

Abstract
Several potential disease-modifying drugs for Alzheimer’s disease (AD) have failed to show any effect on disease progression in clinical trials, conceivably because the AD subjects are already too advanced to derive clinical benefit from treatment and because diagnosis based on clinical criteria alone introduces a high misdiagnosis rate. Thus, well-validated biomarkers for early detection and accurate diagnosis are crucial. Low cerebrospinal fluid (CSF) concentrations of the amyloid-β (Aβ1-42) peptide, in combination with high total tau and phosphorylated tau, are sensitive and specific biomarkers highly predictive of progression to AD dementia in patients with mild cognitive impairment. However, interlaboratory variations in the results seen with currently available immunoassays are of concern. Recent worldwide standardization efforts and quality control programs include standard operating procedures for both preanalytical (e.g., lumbar puncture and sample handling) and analytical (e.g., preparation of calibration curve) procedures. Efforts are also ongoing to develop highly reproducible assays on fully automated instruments. These global standardization and harmonization measures will provide the basis for the generalized international application of CSF bio-markers for both clinical trials and routine clinical diagnosis of AD.
Keywords: Cerebrospinal fluid, Biomarkers, Alzheimer’s disease, β-Amyloid, Tau protein, Mild cognitive impairment
1. Introduction
Alzheimer’s disease (AD) is a complex progressive neurodegenerative disease affecting approximately 14 million people in Europe and the United States [1,2], including almost one-half of the population aged >85 years (43%) [2,3]. In the early stages, the pathologic changes in AD primarily affect the medial temporal lobe, subsequently progressing to neocortical-associated areas [4,5]. The hallmarks of the disease are neuritic plaques composed of the amyloid-β peptide (Aβ) and neurofibrillary tangles (NFTs) composed of hyper-phosphorylated tau protein (P-tau) [5].
The neuropathology of underlying AD starts decades before the appearance of clinical symptoms [6–10], and evidence suggests that AD should be considered as having three main stages: (1) presymptomatic, (2) “prodromal” with mild symptoms (mainly disturbances in episodic memory), and (3) symptomatic with dementia [11]. In many cases, mild cognitive impairment (MCI) can be considered a “transitional zone” between the cognitive decline seen in normal aging and the cognitive dysfunctions of AD dementia. Although as many as 10% to 20% of patients with MCI progress to AD per year [12], other causes of MCI include cerebrovascular disease, polypharmacy, depression, excessive alcohol/drug use, and neurodegeneration unrelated to AD [13].
The diagnosis of mild AD dementia—and especially prodromal AD—remains difficult on purely clinical grounds [11,14], although there is some evidence that specific memory tests identify the amnestic syndrome of the hippocampal type [15,16]. The accuracy of current clinical AD diagnostic methods to predict pathologic diagnoses (in the absence of biomarker information) is generally low; sensitivities have been reported to range from 71% to 88% and specificities from 44% to 71%, depending on the specific histopathologic diagnostic criteria used [17]. In addition, reports from large clinical trials of drug candidates with disease-modifying potential show that 10% to 35% of clinically diagnosed AD cases with mild-to-moderate dementia have negative amyloid positron emission tomography (PET) scans [18]. This means that a large proportion of individuals in these trials have no or little Aβ pathology in the brain and thus do not have the disease for which the drug is to be tested, adversely affecting the ability to identify a beneficial clinical effect of the drug. The accuracy of clinical diagnosis is probably even lower in the very early clinical stages of the disease (i.e., in patients with prodromal AD). This variable and relatively poor performance is particularly troubling given the high level of expertise of the clinicians in the specialized AD centers making the diagnosis, and the diagnostic accuracy in primary or secondary care settings is likely to be even lower.
In the last two decades, there have been intensive efforts to develop disease-modifying drugs to counteract the progression of AD. Because initiating treatment with these agents early in the disease continuum is expected to provide the greatest long-term benefits, there is a critical need for further progress in the development and validation of diagnostic tools to accurately identify patients with early AD dementia (and especially prodromal AD) for inclusion in clinical trials [14]. Aside from the need for biomarkers to identify patients for inclusion in registration trials of disease-modifying agents, once these drugs are approved for widespread use, diagnostic tools will also be required to recognize prodromal AD patients and provide appropriate treatments in routine clinical practice.
The use of magnetic resonance imaging (MRI), 18F-2-fluoro-2-deoxy-D-glucose positron emission tomography (FDG PET), amyloid (Aβ) PET [19,20], and candidate fluid biomarkers for AD has been investigated extensively for a number of years [21]. Like MRI, FDG PET has demonstrated sensitivity for AD identification at the MCI [22] and even the normal stages of cognition [7,23], but these modalities are not pathologically specific. Amyloid PET has demonstrated some specificity for AD lesions [24], but the sensitivity of this modality continues to be investigated.
Although protein content is lower in cerebrospinal fluid (CSF) than in serum, CSF is an ideal source for developing viable biomarkers in AD as it directly interacts with the extracellular space in the brain, thus potentially reflecting the associated biochemical/pathologic changes [25]. As a result, CSF biomarkers have been become accepted and adopted to varying degrees for the clinical diagnosis of AD in different countries. Indeed, the European Federation of Neurological Societies (EFNS) guidelines recommend routine CSF analysis in the differential diagnosis of atypical AD characterized by prominent early deficits rather than episodic memory [1]. Although the International Working Group and, later, the National Institute on Aging and the Alzheimer’s Association (NIA-AA) suggest that CSF bio-markers may add diagnostic value, they do not at present advocate the use of AD biomarker tests for routine diagnosis as they believe that further research, validation, and standardization are required and because access to biomarkers is restricted in some settings [11,26].
An international Expert Committee meeting was held in April 2012 to discuss the development and validation of CSF diagnostic assays and the optimization of their use as in vitro diagnostic tools, with a particular emphasis on the employment of CSF biomarkers to identify patients with prodromal AD. This meeting was held with the unrestricted support of Roche Diagnostics. This article summarizes the conclusions from that meeting and is not meant to be a guideline or a position article. The authors take full responsibility for the manuscript and the industrial partner did not advise or interfere with its content. The article aims to stimulate the international community to focus on the role and utility of CSF biomarkers for the diagnosis of prodromal AD.
2. Which CSF biomarkers are the most appropriate?
Although a multitude of CSF biomarkers for specific pathologic changes and nonspecific markers of oxidative damage or inflammation in AD patients have been proposed, many of them have only been reported in single publications and the results have been difficult to replicate. The most consistent findings have been obtained with the Aβ1-42 peptide (Aβ42), total tau (T-tau), and P-tau [21,27,28].
It is important to determine how the change in the CSF levels of these proteins relates to the pathology or neurochemical disturbances in the AD brain. It has been shown that the level of Aβ42 in postmortem ventricular CSF shows an inverse correlation with plaque load in cortical regions [29], a finding that has been replicated in later studies [30] and in biopsied brain from living subjects [31]. In addition, decreased CSF concentrations of Aβ42 have been found to correlate with high retention of Pittsburgh Compound B using PET [30,32–36]. Taken together, these studies show that in AD patients, the reduction in CSFAβ42 reflects deposition of the peptide in plaques in the cortex. It is likely, however, that other pathologic processes will also generate low CSF levels of Aβ42. In support of this, low levels of CSF Aβ42 are also detected in a proportion of cases with plaque-negative Creutzfeldt-Jakob disease (CJD) and in patients with bacterial meningitis [37,38].
Studies comparing CSF samples taken during life with autopsy measures of tangle density show that CSF P-tau correlates with the amount of neocortical NFT pathology [30,39]. Thus, CSF P-tau likely reflects the phosphorylation state of tau and the formation of tangles in the brain. In contrast, CSF T-tau is a more dynamic biomarker, which reflects the intensity of both acute neuronal damage and chronic neuronal degeneration in the brain. For this reason, high CSF T-tau is associated with faster progression from MCI to AD with a more rapid cognitive decline and high mortality rate in patients with AD dementia [40–42].
Several studies have shown that CSF Aβ42, T-tau, and P-tau show no or minimal change during the clinical phase of AD with dementia [43–45] or during the progression from MCI to AD dementia [46]. Thus, the change from normal to pathologic levels most likely occurs during the preclinical asymptomatic phase of the disease. Because CSF biomarkers are stable during the clinical course, they cannot be used as markers for prognosis or staging of the disease, unlike markers such as serum creatinine, which increases with increasing impairment of renal function to reach very high levels in the final stage of kidney failure.
