

Background
The gangliosidoses (Tay-Sachs disease, Sandhoff disease and GM1-gangliosidosis) are progressive neurodegenerative diseases caused by lysosomal enzyme activity deficiencies and consequent accumulation of gangliosides in the central nervous system (CNS). The infantile forms are distinguished from the juvenile forms by age of onset, rate of disease progression and age of death. There are no approved treatments for the gangliosidoses. In search of potential biomarkers of disease, we quantified 188 analytes in CSF and serum from living human patients with longitudinal (serial) measurements. Notably, several associated with inflammation were elevated in the CSF of infantile gangliosidosis patients, and less so in more slowly progressing forms of juvenile gangliosidosis, but not in MPS disease. Thirteen CSF and two serum biomarker candidates were identified. Five candidate biomarkers were distinguished by persistent elevation in the CSF of patients with the severe infantile phenotype: ENA-78, MCP-1, MIP-1α, MIP-1β, TNFR2. Correspondence of abnormal elevation with other variables of disease --- i.e., severity of clinical phenotype, differentiation from changes in serum, and lack of abnormality in other neurodegenerative lysosomal diseases ---identifies these analytes as biomarkers of neuropathology specific to the gangliosidosis diseases.
Keywords: GM1-gangliosidosis, metabolomic, ganglioside, glycosphingolipid, Tay-Sachs disease, Sandhoff disease
Introduction
The gangliosidoses diseases --- Tay-Sachs disease, Sandhoff disease (GM2-gangliosidoses) and GM1-gangliosidosis) --- are inherited metabolic diseases of lysosomal lipid catabolism in which accumulation of ganglioside, i.e., glycosphingolipids containing one or more sialic acids. Accumulation in the central nervous system is associated with progressive neurodegeneration. GM1-gangliosidosis is caused by mutations to the GLB1 gene, resulting in deficiency of β-galactosidase activity and subsequent accumulation of GM1-ganglioside [1,2]. The GM2-gangliosidoses, Tay-Sachs disease and Sandhoff disease, are caused by mutations of genes encoding the α subunit (HEXA gene) and β subunit (HEXB gene), respectively, of β –hexosaminidase A enzyme, resulting in accumulation of GM2-ganglioside [1,2]. The AB variant form of GM2 gangliosidosis is caused by a genetic defect in the GM2A gene that codes for the GM2-ganglioside activator protein, a co-factor for β-hexosaminidase A enzyme required for activation of β-hexosaminidase A [1,2].
Natural history studies have improved understanding of disease presentation and progression. For the infantile, late-infantile and juvenile forms, death occurs in childhood [1]. The infantile forms of GM1-gangliosidosis and GM2-gangliosidosis share many similarities in their clinical phenotypes, including neurodevelopmental delays appearing within the first 6 months of life, hypotonia, dysphagia, and seizures often occurring within the first year of life, and death often occurring by the third year of life [1-4]. A late-infantile form of GM1-gangliosidosis has also been described, with symptoms appearing between the first 1-2 years of life and is usually classified as a variant of the juvenile form [1-4]. In contrast to the infantile forms of GM1- and GM2-gangliosidosis, the onset of symptoms in the juvenile forms is often between the second and fifth year of life and may present with ataxia and coordination difficulties, progressing to development of dysarthria, dysphagia, hypotonia, and seizures, with death occurring before or during adolescence [1,2,4,5]. Little is understood about the mechanism(s) by which the ganglioside accumulation leads to tissue damage at a molecular level, but research in animal models and human cadavers increasingly points to the role of inflammatory mediators [6,7]. Moreover, mouse models of gangliosidoses have demonstrated expression of inflammatory mediators changing during different stages of disease progression [7].
Hypothesis
For this study, we hypothesized that markers of CNS inflammatory processes would be more highly expressed in patients with the severe infantile gangliosidosis phenotype, compared to patients with late-infantile and juvenile forms, and that such inflammatory markers, when identified will serve as candidate biomarkers for future therapies.
