

Abstract
Background
Nitric oxide (NO) has been largely associated with cardiovascular protection through improvement of endothelial function. Recently, new evidence about modulation of NO release by microRNAs (miRs) has been reported, which could be involved with statin-dependent pleiotropic effects, including anti-inflammatory properties related to vascular endothelium function.
Objective
To evaluate the effects of cholesterol-lowering drugs including the inhibitors of cholesterol synthesis, atorvastatin and simvastatin, and the inhibitor of cholesterol absorption ezetimibe on NO release, NOS3 mRNA expression and miRs potentially involved in NO bioavailability.
Methods
Human umbilical vein endothelial cells (HUVEC) were exposed to atorvastatin, simvastatin or ezetimibe (0 to 5.0 μM). Cells were submitted to total RNA extraction and relative quantification of NOS3 mRNA and miRs -221, -222 and -1303 by qPCR. NO release was measured in supernatants by ozone-chemiluminescence.
Results
Both statins increased NO levels and NOS3 mRNA expression but no influence was observed for ezetimibe treatment. Atorvastatin, simvastatin and ezetimibe down-regulated the expression of miR-221, whereas miR-222 was reduced only after the atorvastatin treatment. The magnitude of the reduction of miR-221 and miR-222 after treatment with statins correlated with the increment in NOS3 mRNA levels. No influence was observed on the miR-1303 expression after treatments.
Conclusion
NO release in endothelial cells is increased by statins but not by the inhibitor of cholesterol absorption, ezetimibe. Our results provide new evidence about the participation of regulatory miRs 221/222 on NO release induction mediated by statins. Although ezetimibe did not modulate NO levels, the down-regulation of miR-221 could involve potential effects on endothelial function.
Keywords: Nitric Oxide, Cholesterol, Hydroxymethylglutaryl-CoA Reductase Inhibitors, Endothelial Cells, microRNAs
Introduction
Dyslipidemia is a key factor in the pathogenesis of atherosclerosis through the relationship of cholesterol excess with the lipid-composition of the plaque. Cholesterol contents are associated with the inflammatory process by modulating inflammatory cytokines, adhesion molecules and other molecules related to the endothelial function in early atherosclerosis1.
Nitric oxide (NO) has been largely associated with cardiovascular protection through improvement of the endothelial function2,3. Several mechanisms have been described to regulate the expression of the endothelial nitric oxide synthase (eNOS) and NO release in the endothelium, including transcriptional and post-transcriptional regulation of the gene encoding eNOS (NOS3), as well as post-translational modifications4,5. Post-transcriptional regulation mediated by non-coding small RNAs has also demonstrated participation in this process and, in this way, new evidence about modulation of NO release by microRNAs (miRs) has been reported. An example of the miR-mediated regulation of NO release is observed for miRs-221/222 which have been related to lower NOS3 mRNA and reduced NO release in endothelial cells6.
Cholesterol-lowering therapies have been largely related with a reduced risk of cardiovascular diseases. Among them, statins are recognized as the main cholesterol-lowering drugs by reducing the cholesterol contents through the inhibition of the 3-hydroxy-3-methyl-glutaryl coA reductase (HMGCR), an enzyme with a key function in the endogenous biosynthesis of the cholesterol7. On the other hand, ezetimibe also reduces cholesterol contents by interacting with the Niemann-Pick C1-like 1 (NPC1L1), which results in a decreased cholesterol absorption8. In addition, a number of pleiotropic effects have been described for statin therapy, including anti-inflammatory properties related to the vascular endothelium function9. Molecular mechanisms involved in statin-related anti‑inflammatory effects and whether or not this depends on intracellular cholesterol reduction remains controversial.
Here, we analyzed the effects of cholesterol-lowering drugs including the inhibitors of cholesterol synthesis, atorvastatin and simvastatin, and the inhibitor of cholesterol absorption ezetimibe on NO release, NOS3 mRNA expression and their effects on the expression of miRs related to NOS3 mRNA modulation: miR-221, miR-222 as well as the miR-1303, a miR predicted to interact with NOS3 mRNA by bioinformatics tools.