Following the hypothetical model of dynamic biomarkers for AD pathology, the aggregation and deposition of Aβ42 is hypothesized to precede clinical symptoms by many years and to occur before biomarker-detectable tau pathology, NFT formation, and neuronal degeneration [3,47,48]. Thus, the drop in CSF Aβ42 should come before the increase in CSF tau proteins (Fig. 1). This is supported in some biomarker studies in sporadic AD that showed that lowering of Aβ42 in CSF is a very early change [49,50], but studies on familial AD suggest that reductions in CSF Aβ42 and elevations in tau compared with normal occur around 10 to 15 years before the expected onset of symptoms [51,52]. However, studies on large neuropathology case series suggest that tau pathology precedes amyloid plaque pathology [6]. Thus, there may be a discrepancy between neuropathologic and clinical biomarker studies [53], which warrants further studies with longitudinal biomarker assessments to validate AD bio-markers as a precise tool to monitor the evolution of pathology during the clinical course of AD.
Fig. 1.
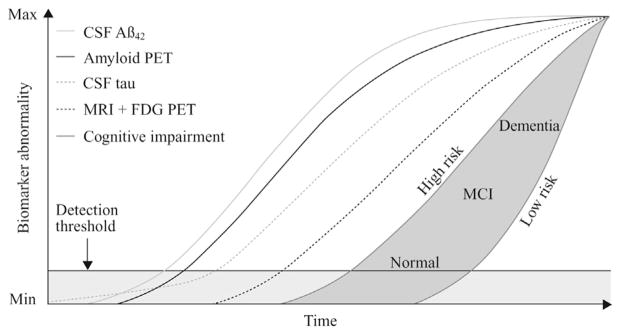
The hypothetical model for biomarkers during the development of AD. The figure shows the relation between time (x-axis) and degree of biomarker abnormality (y-axis). Biomarkers for cortical Aβ deposition include CSF Aβ42 and amyloid PET imaging. Neurodegeneration is measured by FDG PET (lowered glucose metabolism in cortical neurons and glial cells) and structural MRI (atrophy), which are drawn concordantly (dark dashed line). Cognitive impairment is illustrated as a zone (dark gray–filled area) with low- and high-risk borders. By definition, all curves converge at the top right-hand corner of the plot, the point of maximum abnormality. CSF, cerebrospinal fluid; Aβ, amyloid β; PET, positron emission tomography; MRI, magnetic resonance imaging; FDG, fluorodeoxyglucose; MCI, mild cognitive impairment. (Adapted from The Lancet Neurology, Vol. 12, Jack CR Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al., Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers., 207-16,2013, with permission from Elsevier.)
There was a consensus among the Expert Committee that the CSF biomarkers have been validated against neuropathology and other biomarker modalities. The CSF bio-markers show no or minimal change during the clinical course of AD and cannot be used for staging of disease severity. The change from normal to pathologic levels occurs during the preclinical asymptomatic phase of the disease, but whether the changes occur in a temporal order [47,48] or simultaneously, after the spread of Aβ and tau pathology with a continuously increasing number of neurons being affected, needs further study in longitudinal biomarker assessments [53].
3. How well do the CSF biomarkers perform diagnostically?
There are numerous reviews on the diagnostic performance of the CSF biomarkers, including in the early stages of AD. In particular, combining these CSF biomarkers adds to diagnostic accuracy, both for early identification of AD and to differentiate AD from non-AD dementias. Indeed, AD patients typically display low concentrations of Aβ42 and high concentrations of T-tau and P-tau, a pattern commonly referred to as the “AD signature” [27,54–59]. The CSF biomarkers are also highly predictive of progression to AD dementia in MCI patients [56,59–65]. For example, the first study evaluating the power of CSF biomarkers in MCI cases with a long (4–6 year) clinical follow-up showed a 95% sensitivity to identify prodromal AD, with 87% specificity against stable MCI cases and patients with other dementias [62].
Some studies have examined whether CSF biomarkers may predict cognitive decline and dementia in cognitively normal elderly individuals. In 2003, Skoog et al. [50] showed that cognitively normal 85-year-olds who later developed dementia had low CSF Aβ42 but not Aβ40 levels. This finding has been confirmed in a population-based study of normal elderly individuals in which low CSF Aβ42 predicted cognitive decline and AD dementia during the 8-year follow-up period [49]. Some studies suggest that CSF Aβ and tau may predict future cognitive decline in cognitively normal elderly individuals [66], whereas other studies report that the reduction in CSF Aβ42 may be more predictive of future cognitive decline than high CSF tau [67].
Based on the scientific evidence and their own expertise, the Expert Committee agreed that although Aβ42 is an early biomarker when used alone, it is not specific for AD. There was a consensus that although tau is a very sensitive biomarker for AD, it increases during normal aging and has limited ability to discriminate AD from other major forms of dementia, including vascular dementia (VaD) and frontotemporal dementia (FTD). In addition, CSF T-tau shows a very marked increase in CJD, thus having diagnostic value in rapidly progressing dementias. It was also noted by the group that although P-tau in combination with Aβ42 increases the specificity of AD diagnosis, T-tau increases the relative sensitivity. Therefore, although the issues are complex and require careful assessment of sensitivity and specificity, it was concluded that Aβ42 and tau (T-tau and P-tau) should be used in combination and that the evidence for the diagnostic accuracy of the “AD signature” is robust.
4. Are the currently available CSF assays appropriate for widespread use in diagnostic testing?
The key features of a diagnostic biomarker are that it should exhibit high sensitivity/specificity and should be reliable, reproducible, noninvasive, simple to perform, and inexpensive [20,68]. Furthermore, it should be able to identify the clinical disease stage and monitor treatment effects.
Commercial AD biomarker assays are currently used to a greater or lesser extent in various countries as both diagnostic and research tools. However, as CSF biomarkers move toward routine clinical use, it is becoming increasingly evident that there are serious limitations underlying the existing assays for Aβ42, T-tau, and P-tau detection. These assays initially had high diagnostic accuracy and low intralaboratory and intra-assay variabilities [69–73]. Nevertheless, it has been shown that both interassay and interlaboratory variabilities are unacceptably high (20%–35%) [64,73,74]. The large inter- and intralaboratory variations have serious consequences as results attained from different laboratories cannot be directly compared, and the intralaboratory variation also makes it difficult to perform and interpret longitudinal studies. Most importantly, laboratories must establish their own reference ranges using locally collected samples from well-characterized patients: this procedure is difficult, time consuming, and expensive.
As outlined in Table 1, sources of bias may be preanalytical (patient selection, lumbar puncture [LP] procedures, sample handling, and storage), analytical (differences in protocols for sample processing), postanalytical (curve fitting for the standard curve) and include other assay-related factors (within-batch or batch-to-batch variation in manufacturing analytical kits) [75,76]. For these reasons, interlaboratory quality control (QC) surveys are essential [73,76]: in this regard, the Alzheimer’s Association has initiated a worldwide global QC program to identify, quantify, and monitor sources of variability [21]. The aim of the QC program is the standardization of CSF biomarker measurements between laboratories (both research and clinical), allowing direct comparisons of biomarker levels between laboratories and thus providing uniform cutoff levels for diagnosis.
Table 1.
Possible sources of variability between CSF studies (adapted from Mattsson et al. [76])
| Source | Cause | Solution |
|---|---|---|
| Preanalytical | Subject selection and diagnostic criteria | Standardization of clinical procedures and diagnostic criteria |
| LP technique (e.g., time of day/diurnal variation, artifacts due to binding of proteins to collection tube) | Development of SOP for standardization of LP and sample shipment/storage conditions | |
| Specimen shipment or storage (e.g., freeze/thaw effects, artifacts due to oxidation of proteins or binding of proteins to storage tubes) | ||
| Analytical | Assay kit handling and storage | Follow instructions |
| Laboratory equipment (e.g., calibration of pipettes, maintenance of ELISA reader) | Perform CSF biomarker analyses in certified laboratories | |
| Analytical variation (e.g., pipetting errors, use of internal QC samples, acceptance criteria for assay) | Follow kit instructions and SOPs for analytical procedures. Assays performed by certified technicians | |
| Postanalytical | Data-handling differences (e.g., type of curve fitting used, software for data calculation) | Follow SOP for analytical procedures |
| Assay manufacturing | Method optimization (e.g., antibody cross-reactivity, assay linearity, and LLOQ) | Perform assay optimization |
| Within-assay and lot-to-lot variabilities due to reagents and manufacturing (e.g., variation in antibody plate coating, purity of antibodies, quantification, and stability of assay calibrators) | Improve QC procedures during manufacturing and compare lots using human CSF samples |
Abbreviations: CSF, cerebrospinal fluid; LP, lumbar puncture; SOP, standard operating procedure; ELISA, enzyme-linked immunosorbent assay; QC, quality control; LLOQ, lower limit of quantification.
