

Summary
Hepatitis C virus uniquely requires the liver specific microRNA-122 for replication, yet global effects on endogenous miRNA targets during infection are unexplored. Here, high-throughput sequencing and crosslinking immunoprecipitation (HITS-CLIP) experiments of human Argonaute (Ago) during HCV infection showed robust Ago binding on the HCV 5′UTR, at known and predicted miR-122 sites. On the human transcriptome, we observed reduced Ago binding and functional mRNA de-repression of miR-122 targets during virus infection. This miR-122 “sponge” effect was relieved and redirected to miR-15 targets by swapping the miRNA tropism of the virus. Single-cell expression data from reporters containing miR-122 sites showed significant de-repression during HCV infection depending on expression level and site number. We describe a quantitative mathematical model of HCV induced miR-122 sequestration and propose that such miR-122 inhibition by HCV RNA may result in global de-repression of host miR-122 targets, providing an environment fertile for the long-term oncogenic potential of HCV.
Introduction
Hepatitis C virus (HCV) is a hepatotropic positive-strand RNA virus of the flaviviridae family that is a leading cause of liver disease globally, with morbidities such as fibrosis, cirrhosis and hepatocellular carcinoma (Yamane et al., 2013). The long ORF of the ~9.6kb HCV genome encodes a polyprotein processed into ten proteins and is flanked by critical structured untranslated regions (UTRs). Unique to this virus is a dependence on the liver specific microRNA-122 (miR-122) (Jopling et al., 2005). Whereas miRNAs typically interact with the 3′UTRs of messenger RNAs (mRNAs) to promote mRNA destabilization and/or translational repression (Bartel, 2009), the binding of miR-122 to two binding sites (seed site S1 and S2) in the 5′UTR of HCV genomic RNA is critical for viral replication (Jopling et al., 2008; Machlin et al., 2011) by moderately stimulating viral protein translation (Henke et al., 2008) and, in concert with Argonaute (Ago), by stabilizing and protecting the uncapped HCV RNA genome from degradation (Li et al., 2013b; Sedano and Sarnow, 2014; Shimakami et al., 2012). As the predominant miRNA in the liver, miR-122 has multiple roles to regulate lipid metabolism (Esau et al., 2006), iron homeostasis (Castoldi et al., 2011), and circadian rhythms (Gatfield et al., 2009). MiR-122 knockout studies in vivo have revealed potent anti-inflammatory and anti-tumorigenic functions (Hsu et al., 2012; Tsai et al., 2012). Antagonizing miR-122 as an HCV therapeutic is a novel strategy (Lanford et al., 2010) with the first-in-class inhibitor, miravirsen/SPC3649, currently in phase II clinical studies (Janssen et al., 2013).
Studies of miRNA action during virus infections have been enhanced with the advent of high-throughput methods to elucidate genome-wide miRNA:mRNA interaction networks biochemically. Such methods (Chi et al., 2009; Hafner et al., 2010), broadly relying on cross-linking and immunoprecipitation (CLIP) of RNA bound to protein, have been applied to latent Kaposi’s sarcoma-associated herpesvirus (KSHV) (Haecker et al., 2012) and Epstein Barr virus (EBV) infections to uncover miRNA regulatory networks involved in promoting viral latency (Skalsky et al., 2012) and regulating cellular apoptosis (Riley et al., 2012).
In the current study, we elucidated global miRNA:target interaction maps during HCV infection on host and viral RNA. We observed Ago engagement at the HCV 5′UTR miR-122 sites, describe replication-dependent argonaute binding throughout viral genomic RNA, and provide evidence of miR-122 binding on an HCV resistant to miR-122 antagonism. On the host transcriptome, our results revealed globally reduced Ago binding and specific de-repression of miR-122 targets upon virus infection. This surprising systems-level observation suggests that HCV RNA functionally sequesters miR-122, and exhibits a miRNA “sponge” effect analogous to roles proposed for competing endogenous RNAs (ceRNA) (Salmena et al., 2011). Taken together, our results establish an RNA virus as a specific and indirect regulator of miRNA activity in the cell.
Results
Argonaute HiTS-CLIP of HCV infected cells
To study miRNA interactions during HCV infection, we either electroporated RNA or infected Huh-7.5 hepatoma cells with J6/JFH1-Clone2 HCV and after 48–72 hrs, when most cells were infected, performed Ago CLIP and RNA-seq measurements (Figure S1A–C). Ago-CLIP was performed using linker ligation as previously described (Figure S1D–F) (Moore et al., 2014). Alignment statistics for CLIP datasets presented in this paper are summarized in Tables S2–S5.
Due to known linker ligation biases in the preparation of small RNA libraries (Zhuang et al., 2012), we used polyG tailing (adapted from (Ingolia et al., 2009)) to determine miRNA abundance profiles (Figure S1G), and found that miR-122 at ~4.9% is the seventh most abundant miRNA (Figure S1H and Table S1). This correlated with previous data on miR-122 abundance in these cells (Figure S1I–J). No systematic bias from linker ligation was observed on mRNA targets due to the relative heterogeneity of RNAseA cleavage in creating mRNA Ago footprints (Figure S1K–L). For subsequent analysis on mRNA-CLIP clusters, we focused on searching the top 50 seed families derived from poly-G CLIP studies, which constituted over 97% of miRNA identified in Huh-7.5 cells.
An Ago binding map of HCV RNA confirms extensive miR-122 engagement
To define a small RNA interaction map on HCV and human mRNA, CLIP reads were mapped onto the HCV and human genomes. Among the 1–2% of CLIP reads mapping to HCV, Ago binding sites were identified by clustering overlapping reads and identifying statistically significant peaks above a uniformly distributed background (Darnell et al., 2011; Licatalosi et al., 2012). We observed major peaks in the 5′UTR, E1, E2, NS5A and NS5B regions of the genome (Figure 1A, top panel). No significant binding was observed on the negative strand (not shown). Notably, 50% of all Ago binding events on HCV RNA overlapped the known miR-122 seed sites in the 5′UTR (Figure 1A and 1B). Achieving nucleotide precision on the Ago:mRNA crosslink site via Crosslink Induced Mutation Site analysis (CIMS (Zhang and Darnell, 2011)), we observed an enrichment of crosslink sites predominantly within and immediately upstream of S2 (at positions 28 and 35), and, to a lesser degree, S1 base pairing locations (Figure 1B). The second largest peak was observed in the HCV IRES and overlapped the pseudoknot and coding start site (Figure 1D). No canonical 7mer or 8mer binding sites for the top 50 miRNA seeds were noted in this region, however, a putative non-canonical miR-122 site in the IRES (Pang et al., 2012) may explain the observed Ago binding.
Figure 1.