Materials and Methods
This study was conducted with IRB approval and IRB-approved consent of the patients' parents or legal guardians. CSF specimens were collected by lumbar puncture with a 22 gauge (1.5 or 3.5 inch) Quincke spinal tap needle while the patient was under general anesthesia for MRI imaging. CSF was collected in 1 mL aliquots directly into cryovials, quickly labeled, placed on dry ice and then transported to the laboratory for distribution. Subsequent review of cell counts demonstrated that all but one specimen was atraumatic and thus without contamination by significant blood. Blood specimens were obtained concurrently by phlebotomy, taken to the laboratory for centrifugation, and then aliquotted and frozen on dry ice. Cerebral spinal fluid (CSF) and serum samples were tested by immunoassay technique performed by Rules Based Medicine, Inc. (RBM) of Austin, Texas, using the Human Discovery Multi-Analyte Profile (MAP) v1.0 panel before June 2012, and using the Human Discovery MAP 175+ v1.0 panel after June 2012. These 188 analytes were quantified in each specimen. Normal ranges for serum analytes were provided by RBM. For serum, normal ranges were determined from the results of 100 presumably healthy subjects; no assumption was made by RBM about the samples having a normal distribution, so the normal serum range comprises 95% of the samples with the highest and lowest 2.5% of the samples excluded for each given assay. Normal ranges for analytes in CSF were provided by RBM and were calculated using data obtained from Britschgi et al [8] and Craig-Schapiro et al [9]. The low and high values of the normal range were calculated as +/- two standard deviations from the mean. Analytes selected for evaluation were those for which absolute values were measurable in both serum and CSF, and for which reference ranges were also available [6,7]. In total, 72 of the analytes met these criteria. Results were sorted by patient diagnosis and patient age at the time of collection. Inflammatory markers showing elevation in CSF and serum of patients with infantile and/or juvenile gangliosidosis were selected and compared to results in the subjects with MPS diseases. Sequential serum and CSF measurements were obtained whenever possible. KaleidaGraph software was used to create scatterplot graphs depicting analyte amounts and changes over time for analytes whose values fell outside of the normal reference range [8,9].
Results
Serum and CSF samples were obtained concurrently from a total of 21 subjects (Table 1), including eight patients with an infantile gangliosidosis diagnosis (three with infantile Tay-Sachs disease, two with infantile Sandhoff disease and three with infantile GM1 gangliosidosis), four patients with juvenile gangliosidosis diagnosis (two with juvenile Tay-Sachs disease, one with juvenile GM1-gangliosidosis, one with late-infantile GM1-gangliosidosis), and nine patients with mucopolysaccharidosis (MPS) diseases (two with Scheie syndrome (MPS IS), one with Hunter syndrome (MPS II), five with Sanfilippo syndrome type A (MPS IIIA), and one with Maroteaux-Lamy syndrome (MPS VI). At least two sequential measures of serum and CSF were obtained for eight of the 12 patients with gangliosidosis diseases and for four of the nine patients with MPS diseases.
Table 1.