The QC program is run by the Clinical Neurochemistry Laboratory at the University of Gothenburg, Sweden, in conjunction with the Alzheimer’s Association and includes a large number of research, clinical, and pharmaceutical CSF laboratories, together with biotechnology companies and a number of reference laboratories (including the Alzheimer’s Disease Neuroimaging Initiative [ADNI] Biomarker Core). The report for each QC round includes information on the measured biomarker levels for the individual laboratory and, for comparison, the mean and variation in biomarker levels across all laboratories involved in the program. In addition, the longitudinal stability in CSF biomarker levels for the individual laboratory, expressed as percent deviation over time, is reported, with these reports serving as a feedback for the participating laboratories, to identify whether the level of a biomarker is outside an acceptable range and to note sudden changes or longitudinal drifts in CSF biomarker levels [21]. Initial results confirm that interlaboratory bias is high and most likely due to factors related to both analytical procedures and assay production with kit-to-kit variations [76]. It is clear that measures need to be undertaken to optimize biomarker analysis, and to facilitate this, standard operating procedures (SOPs) concerning standardization of sample processing, laboratory techniques, and assay execution have been proposed to reduce the variability and increase the diagnostic utility of AD CSF biomarkers [76,77].
There was a consensus among the Expert Committee that interlaboratory variability will be minimized by the introduction of the worldwide QC programs, SOPs, and the use of high-quality assays that have been produced with rigorous QC measures and that can also be run on fully automated instruments. These measures are required before CSF biomarker assays are appropriate and applicable for widespread clinical use.
5. How should the cutoff levels of biomarkers be defined to ensure acceptable specificity and sensitivity for an AD diagnostic assay?
Sensitivity and specificity are of equal significance in AD diagnosis: a high sensitivity is needed to identify the highest proportion of AD patients and minimize false negatives, whereas a high specificity is necessary for diagnostic accuracy to minimize false positives and exclude non-AD dementias or prodromal stages of non-AD dementias. As early as 1998, an expert committee advised that although conventional diagnostic criteria can provide a sensitivity of around 85% in patients with overt dementia (but much lower in patients with early stage AD), an appropriate biomarker should have sensitivity approaching or greater than this [68]. Although a test with 100% specificity would always differentiate AD from other causes of dementia, a useful biomarker in AD diagnosis was defined as having a specificity of 75% to 85% or more [68].
Table 2 summarizes the performance of Aβ42, T-tau, and P-tau as core CSF biomarkers. It is now the opinion among experts that combined Aβ42, T-tau, and P-tau provide distinctive and reliable biomarkers for AD [21,28,78–80]. It has also been suggested that the combined use of MRI or PET imaging with CSF biomarker analysis [81], with results from specific memory tests and a brief informant-based questionnaire, may further increase diagnostic accuracy, particularly in the prodromal stages [82]. Validation of the AD CSF signature includes defining appropriate cutoff levels to ensure proper specificity and sensitivity.
Table 2.
Performance of core biomarkers in Alzheimer’s disease
| Biomarker | Pathogenic process | Change in biomarker level in AD and prodromal AD | Comment |
|---|---|---|---|
| Aβ42 | Amyloidogenic pathway of APP metabolism | Reduced to around 50% of control levels [27,83] | CSF Aβ42 is the central CSF biomarker for brain Aβ metabolism and plaque formation [29,32,34] |
| P-tau | Tau phosphorylation | Increased to around 200% of control levels [27,83] | High CSF P-tau is specific to AD [83] |
| T-tau | Neuronal degeneration | Increased to around 300% of control levels [27,83] | High CSF T-tau is found in AD but also in other brain disorders with neuronal damage such as stroke and brain trauma non-AD dementias [84,85]; very high CSF T-tau, together with normal P-tau, is found in CJD [86] |
Abbreviations: AD, Alzheimer’s disease; Aβ, amyloid beta; APP, apolipoprotein; CSF, cerebrospinal fluid; P-tau, phosphorylated tau; T-tau, total tau; CJD, Creutzfeldt-Jakob disease.
Although positively correlated, different assay platforms for Aβ42, T-tau, and P-tau generate different absolute values, which pose a challenge for biomarker standardization and the establishment of universal cutoff values to identify groups with or without the underlying disease pathology [57,71]. Because of interlaboratory variation, the reference ranges of these biomarkers must currently be defined by each laboratory as the direct transference of the cutoff values, and reference ranges from one laboratory is not acceptable and leads to variations in diagnostic accuracy. Efforts are ongoing to transfer current manual enzyme-linked immunosorbent assay methods to fully automated instruments or laboratory analyzers and to make validated assay versions with high stability between batches. These assays will, when generally available, most likely show interlaboratory variations low enough to enable the introduction of general cutoff levels for AD diagnosis. In addition, this type of instrument allows direct analysis of single CSF samples, which will substantially shorten turnaround time for results.
In-depth discussions within the Expert Committee regarding the importance of sensitivity and specificity when defining the cutoff for an in vitro diagnostic marker revealed that, in the United States, insurance companies and patients require high specificity to reduce false positives, whereas physicians require high sensitivity to facilitate early treatment. There is a large body of scientific literature showing that the CSF biomarkers Aβ42, T-tau, and P-tau reach sensitivity and specificity figures of 85% to 90%, thus exceeding the 85% sensitivity and 75% specificity recommended by the 1998 Biomarker Working Group [68]. Reasons for not achieving higher performance include that most data come from clinically diagnosed patients who may have mixed pathologies or be misdiagnosed, that a percentage of “controls” may in fact have preclinical AD, and that there is an overlap in pathology between AD and other common dementias such as FTD, VaD, and dementia with Lewy bodies.
6. How should an AD diagnostic CSF biomarker assay be validated?
When validating AD biomarkers, it is essential to have well-matched controls, validation of clinical diagnosis, and confirmation of sensitivity and specificity in large multicenter studies. Sources of bias (at the level of patient inclusion or diagnosis) must be reduced, and retrospective validation studies should be performed using well-classified samples within a stringent trial protocol. Even if studies are large, they may differ in demographics and neuropathology because of the inclusion and exclusion criteria for patients and controls. For example, differences in the education level or percentage of cases with mixed pathologies between cohorts can contribute to study-specific biases. Thus, to validate a biomarker, cutoffs for sensitivity and specificity should be estimated in one study population and validated in another (e.g., European and US cohorts) using the same assay, thus also following the Standards for Reporting Diagnostic Accuracy (STARD) criteria [87]. A large multicenter trial including 750 MCI cases demonstrated good performance with regards to identifying prodromal AD when applying the STARD criteria for validation [64].
The US Food and Drug Administration recently approved 18F florbetapir injection (Amyvid; Lilly USA, Indianapolis, IN, USA) for use in patients being evaluated for AD and other causes of cognitive decline. The 18F florbetapir validation studies were performed in postmortem brain tissues [88], and it has been suggested that the correlation of CSF biomarker levels with neuropathologic findings at autopsy may provide an alternative strategy useful in CSF biomarker validation [54].
Because AD pathology is initiated decades before the clinical symptoms of dementia become apparent, it is possible that, in validation trials, “controls” may have an established but undetected AD pathology. A strategy to avoid the recruitment of asymptomatic patients at risk for AD (i.e., cognitively normal individuals with brain amyloidosis) in the control group is to use an extended observation period (perhaps as long as 6–8 years) and classify patients as AD or controls only when clinical/neuropsychological diagnoses are supported by neuroimaging data (amyloid PET and MRI) and remain aligned with the presence or absence of the AD signature [23,89–91]. In these studies, CSF from normal subjects obtaining a future clinical neurologic diagnosis should not be used as a reference for biomarker validation [90,91] because this later requirement may be seen by some as circular in nature as the predictor and the end point are the same. The Expert Committee also proposed to correlate CSF biomarker results with external end points including amyloid PET and postmortem brain pathology. This approach is superior to the retrospective method as one does not know the time course of the AD signature at an individual level.
The Expert Committee advised that to fully validate an AD biomarker, well-matched samples should be obtained retrospectively from relevant cohorts and that the cutoffs for sensitivity and specificity should be estimated in one study population and validated in another (e.g., European and US cohorts) using the same assay. Furthermore, a large cohort of longitudinal samples would be required, and a reliable control would be vitally important to accomplish appropriate validation.
7. How can the prodromal AD population be identified practically for CSF biomarker testing?
The fact that the primary care physician often is the first point of contact for a patient with cognitive disturbances and possible neurodegenerative disorders [92] may act as a barrier in the identification of prodromal AD patients because when diagnosis occurs, it is typically at a late stage in the disease process [93]. Indeed, there have been reports of delays of up to 4 years from the recording of cognitive symptoms in primary care notes to the diagnosis of dementia being made [94]. The primary care physician may refer the patient to a specialist, but lack of general practitioner awareness may represent a barrier preventing access to a specialized AD service that would be able to determine whether a patient should undergo more rigorous testing.