Argonaute binding maps on HCV RNA. (A) Mock-subtracted binding map of Ago-CLIP reads across HCV genomic RNA in WT or ΔDrosha Huh-7.5 cells. Data were normalized to total cellular and virus read depth for comparison. Significant peaks per track are named by location and indicated by asterisks. Bottom CIMS track shows location of all deletions (gray) and statistically significant CIMS deletions (red) from the WT track. (B) Ago binding in significant peaks from WT Huh-7.5 cells in (A) shown as normalized read densities calculated per dataset. Data were normalized to background read density of non-peak regions (dashed line). Asterisks, **P<0.01, *P<0.05, Student’s t-test. Error bars, ±SD. (C) Schematic of a miR-122 binding model to S1 and S2 highlighting locations of CIMS deletions. (D) Zoom in view of Ago binding from WT cells in (A) across the viral IRES into the coding sequence. IRES domains (II–IV), associated stemloops (a–d) and the pseudoknot (pk) region are indicated. Upper track displays seeds for the top 50 miRNA seeds, previously proposed miR-122 binding (Pang et al., 2012) highlighted in red. (E–F) Ago binding timecourse of WT (E) and replication deficient (GNN) (F) HCV post-electroporation (n=2). (G) Absolute qPCR measurements of miR-122 and HCV RNA levels at indicated time points post electroporation (n=3). Replication-deficient J6/JFH1-GNN and mock controls are shown. Dashed line indicates lower limit of quantitation. Error bars, ±SD. See also Figure S3.
To probe the general miRNA dependence of Ago binding on HCV RNA and to specifically enrich for miR-122 dependent binding, we deleted Drosha from Huh-7.5 cells to globally disrupt most miRNA biogenesis, using a CRISPR based genome editing strategy (Figure S2). From CLIP in ΔDrosha supplemented with miR-122 mimic to support HCV infection (Figure S2G), we observed that binding to S1/S2 and the IRES was maintained, and notably enhanced at NS5B binding sites (Figure 1A and 1C), both of which contain conserved miR-122 sites (Figure S3), of which one was previously shown to be inhibitory (Nasheri et al., 2011). Ago binding to E1 and E2 peaks was reduced in ΔDrosha cells, suggesting that a minor proportion Ago binding on HCV RNA is due to other miRNAs. While reports have suggested that numerous miRNAs interact with HCV RNA (reviewed in (Singaravelu et al., 2014)), among these only let-7 and miR-196 families fell within the top 50 miRNAs expressed, and no seeds from either of these families were observed within significant Ago binding peaks. Taken together, these data suggest that miR-122 constitutes the predominant miRNA interaction with HCV RNA in these cells and is largely confined to the 5′UTR.
Ago binding to miR-122 sites on HCV RNA occurs early and is replication independent
To address the timing of Ago binding to HCV RNA, we performed CLIP over a time course after electroporation of WT or replication defective (GNN) RNA genomes. We observed comparable Ago binding at the S1/S2 sites of WT and GNN mutants as early as 6 hours post electroporation (Figure 1E–F). Ago binding to the 5′UTR in general and to miR-122 sites in particular, remained stable throughout the WT timecourse, but decreased steadily for the GNN mutant. Ago binding to regions outside the 5′UTR emerged after 24 hours, were not observed in the GNN mutant, and correlated with HCV RNA abundance over time (Figure 1E–G). This suggested early Ago binding to the 5′UTR, and additional replication or abundance dependent low-level Ago targeting of the viral ORF.
An HCV resistant to miR-122 antagonism engages miR-122 and Ago
To further dissect the impact of non miR-122 Ago binding on HCV RNA, we focused on an HCV recombinant that is resistant to miR-122 antagonism (Li et al., 2011). This virus, for which the first HCV 5′UTR stem-loop is replaced with cellular U3 snoRNA, lacks the S1 site but contains an intact S2 site. Upon deleting miR-122 from Huh-7.5 cells (ΔmiR-122) using a CRISPR based strategy (Figure S2), we observed that while WT virus replication was abolished in ΔmiR-122 cells, U3 virus replication was largely unaffected (Figure 2A–B). Reintroducing miR-122 completely rescued WT virus replication, and had a small but consistent (2–4 fold) proviral effect on the U3 virus for the last timepoint tested (Figure 2A–B), suggesting that U3 virus replication is largely miR-122 independent. U3 virus replication could also be launched in ΔDrosha cells, yet in WT cells S2 p3 and p3,4 mutants were negative for HCV replication over 3 weeks suggesting perturbation of overlapping functions on the RNA (not shown). In CLIP on U3 infected Huh-7.5 cells, we observed Ago binding and crosslink mapping at the S2 miR-122 site specifically (Figure 2C) mirroring WT virus, and indicating that the U3 viral RNA residually engages miR-122. In the presence of increasing concentrations of miR-122 locked nucleic acid (LNA) inhibitor, we observed a dose-dependent decrease in Ago binding across the viral ORF, and a significant decrease in the S2 and IRES binding locations (Figure 2D–E), consistent with a limited proviral effect of miR-122 and specific miR-122 binding to S2 and the IRES. Furthermore, S2 and IRES binding was lost in U3 infected ΔDrosha and ΔmiR-122 cells (not shown). The striking miR-122 independence of this virus points to a potential avenue of resistance to LNA based therapeutics in the form of recombinant viruses with similarly large 5′UTR stem-loops.
Figure 2.

An HCV mutant resistant to miR-122 antagonism engages Ago and miR-122. (A–B) Timecourse qPCR measurements of WT-Clone2 virus (A) or U3-Clone2 virus (B) in WT cells or in ΔmiR-122 cells with or without 3nM miR-122 supplementation. Error bars, ±SD. (C) Ago binding map (top track) and CIMS locations (bottom track) across the U3 virus 5′UTR corresponding to miR-122 binding at S2. Relevant CIMS deletions are shown in gray (not significant) and red (significant). U3 snoRNA sequence is shown in green. (D) Ago binding map across the U3 virus genome after treatment with increasing doses of LNA122. Significant peaks are named by location and indicated by asterisks. Bottom CIMS track shows location of all deletions (gray) and significant CIMS deletions (red) for the untreated dataset. (E) Ago binding in significant peaks from untreated U3 datasets in (D) shown as normalized read densities calculated per dataset. ****P<0.0001, *P<0.05, one-way ANOVA with bonferroni correction. Error bars, ±SD.
HCV infection functionally reduces argonaute binding on host miR-122 targets
Given the crucial requirement of miR-122 for HCV replication, and in light of the result that HCV RNA levels accumulate to within one log of miR-122 levels (Figure 1G), we hypothesized that the HCV genome may act as a “sponge” for cellular miR-122, where viral replication may exert a broadly de-repressive effect on host miR-122 targets. We reasoned that this effect would be noticeable via CLIP as reduced Ago binding of miR-122 targets upon infection that may result in a specific increase in the expression of these targets measured by mRNA-seq. Indeed, comparing HCV infected to uninfected cells, we observed significantly reduced Ago binding globally for mRNA targets for which a miR-122 seed was present, compared to the combined targets of the miR-15/16 family, as a representative targetome of similar size to the miR-122 target network, and the top ten or the top fifty miRNA families cumulatively (Figure 3A). Significant changes in miR-122 binding were observed for all canonical seed types (as defined in (Bartel, 2009)) (Figure 3B). Additionally, the greatest change in Ago association was observed in CLIP clusters within 3′UTRs, and was less significant in CDS (coding exons), 5′UTRs, and introns (Figure S4A). Notably, the 3′UTR targets of other miRNAs suggested to bind HCV RNA directly were not altered upon HCV infection (Figure S4B). Through RNA-seq measurements we observed functional de-repression of CLIP derived miR-122 3′UTR targets after virus infection such that greater RNA abundance was evident when compared to all miRNA targets (Figure 3C). Likewise, we observed significant expression changes for all miR-122 target seed types (Figure 3D). Compared to bioinformatic prediction using Targetscan6.2 (TS) (Lewis et al., 2005), we found that CLIP largely complemented and expanded upon predicted miR-122 targets (Figure S4C–G). 3′UTR targets identified via CLIP and predicted by TS exhibited the greatest change in Ago binding (Figure S4C) and mRNA de-repression (Figure S4D–E) compared to expressed targets unique to either search modality. Of the expressed 731 miR-122 CLIP targets of all seed types identified via CLIP, 48% and 9% overlapped with non-conserved and conserved TS predictions, respectively (Figure S4F). Focusing on a more stringent set of 7mer and 8mer seeds for CLIP data yielded even greater overlap, such that only 5% of CLIP derived targets were not represented in either TS conservation category (Figure S4G). These results highlight a broad convergence between CLIP and bioinformatic prediction to outline a set of miR-122 targets specifically derepressed upon virus infection.