| Patient (N= 22) | Diagnosis | Gender | Genotype | Age at First Sample Draw (months) | Age at Last Sample Draw (months) | Number of Serum Samples | Number of CSF Samples |
|---|---|---|---|---|---|---|---|
| A | Infantile Sandhoff |
F | c.901G>C (p.G301R) (exon 7) | 34 | 34 | 1 | 1 |
| Unknown | |||||||
| B | Infantile Sandhoff |
F | Unknown | 14 | 14 | 1 | 1 |
| Deletion of exons 1-5 Deletion of exons 4-5 | |||||||
| C | Infantile Tay-Sachs |
M | IVS9+1G>A | 17 | 31 | 5 | 5 |
| IVS9+1G>A | |||||||
| D | Infantile Tay-Sachs |
F | c.986+9A>G (intron 8) | 18 | 18 | 1 | 1 |
| c.1073+1G>A (intron 9) | |||||||
| E | Infantile Tay-Sachs |
F | c.929_930delCT | 22 | 36 | 3 | 3 |
| c.1073+1G>A | |||||||
| F | Juvenile Tay-Sachs |
F | c.1274_1277dupTATC (exon 11) | 85 | 122 | 3 | 5 |
| c.1382G>T(p.G461V) (exon 12) | |||||||
| G | Juvenile Tay-Sachs |
F | c.77G>QA | 103 | 123 | 2 | 3 |
| c.1496G>A | |||||||
| H | Late-Onset Tay-Sachs |
M | G269S | 566 | 566 | 1 | 0 |
| G269S | |||||||
| I | Infantile
GM1 Gangliosidosis |
F | c.1480-2A>G | 10 | 25 | 2 | 3 |
| c.952A>G (p.Asn318Asp) | |||||||
| J | Infantile
GM1 Gangliosidosis |
F | Unknown | 13 | 35 | 3 | 3 |
| Unknown | |||||||
| K | Infantile
GM1 Gangliosidosis |
F | c.377T>C | 13 | 13 | 1 | 1 |
| Partial deletion of HEXA gene, extent of deletion undetermined | |||||||
| L | Late-Infantile GM1 Gangliosidosis |
M | R201C | 29 | 53 | 7 | 6 |
| K578R | |||||||
| M | Juvenile
GM1 Gangliosidosis |
F | c.464T>G | 68 | 68 | 1 | 0 |
| c.464T>G | |||||||
| N | MPS-Is | M | c.1205A>G | 42 | 68 | 1 | 2 |
| c.1139A>G | |||||||
| O | MPS-Is | M | p.Q70X | 45 | 55 | 2 | 2 |
| p.L18P | |||||||
| P | MPS-II | M | Unknown | 47 | 47 | 1 | 1 |
| Unknown | |||||||
| Q | MPS-IIIa | M | p.S298P | 132 | 146 | 2 | 2 |
| Unknown | |||||||
| R | MPS-IIIa | M | c.734G>A | 133 | 133 | 1 | 1 |
| c.1063G>A | |||||||
| S | MPS-IIIa | M | p.S298P | 72 | 85 | 2 | 2 |
| Unknown | |||||||
| T | MPS-IIIa | F | c.1979C>G | 66 | 66 | 1 | 1 |
| c.364G>A | |||||||
| U | MPS-IIIa | F | p.S66W | 44 | 44 | 1 | 1 |
| p.C481X | |||||||
| V | MPS-VI | M | Unknown | 206 | 206 | 0 | 1 |
| Unknown |
Among gangliosidosis patients, 13 of the 72 analytes were found to have elevations in CSF (Table 2). Two analytes, osteopontin and insulin-like growth factor-binding protein (IGFBP-2), showed elevation in serum samples. Five analytes were distinguished by persistent elevation in the infantile gangliosidosis phenotype: epithelial-derived neutrophil-activating protein 78 (ENA-78), monocyte chemotactic protein 1 (MCP-1), macrophage inflammatory protein-1 alpha (MIP-1α), macrophage inflammatory protein-1 beta (MIP-1β), tumor necrosis factor receptor 2 (TNFR2). This was in contrast to findings in patients with more attenuated forms of gangliosidosis and those with MPS diseases.
