

Abstract
Nucleophosmin-anaplastic lymphoma kinase (NPM-ALK) is an aberrant fusion gene product with tyrosine kinase activity and is expressed in substantial subset of anaplastic large cell lymphomas (ALCL). It has been shown that NPM-ALK binds to and activates signal transducer and activator of transcription 3 (STAT3). Although NPM-ALK+ ALCL overall shows a better prognosis, there is a sub-group of patients who relapses and is resistant to conventional chemotherapeutic regimens. NPM-ALK is a potential target for small molecule kinase inhibitors. Crizotinib (PF-2341066) is a small, orally bioavailable molecule that inhibits growth of tumors with ALK activity as shown in a subgroup of non-small lung cancer patients with EML4-ALK expression. In this study, we have investigated the in vitro effects of Crizotinib in ALCL cell line with NPM-ALK fusion. Crizotinib induced marked downregulation of STAT3 phosphorylation, which was associated with significant apoptotic cell death. Apoptosis induction was attributed to caspase-3 cleavage and marked downregulation of the Bcl-2 family of proteins including MCL-1. These findings implicate that Crizotinib has excellent potential to treat patients with NPM-ALK+ ALCL through induction of apoptotic cell death and downregulation of major oncogenic proteins in this aggressive lymphoma.
Keywords: Crizotinib, STAT3, ALCL, NPM-ALK, MCL-1
1. INTRODUCTION
Anaplastic large cell lymphoma (ALCL) is an aggressive type of non-Hodgkin lymphoma that was first described by Stein et al in 1985 as characteristically showing large pleomorphic lymphocytes with abundant cytoplasm, occasional horseshoe nuclei, and tendency for sinusoidal infiltration.[1] ALCL tumor cells universally express CD30 and predominantly demonstrate T cell immunophenotype.[1–3]
A substantial group of ALCL cases (60–80%) [2] harbors a fusion gene product that results from a chromosomal translocation t (2, 5) (p23, q35). This translocation causes juxtaposition of the nucleophosmin (NPM) gene at 5q35 with the anaplastic lymphoma kinase (ALK) gene at 2p23 [4, 5]. Whereas wild-type ALK is a receptor tyrosine kinase [6], NPM is a non-ribosomal RNA-binding protein that functions in bidirectional shuttling of proteins between the nucleus and cytoplasm [7]. NPM oligomerizes ALK and leads to auto-phosphorylation and activation of the ALK tyrosine kinase [8].
NPM-ALK is an oncogenic fusion protein which leads to tyrosine phosphorylation and activation of various downstream targets essential for cellular survival. One of the most important targets of NPM-ALK is signal transducer and activator of transcription 3 (STAT3) [9–11] and it’s down regulation results in apoptosis and cell cycle arrest.[10] Although ALK+ ALCL patients have a better prognosis compared to ALK− ALCL ones, some patients are found to be resistant to conventional chemotherapeutic regimens; therefore, this warrants development of novel therapeutic strategies with much more selective inhibitory effects against NPM-ALK.[12–14]
Crizotinib is a small orally bioavailable molecule that potently inhibits ALK kinase activity [15, 16]. This agent has already shown promising results in patients with ALK positive non-small cell lung cancers [17]. In this study, we assessed in vitro effects of Crizotinib in ALCL with NPM-ALK fusion, and analyzed modifications of apoptotic regulatory proteins.
2. MATERIALS AND METHODS
2.1. Cell Line and Culture
An ALK+ ALCL cell line, SUDHL-1, was obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSMZ, Germany)[18]. SUDHL-1 cell line was grown in Cellgro® RPMI 1640 medium supplemented with L-Glutamine and 25mM HEPES (Mediatech, Manassas, VA, USA) and also 10% Cellgro® heat-inactivated fetal bovine serum (Mediatech), 10 000 U/ml penicillin (Sigma, St Louis, MO, USA), 10 mg/ml streptomycin (Sigma) and extra 1xL-glutamine (Life Technologies, NY, USA), and incubated at 37°C in 95% air and 5% CO2. Standard passage techniques were used.
2.2. Reagents
Crizotinib (PF-2341066) was obtained from Selleck Chemicals LLC, (Houston, TX) and diluted in DMSO to prepare 10 mM stock solution and stored in −80°C. Antibodies used were purchased from Santa Cruz biotechnology (Santa Cruz, CA, USA) and included: pSTAT3 (Tyr705; clone: B-7), BCL-XL (H-5), BAX (B-9), MCL-1 (H-260) and goat anti-rabbit IgG-PE. ALK antibody (Clone ALK1) for IHC was obtained from DAKO Corporation (Carpenteria, CA). Cleaved caspase-3 (Asp175) was obtained from Cell Signaling (Danvers, MA).