Figure 3.

HCV infection de-represses endogenous miR-122 targets. (A) Cumulative density function (CDF) of the log2 fold change in CLIP binding between infected and uninfected cells for all 3′UTR clusters containing indicated 7–8mer seeds by family, from triplicate experiments. “Top” refers to the top 10 miRNA families, exclusive of miR-122. “All” refers to the top 50 miRNA families, inclusive of miR-122. Two-sided K-S test p-value between miR-122 and all targets shown. (B) The mean log2 fold change (± ranges) in CLIP binding on miR-122 3′UTR targets versus all targets during HCV infection broken down by seed type. (C) A CDF plot during HCV infection as in (A) but measuring target mRNA expression via RNA-Seq, from duplicate experiments at 72hrs post-infection. Targets with more than one miRNA binding site were collapsed such that no gene is represented more than once per category. (D) The mean log2 fold change (± ranges) in mRNA expression of CLIP targets during HCV infection broken down by seed type. (E–G) CDF plot as in (A), between treatment over control cells with LNA122 (E) or miravirsen (F) at 30nM or genetic deletion (G) of miR-122 (ΔmiR-122), each from triplicate experiments. (H) Proportional Venn diagram showing the overlap of miR-122 targets with reduced CLIP binding across ΔmiR-122, LNA or miravirsen treatment, and HCV infection conditions. Hypergeometic p-value of overlap shown. Asterisks: ****P<0.0001, ***P<0.001, **P<0.01, *P<0.05, two-sided Mann-Whitney U-test. See also Figures S2 and S4.
To further corroborate our CLIP observations with HCV infection, we performed CLIP after pharmacologic inhibition of miR-122 and in ΔmiR-122 Huh-7.5 cells. The reduced Ago binding on miR-122 3′UTR targets during HCV infection was similar to 30nM LNA122 or miravirsen treatment (Figure 3E–F) and to ΔmiR-122 cells compared to unedited controls (Figure 3G). Interrogating the list of 3′UTR targets exhibiting reduced Ago binding across these three conditions revealed highly significant overlap (Figure 3H) suggesting that the effect of HCV replication on lowering functional miR-122 levels is functionally similar to antagonizing miR-122. The full complement of miRNA targets identified in these studies is presented in Table S6.
Transcriptome regulation by miR-122 sequestration in vitro is predictive of sequestration in vivo
As the global effect of HCV replication on host miR-122 usage mirrored LNA inhibition, and more profoundly miR-122 deletion, we hypothesized that miR-122 targets as a class might be de-repressed in human livers as a result of HCV infection. To this end, we performed a meta-analysis of published liver biopsy microarray data related to miR-122 inhibition or HCV infection. Comparing TS predictions for expressed miR-122 or miR-15 targets, as well as CLIP-identified miR-122 targets to all expressed genes in microarray data from miravirsen treated chimpanzee livers (Lanford et al., 2010), we noted a significant de-repression for TS or CLIP miR-122 targets compared to all genes or to predicted miR-15 targets (Figure 4A). In this data set, the CLIP-identified miR-122 targetome as a group was broadly more de-repressed than TS predictions. We performed the same analysis comparing HCV infected versus uninfected samples from two array datasets (Mas et al., 2009; Peng et al., 2009) and, despite the unknown proportion of infected cells, found in both that miR-122 target predictions were significantly de-repressed compared to all genes or to miR-15 target predictions in both datasets (Figure 4B–C). While these results cannot directly confirm an HCV sponge effect in vivo, they do emphasize that the overlap between CLIP results in vitro and expression results in vivo may indicate specific de-repression of miR-122 targets during HCV infection.
Figure 4.
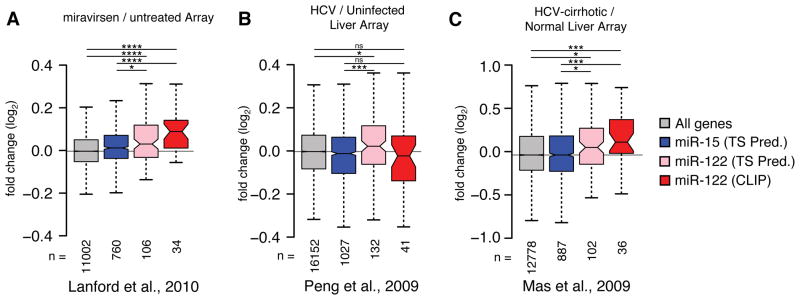
Meta-analysis of published array data suggests HCV induced changes on the miR-122 target network. (A) Miravirsen pre- and post-treatment array data from four HCV infected chimpanzees (Lanford et al., 2010) was binned according to conserved 7–8mer TargetScan (TS) predictions for miR-15 or miR-122, or from miR-122 targets with CLIP support from the current study. Boxplot whiskers denote 1.5 times the inter-quartile distance from the nearest quartile. The mean fold change in expression for miR-122 targets was compared to miR-15 targets or all genes represented on the array, where the number of genes in each bin (n) is indicated. (B) Analysis as in (A) comparing 24 HCV positive to 5 negative liver biopsies (Peng et al., 2009). (C) Analysis as in (A) comparing 41 HCV positive with cirrhosis samples to 19 normal livers (Mas et al., 2009). Asterisks: ****P<0.0001, ***P<0.001, ** P<0.01, *P<0.05, ns P>0.05, two-sided Mann-Whitney U-test.
Validation of HCV induced miR-122 sequestration in bulk and single cells
The results thus far describe the global characteristics of the HCV induced miR-122 sponge effect on the host transcriptome. We next used luciferase and fluorescent reporters of miRNA activity to validate the HCV miR-122 sponge on individual 3′UTRs. The endogenous repression of luciferase reporters containing one miR-122 seed, as well as 3′UTRs of CLIP derived miR-122 targets was enhanced upon adding additional miR-122, and was reversed upon LNA mediated miR-122 inhibition (Figure 5A). We also observed seed-dependent statistically significant de-repression of these reporters upon virus infection (Figure 5A).
Figure 5.