When considering the role of CSF biomarkers in identifying AD in the predementia or prodromal stage of the disease, one of the most controversial issues is the collection procedure. Clinically, LPs are routinely performed for diagnosis of many other brain disorders (e.g., meningitis, encephalitis, multiple sclerosis) and for the administration of spinal anesthesia and chemotherapy. However, there are many challenges associated with the perception and acceptance of LP and CSF analysis, among both patients and physicians, and currently, there is no common consensus on the use of LP in this setting. Possible reasons for not performing LP include fear of complications, lack of training, time constraints, and low reimbursement related to the absence of an approved test. Additionally, because analysis may not be performed at a local laboratory, CSF samples should be frozen and transported on dry ice, and tests may be associated with high costs and a long turnaround time.
Although the invasive nature of LP may be a factor limiting its use and restricting the identification of prodromal patients, it is important to note that imaging techniques such as PET can also be considered minimally invasive as they require the intravenous administration of a radioactive tracer. Although severe complications associated with LP are rare or absent, fear of side effects, primarily the well-publicized post-LP headache (PLPH), has added to the challenges associated with the implementation of CSF analyses in routine AD diagnostics [95]. In reality, prospective studies of patients admitted for cognitive symptoms report no serious complications, and the incidence of PLPH in patients with cognitive disturbances is low (<2%) and comparable with the risk of headache associated with amyloid PET (1.8%) [95,96]. The frequency of LPs in the clinical diagnostic assessment of patients with cognitive symptoms varies between countries, including in the Western world. Although high in northern Europe (e.g., Sweden and Finland), LP frequency has been very low in the United States and in Canada. The reason for this is unknown and most likely multifactorial in nature. Education programs for clinicians have helped to promote the procedure in Sweden. It is noteworthy that the ADNI study has had an impact on the situation in the United States: from being very rarely performed, the frequency of LPs was very high (>50%) in the first ADNI study [59] and the acceptance rate is >85% in ADNI-2 [97], which suggests both that patients have a much more confident or relaxed attitude about LP than may have been assumed and that the attitude of the clinicians toward the procedure is very important. Indeed, an increasing number of memory clinics in several countries are starting CSF biomarker testing in clinical diagnostic routine.
It is highly pertinent to note that in 2011, the European Medicines Agency determined that the AD CSF biomarker signature is useful for prodromal enrichment of clinical trial populations [98]. However, the Expert Committee observed that the acceptance and positioning of LP procedures and the use of CSF biomarkers vary significantly between countries and that measures are required to overcome the obstacles discussed previously before the use of LP in the “at-risk” prodromal AD population becomes routine. Thus, a global standardization of LP procedures for CSF biomarker analysis is needed, and the appropriate setting (primary care vs. specialist center and neurologist vs. geriatrician vs. psychiatrist) has to be established. As a guide to facilitate this, the Alzheimer’s Association QC program published a standardized flow chart for LP and CSF processing [21].
8. How should CSF biomarkers and imaging techniques be used together to diagnose AD?
Although it has been established that the patterns of regional cerebral atrophy and glucose hypometabolism characteristic of AD can be detected by MRI and FDG PET [23,24,99], only Aβ PET imaging is specific for the amyloid pathology of AD [24]. And although clinical studies demonstrate that baseline imaging and CSF bio-markers are independently associated with cognitive decline [100], the use of imaging techniques such as MRI or FDG PET in combination with the AD biomarker signature increases the sensitivity of AD and prodromal AD diagnosis [100–104]. Moreover, some studies have suggested that the use of MRI estimates of CSF space may afford a correction for the dilution of CSF-derived proteins that are uniquely produced in the brain such as T-tau and P-tau [105,106].
There have been discussions in the literature as to whether a one-step testing strategy with high sensitivity and specificity should be adopted in AD diagnosis or whether it would be more appropriate to use a stepwise approach with relatively inexpensive and highly sensitive core CSF biomarkers followed by more rigorous imaging-based testing to increase diagnostic specificity [19,20]. Because the full spectrum of imaging and CSF analysis methods is seldom used, knowledge on how they may best be combined remains limited, and there are currently no approved international guidelines defining how CSF biomarkers and imaging techniques should be used together to diagnose AD.
As reviewed previously, several studies have shown that low CSF Aβ42 correlates tightly with a high global cortical amyloid PET signal. In other words, the vast majority of AD patients (85%–100%) show either normal or positive results by amyloid PET scanning and CSFAβ42 measurements [32–36]. A small percentage of patients show discordant amyloid biomarker results, either positive CSF Aβ42 but normal amyloid PET or normal CSF Aβ42 but positive amyloid PET. One study contained a large enough number of patients to allow comparisons of these groups [107]. In this study, the CSF-positive and PET-negative group showed more cognitive impairment and higher CSF tau levels, whereas the CSF-negative and PET-positive group consisted of persons with normal cognition and those with only minimal cognitive impairment [107]. It is too early to conclude that there is any true discrepancy between the two techniques that allows identification of subgroups of patients or individuals. Instead, we need further studies to understand the rationale for the discordant cases.
However, the EFNS recommends the use of CSF biomarkers, computed tomography, MRI/FDG PET, and cognitive assessment for differential AD diagnosis [1]; the NIA-AA describes a diagnostic model that incorporates CSF Aβ42 or amyloid PET and CSF tau, structural MRI, or FDG PET [26], and the “Dubois diagnostic criteria” stipulate that in addition to memory impairment, there must also be at least one or more abnormal biomarkers (MRI, PET, and Aβ/tau proteins) for a diagnosis of AD [11].
9. What are the ethical considerations associated with a diagnosis of AD before the advent of disease-modifying drugs?
Diagnosis of prodromal AD before the availability of disease-modifying drugs raises ethical concerns, and the debate often focuses on the potentially harmful psychological consequences of early diagnosis to the patient. However, early diagnosis does not prompt a severe emotional response in most patients and may, in fact, provide some relief once an explanation for the cognitive symptoms is known [108]. Early diagnosis can be thought of as an opportunity for patients to plan for the future in an informed manner and may help identify patients with cognitive dysfunction secondary to other diseases that may be responsive to available treatments [109].
However, the ethical implications of a misdiagnosis of AD should not be ignored; if a false-positive diagnosis results in treatment, it is important to consider any potential and harmful side effects. The risk of serious adverse effects from investigative treatments should not be underestimated [110], and although rigorous toxicity studies are performed on all new drugs, rare side effects are often detected only after widespread use [109].
Although no cure for AD is currently available, ongoing development of disease-modifying drugs will substantially alter the meaning of a diagnosis of AD, moving it into the domain of potentially treatable illnesses. Thus, it is anticipated that ethical implications will change dramatically when disease-modifying drugs become available.
10. Conclusions
The evidence for the benefit of CSF biomarkers in AD research is highly compelling as many studies have shown the utility of the AD signature (low Aβ42 together with high T-tau and P-tau) for AD diagnosis and have demonstrated its use as a predictor of disease progression [56,59,60,62,103,111,112]. Indeed, the high sensitivity/specificity of the AD signature could lead to its development as the “gold standard” in early/prodromal AD diagnostics. However, because no universally validated test is currently available, it is essential to further accelerate the validation process of the AD signature through data pooling and well-designed longitudinal studies [25].
The significance of interlaboratory variations in diagnostic sensitivity and specificity must not be underestimated, and it is vitally important to establish SOPs with reference to sample collection, storage and processing, assay format, and data interpretation. This can be met by applying a strict clinical trial framework (standardization of design, efficacy criteria, statistical methodology, etc.), although in the routine diagnostic laboratory, it is important to have established and well-validated cutoff values and rigorous QC procedures. Development of assays for fully automated analytical instruments would also be instrumental in reducing both analytical and batch-to-batch variation for these biomarkers. The relevance of an early diagnosis relies on the hypothesis that pharmacologic interventions with disease-modifying drugs are likely to produce clinically relevant benefits if started early in the AD continuum. CSF biomarkers have clinical utility in the differentiation between AD dementia, MCI due to normal aging, and non-AD dementias [113], and experts agree that the further discovery and validation of CSF biomarkers is essential to improve early diagnosis and accelerate the development of new therapies.
RESEARCH IN CONTEXT.
Systematic review: Numerous research articles show that the Alzheimer’s disease (AD) cerebrospinal fluid (CSF) biomarkers amyloid-β (Aβ1-42), total tau, and phosphorylated tau are sensitive and specific to identify prodromal AD patients.
Interpretation: Because clinical trials show that 15% to 30% of clinically diagnosed AD patients do not have amyloid pathology, these biomarkers would be highly valuable to enrich trials with true AD patients. Recent data showing large between-laboratory variations for current immunoassays are of concern because this will impede general use of CSF biomarkers in clinical practice.
Interpretation: Worldwide efforts are ongoing for standardization of CSF biomarkers, from preanalytical sample handling to laboratory procedures. A quality control program is running to monitor between-laboratory and longitudinal performance. Biotechnology companies are also working on developing validated and highly reproducible assays on fully automated instruments. These global standardization efforts will facilitate a general application of CSF biomarkers, both for clinical diagnosis and in drug development.