2.3. Methods
2.3.1. Immunohistochemistry
After Institutional Review Board approval, 19 cases of ALCL were retrieved from archives of pathology and laboratory medicine at Cedars-Sinai Medical Center, Los Angeles, California. Paraffin-embedded tissue specimens were sectioned into 4 μm thickness, deparaffinized, and rehydrated. Antigen retrieval was performed by heating at 95°C to 100°C in 10 mmol/L sodium citrate buffer (pH 6.0) for 20 minutes. Sections were then blocked in 5% bovine serum albumin for 10 minutes and then incubated with primary antibodies according to antibody manufacture’s protocol. After washing off the primary pSTAT3 antibody (Tyr705) (M9C6) from Cell Signaling (Danvers, MA) with tris-buffered saline Tween-20, the sections were incubated with secondary antibodies according to manufacturer’s datasheet. For immunohistochemistry, DAB solution (DAKO, Carpinteria, CA) was applied, and 50% hematoxylin was used for counter stain.
2.3.2. Proliferation assay
Cell viability of cultured SUDHL-1 cells was determined by the MTS [3-(4, 5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] assay (CellTiter 96 One Solution Aqueous kit; Promega, Madison, WI). Cells were cultured in a flat-bottom 96-well tissue plates at 0.5 × 105 cells per well in a total volume of 100 μL. After incubation with various concentrations of Crizotinib, 20 μL of MTS was dispensed into each well and cells were incubated for an additional hour at 37 °C and 5% CO2. Cytotoxicity was assessed by determination of reduced tetrazolium (formazan) created by metabolically active cells and detected at a 490 nm absorbance by a spectrophotometer (SpectraMax384, Molecular Devices, Sunnyvale, CA). Each condition was performed in triplicates with results reported as mean value. Concentration-response analyses were performed to establish the optimum concentration of Crizotinib and statistical analysis was performed with paired t-test using Microsoft Excel software.
2.3.3. Apoptosis assays
For morphologic analysis of apoptosis, cells were washed twice in PBS without Ca2+ or Mg2+. Cytospin slides were then prepared, stained with Wright-Giemsa, and examined by light microscopy. The morphologic features considered consistent with apoptosis included cell shrinkage, nuclear condensation, nuclear fragmentation, and formation of apoptotic bodies.
For flow cytometric analysis of apoptosis, cells were centrifuged at 370 × g for 5 minutes, washed with PBS × 2, and then incubated at 4°C in 600 μL of ethanol and 100 μL of FBS (Mediatech, Manassas, VA, USA) for 30 minutes. Cells were washed in PBS, and then centrifuged at 370 × g for 5 minutes. The cell pellet was reconstituted with 5 μg of RNAse (Roche, Indianapolis, IN) and incubated at 37 °C for 15 min. After cooling at room temperature for 5 minutes, cells were incubated with 50 μg of Propidium Iodide (BD Pharmingen) for 60 minutes at room temperature in the dark. The cells were washed with PBS and suspended in ice-cold PBS. Cells were analyzed on an FACS 500 (Beckman Coulter, Indianapolis, IN) flow cytometer.
2.3.4. Flow cytometry detection of apoptotic proteins and STAT3
Cultured SUDHL-1 cells were incubated with different concentrations of Crizotinib for 24 hours for apoptotic molecules and 16 hours for pSTAT3. Cells were centrifuged at 370 × g for 5 minutes, washed with PBS × 2 times, then fixed and permeabilized using a Fix & Perm kit (Caltag/Invitrogen, Carlsbad, CA) following manufacturer’s protocol. First cell pellet were fixed using 100 μl of solution A for 15 minutes in room temperature, then cells were washed with PBS. Secondly, cell pellet was incubated with proper concentration of fluorescent antibody and 100 μL of solution B for 30 minutes in room temperature in the dark. Cell suspension washed with PBS and then suspended in 1 ml of ice-cold PBS without Ca+ and Mg+ and then acquired using flow cytometer device. In case of MCL-1 antibody since it wasn’t fluorescent by itself the second step with solution B was repeated with fluorescent secondary antibody.