Validation of HCV induced de-repression of miR-122 targets in bulk and single-cell resolution. (A) Luciferase reporter measurements for synthetic miR-122, miR-17, or cellular 3′UTR target constructs. Data were normalized to “no oligo” p3,4 mutant conditions. Significance testing was performed relative to endogenous “no oligo” repression for each tested construct. Asterisks: *** P<0.001, ** P<0.01, * P<0.05, ANOVA with bonferroni correction. (B) Two-color fluorescent reporter containing a bidirectional Tet promoter that drives expression of blue and red fluorescent proteins (TagBFP and TagRFP). Each fluorescent protein is tagged with a nuclear localization sequence (NLS) to aid in flow cytometric analysis. The 3′UTR of TagRFP is engineered to contain N binding sites for miR-122, or full 3′UTRs of miR-122 targets. (C–F) Log-log transfer functions for N=1 (C), ALDOA 3′UTR (D), N=4 (E) or one perfectly complementary (F) miR-122 site in the presence or absence of 30nM miRNA mimic and/or HCV infection.
As cellular mRNA and HCV RNA expression levels can vary widely between individual cells (Kandathil et al., 2013; Sheahan et al., 2014), we sought to achieve a more thorough understanding of the HCV miRNA sponge on host miRNA targets at a quantitative single-cell level. Previous work demonstrated that miRNAs generate thresholds of gene expression such that miRNA repression can be highest on low abundance targets, and can be virtually non-existent on high abundance targets. Furthermore, these thresholds can be altered upon manipulating miRNA levels (Mukherji et al., 2011). To test whether HCV replication could broadly impact functional miR-122 levels, we adapted the strategy used by Mukherji et al. to construct two-color tet-inducible fluorescent reporters of miRNA activity amenable to flow cytometry (Mukherji et al., 2011)(Figure 5B).
Testing reporters with N=1 and 6 miR-122 binding sites in the presence of miR-122 mimic, we observed miR-122 repression that increased with N, as expected, whereas adding LNA122 decreased repression (Figure S5 A–D). HCV infection in both contexts resembled LNA inhibition where de-repression was notably more pronounced in cells expressing low amounts of reporter, demonstrating previously reported miRNA thresholding effects (Mukherji et al., 2011) (Figure S5A and C). Importantly, no such changes were observed for a reporter with a p3,4 miR-122 seed (“N1m”). (Figure S5E–F). Additionally, we tested a reporter with a perfectly complementary miR-122 site, thus making the miRNA behave as a siRNA. This reporter exhibited no thresholding such that mimic repression, or LNA and HCV de-repression was observed at all expression levels (Figure S5G–H). These data suggest that HCV infection modulates functional miR-122 levels to relieve endogenous repression on host targets in a stoichiometric manner, governed by target expression level and the number of miRNA binding sites.
As miR-122 levels in Huh7 derived cells are estimated to be 10-fold lower than primary adult liver tissue (Chang et al., 2004), we next explored the HCV sponge effect in the presence of excess miR-122. Exogenous miR-122 addition increaseed intracellular miR-122 by up to 10-fold in Huh-7.5 cells, within the range of miR-122 levels measured from patient liver biopsies (Figure S6A–B). As no changes in HCV RNA levels were observed, the resulting miR-122:HCV ratio went from ~15:1 fold at the lowest, to over 100:1 with 30nM of miR-122 mimic added (Figure S6C). Testing N=1 or AldoA 3′UTR reporter constructs in this in vivo-like context, we observed that HCV infection was able to relieve 30nM of mimic repression to untreated levels for low but not high abundance targets (Figure 5C–D). The ability for HCV to rescue excess miR-122 repression was not as pronounced for the N=6 construct (Figure 5E) whearas a reporter containing a perfectly complementary miR-122 site was particularly sensitive to rescue by HCV replication (Figure 5F). Similar, dose-dependent results were obtained under 0.3 or 3nM mimic treatment for all constructs (Figure S6D–G). Taken together, these results suggest that miR-122 sponging by HCV can exist in more physiologic miR-122 concentration settings.
A quantitative model of miR-122 sponging by HCV RNA
To achieve a more quantitative understanding of the HCV sponge, we used our dose-dependent mimic and LNA reporter system measurements to expand the miRNA model of gene regulation presented by Mukherji et al. to incorporate a competing self-replicating viral target (Figure 6A). Here, if HCV RNA is present at sufficiently high numbers or has relatively high binding strengths compared to other miR-122 targets, it acts to reduce the available miR-122 pool, and de-represses miR-122 targets (r, measured as TagRFP fluorescence) relative to non-targets (r0, measured as TagBFP fluorescence) (Figure 6B). We developed a quantitative formula for HCV induced reduction of the miR-122 pool in this scenario (see Supplementary Information). Assuming steady state levels of HCV RNA at the time of measurements resulted in a decrease of the model parameter θ, which governs the amount of free miRNA in the system. The number of miR-122 sites is estimated by the model parameter λ that is related to the total binding strength of miR-122 to a particular site. By tuning these parameters, we accurately fitted experimental data of endogenous miR-122 repression of reporters with increasing numbers of miR-122 sites (Figure 6C).
Figure 6.

Quantitative modeling of miR-122 sequestration by HCV. (A) Illustration of model reactions for miR-122 dynamics, including transcription and translation of a target mRNA, binding to miR-122 and decay of mRNA species. HCV RNA can replicate, be degraded, or bind miR-122, functionally sequestering miR-122 and leading to de-repression of mRNA targets. (B) Increasing amounts of HCV (or a relative increase in binding strength at miR-122 sites) leads to changes in single-cell gene expression as compared to unregulated targets, with stronger effects at the low mRNA expression levels. Parameters used are fitted from data in (C). Each curve, from top to bottom, represents a reduction in the miRNA pool by 20%. Inset displays model on a linear scale. (C) Model fitting of the steady state approximation to experimental data while increasing the number of binding sites corresponding to changes in total binding strength. (D) Model fitting for the N=4 case showing a 50% reduction in the miRNA pool by HCV modeled by a proportional change in the theta parameter. (E) Model fitting for the N=4 construct under 30nM miR-122 mimic addition ± HCV infection. (F) Increasing HCV:miR-122 binding strength or HCV RNA abundance results in functional de-repression of miR-122 targets. The curves (top to bottom) represent 10 percentage-points increases in the available miR-122 pool (10% to 100% availability). (G–H) Experimental HCV induced derepression of synthetic miR-122 binding site constructs (G) or endogenous 3′ UTRs with miR-122 binding sites (H). See also Figure S5 and S6.
To explore the effect of HCV on the miR-122 pool, we fitted the model to experimental data with four miR-122 sites during infection (Figure 6D), and estimated the change to parameter θ to correspond to an approximate 50% reduction in available miR-122. A similar result was obtained for the N=4 construct in the presence of HCV and 30nM miR-122 mimic (Figure 6E). The model estimated that the highest theoretical HCV levels reducing the miR-122 pool by 90% could de-repress mRNA targets by up to 4.5-fold for low-expressed mRNAs (Figure 6F). Synthetic reporter measurements agreed with model predictions for 50% reductions in miR-122 levels, where de-repression was most drastic for low expressed targets harboring multiple miR-122 sites (Figure 6G).