Methods
Cell culture, treatments and cell viability analysis
Human umbilical vein endothelial cells (HUVEC) were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum, 44 mmol/L sodium bicarbonate, 100 µg/mL streptomycin and 100 U/mL penicillin. Cells were grown at 37°C in a humidified atmosphere containing 5% CO2.
Cells at passage 4-8 were treated with atorvastatin (kindly provided by Pfizer Pharmaceuticals Ltd., Guarulhos, SP, Brazil), simvastatin (Sigma, St. Louis, MO, USA) or ezetimibe (kindly provided by Merck/Schering-Plough, NJ, USA). Atorvastatin was dissolved in methanol whereas simvastatin and ezetimibe were dissolved in ethanol. Simvastatin was activated by incubation with 0.1N NaOH solution at 50°C for 2 h, followed by neutralization at pH 7.0 and adjustment of the concentration to 5.6 mM. The final concentration of methanol or ethanol in the culture medium did not exceed 0.1% and 0.2%, respectively.
Toxicity of the drugs was evaluated by measuring the percentage of cells with a loss of membrane integrity and DNA fragmentation. Regarding membrane integrity, cells (5 x 105) were treated with each drug and the percentage of viable cells was determined by flow cytometry using propidium iodide solution (50 mg/mL in phosphate buffer saline), which intercalates with DNA and is unable to pass through intact membranes. For the DNA fragmentation assay, the percentage of fragmented DNA was detected by flow cytometry after cells (5 x 105) were incubated for 2 h with a hypotonic solution (0.1% sodium citrate and 0.1% Triton X-100) containing 50 mg/mL of propidium iodide.
Cells (numbering 10,000) were analysed in a FACSCanto II flow cytometer (Becton Dickinson, San Jose, CA, USA). Red (propidium iodide) fluorescence was evaluated using a585 nm filter. Data were acquired and analyzed using the FACS/Cell Quest software (Becton Dickinson, San Jose, CA, USA).
HUVEC (1.0 x 106 cells/mL) were treated with atorvastatin, simvastatin or ezetimibe for 24 h. Cells were incubated for 6 h (18 h after starting drug treatment) with 10 ng/mL of human TNFα (Invitrogen – Life Technologies, CA, USA) in order to stimulate an inflammatory response simulating the chronic inflammation observed in early atherosclerosis. After incubations, supernatants of cell cultures were separated and cells were submitted to RNA extraction.
Nitric oxide measurement
NO concentration in supernatants from HUVEC treated with cholesterol-lowering drugs was determined by ozone-chemiluminescence using the Sievers NO Analyzer 280 (GE Analytical instruments, CO, USA).
Total RNA isolation
RNA extraction from HUVEC was performed using the mirVvana miRNA isolation kit (Ambion Inc., TX, USA), which separates total RNA including miRNA fraction, following the manufacturer's suggested protocol. RNA concentration was measured by spectrophotometry using NanoDrop ® (NanoDrop Technologies INC., DE, USA) and RNA integrity was further evaluated using the Bioanalyzer®2100 (Agilent Technologies, CA, USA). Samples with RNA integrity number (RIN) lower than 5 were not used for mRNA experiments.
NOS3 mRNA quantification
cDNA for NOS3 (NM_000603.4) relative quantification was produced from 1 μg of total RNA with Superscript™ II Reverse Transcriptase (Life Technologies, CA, USA). NOS3 mRNA expression was measured by quantitative real-time PCR (qPCR) using the SYBR® Green system. The sequences of primers for NOS3 transcript are described as follows: forward, 5'-TCCCCGGGTCCTGTGTATG-3'; and reverse, 5'-TCCATCAGGGCAGCTGCAAA-3'. The qPCR assays were carried out in 96-well plates using a 7500 Fast Real-Time PCR system (Life Technologies, CA, USA).