3. RESULTS
3.1. Immunohistochemical Analysis of pSTAT3 and ALK in ALCL
Nineteen ALCL cases were retrieved and studied for ALK and pSTAT3 positivity. Seven of the cases showed positivity for ALK (both cytoplasmic and nuclear noted in all seven cases) and twelve were negative. All seven ALK positive ALCL cases showed positivity for pSTAT3 with intensity varying from 1+ to 4+ positivity (20–90% lymphoma cells staining). Seven of twelve cases of ALK negative ALCL showed positive staining for pSTAT3 with intensity varying from 1+ to 4+ (30–100%), (Figure 1). All of the positive cases revealed unequivocal primarily nuclear staining of pSTAT3 in lymphoma cells.
Fig. 1.
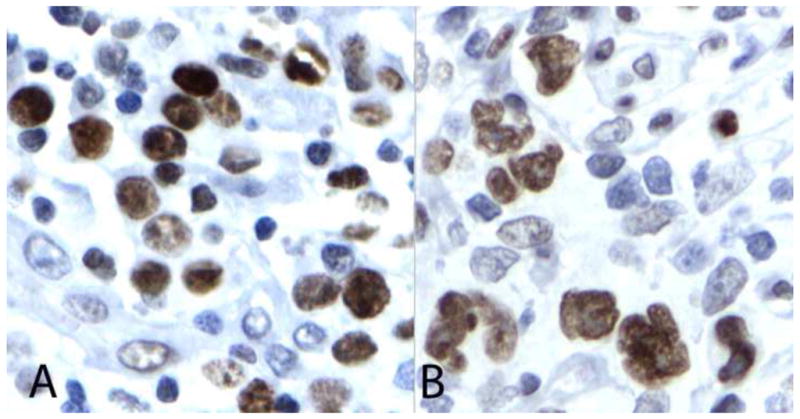
Strong nuclear expression of pSTAT3 is demonstrated in the nuclei of the lymphoma cells in representative cases of ALK positive (A) and ALK negative (B) tumors.
3.2. Crizotinib Inhibits Cell Proliferation in SUDHL-1 Cells
In order to study potential inhibitory effects of Crizotinib on ALK+ ALCL, SUDHL-1 cells were treated for 72 hours with varying concentrations of Crizotinib (0, 0.001, 0.01, 0.1, 1 and 10μM) and subjected to a colorimetric cell viability assay. As seen in Figure 2, there was a significant decrease in light absorption at 490 nm between 0.1 and 1μM.
Fig. 2.
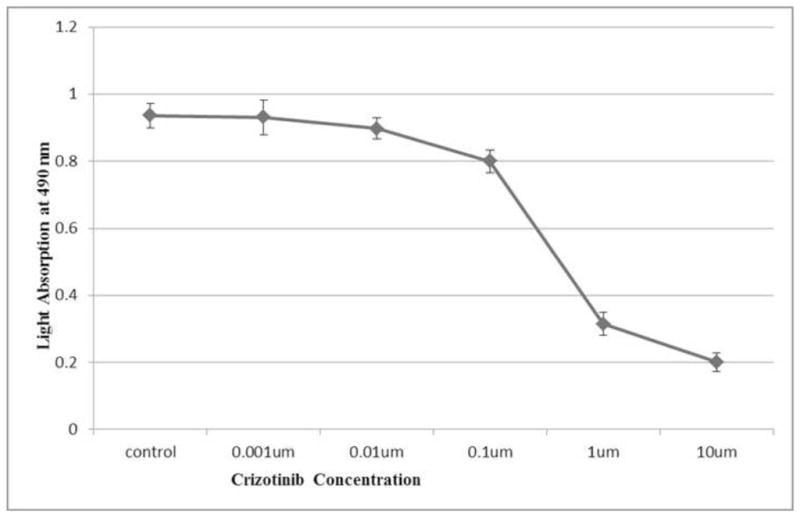
Concentration-dependent cytotoxic effects of Crizotinib on SUDHL-1 cells: SUDHL-1 cells were cultured in suspension and treated with Crizotinib at various concentrations as indicated above (X-axis). Cellular viability of samples was determined by the MTS assay with absorbance at 490 nm determined after each treatment (Y-axis). Data points represent the mean numbers of triplicate results. Overall, a dose-dependent decrease in cell viability with Crizotinib was seen.