Table 2.
| Elevated Inflammatory Markers in CSF of Specific Patient Groups | Elevated
Inflammatory Markers in Serum of Specific Patient Groups |
||||||
|---|---|---|---|---|---|---|---|
| All Patients | All patients except MPS-IS and Sanfilippo syndrome type A |
All patients except MPS-IS |
Only elevated
in: Infantile gangliosidosis and Late-infantile GM1- gangliosidosis |
Only elevated
in: Infantile gangliosidosis |
Mixed
results across patient groups |
All patients | Infantile GM1 gangliosidosis |
| Heparin-Binding EGF-Like Growth Factor (HB-EGF) | Alpha-1-Antichymotrypsin (AACT) | Hepatocyte Growth Factor (HGF) | Macrophage Migration Inhibitory Factor (MIF) | Epithelial-Derived Neutrophil-Activating Protein 78 (ENA-78) | Apolipoprotein A-I (Apo A-I) | Osteopontin | Insulin-like Growth Factor-Binding Protein 2 (IGFBP-2) |
| Tamm-Horsfall Urinary Glycoprotein (THP) | Sex Hormone-Binding Globulin (SHBG) | Matrix Metalloproteinase-3 (MMP-3) | Monocyte Chemotactic Protein 1 (MCP-1) | ||||
| Macrophage Inflammatory Protein-1 alpha (MIP-1 alpha) | |||||||
| Macrophage Inflammatory Protein-1 beta (MIP-1 beta) | |||||||
| Tumor Necrosis Factor Receptor 2 (TNFR2) | |||||||
| Inflammatory Markers Not Elevated in CSF or Serum | |||||||
| Alpha-1-Microglobulin (A1Micro) | Apolipoprotein C-I (Apo C-I) | Cystatin-C | Immunoglobulin M (IGM) | Placenta Growth Factor (PLGF) | Tumor Necrosis Factor alpha (TNF-alpha) | ||
| Alpha-2-Macroglobulin (A2Macro) | Apolipoprotein C-III (Apo C-III) | Epidermal Growth Factor Receptor (EGFR) | Interleukin-6 receptor (IL-6r) | T-Cell-Specific Protein RANTES (RANTES) | Serotransferrin (Transferrin) | ||
| Alpha-1-Antitrypsin (AAT) | Apolipoprotein D (Apo D) | EN-RAGE | Interleukin-8 (IL-8) | Resistin | Thyroid-Stimulating Hormone (TSH) | ||
| Angiotensin-Converting Enzyme (ACE) | Apolipoprotein E (Apo E) | Fetuin-A | Interferon gamma Induced Protein 10 (IP-10) | S100 calcium-binding protein B (S100-B) | Vascular Cell Adhesion Molecule-1 (VCAM-1) | ||
| Adiponectin | Apolipoprotein H (Apo H) | Ferritin (FRTN) | Leptin | Serum Amyloid P-Component (SAP) | Vitronectin | ||
| Alpha-Fetoprotein (AFP) | Beta-2-Microglobulin (B2M) | Follicle-Stimulating Hormone (FSH) | Lectin-Like Oxidized LDL Receptor 1 (LOX-1) | Stem Cell Factor (SCF) | von Willebrand Factor (vWF) | ||
| Angiopoietin-2 (ANG-2) | Complement C3 (C3) | Growth Hormone (GH) | Monokine Induced by Gamma Interferon (MIG) | Superoxide Dismutase 1, Soluble (SOD-1) | |||
| Apolipoprotein A-II (Apo A-II) | CD5 (CD5L) | Haptoglobin | Neuronal Cell Adhesion Molecule (Nr-CAM) | Thyroxine-Binding Globulin (TBG) | |||
| Apolipoprotein A-IV (Apo A-IV) | Complement Factor H – Related Protein 1 (CFHR1) | Intercellular Adhesion Molecule 1 (ICAM-1) | N-terminal prohormone of brain natriuretic peptide (NT roBNP) | Trefoil Factor 3 (TFF3) | |||
| Apolipoprotein B (Apo B) | C-Reactive Protein (CRP) | Immunoglobulin A (IgA) | Plasminogen Activator Inhibitor 1 (PAI-1) | Tissue Inhibitor of Metalloproteinases 1 (TIMP-1) | |||
Caveats
In this study, we were unable to obtain precise measurements in either serum or CSF for several inflammatory markers that were of interest and thus these analytes were excluded from the final analyses. Included in this group were prolactin, for which elevated levels were found in CSF of patients with infantile GM1-gangliosidosis, but for which serum values were unavailable. The tyrosine kinase, Axl, and inflammatory marker CCL18/PARC were found to be elevated in serum samples of GM1-gangliosidosis, but CSF values were unavailable. Interleukin 16 (IL-16) was elevated in CSF of infantile GM1- and GM2- gangliosidosis, but CSF results for patients with a juvenile phenotype were not available. Evaluation of these analytes would be interesting in the future.