Six reference genes [ubiquitin C (UBC), glyceraldehyde-3-phosphate dehydrogenase (GAPD), beta-2-microglobulin (B2M), hypoxanthine phosphoribosyl-transferase I (HPRTI), succinate dehydrogenase complex, subunit A (SDHA) and hydroxymethyl-bilane synthase (HMBS)] were tested and analyzed using the geNorm software10, in order to choose the gene with lower variability for our cellular model under each experimental condition.The most stable gene in experimental conditions was HMBS, and the sequences of primers used for this endogenous gene are described as follows: forward, 5'-GGCAATGCGGCTGCAA-3'; and reverse, 5'-GGGTACCCACGCGAATCAC-3'.
All samples were assayed in duplicate. The relative quantification of gene expression was analyzed using the comparative Ct method using the formula 2-ΔΔCt 11 using cells treated with the vehicle stimulated for 6 h of TNFα (10 ng/mL) as a calibrator.
microRNA analysis
miR-221 and miR-222 (MI00000298 and MI00000299, respectively) were selected from previous reports from the literature whereas miR-1303 (MI0006370) was chosen using bioinformatics tools. Predictive analysis using TargetScan (www.targetscan.org) showed that miR-1303 is a conserved miR with a high score for mRNA-miR interaction (context score percentile: 99) and this prediction was further confirmed using the miRNA target prediction databases miRDB (www.mirdb.org) and Target Miner (www.isical.ac.in/~bioinfo_miu).
miR relative quantification was performed by stem loop TaqMan® RT-qPCR (Life Technologies, CA, USA). Specific stem loop primers were used to synthetize the cDNA from 10 ng of total RNA using the micro-RNA transcription kit (Life Technologies, CA, USA). Predesigned assays were purchased from Life Technologies to perform the relative quantification using the RNU24 as an endogenous reference. RNU24 was selected as the most stable endogenous reference among 4 small nucleolar RNAs (RNU24, RNU6B, RNU58, RNU44), which were evaluated by using the geNorm software10.
Statistical analysis
Statistical analyses were performed using Prism v.5.0 for Windows (Graph Pad Software Inc., CA, USA) and SPSS v.15 for Windows (SPSS Inc., Madrid, Spain). Data are presented as mean ± SEM and differences among means were tested by one-way ANOVA followed by the Tukey post-test comparing each treatment with control. Statistical significance was set for p < 0.05.
Results
After 24h of treatment with statins, HUVEC had increased loss of membrane integrity (11% and 12% for atorvastatin and simvastatin, respectively) and increased DNA fragmentation (31% and 28% for atorvastatin and simvastatin, respectively) when using concentrations equal or higher than 10 µM. Cell viability was not affected by any treatment – atorvastatin, simvastatin or ezetimibe – after 24 h exposure with up to 5 µM of each drug (data not shown).
Stimulation of inflammatory response by TNFα did not modify NOS3 mRNA expression or NO release in endothelial cells, as observed in Figures 1 and 2, respectively. Atorvastatin and simvastatin increased NOS3 mRNA at 5 µM after 24h (p < 0.05), whereas no effect on NOS3 mRNA was observed for ezetimibe (Figure 1). Both statins induced the release of NO from HUVEC when incubated for 24 h with 5 µM (p < 0.05) whereas ezetimibe did not modify the NO concentration in supernatants (Figure 2).
Figure 1.
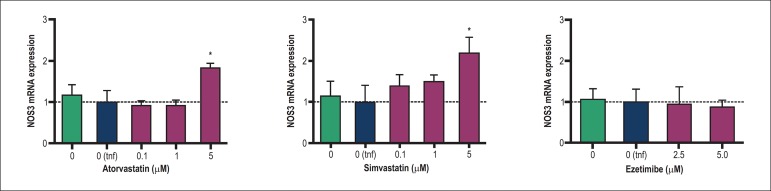
Influence of atorvastatin, simvastatin and ezetimibe on NOS3 mRNA expression in HUVEC. Columns and error bars represents mean and SEM and were compared with the vehicle control stimulated with TNFα [(0 tnf); dotted line)] by one-way ANOVA followed by Tukey post-test. HUVEC were treated for 24 h and stimulated with human TNFα (10 ng/mL) 6 h before RNA extraction. Data were obtained from 4-6 independent experiments and mRNA expression was calculated using the comparative Ct method using the HMBS as endogenous reference. (*), p < 0.05.