In similar measurements with reporters containing full 3′UTRs, we observed modest de-repression upon HCV infection with an average change of 25% across all expression levels for the previously known targets with one miR-122 site, AldoA, PKM2, and P4HA1, but not for CS 3′UTRs (Figure 6H and S5H–L). The CLIP-identified targets CTDNEP1, SFT2D1, MASP1 and MAL2 behaved similarly, with all four tested being reduced upon miR-122 mimic addition and all except MAL2 de-repressed upon adding virus (Figure S5M–P). Our quantitative model outlines several factors controlling HCV induced de-repression of host mRNA targets, namely, the expression level of the target mRNA, mRNA-miR-122 binding strength, and the number of sites on the target mRNA.
A miR-15 dependent HCV redirects miRNA sequestration
To go beyond the correlative connection between our CLIP, RNA-seq, and modeling results, we set out to confirm the sponge effect by swapping the miRNA tropism of the virus to determine if the miR-122 sponge could be redirected to the targets of another miRNA. We selected miR-15a/b, as these miRNAs had a sufficiently altered but GC rich seed, and maintained the auxiliary pairing at nucleotides 2–3 and 30–31 of the viral genome (Machlin et al., 2011) (Figure 7A and S7A). In total, miR-15a/b constituted 5.3±1.2% of miRNA identified via miRNA-CLIP, compared to 4.9±2.0% for miR-122 (Table S1 and Figure S7A). An electroporated miR-15 variant HCV luciferase reporter virus (m15) was viable, and replicated to within one log of the WT virus after 72hrs (Figure 7B). Notably, the miR-15 virus was resistant to increasing concentrations of LNA122 (IC50 > 50nM) but susceptible to LNA15a/b (IC50 = 10nM) (Figure 7C). Similar results were obtained with non-reporter viruses through measuring HCV replication, spread of infection and virus titers (Figure S7B–G). Unlike WT virus, the m15 virus was viable in ΔmiR-122 cells (Figure 7D), thus demonstrating complete HCV replication independent of miR-122.
Figure 7.

Exchanging HCV miRNA tropism redirects functional miRNA sequestration. (A) Base pairing diagram of miR-15a onto the mutated m15 HCV RNA. Base changes from the WT at S1 and S2 are highlighted in blue. (B) Luciferase measurements of supernatants from WT and m15 HCV reporter virus electroporations (±SD). Non-replicating GNN control is shown. (C) Dose response of WT and m15 reporter viruses following pre-treatment with LNA inhibitors of miR-122 or miR-15a/b at indicated concentrations, measured at 96 hours post-infection (±SD). (D) Time course post infection of ΔmiR-122 Huh-7.5 cells of indicated viruses (±SD). (E) Ago binding map of m15 virus infection in WT Huh-7.5 (top panel) or ΔmiR-122 Huh-7.5 cells (bottom panel). Data were normalized to total cellular read depth for cross track comparison. Statistically significant peaks per track are named by location and are indicated by asterisks. (F) Ago binding in significant peaks from (E) shown as normalized read densities calculated per dataset. Two-sided Student’s t-test used. Error bars, ±SD.. (G) CDF plot of the log2 fold change in mRNA expression between HCV m15 infected and uninfected cells for all 3′UTR clusters containing indicated 7–8mer seeds by family, from duplicate experiments at 96hrs post infection. “Top” refers to the top 10 miRNA families, exclusive of miR-122 and miR-15. “All” refers to the top 50 miRNA families, inclusive of miR-122 and miR-15. Two-sided K-S test P-value comparing miR-15 (blue) or miR-122 (red) clusters to “All” is shown. (H) Infectivity titers of m15 virus in ΔmiR-122 Huh-7.5 complemented with exogenous miR-122 at indicated concentrations. One-way ANOVA with Bonferroni correction, whiskers, ± ranges. Asterisks: ****P<0.0001, ***P<0.001, **P<0.01, *P<0.05. See also Figure S7.
Ago binding on the m15 virus in Huh-7.5 cells largely mirrored results observed with WT virus (Figure 7E, top panel). Interestingly, we observed reduced Ago binding in ΔmiR-122 cells for the m15 virus in IRES and NS5B peaks harboring conserved miR-122 seeds, further suggesting miR-122 dependence for Ago binding in these regions (Figure 7E–F).
Turning our attention to the host, we measured the effect on the miR-15 targetome due to m15 virus replication via RNA-seq and confirmed de-repression specific to miR-15 targets while no longer observing effects on the miR-122 target network (Figure 7G). These results highlight the causal nature of an HCV induced miRNA sponge as both functional and somewhat modular. We note that the m15 virus sponge effect was generally weaker than for the WT virus, likely due to the lower replication level observed and possibly to binding of the miR-15 family member, miR-16, which shares the seed site but may not be able to engage the m15 genome due to lack of auxiliary pairing (Figure S7A). Modification of the HCV 5′UTR could have direct effects on viral replication, and may explain the slight attenuation observed for the m15 virus (Figure 7B).
To further evaluate pro- or anti-viral effects of miR-122 abundance on the m15 virus, we measured m15 virus replication in ΔmiR-122 cells after re-introducing miR-122 at various concentrations. We observed a slight but significant increase in secreted m15 virus levels upon re-introducing miR-122 (Figure 7H). RNA levels and percent of infected cells were also increased in m15 infected ΔmiR-122 cells upon restoring miR-122 levels (Figure S7H–I). Conversely, we observed a significant reduction in viral titers after LNA-122 treatment of m15 infected Huh-7.5 cells (Figure S7G). Taken together, these results suggest that miR-122 sequestration by HCV replication confers a slightly less pro-viral cellular environment; an effect that may be a small trade-off for the hugely beneficial role miR-122 plays directly in the viral life-cycle.
Discussion
The aim of this study was to obtain an unbiased view of the miRNA interactome during HCV infection on both viral and cellular targets. We provide direct biochemical evidence of replication independent Ago association with HCV RNA at the 5′UTR in a number of infection contexts. Our results chiefly establish HCV RNA may act as a competitive inhibitor of miR-122 activity, an idea closely aligned with proposed roles for ceRNAs (Salmena et al., 2011). While both HCV RNA and ceRNAs share the theme of de-repressing a miRNA regulated network by increasing the pool of available targets through RNA expression, they differ in their mode of interaction with miRNAs. HCV genomic RNA critically requires miR-122 interaction to stabilize the viral genome and stimulate translation and replication, while most cellular miRNA targets are degraded upon encountering a miRNA. Moreover, unlike cellular mRNA targets, HCV genomic RNA is its own substrate for replication, and thus constitutes a positive feedback loop to sequester additional miR-122. These distinguishing features suggest different parameters for HCV versus ceRNA based sponge effects on a miRNA target network. This is relevant in light of recent findings showing that endogenous miR-122 repression is only relieved when ceRNAs are forcibly expressed at super-physiological levels (Denzler et al., 2014). As HCV RNA represented at most between 1–2% of CLIP reads following infection, our data suggest ceRNA activity may occur naturally at lower expression levels. Indeed, single-cell reporter measurements and mathematical modeling of the mRNA, HCV and miRNA interplay suggest that HCV is able to de-repress host miR-122 targets, due to an approximate 2-fold reduction in available miR-122 under our experimental levels of HCV replication. Natural viral derived miRNA sponges have been described previously (Cazalla et al., 2010; Lee et al., 2013) though no examples are currently known from RNA viruses. Whether this systems-level phenomenon occurs with other, more robust RNA virus infections remains to be explored.