It is important to note that, one patient with MPS IS (patient “M”), and the patients with MPS II and MPS VI were receiving treatment with intravenous enzyme replacement therapy (ERT) for at least 2 years prior to CSF samples being obtained. Patient “N”, began ERT after the first CSF sample was obtained. None of the patients with MPS IIIA had received treatment at the time these samples were drawn, as there were no FDA-approved or experimental treatments for MPS IIIA at the time of this study. None of the 22 subjects had received a hematopoietic cell transplant.
Discussion
As recently emphasized by Christopher Austin (Director, National Center for Advancing Translational Sciences, NIH; RDCRN3 Steering Committee, Rockville, MD, October 1, 2014) focusing on common metabolic pathways shared by different diseases is an important strategy for understanding the pathophysiology of disease, and might be an efficient approach to identify therapies useful for all. Toward understanding the pathophysiologic mechanisms and potential treatment of these diseases, we have been employing this approach for the gangliosidosis diseases, i.e., Tay-Sachs disease, Sandhoff disease, and GM1-gangliosidosis.
Inflammation
This is the first study to broadly screen living human patients for potential biomarkers of gangliosidosis disease by obtaining sequential measurements of a large battery of analytes. Several observations point to the role of inflammatory mediators as active agents in CNS pathology. Of the 188 metabolites tested, abnormalities of analytes associated with inflammation were prominent. In this study, five markers of inflammation, ENA-78, MCP-1, MIP-1α, MIP-1β, TNFR2 were distinguished by their persistent elevation in the CSF of patients with infantile gangliosidosis phenotype, but not in the other patient groups. To our knowledge, ENA-78, a neutrophil chemokine, MIP-1β and TNFR2 have not been previous implicated as elevated inflammatory mediators associated with gangliosidosis pathology. TNFR2 has been found to act as a neuroprotective agent in neurodegenerative disorders and is important for remyelination [10,11], and may be a response to neuroinflammation in these patients. MCP-1 and MIP-1α have been shown to be elevated in the CNS of murine Sandhoff disease in association with microglial activation; MIP-1α levels correlated with substrate accumulation, especially N-acetylglucosaminyl (GlcNAc)-oligosaccharide [12,13]. In sum, these elevated analytes are all cytokines associated with glial cell activation, and point to a mechanism of neuroinflammation [7-13]. We presume that these cytokines are derived from microglial cells in the central nervous system, and reflect primary pathology in nervous system tissues. It seems unlikely that these reflect more systemic abnormalities in that serum levels were normal in both infantile and juvenile patients. This CNS localization further emphasizes the concept that disease is primarily central nervous system disease, and not systemic.
Severity
Notably, the analytes that were consistently abnormal among patients with gangliosidosis diseases were more markedly aberrant among those with the more severe infantile-onset forms in comparison to those with more slowly progressing or attenuated juvenile-onset phenotypes. This relationship of severity of disease to degree of analyte elevation is important; this correspondence enhances the view that the rate of disease progression is associated with the level of inflammatory molecule abnormality.
CNS association
Of the abnormal inflammatory analytes, 13 were significantly elevated in CSF while only two of these were elevated in serum. This differentiation of body compartment corresponds to the somatic distribution of disease, with major pathology in the brain in comparison to phenotypic features that are outside of the blood brain barrier. Thus, abnormalities in the CSF parallel the disease pathology in physical localization. Elevated CSF inflammatory analytes would thus seem to be directly indicative of CNS pathology with relative sparing of non-CNS disease. These abnormalities reflect CNS disease specifically, and are not a general indicator of lysosomal substrate accumulation or dysfunction.