Figure 2.
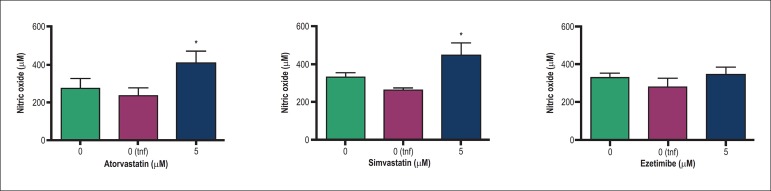
Influence of atorvastatin, simvastatin and ezetimibe on nitric oxide release in HUVEC. Columns and error bars represent mean and SEM and were compared by one-way ANOVA followed by Tukey post-test. HUVEC cells were treated for 24 h and stimulated with human TNFα [10 ng/mL; 0(tnf)] 6 h before supernatant isolation for NO measurements. Data were obtained from 5 independent experiments and NO was measured by ozone-chemiluminescence. (*), p < 0.05.
The effects of atorvastatin, simvastatin and ezetimibe on the expression of miR-221, miR-222 and the miR-1303 are shown in Figure 3. Atorvastatin reduced the expression of miR-221 at 1 and 5 µM and the expression of the miR-222 at 5 µM. Simvastatin down-regulated only miR-221 at 5 µM after 24 h of treatment whereas ezetimibe also reduced the expression of miR-221 at 5 µM. Moreover, the variation of miR-221 after 5 µM treatment with atorvastatin and simvastatin correlated negatively with NOS3 mRNA expression (r2: -0.989 and 0.915 for atorvastatin and simvastatin, respectively; p < 0.05; data not shown). The magnitude of the variation of NOS3 also correlated negatively with miR-222 change after statin treatment (r2: -0.992 and 0.890 for atorvastatin and simvastatin, respectively; p < 0.05; data not shown). On the other hand, miR-1303 did not show statistical differences after any treatment.
Figure 3.

Influence of atorvastatin, simvastatin and ezetimibe on expressions of miR-221, miR-222 and miR-1303 in HUVEC. Columns and error bars represent mean and SEM and were compared with the vehicle control stimulated with TNFα (0 tnf; dotted line) by one-way ANOVA followed by Tukey post-test. HUVEC were treated for 24 h and stimulated with human TNFα (10 ng/mL) 6 h before RNA extraction. Data were obtained from 4 independent experiments and mRNA expression was calculated using the comparative Ct method using the RNU24 as endogenous reference. (*), p < 0.05.
Discussion
In this study, the inhibition of cholesterol synthesis by statins, but not the inhibition of cholesterol absorption by ezetimibe, modulated the NO release in endothelial cells. Our results showed a correlation between NOS3 mRNA expression and NO release in HUVEC treated with statins, as previously observed in cultured endothelial cells exposed to fluvastatin treatment12. Moreover, previous works have also suggested that the regulation of eNOS and the NO release by statins is also dependent on posttranslational modifications. For example, Aoki et al. reported that, besides the NOS3 mRNA up-regulation, fluvastatin also modified the phosphorylation pattern of endothelial cells12. A similar posttranslational modification was observed in isolated rat arteries treated with simvastatin13, which also caused an enhanced phosphorylation of eNOS at the Ser 1177 through regulation of the PKB/Akt, a process that leads to a higher enzyme activity and increased NO release14.