Acknowledgments
K.B., B.D., and H.H. receive research support from Bio-markers for Alzheimer’s disease and Parkinson’s disease (BIOMARKAPD). H.H. was supported by the Katharina-Hardt-Foundation, Bad Homburg, Germany. H.H. and B.D. thank the Fondation Pour La Recherche Sur Alzheimer (FRA), Paris, France, for support. A.M.F. receives research support from the National Institute on Aging (grant P01AG026276) and the Hope Centre for Neurological Disorders. P.L. is supported by the German Bundesministerium für Bildung und Forschung (grant 01ED1203D) within the BiomarkAPD Project of the JPND. M.J.de.L. is supported by the NIH-NIA. We acknowledge the role of Elizabeth Davison, PhD, and Sue Laing, PhD (Elements Communications Ltd, Westerham, UK), in providing writing and editorial support funded by Roche Diagnostics International Ltd.
Footnotes
Conflicts of interest: All authors participated in the international Expert Committee meeting (held in April 2012) and were involved in the discussions regarding development and validation of CSF diagnostic assays, which form the basis of this publication. This meeting was held with the unrestricted support of Roche Diagnostics International Ltd. The authors take full responsibility for the manuscript and the industrial partner did not advise or interfere with its content. Each of the authors contributed to the writing, critical review and revision of the article, and approved the final version. K.B. has served as a consultant for Innogenetics, Kyowa Hakko Kirin Pharma, Pfizer, and Roche. H.H. declares no competing financial interests related to the present article. During the last 2 years (2011–2013), he has received lecture honoraria, research grants, and/or travel funding, participated in scientific advisory boards and/or as a consultant to diagnostic and biotechnology companies involved in the manufacture and marketing of biomarkers and/or diagnostics for cognitive impairment and Alzheimer’s disease including Roche Diagnostics, GE Healthcare, Avid, Eli Lilly and Company, GlaxoSmithKline-Biologicals, Jung-Diagnostics, Thermo Fisher Scientific Clinical Diagnostics, and Cytox. H.H. is the coinventor in pending patent submissions relating to biological markers and/or diagnostics and has not received any royalties. A.M.F. is a member of the US Alzheimer’s Disease Advisory Board for Lilly USA. P.L. has received consultation honoraria from Innogenetics and IBL International. Through collaboration with New York University, M.J.de.L. holds patents on PETand MRI image analysis techniques that have been commercially licensed.
References
- 1.Hort J, O’Brien JT, Gainotti G, Pirttila T, Popescu BO, Rektorova I, et al. EFNS guidelines for the diagnosis and management of Alzheimer’s disease. Eur J Neurol. 2010;17:1236–48. doi: 10.1111/j.1468-1331.2010.03040.x. [DOI] [PubMed] [Google Scholar]
- 2.Thies W, Bleiler L. 2011 Alzheimer’s disease facts and figures. Alzheimers Dement. 2011;7:208–44. doi: 10.1016/j.jalz.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 3.Trojanowski JQ, Hampel H. Neurodegenerative disease biomarkers: guideposts for disease prevention through early diagnosis and intervention. Prog Neurobiol. 2011;95:491–5. doi: 10.1016/j.pneurobio.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Leon MJ, Golomb J, George AE, Convit A, Tarshish CY, McRae T, et al. The radiologic prediction of Alzheimer disease: the atrophic hippocampal formation. AJNR Am J Neuroradiol. 1993;14:897–906. [PMC free article] [PubMed] [Google Scholar]
- 5.Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- 6.Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997;18:351–7. doi: 10.1016/s0197-4580(97)00056-0. [DOI] [PubMed] [Google Scholar]
- 7.de Leon MJ, Convit A, Wolf OT, Tarshish CY, DeSanti S, Rusinek H, et al. Prediction of cognitive decline in normal elderly subjects with 2-[(18)F]fluoro-2-deoxy-D-glucose/poitron-emission tomography (FDG/PET) Proc Natl Acad Sci U S A. 2001;98:10966–71. doi: 10.1073/pnas.191044198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hulette CM, Welsh-Bohmer KA, Murray MG, Saunders AM, Mash DC, McIntyre LM. Neuropathological and neuropsychological changes in “normal” aging: evidence for preclinical Alzheimer disease in cognitively normal individuals. J Neuropathol Exp Neurol. 1998;57:1168–74. doi: 10.1097/00005072-199812000-00009. [DOI] [PubMed] [Google Scholar]
- 9.Morris JC, Storandt M, McKeel DW, Jr, Rubin EH, Price JL, Grant EA, et al. Cerebral amyloid deposition and diffuse plaques in “normal” aging: evidence for presymptomatic and very mild Alzheimer’s disease. Neurology. 1996;46:707–19. doi: 10.1212/wnl.46.3.707. [DOI] [PubMed] [Google Scholar]
- 10.Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann Neurol. 1999;45:358–68. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 11.Dubois B, Feldman HH, Jacova C, DeKosky ST, Barberger-Gateau P, Cummings J, et al. Research criteria for the diagnosis of Alzheimer’s disease: revising the NINCDS-ADRDA criteria. Lancet Neurol. 2007;6:734–46. doi: 10.1016/S1474-4422(07)70178-3. [DOI] [PubMed] [Google Scholar]
- 12.Petersen RC, Aisen PS, Beckett LA, Donohue MC, Gamst AC, Harvey DJ, et al. Alzheimer’s Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology. 2010;74:201–9. doi: 10.1212/WNL.0b013e3181cb3e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Torpy JM, Lynm C, Glass RM. JAMA patient page. Mild cognitive impairment. JAMA. 2009;302:452. doi: 10.1001/jama.302.4.452. [DOI] [PubMed] [Google Scholar]
- 14.Aluise CD, Sowell RA, Butterfield DA. Peptides and proteins in plasma and cerebrospinal fluid as biomarkers for the prediction, diagnosis, and monitoring of therapeutic efficacy of Alzheimer’s disease. Biochim Biophys Acta. 2008;1782:549–58. doi: 10.1016/j.bbadis.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarazin M, Berr C, De Rotrou J, Fabrigoule C, Pasquier F, Legrain S, et al. Amnestic syndrome of the medial temporal type identifies prodromal AD: a longitudinal study. Neurology. 2007;69:1859–67. doi: 10.1212/01.wnl.0000279336.36610.f7. [DOI] [PubMed] [Google Scholar]
- 16.Wagner M, Wolf S, Reischies FM, Daerr M, Wolfsgruber S, Jessen F, et al. Biomarker validation of a cued recall memory deficit in prodromal Alzheimer disease. Neurology. 2012;78:379–86. doi: 10.1212/WNL.0b013e318245f447. [DOI] [PubMed] [Google Scholar]
- 17.Beach TG, Monsell SE, Phillips LE, Kukull W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005–2010. J Neuropathol Exp Neurol. 2012;71:266–73. doi: 10.1097/NEN.0b013e31824b211b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370:322–33. doi: 10.1056/NEJMoa1304839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hampel H, Shen Y, Walsh DM, Aisen P, Shaw LM, Zetterberg H, et al. Biological markers of amyloid beta-related mechanisms in Alzheimer’s disease. Exp Neurol. 2010;223:334–46. doi: 10.1016/j.expneurol.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hampel H, Frank R, Broich K, Teipel SJ, Katz RG, Hardy J, et al. Biomarkers for Alzheimer’s disease: academic, industry and regulatory perspectives. Nat Rev Drug Discov. 2010;9:560–74. doi: 10.1038/nrd3115. [DOI] [PubMed] [Google Scholar]
- 21.Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. 2010;6:131–44. doi: 10.1038/nrneurol.2010.4. [DOI] [PubMed] [Google Scholar]
- 22.De Santi S, de Leon MJ, Rusinek H, Convit A, Tarshish CY, Roche A, et al. Hippocampal formation glucose metabolism and volume losses in MCI and AD. Neurobiol Aging. 2001;22:529–39. doi: 10.1016/s0197-4580(01)00230-5. [DOI] [PubMed] [Google Scholar]
- 23.Mosconi L, Tsui WH, Pupi A, De Santi S, Drzezga A, Minoshima S, et al. (18)F-FDG PET database of longitudinally confirmed healthy elderly individuals improves detection of mild cognitive impairment and Alzheimer’s disease. J Nucl Med. 2007;48:1129–34. doi: 10.2967/jnumed.107.040675. [DOI] [PubMed] [Google Scholar]