Clinical application
These studies suggest that elevation of CSF inflammatory markers may be important, indicating the severity of clinical disease. As illustrated here, markedly elevated inflammatory markers are closely associated with more rapid and severe infantile disease while low, normal levels are found in patients with the slower, late-onset and attenuated brain damage of juvenile-onset gangliosidosis. Thus, regular quantification of CSF inflammatory markers in these diseases is of immediate clinical application and may be key to understanding the response to treatments. CSF inflammatory biomarkers should be monitored regularly in every patient.
Different from other ‘neurologic’ lysosomal disease
Lysosomal diseases other than the gangliosidoses have neurologic manifestations including cognitive loss, hydrocephalus, and other forms of progressive neurodegeneration. For example, the MPS conditions, Hurler syndrome and Sanfilippo syndrome, are characterized by such progressive neurodegeneration with inevitable cognitive declines, cerebral atrophy, seizures and hydrocephalus. However, comparison of inflammatory biomarkers from patients with these diseases failed to show the same abnormalities in either serum or CSF. This distinction — differences in elevated inflammatory biomarkers between gangliosidoses and mucopolysaccharidoses — provides important information pointing to different pathophysiologic mechanisms. Importantly, the CSF inflammatory biomarker differences between gangliosidosis disease and MPS disease emphasizes their value as ganglioside-specific biomarkers.
Conclusions
The observation of significant elevations of specific analytes associated with inflammation in CSF identifies these analytes as potential biomarkers of the underlying pathology of the gangliosidoses diseases. The correspondence of abnormal elevation with other variables of disease --- i.e., severity of clinical phenotype, differentiation from levels in serum, and lack of abnormality in other neurodegenerative lysosomal diseases --- identifies these analytes as biomarkers of neuropathology that is specific to the gangliosidoses. These observations should unveil the underlying molecular and cellular pathologic mechanisms of gangliosidosis disease and help evaluate response to therapy.
Summary
This study conducted a longitudinal evaluation of patients with infantile- and juvenile-onset gangliosidosis, comparing analytes in CSF and serum in search of potential biomarkers of disease. We found consistent elevations of analytes associated with inflammation. Thirteen CSF and two serum biomarker candidates were identified, with five CSF biomarkers showing exclusivity for infantile GM1- and GM2-gangliosidosis. Further study of these biomarkers advances understanding of infantile and juvenile gangliosidoses and will impact the development of potential treatments and treatment goals.