The modulation of NO by statins has been proposed as a central mechanism by which these drugs can exert their anti‑inflammatory effects on the vascular endothelium. Due to this hypothesis, previous works using an animal model knockout for NOS3 have been performed to evaluate whether pleiotropic effects of statins are dependent on eNOS modulation. Atorvastatin did not reduce NFκB activation or infarct size in eNOS-/- mice undergoing coronary artery occlusion as compared with wild-type mice15. Similarly, rosuvastatin failed to attenuate the leucocyte-endothelium interactions in peri-intestinal venules of eNOS-/- mice16. These data suggest that anti-inflammatory effects observed for statins require the release of NO by the vascular endothelium; however recent data have shown that anti-inflammatory effects of statins were also present in eNOS deficient animals, as in eNOS-/- mice treated with atorvastatin that had similar platelet activation compared with wild-type mice17.
Despite the controversial results about the role of eNOS in statins-induced anti-inflammatory effects, it is indubitable that an increased release of NO in the vascular endothelium by these or other treatments will be beneficial in reducing the risk of cardiovascular diseases. The emergence of new molecular mechanisms regulating the gene expression of NOS3 and the release of NO, as the posttranscriptional regulation by miRs, are very interesting for the understanding of this process and for exploring new therapeutic targets.
Here, we evaluated the effect of statins – atorvastatin and simvastatin – as well as the effect of the cholesterol absorption inhibitor ezetimibe on the expression of miR‑221, miR-222 and miR-1303. Atorvastatin reduced the expression of miR‑221 and miR-222, whereas simvastatin down-regulated only miR-221. Interestingly, ezetimibe also reduced the expression of miR-221 showing that the expression of this miR could be dependent on the regulation of intracellular cholesterol, regardless of the mechanism by which cholesterol content is modulated. On the other hand, none of the treatments modified the expression of the miR-1303, demonstrating that the modulation of NOS3 and release of NO by statins are not dependent on this miR, although a miR:mRNA interaction was predicted by bioinformatics tools. A limitation of our study is that we focused only on the effects of cholesterol-lowering therapies on the proposed miRNA‑mediated NO modulation and we did not evaluate the effects of the drugs on eNOS protein level or its posttranscriptional modifications. Here, we propose a mechanism by which statins contribute to the modulation of NO release in vascular endothelium. However, this mechanism should be considered one of a number of actions mediated by statins to improve endothelial function.
The miRs -221 and -222 have demonstrated their participation in the progression of cardiovascular diseases18, as observed in rat carotid artery lesions after angioplasty in which their expression was increased19. In the same way, miR-221 and miR-222 were increased in endothelial progenitor cells from patients with coronary artery disease (CAD) and in those patients, 12 months of treatment with atorvastatin, but not with pravastatin, diminished the expression of miRs-221/22220. Those results are in agreement with the reduction of these miRs mediated by statins in our study.
Endothelial cells transfected with miRs-221/222 showed a reduction in both eNOS protein level and release of NO21. However, the direct mechanism by which these miRs may exert that effect is unknown because there is no target region for miRs-221/222 in the NOS3 mRNA molecule generating a mRNA:miR interaction according to the predictive analysis by bioinformatics tools. Therefore, it is important to highlight that the modulation of eNOS by miRs-221/222 lacks a molecular explanation, although it was demonstrated empirically. On the other hand, Liu et al. proposed that miRs-221/222 might modulate the proliferation of smooth muscle cells in the endothelium by targeting p27 (Kip1) and p57 (Kip2)19 However, no relation of miRs-221/222 with NOS3 modulation and NO release was related in that work.