The establishment of a miR-15 dependent HCV suggests that the miR-122 sponge effect is largely dispensable for the virus in the Huh-7.5 cell context. Indeed, we observed that LNA-122 slightly reduced m15 virus titers and that restoring miR-122 in ΔmiR-122 cells increased titers, suggesting that the miR-122 sponge may reflect a tradeoff for the large, positive and direct impact of miR-122 on WT HCV replication. Conceivably, HCV replication may exert enough pressure on miR-122 levels to de-repress targets such that the cellular environment is passively altered to negatively impact viral replication. Or more actively, there may exist miR-122 targets that act as sensors for low miR-122 levels, and by extension, the health of the hepatocyte. While future work will be needed to shed light on specific players involved in this process, our data suggest that viral replication faces a ceiling by reducing levels of an otherwise pro-viral miRNA.
How might the HCV miR-122 sponge impact a hepatocyte? Work with miR-122 knockout mice, which develop progressive liver disease that spontaneously results in HCC (Hsu et al., 2012; Tsai et al., 2012) suggests that miR-122 tumor suppressor activity is essential for long-term liver homeostasis. It is tantalizing to speculate that miR-122 sequestration in a chronic HCV infection may be a molecular link to the heterogeneous liver dysfunction that characterizes HCV induced disease. Indeed, a number of miR-122 targets that we confirm or establish via CLIP and reporter measurements, such as P4HA1, PKM2, and MASP1 are known to be upregulated in fibrosis or HCC (Jung et al., 2011; Li et al., 2013a), with MASP1 notable for being specifically linked with HCV-associated HCC (Saeed et al., 2013). Still, there are challenges for a direct demonstration of an HCV miR-122 sponge in vivo. Our bulk cell measurements estimate ten-fold higher miR-122 levels in primary liver tissue versus hepatoma cell line derivatives (105 vs 104 estimate ranges per cell), while HCV levels per cell are estimated to range from 1 to 102 copies per hepatocyte (Kandathil et al., 2013), in contrast to Huh7 derivatives with 103 copies per cell. Yet, we observed functional HCV-induced de-repression in our single-cell reporter assay after addition of miR-122 in Huh-7.5 cells to mimic levels in vivo. Furthermore, we found specific de-repression of miR-122 targets in microarrays from HCV infected livers and from miravirsen treated chimpanzees, supporting the existence of an HCV sponge effect in vivo. Unlike the >90% infection frequencies in Huh-7.5 cells, the percentage of infected hepatocytes in chronically infected patients based on in situ hybridization of liver biopsy tissue, ranges from as low as 0.07% to as high as 100%, with medians in the 20–40% range (Liang et al., 2009; Pal et al., 2006). Combined with HCV genotype, dynamic replication variation within the liver, and host variability in innate immune responses (Sheahan et al., 2014), a complex picture of HCV infection emerges that would largely mask observations of HCV sponge effects in bulk cell or tissue Ago-CLIP measurements. As it remains possible that functional effects of such a sponge may impact highly infected cells, our data highlight the possibility of searching for transcriptome level changes to the miR-122 target network in response to HCV infection in individual cells. The extension of CLIP and RNA-seq in single-cell and primary contexts provides a compelling platform to address these and other long-term disease driven changes to a miRNA target network.
Experimental Procedures
Culture of cell lines, generation and characterization of WT and m15 HCV
Cell lines were cultured and generated as described in Supplemental Information. pJ6/JFH1-Clone2, pJ6/JFH-Clone2-5AB-Ypet, and pJc1FLAG(p7-nsGluc2A) are fully-infectious HCV non-reporter and reporter viruses respectively, that have been previously described (Catanese et al., 2013; Horwitz et al., 2013; Marukian et al., 2008). To construct miR-15 dependent viruses in both backgrounds, we used an overlap PCR mutagenesis strategy. HCV cloning, RNA transcription, electroporation, infection and related virus assays are described in detail in the Supplemental Information.
Argonaute HiTS-CLIP Analysis
Argonaute CLIP was performed generally following previous work (Chi et al., 2009), (Moore et al., 2014). Poly-G CLIP is a direct adaptation of the single linker ligation BrdU CLIP protocol (Weyn-Vanhentenryck et al., 2014). Relevant details pertaining to the CLIP protocol, multiplexed library preparation, and bioinformatic analysis are described in full in the Supplemental Information.
mRNA-seq library construction
mRNA-seq libraries were prepared from Trizol extracted RNA following Illumina TruSeq protocols for poly-A selection, fragmentation, and adapter ligation. Multiplexed libraries were sequenced as 100nt single-end runs on either GAII or HiSeq-2000 platforms.
Luciferase reporter assays
Luciferase reporter vectors were cloned by inserting short oligonucleotides or PCR amplified target 3′UTRs into psiCHECK-2. The sequence of DNA oligonucleotides used for cloning are found in Table S7. Huh-7.5 cells were transfected over night with 2.56nM final concentration LNA122 (Exiqon) or miR-122 mimic (Thermo Fisher) using RNAi/MAX (Invitrogen). Alternatively, cells were infected with HCV (J6/JFH1-clone2), MOI=3 over night. 24 hrs later, cells were transfected with 1ng/well psiCHECK-2 reporter plasmid using Lipofectamine2000 (Invitrogen) and incubated over night before lysis in Passive Lysis Buffer and evaluation of luciferase levels using the Dual Luciferase Reporter Assay (Promega) on a Omega Fluorostar reader (BMG Labtech).
Single-cell reporter measurements
Construction of miR-122 fluorescent reporters largely mirrored previous work with miR-20 (Mukherji et al., 2011). For a list of primers used in plasmid construction, please refer to Table S7. For flow cytometry, cells were run on a MACSQuant VYB flow cytometer (Miltenyi Biotec) after fixation to detect TagBFP, TagRFP, and Ypet signals. The raw FACS data were analyzed with FlowJo software to gate single, intact cells according to their forward (FSC-A) and side (SSC-A) scatter profiles. HCV positive cells were gated on the basis of Ypet signal above uninfected background. Untransfected cells were used to characterize the cellular autoflourescence in BFP and RFP channels, from which we subtracted the mean plus two standard deviations of the autofluorescent signal for each channel in transfected cells. Cells with BFP and RFP fluorescence levels less than 0 after background subtraction were excluded from further analyses. Data were log-transformed and binned according to BFP levels, and the mean RFP signal was calculated for each BFP bin.