- 24.Johnson KA, Fox NC, Sperling RA, Klunk WE. Brain imaging in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006213. doi: 10.1101/cshperspect.a006213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hampel H, Lista S, Khachaturian ZS. Development of biomarkers to chart all Alzheimer’s disease stages: the royal road to cutting the therapeutic Gordian Knot. Alzheimers Dement. 2012;8:312–36. doi: 10.1016/j.jalz.2012.05.2116. [DOI] [PubMed] [Google Scholar]
- 26.McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Jr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263–9. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blennow K, Hampel H. CSF markers for incipient Alzheimer’s disease. Lancet Neurol. 2003;2:605–13. doi: 10.1016/s1474-4422(03)00530-1. [DOI] [PubMed] [Google Scholar]
- 28.Blennow K, Zetterberg H, Fagan AM. Fluid biomarkers in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006221. doi: 10.1101/cshperspect.a006221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strozyk D, Blennow K, White LR, Launer LJ. CSF Abeta 42 levels correlate with amyloid-neuropathology in a population-based autopsy study. Neurology. 2003;60:652–6. doi: 10.1212/01.wnl.0000046581.81650.d0. [DOI] [PubMed] [Google Scholar]
- 30.Tapiola T, Alafuzoff I, Herukka SK, Parkkinen L, Hartikainen P, Soininen H, et al. Cerebrospinal fluid {beta}-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch Neurol. 2009;66:382–9. doi: 10.1001/archneurol.2008.596. [DOI] [PubMed] [Google Scholar]
- 31.Seppala TT, Nerg O, Koivisto AM, Rummukainen J, Puli L, Zetterberg H, et al. CSF biomarkers for Alzheimer disease correlate with cortical brain biopsy findings. Neurology. 2012;78:1568–75. doi: 10.1212/WNL.0b013e3182563bd0. [DOI] [PubMed] [Google Scholar]
- 32.Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006;59:512–9. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- 33.Fagan AM, Mintun MA, Shah AR, Aldea P, Roe CM, Mach RH, et al. Cerebrospinal fluid tau and ptau(181) increase with cortical amyloid deposition in cognitively normal individuals: implications for future clinical trials of Alzheimer’s disease. EMBO Mol Med. 2009;1:371–80. doi: 10.1002/emmm.200900048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Forsberg A, Engler H, Almkvist O, Blomquist G, Hagman G, Wall A, et al. PET imaging of amyloid deposition in patients with mild cognitive impairment. Neurobiol Aging. 2008;29:1456–65. doi: 10.1016/j.neurobiolaging.2007.03.029. [DOI] [PubMed] [Google Scholar]
- 35.Jagust WJ, Landau SM, Shaw LM, Trojanowski JQ, Koeppe RA, Reiman EM, et al. Relationships between biomarkers in aging and dementia. Neurology. 2009;73:1193–9. doi: 10.1212/WNL.0b013e3181bc010c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tolboom N, van der Flier WM, Yaqub M, Boellaard R, Verwey NA, Blankenstein MA, et al. Relationship of cerebrospinal fluid markers to 11C-PiB and 18F-FDDNP binding. J Nucl Med. 2009;50:1464–70. doi: 10.2967/jnumed.109.064360. [DOI] [PubMed] [Google Scholar]
- 37.Sjogren M, Gisslen M, Vanmechelen E, Blennow K. Low cerebrospinal fluid beta-amyloid 42 in patients with acute bacterial meningitis and normalization after treatment. Neurosci Lett. 2001;314:33–6. doi: 10.1016/s0304-3940(01)02285-6. [DOI] [PubMed] [Google Scholar]
- 38.Wiltfang J, Esselmann H, Smirnov A, Bibl M, Cepek L, Steinacker P, et al. Beta-amyloid peptides in cerebrospinal fluid of patients with Creutzfeldt-Jakob disease. Ann Neurol. 2003;54:263–7. doi: 10.1002/ana.10661. [DOI] [PubMed] [Google Scholar]
- 39.Buerger K, Ewers M, Pirttila T, Zinkowski R, Alafuzoff I, Teipel SJ, et al. CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer’s disease. Brain. 2006;129:3035–41. doi: 10.1093/brain/awl269. [DOI] [PubMed] [Google Scholar]
- 40.Blom ES, Giedraitis V, Zetterberg H, Fukumoto H, Blennow K, Hyman BT, et al. Rapid progression from mild cognitive impairment to Alzheimer’s disease in subjects with elevated levels of tau in cerebrospinal fluid and the APOE epsilon4/epsilon4 genotype. Dement Geriatr Cogn Disord. 2009;27:458–64. doi: 10.1159/000216841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Samgard K, Zetterberg H, Blennow K, Hansson O, Minthon L, Londos E. Cerebrospinal fluid total tau as a marker of Alzheimer’s disease intensity. Int J Geriatr Psychiatry. 2010;25:403–10. doi: 10.1002/gps.2353. [DOI] [PubMed] [Google Scholar]
- 42.Wallin AK, Hansson O, Blennow K, Londos E, Minthon L. Can CSF biomarkers or pre-treatment progression rate predict response to cholinesterase inhibitor treatment in Alzheimer’s disease? Int J Geriatr Psychiatry. 2009;24:638–47. doi: 10.1002/gps.2195. [DOI] [PubMed] [Google Scholar]
- 43.Blennow K, Zetterberg H, Minthon L, Lannfelt L, Strid S, Annas P, et al. Longitudinal stability of CSF biomarkers in Alzheimer’s disease. Neurosci Lett. 2007;419:18–22. doi: 10.1016/j.neulet.2007.03.064. [DOI] [PubMed] [Google Scholar]
- 44.Mattsson N, Portelius E, Rolstad S, Gustavsson M, Andreasson U, Stridsberg M, et al. Longitudinal cerebrospinal fluid biomarkers over four years in mild cognitive impairment. J Alzheimers Dis. 2012;30:767–78. doi: 10.3233/JAD-2012-120019. [DOI] [PubMed] [Google Scholar]
- 45.Zetterberg H, Pedersen M, Lind K, Svensson M, Rolstad S, Eckerstrom C, et al. Intra-individual stability of CSF biomarkers for Alzheimer’s disease over two years. J Alzheimers Dis. 2007;12:255–60. doi: 10.3233/jad-2007-12307. [DOI] [PubMed] [Google Scholar]
- 46.Andreasen N, Minthon L, Vanmechelen E, Vanderstichele H, Davidsson P, Winblad B, et al. Cerebrospinal fluid tau and Abeta42 as predictors of development of Alzheimer’s disease in patients with mild cognitive impairment. Neurosci Lett. 1999;273:5–8. doi: 10.1016/s0304-3940(99)00617-5. [DOI] [PubMed] [Google Scholar]
- 47.Jack CR, Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9:119–28. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jack CR, Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic bio-markers. Lancet Neurol. 2013;12:207–16. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gustafson DR, Skoog I, Rosengren L, Zetterberg H, Blennow K. Cerebrospinal fluid beta-amyloid 1-42 concentration may predict cognitive decline in older women. J Neurol Neurosurg Psychiatry. 2007;78:461–4. doi: 10.1136/jnnp.2006.100529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Skoog I, Davidsson P, Aevarsson O, Vanderstichele H, Vanmechelen E, Blennow K. Cerebrospinal fluid beta-amyloid 42 is reduced before the onset of sporadic dementia: a population-based study in 85-year-olds. Dement Geriatr Cogn Disord. 2003;15:169–76. doi: 10.1159/000068478. [DOI] [PubMed] [Google Scholar]
- 51.Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ringman JM, Coppola G, Elashoff D, Rodriguez-Agudelo Y, Medina LD, Gylys K, et al. Cerebrospinal fluid biomarkers and proximity to diagnosis in preclinical familial Alzheimer’s disease. Dement Geriatr Cogn Disord. 2012;33:1–5. doi: 10.1159/000335729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Braak H, Zetterberg H, Del Tredici K, Blennow K. Intraneuronal tau aggregation precedes diffuse plaque deposition, but amyloid-beta changes occur before increases of tau in cerebrospinal fluid. Acta Neuropathol. 2013;126:631–41. doi: 10.1007/s00401-013-1139-0. [DOI] [PubMed] [Google Scholar]
- 54.Clark CM, Xie S, Chittams J, Ewbank D, Peskind E, Galasko D, et al. Cerebrospinal fluid tau and beta-amyloid: how well do these bio-markers reflect autopsy-confirmed dementia diagnoses? Arch Neurol. 2003;60:1696–702. doi: 10.1001/archneur.60.12.1696. [DOI] [PubMed] [Google Scholar]