An important extension of our work was the evaluation of the effect on the expression of the studied miRs of a non‑statin cholesterol-lowering drug, the ezetimibe. Although most beneficial effects of ezetimibe in humans relate to its capacity to reduce cholesterol absorption in the intestine, the influence of this drug in other tissues, as the vascular endothelium, is an important issue to be investigated. In this way, the reduction of the expression of miR-221 could be considered an important indicator of possible pleiotropic effects of the ezetimibe therapy. A recent study also evaluated the effect of ezetimibe in an extra‑intestinal tissue and showed that the ezetimibe inhibited the PMA-induced monocyte to macrophage differentiation, and the authors suggested that one of the mechanisms involved in the process could be the regulation of the miRs -155, -222, -424 and -503 by the ezetimibe in that cellular model22. Although miR-222 expression was not modified by ezetimibe in our work, the differences in the cellular models should be considered in this divergence. Moreover, the reduction of isoprenoids synthesis by statins – described as the main mechanism by which these drugs could exert their anti-inflammatory effects9 - could also influence the differences observed between inhibitors of synthesis and absorption of cholesterol on endothelial NO modulation observed in this study. On the other hand, the observed reduction of miR-221 without an increment of NO release by ezetimibe led us to think that the reduction of this miR is not necessarily the only mechanism modulating the NO pathway. However, it is feasible to propose that using ezetimibe in a combination therapy with a statin could result in more benefits regarding endothelial function; however other studies are needed to elucidate this hypothesis.
Conclusion
Statin-induced down-regulation of miRs-221/222 is a new potential pathway by which the cholesterol-lowering treatment may exert its beneficial effects on NO release in human endothelium. Moreover, the observed reduction of miR-221 by ezetimibe should be investigated in the future as a potential contribution of this drug in endothelial function when used in a combination therapy.
Acknowledgements
The present study was supported by the grant # 2010/15353-8, São Paulo Research Foundation (FAPESP), São Paulo, Brazil. A. Cerda is a recipient of a fellowship from CAPES-Brazil. R.G. Basso, M.H. Hirata and R.D.C. Hirata are recipients of fellowships from CNPq-Brazil.
Footnotes
Author contributions
Conception and design of the research, analysis and interpretation of the data and critical revision of the manuscript for intellectual content: Cerda A, Hirata MH, Hirata RDC; acquisition of data: Cerda A, Moreno FC, Basso RG; statistical analysis and writing of the manuscript: Cerda A; obtaining financing: Cerda A, Hirata RDC.
Potential Conflict of Interest
No potential conflict of interest relevant to this article was reported.
Sources of Funding
This study was funded by Fundação de Amparo à Pesquisa do Estado de São Paulo.
Study Association
This article is part of the thesis of Doctoral submitted by Alvaro Cerda, from Universidade de São Paulo.
References
- 1.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105(9):1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 2.Förstermann U, Münze T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113(13):1708–1714. doi: 10.1161/CIRCULATIONAHA.105.602532. [DOI] [PubMed] [Google Scholar]
- 3.Lei J, Vodovotz Y, Tzeng E, Billiar TR. Nitric oxide, a protective molecule in the cardiovascular system. Nitric Oxide. 2013;35:135–185. doi: 10.1016/j.niox.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 4.Searles CD. Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression. Am J Physiol Cell Physiol. 2006;291(5):C803–C816. doi: 10.1152/ajpcell.00457.2005. [DOI] [PubMed] [Google Scholar]
- 5.Qian J, Fulton D. Post-translational regulation of endothelial nitric oxide synthase in vascular endothelium. Front Physiol. 2013;4:347–347. doi: 10.3389/fphys.2013.00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suárez Y, Fernández-Hernando C, Pober JS, Sessa WC. Dicer dependent microRNAs regulate gene expression and functions in human endothelial cells. Circ Res. 2007;100(8):1164–1173. doi: 10.1161/01.RES.0000265065.26744.17. [DOI] [PubMed] [Google Scholar]