3.3. Crizotinib Induces Apoptosis in SUDHL-1 Cells
In order to determine if the cytotoxic effects of Crizotinib involves cellular apoptotic pathways, morphologic assessment by staining and cell cycles analysis of SUDHL-1 cells by flow cytometry were performed after 72 hours treatment with 0.1 and 1μM Crizotinib. At 1μM concentration, Crizotinib induced noticeable morphologic changes in SUDHL-1 cells consistent with apoptosis (e.g. cytoplasmic vacuolization, nuclear shrinkage and fragmentation, chromatin condensation and cellular disintegration) as seen in Figure 3. To further confirm that these changes were indeed due to apoptosis, cells were analyzed with propidiumiodide staining (PI). As seen in Figure 4, 1μM Crizotinib increased the sub G0/G1 (apoptotic) population.
Fig. 3.
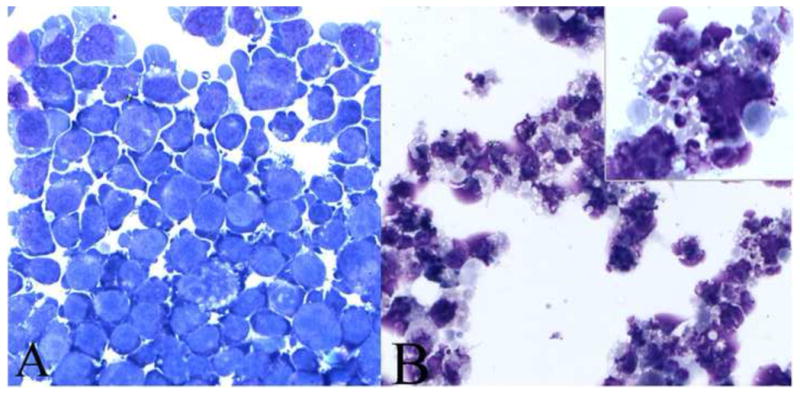
Morphologic hallmarks of apoptosis are evident in Crizotinib treated SUDHL-1 cells: SUDHL-1 cells were incubated with 1 μM of Crizotinib for 72 hrs and morphologically examined after cytospin preparation. (A) Untreated control cells. (B) Treated cells showing extensive cell nuclear shrinkage, fragmentation of chromatin and vacuolization (see inset) consistent with apoptosis (1000×).
Fig. 4.
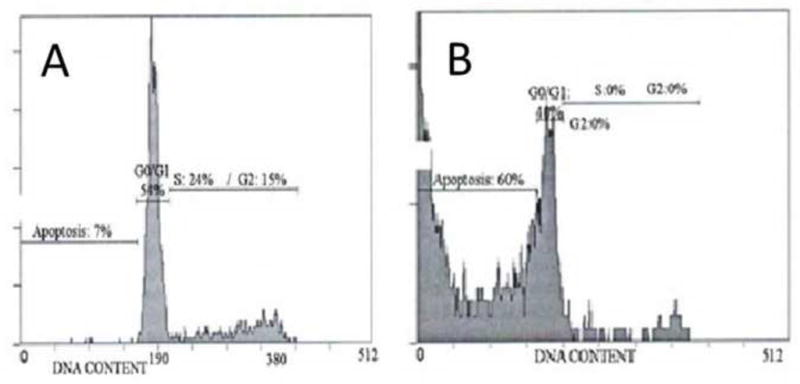
Representative histograms of cell cycle analysis by PI after 72 hours treatment with 0 and 1 μM Crizotinib, performed in triplicate. (A) The untreated cell suspension showed minimum of apoptosis with 7% of the population in sub-G0/G1. (B) Crizotinib treated cells show marked apoptosis by observation of significant increase in sub-G0/G1 population (60%).
3.4. Crizotinib Induces Significant Reduction of STAT3 Phosphorylation
Since STAT3 phosphorylation has been implicated for cell survival in ALK ALCL, we decided to assess modification in STAT3 with Crizotinib treatment. In an attempt to show if the cytotoxic effects of Crizotinib is associated with pSTAT3 down regulation, SUDHL-1 cell suspension was treated with different concentrations of Crizotinib for 16 hours. Flow cytometry evaluation of intracellular expression of pSTAT3 showed significant reduction of STAT3 phosphorylation after treatment with 1μM Crizotinib for 16 hours. (Figure 5)
Fig. 5.