- 55.de Leon MJ, DeSanti S, Zinkowski R, Mehta PD, Pratico D, Segal S, et al. Longitudinal CSF and MRI biomarkers improve the diagnosis of mild cognitive impairment. Neurobiol Aging. 2006;27:394–401. doi: 10.1016/j.neurobiolaging.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 56.Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol. 2007;64:343–9. doi: 10.1001/archneur.64.3.noc60123. [DOI] [PubMed] [Google Scholar]
- 57.Fagan AM, Shaw LM, Xiong C, Vanderstichele H, Mintun MA, Trojanowski JQ, et al. Comparison of analytical platforms for cerebrospinal fluid measures of beta-amyloid 1-42, total tau, and p-tau181 for identifying Alzheimer disease amyloid plaque pathology. Arch Neurol. 2011;68:1137–44. doi: 10.1001/archneurol.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Galasko D, Chang L, Motter R, Clark CM, Kaye J, Knopman D, et al. High cerebrospinal fluid tau and low amyloid beta42 levels in the clinical diagnosis of Alzheimer disease and relation to apolipoprotein E genotype. Arch Neurol. 1998;55:937–45. doi: 10.1001/archneur.55.7.937. [DOI] [PubMed] [Google Scholar]
- 59.Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, et al. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol. 2009;65:403–13. doi: 10.1002/ana.21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brys M, Pirraglia E, Rich K, Rolstad S, Mosconi L, Switalski R, et al. Prediction and longitudinal study of CSF biomarkers in mild cognitive impairment. Neurobiol Aging. 2009;30:682–90. doi: 10.1016/j.neurobiolaging.2007.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Diniz BS, Pinto JA, Junior, Forlenza OV. Do CSF total tau, phosphor-ylated tau, and beta-amyloid 42 help to predict progression of mild cognitive impairment to Alzheimer’s disease? A systematic review and meta-analysis of the literature. World J Biol Psychiatry. 2008;9:172–82. doi: 10.1080/15622970701535502. [DOI] [PubMed] [Google Scholar]
- 62.Hansson O, Zetterberg H, Buchhave P, Londos E, Blennow K, Minthon L. Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol. 2006;5:228–34. doi: 10.1016/S1474-4422(06)70355-6. [DOI] [PubMed] [Google Scholar]
- 63.Li G, Sokal I, Quinn JF, Leverenz JB, Brodey M, Schellenberg GD, et al. CSF tau/Abeta42 ratio for increased risk of mild cognitive impairment: a follow-up study. Neurology. 2007;69:631–9. doi: 10.1212/01.wnl.0000267428.62582.aa. [DOI] [PubMed] [Google Scholar]
- 64.Mattsson N, Zetterberg H, Hansson O, Andreasen N, Parnetti L, Jonsson M, et al. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA. 2009;302:385–93. doi: 10.1001/jama.2009.1064. [DOI] [PubMed] [Google Scholar]
- 65.Snider BJ, Fagan AM, Roe C, Shah AR, Grant EA, Xiong C, et al. Cerebrospinal fluid biomarkers and rate of cognitive decline in very mild dementia of the Alzheimer type. Arch Neurol. 2009;66:638–45. doi: 10.1001/archneurol.2009.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vos SJ, Xiong C, Visser PJ, Jasielec MS, Hassenstab J, Grant EA, et al. Preclinical Alzheimer’s disease and its outcome: a longitudinal cohort study. Lancet Neurol. 2013;12:957–65. doi: 10.1016/S1474-4422(13)70194-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stomrud E, Hansson O, Blennow K, Minthon L, Londos E. Cerebrospinal fluid biomarkers predict decline in subjective cognitive function over 3 years in healthy elderly. Dement Geriatr Cogn Disord. 2007;24:118–24. doi: 10.1159/000105017. [DOI] [PubMed] [Google Scholar]
- 68.No authors listed Consensus report of the Working Group on: molecular and biochemical markers of Alzheimer’s disease. Neurobiol Aging. 1998;19:109–16. [PubMed] [Google Scholar]
- 69.Andreasen N, Hesse C, Davidsson P, Minthon L, Wallin A, Winblad B, et al. Cerebrospinal fluid beta-amyloid(1-42) in Alzheimer disease: differences between early- and late-onset Alzheimer disease and stability during the course of disease. Arch Neurol. 1999;56:673–80. doi: 10.1001/archneur.56.6.673. [DOI] [PubMed] [Google Scholar]
- 70.Olsson A, Vanderstichele H, Andreasen N, de Meyer G, Wallin A, Holmberg B, et al. Simultaneous measurement of beta-amyloid(1-42), total tau, and phosphorylated tau (Thr181) in cerebrospinal fluid by the xMAP technology. Clin Chem. 2005;51:336–45. doi: 10.1373/clinchem.2004.039347. [DOI] [PubMed] [Google Scholar]
- 71.Reijn TS, Rikkert MO, van Geel WJ, de Jong D, Verbeek MM. Diagnostic accuracy of ELISA and xMAP technology for analysis of amyloid beta(42) and tau proteins. Clin Chem. 2007;53:859–65. doi: 10.1373/clinchem.2006.081679. [DOI] [PubMed] [Google Scholar]
- 72.Andreasen N, Vanmechelen E, Vanderstichele H, Davidsson P, Blennow K. Cerebrospinal fluid levels of total-tau, phospho-tau and A beta 42 predicts development of Alzheimer’s disease in patients with mild cognitive impairment. Acta Neurol Scand Suppl. 2003;179:47–51. doi: 10.1034/j.1600-0404.107.s179.9.x. [DOI] [PubMed] [Google Scholar]
- 73.Lewczuk P, Beck G, Ganslandt O, Esselmann H, Deisenhammer F, Regeniter A, et al. International quality control survey of neurochemical dementia diagnostics. Neurosci Lett. 2006;409:1–4. doi: 10.1016/j.neulet.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 74.Verwey NA, van der Flier WM, Blennow K, Clark C, Sokolow S, De Deyn PP, et al. Aworldwide multicentre comparison of assays for cerebrospinal fluid biomarkers in Alzheimer’s disease. Ann Clin Biochem. 2009;46:235–40. doi: 10.1258/acb.2009.008232. [DOI] [PubMed] [Google Scholar]
- 75.Teunissen CE, Verwey NA, Kester MI, van Uffelen K, Blankenstein MA. Standardization of assay procedures for analysis of the CSF biomarkers amyloid beta(1-42), tau, and phosphorylated tau in Alzheimer’s disease: report of an International Workshop. Int J Alzheimers Dis. 2010 doi: 10.4061/2010/635053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mattsson N, Andreasson U, Persson S, Arai H, Batish SD, Bernardini S, et al. The Alzheimer’s Association external quality control program for cerebrospinal fluid biomarkers. Alzheimers Dement. 2011;7:386–95. e6. doi: 10.1016/j.jalz.2011.05.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.del Campo M, Mollenhauer B, Bertolotto A, Engelborghs S, Hampel H, Simonsen AH, et al. Recommendations to standardize preanalytical confounding factors in Alzheimer’s and Parkinson’s disease cerebrospinal fluid biomarkers: an update. Biomark Med. 2012;6:419–30. doi: 10.2217/bmm.12.46. [DOI] [PubMed] [Google Scholar]
- 78.Dubois B, Feldman HH, Jacova C, Cummings JL, DeKosky ST, Barberger-Gateau P, et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol. 2010;9:1118–27. doi: 10.1016/S1474-4422(10)70223-4. [DOI] [PubMed] [Google Scholar]
- 79.Fagan AM, Holtzman DM. Cerebrospinal fluid biomarkers of Alzheimer’s disease. Biomark Med. 2010;4:51–63. doi: 10.2217/BMM.09.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lewczuk P, Zimmermann R, Wiltfang J, Kornhuber J. Neurochemical dementia diagnostics: a simple algorithm for interpretation of the CSF biomarkers. J Neural Transm. 2009;116:1163–7. doi: 10.1007/s00702-009-0277-y. [DOI] [PubMed] [Google Scholar]
- 81.Teipel SJ, Sabri O, Grothe M, Barthel H, Prvulovic D, Buerger K, et al. Perspectives for multimodal neurochemical and imaging bio-markers in Alzheimer’s disease. J Alzheimers Dis. 2013;33:S329–47. doi: 10.3233/JAD-2012-129030. [DOI] [PubMed] [Google Scholar]
- 82.Galvin JE, Fagan AM, Holtzman DM, Mintun MA, Morris JC. Relationship of dementia screening tests with biomarkers of Alzheimer’s disease. Brain. 2010;133:3290–300. doi: 10.1093/brain/awq204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Blennow K. Cerebrospinal fluid protein biomarkers for Alzheimer’s disease. NeuroRx. 2004;1:213–25. doi: 10.1602/neurorx.1.2.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hesse C, Rosengren L, Andreasen N, Davidsson P, Vanderstichele H, Vanmechelen E, et al. Transient increase in total tau but not phospho-tau in human cerebrospinal fluid after acute stroke. Neurosci Lett. 2001;297:187–90. doi: 10.1016/s0304-3940(00)01697-9. [DOI] [PubMed] [Google Scholar]