Supplementary Material
Acknowledgments
We thank C. Zhang for bioinformatics support; M. Moore for flow cytometry assistance; and M. E. Castillo, S. M. Pecoraro Di Vittorio, J. Palarca, J. Sable, B. Hough-Loomis, and J. Smith for excellent technical and administrative assistance. Lastly, we thank members of the Darnell and Rice labs for invaluable advice. This study was supported by grants from the Public Health Service, NIH, NIAID (AI099284, AI072613, AI075099, AI091707, AI090055), Office of the Director through the NIH Roadmap for Medical Research (DK085713), NCI (CA057973), The Rockefeller University Center for Clinical and Translational Science (UL1RR024143), the Center for Basic and Translational Research on Disorders of the Digestive System through the generosity of the Leona M. and Harry B. Helmsley Charitable Trust, the Greenberg Medical Research Institute, and the Starr Foundation. J.M.L. was supported by a David Rockefeller Graduate Student Fellowship. T.K.H.S. was supported by a Postdoctoral Fellowship and a Sapere Aude Research Talent Award from The Danish Council for Independent Research. C.N.T. was supported by an HHMI International Predoctoral Fellowship. M.T.C. was supported by the Rockefeller University Women and Science Fellowship. R.B.D. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Accession numbers
HiTS-CLIP and RNA-seq data has been deposited in the GEO under accession number GSE64680.
Author contributions. J.M.L. conceived the project; J.M.L., T.K.H.S., C.M.R. and R.B.D. designed the experiments; J.M.L., T.K.H.S, K.S.S., A.M., J.J.F., E.N. and M.T.C. performed the experiments; C.N.T. constructed Huh-7.5 TetON cells; Y.P.J and I.M.J provided clinical samples; J.M.L and T.D. developed the mathematical model; J.M.L, T.K.H.S, and T.D. analyzed the data; J.M.L, T.K.H.S, T.D., C.M.R. and R.B.D. wrote the manuscript, with input from all authors.
References
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castoldi MM, Spasic MMV, Altamura SS, Elmén JJ, Lindow MM, Kiss JJ, Stolte JJ, Sparla RR, D’Alessandro LAL, Klingmüller UU, et al. The liver-specific microRNA miR-122 controls systemic iron homeostasis in mice. J Clin Invest. 2011;121:1386–1396. doi: 10.1172/JCI44883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catanese MT, Loureiro J, Jones CT, Dorner M, von Hahn T, Rice CM. Different Requirements for Scavenger Receptor Class B Type I in Hepatitis C Virus Cell-Free versus Cell-to-Cell Transmission. Journal of Virology. 2013;87:8282–8293. doi: 10.1128/JVI.01102-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazalla D, Yario T, Steitz JA, Steitz J. Down-regulation of a host microRNA by a Herpesvirus saimiri noncoding RNA. Science. 2010;328:1563–1566. doi: 10.1126/science.1187197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Nicolas E, Marks D, Sander C, Lerro A, Buendia MA, Xu C, Mason WS, Moloshok T, Bort R, et al. miR-122, a mammalian liver-specific microRNA, is processed from hcr mRNA and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol. 2004;1:106–113. doi: 10.4161/rna.1.2.1066. [DOI] [PubMed] [Google Scholar]
- Chi S, Zang J, Mele A, Darnell R. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009 doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KYS, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, et al. FMRP Stalls Ribosomal Translocation on mRNAs Linked to Synaptic Function and Autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denzler R, Agarwal V, Stefano J, Bartel DP, Stoffel M. Assessing the ceRNA Hypothesis with Quantitative Measurements of miRNA and Target Abundance. Mol Cell. 2014;54:766–776. doi: 10.1016/j.molcel.2014.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, Watts L, Booten SL, Graham M, McKay R, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metabolism. 2006;3:87–98. doi: 10.1016/j.cmet.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Gatfield D, Le Martelot G, Vejnar CE, Gerlach D, Schaad O, Fleury-Olela F, Ruskeepaa AL, Oresic M, Esau CC, Zdobnov EM, et al. Integration of microRNA miR-122 in hepatic circadian gene expression. Genes Dev. 2009;23:1313–1326. doi: 10.1101/gad.1781009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haecker II, Gay LAL, Yang YY, Hu JJ, Morse AMA, McIntyre LML, Renne RR. Ago HITS-CLIP expands understanding of Kaposi’s sarcoma-associated herpesvirus miRNA function in primary effusion lymphomas. PLoS Pathog. 2012;8:e1002884–e1002884. doi: 10.1371/journal.ppat.1002884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner MM, Landthaler MM, Burger LL, Khorshid MM, Hausser JJ, Berninger PP, Rothballer AA, Ascano MM, Jungkamp ACA, Munschauer MM, et al. Transcriptome-wide Identification of RNA-Binding Protein and MicroRNA Target Sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henke JI, Goergen D, Zheng J, Song Y, Schüttler CG, Fehr C, Jünemann C, Niepmann M. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J. 2008;27:3300–3310. doi: 10.1038/emboj.2008.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz JA, Dorner M, Friling T, Donovan BM, Vogt A, Loureiro J, Oh T, Rice CM, Ploss A. Expression of heterologous proteins flanked by NS3-4A cleavage sites within the hepatitis C virus polyprotein. Virology. 2013;439:23–33. doi: 10.1016/j.virol.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu S-H, Wang B, Kota J, Yu J, Costinean S, Kutay H, Yu L, Bai S, La Perle K, Chivukula RR, et al. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J Clin Invest. 2012;122:2871–2883. doi: 10.1172/JCI63539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen HLA, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, van der Meer AJ, Patick AK, Chen A, Zhou Y, et al. Treatment of HCV Infection by Targeting MicroRNA. New England Journal of Medicine. 2013 doi: 10.1056/NEJMoa1209026. [DOI] [PubMed] [Google Scholar]
- Jopling CL, Schütz S, Sarnow P. Position-dependent function for a tandem microRNA miR-122-binding site located in the hepatitis C virus RNA genome. Cell Host Microbe. 2008;4:77–85. doi: 10.1016/j.chom.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005;309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- Jung CJC, Iyengar SS, Blahnik KRK, Ajuha TPT, Jiang JXJ, Farnham PJP, Zern MM. Epigenetic modulation of miR-122 facilitates human embryonic stem cell self-renewal and hepatocellular carcinoma proliferation. PLoS ONE. 2011;6:e27740–e27740. doi: 10.1371/journal.pone.0027740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandathil AJ, Graw F, Quinn J, Hwang HS, Torbenson M, Perelson AS, Ray SC, Thomas DL, Ribeiro RM, Balagopal A. Gastroenterology. 2013;145:1404–1413. e1410. doi: 10.1053/j.gastro.2013.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, Kauppinen S, Orum H. Therapeutic Silencing of MicroRNA-122 in Primates with Chronic Hepatitis C Virus Infection. Science. 2010;327:198–201. doi: 10.1126/science.1178178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Song J, Kim S, Kim J, Hong Y, Kim Y, Kim D, Baek D, Ahn K. Selective Degradation of Host MicroRNAs by an Intergenic HCMV Noncoding RNA Accelerates Virus Production. Cell Host Microbe. 