Fig. 1.
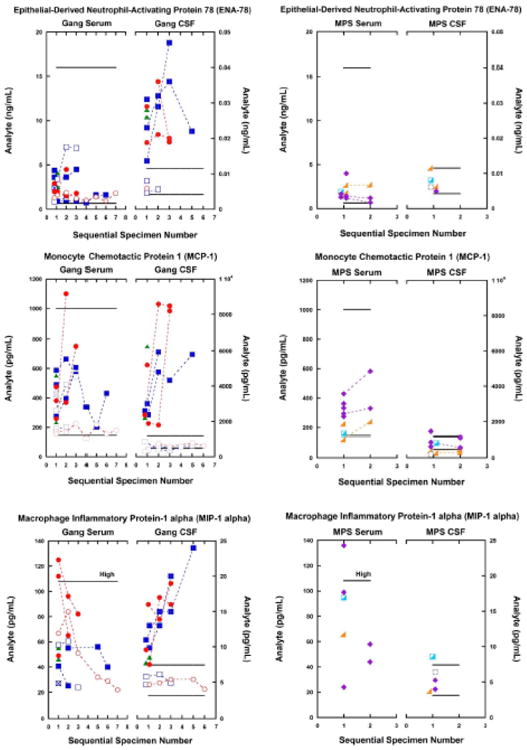
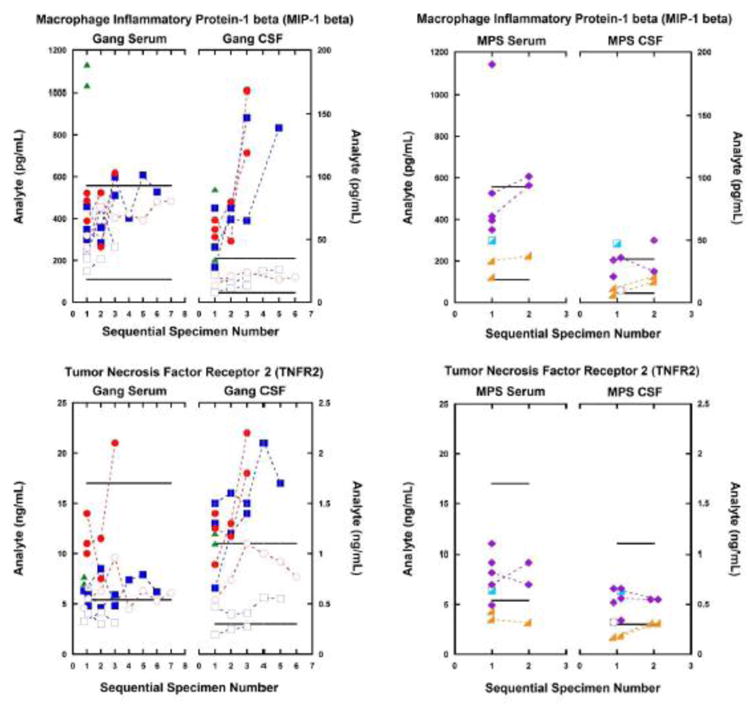
Among gangliosidosis (Gang) patients (left-side charts), five CSF analytes of inflammation were consistently elevated in patients with the infantile-onset disease in contrast to the normal levels found in patients with juvenile-onset disease. This relationship of severity of disease to analyte elevation is important, this correspondence reflecting the rate of disease progression.
CSF and serum inflammatory biomarkers from patients with MPS diseases (right-side charts) showed no abnormalities in either serum or CSF. The analyte differences between gangliosidosis disease and MPS disease emphasize the value of these inflammatory biomarkers, and illustrate that the analyte abnormalities are not general indicators of lysosomal storage or dysfunction, but reflect some other pathologic mechanism of gangliosidosis disease in the central nervous system.

Highlights.
Of 188 analytes in gangliosidosis patients, 13 inflammatory biomarkers were found
Thirteen CSF and 2 serum biomarker candidates were identified
Five CSF biomarkers were elevated in infantile gangliosidosis only
Elevated CSF cytokines in gangliosidosis are biomarkers for glial activation
Elevated biomarkers are indicators of inflammation in gangliosidoses
Acknowledgments
The Lysosomal Disease Network (U54NS065768) is a part of the National Institutes of Health (NIH) Rare Diseases Clinical Research Network (RDCRN), supported through collaboration between the NIH Office of Rare Diseases Research (ORDR) at the National Center for Advancing Translational Science (NCATS), the National Institute of Neurological Disorders and Stroke (NINDS) and National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sarafoglou K, Hoffmann GF, Roth KS. Pediatric Endocrinology and Inborn Errors of Metabolism. McGraw-Hill Company; New York: 2009. pp. 721–725.pp. 738–740.pp. 744–745. [Google Scholar]
- 2.Barranger JA, Cabrera-Salazar MA, editors. Lysosomal Storage Disorders. Springer; 2007. [PubMed] [Google Scholar]
- 3.Bley AE, Giannikopoulos OA, Hayden D, Kubilus K, Tifft CJ, Eichler FS. Natural history of infantile G(M2) gangliosidosis. Pediatrics. 2011;128:e1233–1241. doi: 10.1542/peds.2011-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith NJ, Winstone AM, Stellitano L, Cox TM, Verity CM. GM2-gangliosidosis in a UK study of children with progressive neurodegeneration: 73 cases reviewed. Dev Med Child Neurol. 2012;54:176–182. doi: 10.1111/j.1469-8749.2011.04160.x. [DOI] [PubMed] [Google Scholar]