- 85.Ost M, Nylen K, Csajbok L, Ohrfelt AO, Tullberg M, Wikkelso C, et al. Initial CSF total tau correlates with 1-year outcome in patients with traumatic brain injury. Neurology. 2006;67:1600–4. doi: 10.1212/01.wnl.0000242732.06714.0f. [DOI] [PubMed] [Google Scholar]
- 86.Riemenschneider M, Wagenpfeil S, Vanderstichele H, Otto M, Wiltfang J, Kretzschmar H, et al. Phospho-tau/total tau ratio in cerebrospinal fluid discriminates Creutzfeldt-Jakob disease from other dementias. Mol Psychiatry. 2003;8:343–7. doi: 10.1038/sj.mp.4001220. [DOI] [PubMed] [Google Scholar]
- 87.Bossuyt PM, Reitsma JB, Bruns DE, Gatsonis CA, Glasziou PP, Irwig LM, et al. Towards complete and accurate reporting of studies of diagnostic accuracy: the STARD initiative. BMJ. 2003;326:41–4. doi: 10.1136/bmj.326.7379.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Choi SR, Schneider JA, Bennett DA, Beach TG, Bedell BJ, Zehntner SP, et al. Correlation of amyloid PET ligand florbetapir F 18 binding with Abeta aggregation and neuritic plaque deposition in postmortem brain tissue. Alzheimer Dis Assoc Disord. 2012;26:8–16. doi: 10.1097/WAD.0b013e31821300bc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.De Santi S, Pirraglia E, Barr W, Babb J, Williams S, Rogers K, et al. Robust and conventional neuropsychological norms: diagnosis and prediction of age-related cognitive decline. Neuropsychology. 2008;22:469–84. doi: 10.1037/0894-4105.22.4.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lewczuk P, Kornhuber J, Vanmechelen E, Peters O, Heuser I, Maier W, et al. Amyloid beta peptides in plasma in early diagnosis of Alzheimer’s disease: a multicenter study with multiplexing. Exp Neurol. 2010;223:366–70. doi: 10.1016/j.expneurol.2009.07.024. [DOI] [PubMed] [Google Scholar]
- 91.Lewczuk P, Kamrowski-Kruck H, Peters O, Heuser I, Jessen F, Popp J, et al. Soluble amyloid precursor proteins in the cerebrospinal fluid as novel potential biomarkers of Alzheimer’s disease: a multi-center study. Mol Psychiatry. 2010;15:138–45. doi: 10.1038/mp.2008.84. [DOI] [PubMed] [Google Scholar]
- 92.Galvin JE, Sadowsky CH. Practical guidelines for the recognition and diagnosis of dementia. J Am Board Fam Med. 2012;25:367–82. doi: 10.3122/jabfm.2012.03.100181. [DOI] [PubMed] [Google Scholar]
- 93.Wilkinson D, Sganga A, Stave C, O’Connell B. Implications of the Facing Dementia Survey for health care professionals across Europe. Int J Clin Pract Suppl. 2005:27–31. doi: 10.1111/j.1368-504x.2005.00484.x. [DOI] [PubMed] [Google Scholar]
- 94.Bamford C, Eccles M, Steen N, Robinson L. Can primary care record review facilitate earlier diagnosis of dementia? Fam Pract. 2007;24:108–16. doi: 10.1093/fampra/cml068. [DOI] [PubMed] [Google Scholar]
- 95.Zetterberg H, Tullhog K, Hansson O, Minthon L, Londos E, Blennow K. Low incidence of post-lumbar puncture headache in 1,089 consecutive memory clinic patients. Eur Neurol. 2010;63:326–30. doi: 10.1159/000311703. [DOI] [PubMed] [Google Scholar]
- 96.Peskind E, Nordberg A, Darreh-Shori T, Soininen H. Safety of lumbar puncture procedures in patients with Alzheimer’s disease. Curr Alzheimer Res. 2009;6:290–2. doi: 10.2174/156720509788486509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. [last accessed 4 February 2014];Alzheimer’s Disease Neuroimaging Initiative. Available at: http://adni.loni.usc.edu.
- 98.European Medicines Agency. Qualification opinion of Alzheimer’s disease. [last accessed 4 February 2014];Novel methodologies/biomarkers for BMS-708163. 2011 Available at: http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2011/02/WC500102018.pdf.
- 99.Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:280–92. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Vemuri P, Wiste HJ, Weigand SD, Shaw LM, Trojanowski JQ, Weiner MW, et al. MRI and CSF biomarkers in normal, MCI, and AD subjects: predicting future clinical change. Neurology. 2009;73:294–301. doi: 10.1212/WNL.0b013e3181af79fb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Walhovd KB, Fjell AM, Brewer J, McEvoy LK, Fennema-Notestine C, Hagler DJ, Jr, et al. Combining MR imaging, positron-emission tomography, and CSF biomarkers in the diagnosis and prognosis of Alzheimer disease. AJNR Am J Neuroradiol. 2010;31:347–54. doi: 10.3174/ajnr.A1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Vos S, van Rossum I, Burns L, Knol D, Scheltens P, Soininen H, et al. Test sequence of CSF and MRI biomarkers for prediction of AD in subjects with MCI. Neurobiol Aging. 2012;33:2272–81. doi: 10.1016/j.neurobiolaging.2011.12.017. [DOI] [PubMed] [Google Scholar]
- 103.Brys M, Glodzik L, Mosconi L, Switalski R, De Santi S, Pirraglia E, et al. Magnetic resonance imaging improves cerebrospinal fluid bio-markers in the early detection of Alzheimer’s disease. J Alzheimers Dis. 2009;16:351–62. doi: 10.3233/JAD-2009-0968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.de Leon MJ, Mosconi L, Blennow K, DeSanti S, Zinkowski R, Mehta PD, et al. Imaging and CSF studies in the preclinical diagnosis of Alzheimer’s disease. Ann N Y Acad Sci. 2007;1097:114–45. doi: 10.1196/annals.1379.012. [DOI] [PubMed] [Google Scholar]
- 105.de Leon MJ, Segal S, Tarshish CY, DeSanti S, Zinkowski R, Mehta PD, et al. Longitudinal cerebrospinal fluid tau load increases in mild cognitive impairment. Neurosci Lett. 2002;333:183–6. doi: 10.1016/s0304-3940(02)01038-8. [DOI] [PubMed] [Google Scholar]
- 106.Quinn JF, Montine KS, Moore M, Morrow JD, Kaye JA, Montine TJ. Suppression of longitudinal increase in CSF F2-isoprostanes in Alzheimer’s disease. J Alzheimers Dis. 2004;6:93–7. doi: 10.3233/jad-2004-6110. [DOI] [PubMed] [Google Scholar]
- 107.Landau SM, Lu M, Joshi AD, Pontecorvo M, Mintun MA, Trojanowski JQ, et al. Comparing positron emission tomography imaging and cerebrospinal fluid measurements of beta-amyloid. Ann Neurol. 2013;74:826–36. doi: 10.1002/ana.23908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Carpenter BD, Xiong C, Porensky EK, Lee MM, Brown PJ, Coats M, et al. Reaction to a dementia diagnosis in individuals with Alzheimer’s disease and mild cognitive impairment. J Am Geriatr Soc. 2008;56:405–12. doi: 10.1111/j.1532-5415.2007.01600.x. [DOI] [PubMed] [Google Scholar]
- 109.Mattsson N, Brax D, Zetterberg H. To know or not to know: ethical issues related to early diagnosis of Alzheimer’s disease. Int J Alzheimers Dis. 2010 doi: 10.4061/2010/841941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bayer AJ, Bullock R, Jones RW, Wilkinson D, Paterson KR, Jenkins L, et al. Evaluation of the safety and immunogenicity of synthetic Abeta42 (AN1792) in patients with AD. Neurology. 2005;64:94–101. doi: 10.1212/01.WNL.0000148604.77591.67. [DOI] [PubMed] [Google Scholar]
- 111.Glodzik L, De Santi S, Tsui WH, Mosconi L, Zinkowski R, Pirraglia E, et al. Phosphorylated tau 231, memory decline and medial temporal atrophy in normal elders. Neurobiol Aging. 2011;32:2131–41. doi: 10.1016/j.neurobiolaging.2009.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Visser PJ, Verhey F, Knol DL, Scheltens P, Wahlund LO, Freund-Levi Y, et al. Prevalence and prognostic value of CSF markers of Alzheimer’s disease pathology in patients with subjective cognitive impairment or mild cognitive impairment in the DESCRIPA study: a prospective cohort study. Lancet Neurol. 2009;8:619–27. doi: 10.1016/S1474-4422(09)70139-5. [DOI] [PubMed] [Google Scholar]
- 113.Blennow K, Vanmechelen E, Hampel H. CSF total tau, Abeta42 and phosphorylated tau protein as biomarkers for Alzheimer’s disease. Mol Neurobiol. 2001;24:87–97. doi: 10.1385/MN:24:1-3:087. [DOI] [PubMed] [Google Scholar]