2013;13:678–690. doi: 10.1016/j.chom.2013.05.007. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Li J, Ghazwani M, Zhang Y, Lu J, Li J, Fan J, Gandhi CR, Li S. miR-122 regulates collagen production via targeting hepatic stellate cells and suppressing P4HA1 expression. J Hepatol. 2013a;58:522–528. doi: 10.1016/j.jhep.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y-P, Gottwein JM, Scheel TK, Jensen TB, Bukh J. MicroRNA-122 antagonism against hepatitis C virus genotypes 1–6 and reduced efficacy by host RNA insertion or mutations in the HCV 5′ UTR. Proc Natl Acad Sci. 2011;108:4991–4996. doi: 10.1073/pnas.1016606108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Masaki T, Yamane D, McGivern DR, Lemon SM. Competing and noncompeting activities of miR-122 and the 5′ exonuclease Xrn1 in regulation of hepatitis C virus replication. Proc Natl Acad Sci. 2013b;110:1881–1886. doi: 10.1073/pnas.1213515110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Shilagard T, Xiao SY, Snyder N, Lau D, Cicalese L, Weiss H, Vargas G, Lemon SM. Visualizing Hepatitis C Virus Infections in Human Liver by Two-Photon Microscopy. Gastroenterology. 2009;137:1448–1458. doi: 10.1053/j.gastro.2009.07.050. [DOI] [PubMed] [Google Scholar]
- Licatalosi DD, Yano M, Fak JJ, Mele A, Grabinski SE, Zhang C, Darnell RB. Ptbp2 represses adult-specific splicing to regulate the generation of neuronal precursors in the embryonic brain. Genes Dev. 2012;26:1626–1642. doi: 10.1101/gad.191338.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machlin ES, Sarnow P, Sagan SM. Masking the 5′ terminal nucleotides of the hepatitis C virus genome by an unconventional microRNA-target RNA complex. Proc Natl Acad Sci. 2011;108:3193–3198. doi: 10.1073/pnas.1012464108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marukian S, Jones CT, Andrus L, Evans MJ, Ritola KD, Charles ED, Rice CM, Dustin LB. Cell culture-produced hepatitis C virus does not infect peripheral blood mononuclear cells. Hepatology. 2008;48:1843–1850. doi: 10.1002/hep.22550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mas VR, Maluf DG, Archer KJ, Yanek K, Kong X, Kulik L, Freise CE, Olthoff KM, Ghobrial RM, McIver P, et al. Genes involved in viral carcinogenesis and tumor initiation in hepatitis C virus-induced hepatocellular carcinoma. Mol Med. 2009;15:85–94. doi: 10.2119/molmed.2008.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Zhang C, Gantman EC, Mele A, Darnell JC, Darnell RB. Mapping Argonaute and conventional RNA-bindingprotein interactions with RNA at single-nucleotideresolution using HITS-CLIP and CIMS analysis. Nat Protoc. 2014;9:263–293. doi: 10.1038/nprot.2014.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherji S, Ebert MS, Zheng GXY, Tsang JS, Sharp PA, van Oudenaarden A. MicroRNAs can generate thresholds in target gene expression. Nat Genet. 2011;43:854–859. doi: 10.1038/ng.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasheri N, Singaravelu R, Goodmurphy M, Lyn RK, Pezacki JP. Competing roles of microRNA-122 recognition elements in hepatitis C virus RNA. Virology. 2011;410:336–344. doi: 10.1016/j.virol.2010.11.015. [DOI] [PubMed] [Google Scholar]
- Pal S, Shuhart MC, Thomassen L, Emerson SS, Su T, Feuerborn N, Kae J, Gretch DR. Intrahepatic hepatitis C virus replication correlates with chronic hepatitis C disease severity in vivo. Journal of Virology. 2006;80:2280–2290. doi: 10.1128/JVI.80.5.2280-2290.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang PS, Pham EA, Elazar M, Patel SG, Eckart MR, Glenn JS. Structural Map of a MicroRNA-122:Hepatitis C Virus Complex. Journal of Virology. 2012;86:1250–1254. doi: 10.1128/JVI.06367-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X, Li Y, Walters KA, Rosenzweig ER, Lederer SL, Aicher LD, Proll S, Katze MG. Computational identification of hepatitis C virus associated microRNA-mRNA regulatory modules in human livers. BMC Genomics. 2009;10:373. doi: 10.1186/1471-2164-10-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley KJ, Rabinowitz GS, Yario TA, Luna JM, Darnell RB, Steitz JA. EBV and human microRNAs co-target oncogenic and apoptotic viral and human genes during latency. EMBO J. 2012;31:2207–2221. doi: 10.1038/emboj.2012.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed A, Baloch K, Brown RJP, Wallis R, Chen L, Dexter L, McClure CP, Shakesheff K, Thomson BJ. Mannan binding lectin-associated serine protease 1 is induced by hepatitis C virus infection and activates human hepatic stellate cells. Clin Exp Immunol. 2013:265–273. doi: 10.1111/cei.12174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language? Cell. 2011;146:353–358. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedano CD, Sarnow P. Short Article. Cell Host Microbe. 2014;16:257–264. doi: 10.1016/j.chom.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheahan T, Imanaka N, Marukian S, Dorner M, Liu P, Ploss A, Rice CM. Interferon Lambda Alleles Predict Innate Antiviral Immune Responses and Hepatitis C Virus Permissiveness. Cell Host Microbe. 2014;15:190–202. doi: 10.1016/j.chom.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimakami T, Yamane D, Jangra RK, Kempf BJ, Spaniel C, Barton DJ, Lemon SM. Stabilization of hepatitis C virus RNA by an Ago2-miR-122 complex. Proc Natl Acad Sci. 2012;109:941–946. doi: 10.1073/pnas.1112263109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singaravelu R, Russell RS, Tyrrell DL, Pezacki JP. Hepatitis C virus and microRNAs: miRed in a host of possibilities. Current Opinion in Virology. 2014;7:1–10. doi: 10.1016/j.coviro.2014.03.004. [DOI] [PubMed] [Google Scholar]
- Skalsky RLR, Corcoran DLD, Gottwein EE, Frank CLC, Kang DD, Hafner MM, Nusbaum JDJ, Feederle RR, Delecluse HJH, Luftig MAM, et al. The viral and cellular microRNA targetome in lymphoblastoid cell lines. PLoS Pathog. 2012;8:e1002484–e1002484. doi: 10.1371/journal.ppat.1002484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai W-C, Hsu S-D, Hsu C-S, Lai T-C, Chen S-J, Shen R, Huang Y, Chen H-C, Lee C-H, Tsai T-F, et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J Clin Invest. 2012;122:2884–2897. doi: 10.1172/JCI63455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyn-Vanhentenryck SM, Mele A, Yan Q, Sun S, Farny N, Zhang Z, Xue C, Herre M, Silver PA, Zhang MQ, et al. HITS-CLIP and Integrative ModelingDefine the Rbfox Splicing-Regulatory Network Linked to Brain Development and Autism. Cell Reports. 2014;6:1139–1152. doi: 10.1016/j.celrep.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane D, McGivern DR, Masaki T, Lemon SM. Current Topics in Microbiology and Immunology. Berlin, Heidelberg: Springer Berlin Heidelberg; 2013. Liver Injury and Disease Pathogenesis in Chronic Hepatitis C; pp. 263–288. [DOI] [PubMed] [Google Scholar]
- Zhang C, Darnell RB. Mapping in vivo protein-RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nat Biotechnol. 2011;29:607–614. doi: 10.1038/nbt.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.