- 5.Maegawa GH, Stockley T, Tropak M, Banwell B, Blaser S, Kok F, Giugliani R, Mahuran D, Clarke JT. The natural history of juvenile or subacute GM2-gangliosidosis: 21 new cases and literature review of 134 previously reported. Pediatrics. 2006;118:e1550–1562. doi: 10.1542/peds.2006-0588. Erratum in: Pediatrics 120 [2007] 936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Myerowitz R, Lawson D, Mizukami H, Mi Y, Tifft CJ, Proia RL. Molecular pathophysiology in Tay-Sachs and Sandhoff diseases as revealed by gene expression profiling. Hum Mol Genet. 2002;11:1343–1350. doi: 10.1093/hmg/11.11.1343. [DOI] [PubMed] [Google Scholar]
- 7.Jeyakumar M, Thomas R, Elliot-Smith D, Smith DA, van der Spoel AC, d'Azzo A, Perry VH, Butters TD, Dwek RA, Platt FM. Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2-gangliosidosis. Brain. 2003;126:974–987. doi: 10.1093/brain/awg089. [DOI] [PubMed] [Google Scholar]
- 8.Britschgi M, Rufibach K, Huang SL, Clark CM, Kaye JA, Li G, Peskind ER, Quinn JF, Galasko DR, Wyss-Coray T. Modeling of pathological traits in Alzheimer's disease based on systemic extracellular signaling proteome. Mol Cell Proteomics. 2011;10:M111 008862. doi: 10.1074/mcp.M111.008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Craig-Schapiro R, Kuhn M, Xiong C, Pickering EH, Liu J, Misko TP, Perrin RJ, Bales KR, Soares H, Fagan AM, et al. Multiplexed immunoassay panel identifies novel CSF biomarkers for Alzheimer's disease diagnosis and prognosis. PLoS One. 2011;6:e18850. doi: 10.1371/journal.pone.0018850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fischer R, Wajant H, Kontermann R, Pfizenmaier K, Maier O. Astrocyte-specific activation of TNFR2 promotes oligodendrocyte maturation by secretion of leukemia inhibitory factor. Glia. 2014 Feb;62(2):272–83. doi: 10.1002/glia.22605. Epub 2013 Dec 6. [DOI] [PubMed] [Google Scholar]
- 11.Patel JR, Williams JL, Muccigrosso MM, Liu L, Sun T, Rubin JB, Klein RS. Astrocyte TNFR2 is required for CXCL12-mediated regulation of oligodendrocyte progenitor proliferation and differentiation within the adult CNS. Acta Neuropathol. 2012 Dec;124(6):847–60. doi: 10.1007/s00401-012-1034-0. Epub 2012 Aug 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsuji D, Kuroki A, Ishibashi Y, Itakura T, Kuwahara J, Yamanaka S, Itoh K. Specific induction of macrophage inflammatory protein 1-alpha in glial cells of Sandhoff disease model mice associated with accumulation of N-acetylhexosaminyl glycoconjugates. J Neurochem. 2005 Mar;92(6):1497–507. doi: 10.1111/j.1471-4159.2005.02986.x. [DOI] [PubMed] [Google Scholar]
- 13.Kawashita E, Tsuji D, Kawashima N, Nakayama K, Matsuno H, Itoh K. Abnormal production of macrophage inflammatory protein-1alpha by microglial cell lines derived from neonatal brains of Sandhoff disease model mice. J Neurochem. 2009 Jun;109(5):1215–24. doi: 10.1111/j.1471-4159.2009.06041.x. Epub 2009 Mar 19. [DOI] [PubMed] [Google Scholar]