun. 2009;20:2902–2904. doi: 10.1039/b904550c. [DOI] [PubMed] [Google Scholar]
- 24.Steel AB, Herne TM, Tarlov MJ. Anal Chem. 1998;70:4670–4677. doi: 10.1021/ac980037q. [DOI] [PubMed] [Google Scholar]
- 25.Levicky R, Herne TM, Tarlov MJ, Satija SK. J Am Chem Soc. 1998;120:9787–9792. [Google Scholar]
- 26.Lao RJ, Song SP, Wu HP, Wang LH, Zhang ZZ, He L, Fan CH. Anal Chem. 2005;77:6475–6480. doi: 10.1021/ac050911x. [DOI] [PubMed] [Google Scholar]
- 27.Keighley SD, Li P, Estrela P, Mighorato P. Biosens Bioelectron. 2008;23:1291–1297. doi: 10.1016/j.bios.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 28.Boozer C, Chen SF, Jiang SY. Langmuir. 2006;22:4694–4698. doi: 10.1021/la052908e. [DOI] [PubMed] [Google Scholar]
- 29.Dharuman V, Hahn JH. Biosens Bioelectron. 2008;23:1250–1258. doi: 10.1016/j.bios.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 30.Dharuman V, Chang BY, Park SM, Hahn JH. Biosens Bioelectron. 2010;25:2129–2134. doi: 10.1016/j.bios.2010.02.019. [DOI] [PubMed] [Google Scholar]
- 31.Campuzano S, Galvez R, Pedrero M, de Villena FJM, Pingarron JM. J Electroanal Chem. 2002;526:92–100. [Google Scholar]
- 32.Liao JC, Mastali M, Gau V, Suchard MA, Møller AK, Bruckner DA, Babbitt JT, Li Y, Gornbein J, Landaw EM, McCabe ERB, Churchill BM, Haake DA. J Clin Microbiol. 2006;44:561–570. doi: 10.1128/JCM.44.2.561-570.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bin X, Sargent EH, Kelley SO. Anal Chem. 2010;82:5928–5931. doi: 10.1021/ac101164n. [DOI] [PubMed] [Google Scholar]
- 34.Campuzano S, Pedrero M, Montemayor C, Fatás E, Pingarron JM. J Electroanal Chem. 2006;586:112–121. [Google Scholar]
- 35.Janek RP, Fawcett WR, Ulman A. Langmuir. 1998;14:3011–3018. [Google Scholar]
- 36.Patterson A, Caprio F, Vallee-Belisle A, Moscone D, Plaxco KW, Palleschi G, Ricci F. Anal Chem. 2010;82:9109–9115. doi: 10.1021/ac1024528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pei H, Lu N, Wen Y, Song S, Liu Y, Yan H, Fan C. Adv Mater. 2010;22:4754–4758. doi: 10.1002/adma.201002767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park S, Brown KA, Hamad-Schifferli K. Nano Lett. 2004;4:1925–1929. [Google Scholar]
- 39.Arinaga K, Rant U, Tornow M, Fujita S, Abstreiter G, Yokoyama N. Langmuir. 2006;22:5560–5562. doi: 10.1021/la060324m. [DOI] [PubMed] [Google Scholar]
- 40.Carrascosa LG, Martínez L, Huttel Y, Román E, Lechuga LM. Eur Biophys J. 2010;39:1433–1444. doi: 10.1007/s00249-010-0599-6. [DOI] [PubMed] [Google Scholar]
- 41.Neidhardt F, Umbarger H. In: Escherichia coli and Salmonella typhimurium. 2nd. Neidhardt F, et al., editors. I. ASM Press; Washington, DC: 1996. pp. 13–16. [Google Scholar]
- 42.LaGier MJ, Scholin CA, Fell JW, Wang J, Goodwin KD. Mar Pollut Bull. 2005;50:1251–1261. doi: 10.1016/j.marpolbul.2005.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.