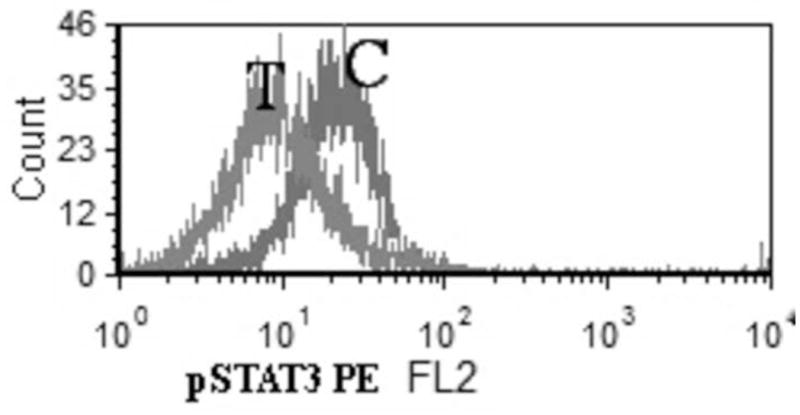
Down regulation of p-STAT3 in SUDHL-1 cells after 16 hours of treatment with 1 μM Crizotinib (T: treated with 1 μM Crizotinib for 16 hours C: Control, untreated SUDHL-1 cells)
3.5. Crizotinib Induces Down Regulation of Apoptotic Molecules
Number of mitochondrial BCL-2 family proteins had been implicated in the regulation of ALCL cell survival. Therefore, we also examined changes in the mitochondrial-associated anti and pro apoptotic proteins including BCL-2, BCL-XL, MCL-1 and BAX as well as changes in caspase-3 cleavage in response to Crizotinib treatment. After 24 hours treatment of SUDHL-1 cell suspension with different concentrations of Crizotinib, expression of these molecules was assessed by flow cytometry. Among the mentioned molecules MCL-1 had the most significant down regulation after 24 hours of treatment with Crizotinib. BCL-2 and BCL-XL were reduced moderately after treatment with Crizotinib. However, BAX pro-apoptotic molecule did not change after treatment with Crizotinib (data not shown). At 24 hours after treatment, Crizotinib (1μM) caused significant increase in caspase-3 cleavage, further supporting apoptosis occurrence. (Figure 6)
Fig. 6.
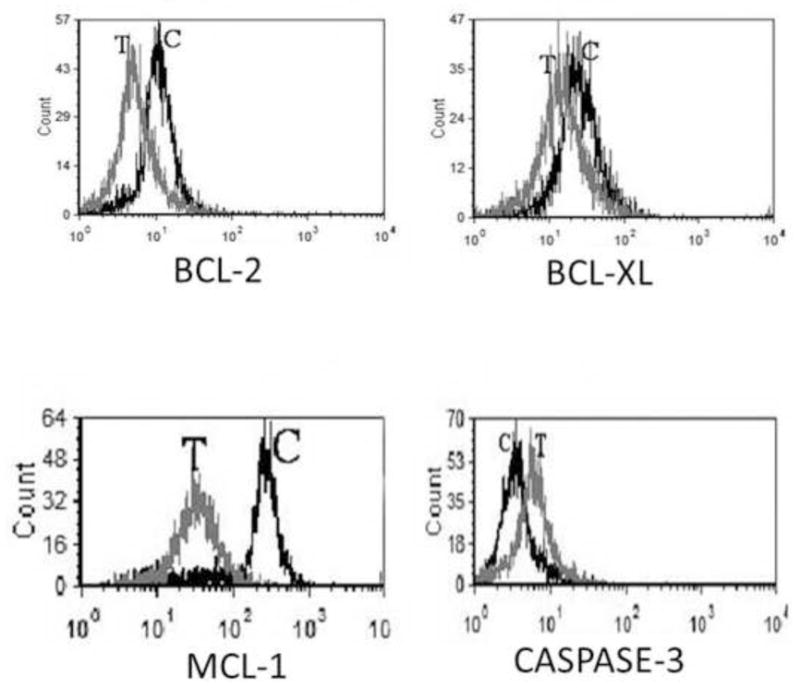
Representative histograms of flow cytometry assessment of apoptotic molecules in SUDHL-1 cell line after 24 hour treatment with 1 μM Crizotinib (T: treated with 1 μM Crizotinib for 24 hours C: Control, untreated SUDHL-1 cells). As noted BCL-2, BCL-XL and MCL-1 are downregulated while there is increased caspase-3 activity.