- 7.Mangravite LM, Thorn CF, Krauss RM. Clinical implications of pharmacogenomics of statin treatment. Pharmacogenomics J. 2006;6(6):360–374. doi: 10.1038/sj.tpj.6500384. [DOI] [PubMed] [Google Scholar]
- 8.Bays HE, Neff D, Tomassini JE, Tershakovec AM. Ezetimibe: cholesterol lowering and beyond. Expert Rev Cardiovasc Ther. 2008;6(4):447–470. doi: 10.1586/14779072.6.4.447. [DOI] [PubMed] [Google Scholar]
- 9.Mihos CG, Salas MJ, Santana O. The pleiotropic effects of the hydroxymethyl- glutaryl-CoA reductase inhibitors in cardiovascular disease: a comprehensive review. Cardiol Rev. 2010;18(6):298–304. doi: 10.1097/CRD.0b013e3181f52a7f. [DOI] [PubMed] [Google Scholar]
- 10.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3(7): doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 12.Aoki C, Nakano A, Tanaka S, Yanagi K, Ohta S, Jojima T, et al. Fluvastatin upregulates endothelial nitric oxide synthase activity via enhancement of its phosphorylation and expression and via an increase in tetrahydrobiopterin in vascular endothelial cells. Int J Cardiol. 2012;156(1):55–61. doi: 10.1016/j.ijcard.2010.10.029. [DOI] [PubMed] [Google Scholar]
- 13.Rossoni LV, Wareing M, Wenceslau CF, Al-Abri M, Cobb C, Austin C. Acute simvastatin increases endothelial nitric oxide synthase phosphorylation via AMP-activated protein kinase and reduces contractility of isolated rat mesenteric resistance arteries. Clin Sci (Lond) 2011;121(10):449–458. doi: 10.1042/CS20110259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh R, et al. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399(6736):597–601. doi: 10.1038/21218. Erratum in: Nature 1999;400(6746):792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ye Y, Martinez JD, Perez-Polo RJ, Lin Y, Uretsky BF, Birnbaum Y. The role of eNOS, iNOS, and NF-kappaB in upregulation and activation of cyclooxygenase-2 and infarct size reduction by atorvastatin. Am J Physiol Heart Circ Physiol. 2008;295(1):H343–H351. doi: 10.1152/ajpheart.01350.2007. [DOI] [PubMed] [Google Scholar]
- 16.Stalker TJ, Lefer AM, Scalia R. A new HMG-CoA reductase inhibitor, rosuvastatin, exerts anti-inflammatory effects on the microvascular endothelium: the role of mevalonic acid. Br J Pharmacol. 2001;133(3):406–412. doi: 10.1038/sj.bjp.0704070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ni R, Peleg T, Gross PL. Atorvastatin delays murine platelet activation in vivo even in the absence of endothelial NO synthase. Arterioscler Thromb Vasc Biol. 2012;32(11):2609–2615. doi: 10.1161/ATVBAHA.112.300090. [DOI] [PubMed] [Google Scholar]
- 18.Pan ZW, Lu YJ, Yang BF. MicroRNAs: a novel class of potential therapeutic targets for cardiovascular diseases. Acta Pharmacol Sin. 2010;31(1):1–9. doi: 10.1038/aps.2009.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu X, Cheng Y, Zhang S, LinY, Yang J, Zhang C. A necessary role of miR-221 and miR-222 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ Res. 2009;104(4):476–487. doi: 10.1161/CIRCRESAHA.108.185363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Minami Y, Satoh M, Malsawa C, Takahashi Y, Tabuchi T, Itoh T, et al. Effect of atorvastatin on microRNA 221 / 222 expression in endothelial progenitor cells obtained from patients with coronary artery disease. Eur J Clin Invest. 2009;39(5):359–367. doi: 10.1111/j.1365-2362.2009.02110.x. [DOI] [PubMed] [Google Scholar]
- 21.Rippe C, Blimline M, Magerko KA, Lawson BR, LaRocca TJ, Donato AJ, et al. MicroRNA changes in human arterial endothelial cells with senescence: relation to apoptosis, eNOS and inflammation. Exp Gerontol. 2012;47(1):45–51. doi: 10.1016/j.exger.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muñoz-Pacheco P, Ortega-Hernández A, Miana M, Cachofeiro V, Fernández-Cruz A, Gómez-Garre D. Ezetimibe inhibits PMAinduced monocyte/macrophage differentiation by altering microRNA expression: a novel anti-atherosclerotic mechanism. Pharmacol Res. 2012;66(6):536–543. doi: 10.1016/j.phrs.2012.09.005. [DOI] [PubMed] [Google Scholar]