4. DISCUSSION
ALCL patients with NPM-ALK expression are predominantly children and young adults who usually show favorable clinical response to standard therapeutic regimens. Nonetheless, not uncommonly these patients relapse, demonstrate notable resistance to conventional therapies and eventually succumb [19, 20]. The chromosomal translocation that results in ALK overexpression occurs in 40–60% of the ALCL cases and presents a unique opportunity for targeted therapies against ALCL [2]. Deregulation of ALK occurs as a consequence of number of translocations that result in ligand independent activation of ALK protein. The great majority of ALK activation (more than 80% of the cases) occurs secondary to t (2; 5) (p23; q35) leading to NPM-ALK fusion; however, a number of alternate translocations also induce ALK constitutive kinase activity. [2, 3] In the case of ALK activation secondary to NPM-ALK fusion most frequently seen in common types of ALCL, NPM encodes the nuclear localization signal that is believed to cause the nuclear localization of NPM-ALK. Activation of ALK kinase leads to activation of number of complex signaling networks, including STAT3 [21], which is also required for ALK-mediated lymphoma genesis and provides a possible therapeutic target.[11]
Our investigation was focusing on STAT3 lead to observation of pSTAT3 positivity in all of our seven ALK positive cases while only 7/12 of our ALK negative cases also showed presence of pSTAT3. STAT3 activation status in ALK positive and negative ALCL cases appears to be inconsistent in various studies. Our findings is in contrast with Zamo et al study that has shown 1/43 pSTAT3 positivity in ALK negative cases.[9] Nasr et al have shown pSTAT3 expression in 25/36 (69%) of ALK positive ALCL patient samples.[22] Our findings are similar to a study by Khoury et al in which STAT3 activation was observed in (61%) of ALK positive ALCL tumors and in 47% of their ALK negative patient samples. These studies indicate that expression of STAT3 is not strictly dependent to ALK expression and alternate pathways likely exist for STAT3 activation and may have ramifications for treatment of ALK negative ALCL patients.[23]
Crizotinib with its inhibitory properties on ALK tyrosine kinase phosphorylation has been suggested to be effective in inducing cell death in NPM-ALK+ ALCL cell lines and tumor regression in vivo xenograft of ALCL in mice models.[15] Our observation of pSTAT3 down regulation by Crizotinib is consistent with that investigation. Caspase-3 cleavage in Crizotinib treated NPM-ALK+ ALCL cell line was observed, as it was previously noted after STAT3 down-regulation.[10]
We extended our investigation to study possible modifications of STAT3 downstream molecules specifically the BCL-2 family of apoptosis regulatory proteins. It has been previously shown that BCL-2 family proteins are important in NPM-ALK+ ALCL survival and resistance to therapies.[24] Our results demonstrate that ALK/pSTAT3 downregulation by Crizotinib alters BCL-2 and BCL-XL. However, we did not observe upregulation of the anti-apoptotic protein BAX after treatment with Crizotinib. We have shown MCL-1 is very likely one of the most important downstream targets of Crizotinib as it revealed very significant decrease in comparison to other BCL-2 proteins. Desjobert et al, utilizing mir29a microRNA, have also suggested the importance of MCL-1 by inhibition of STAT3.[25] Similarly, Amin et al have observed MCL-1 downregulation after inhibition of STAT3 by transfection of Karpas 299 cell line with STAT3 dominant negative adenoviral vector.[10] Rust et al has also noted frequent MCL-1 positivity in 100% of their ALCL patient samples (23/23) [26]; however, since MCL-1 was also positive in all of their ALK negative patient samples (9/9) authors suggested possibility of alternate signal transcription pathways besides pSTAT3 in its modification and up regulation especially in ALK negative ALCL. [26]
Several lines of evidence suggest that MCL-1 plays a very important role in maintaining cellular survival, but we also observed decreased BCL-2 and BCL-XL expression levels after treating SUDHL-1 cell line with Crizotinib. Rassidakis et al have shown that none of their ALK positive ALCL samples were BCL-2 positive (0/21); however, significant number of their ALK negative ALCL cases was positive for BCL-2 [27, 28]. In our study, both BCL-2 and BCL-XL were down regulated after treatment with Crizotinib. BCL-XL positivity in ALK positive and negative patient samples was also observed in a study by Rassidakis et al study but not by Rust et al study.[26, 27] Zamo et al and Chiarle et al showed that constitutive activation of STAT3 via ALK leads to BCL-XL up regulation and increasing of cell survival.[9, 11] However, interestingly they showed ALK expression did not modulate the levels of BCL-2 and MCL-1 [9] which is in contrast to our results or Amin et al results [10]. A possible explanation for this could be related to use of a myeloma cell line U266 that was transfected with NPM-ALK to assess modification of BH3 proteins.[9] U266 cell line overexpresses MCL-1, which most likely modulates the apoptotic signaling.
In conclusion, our study provides a proof-of-principle of how the ALK inhibitor Crizotinib induces its inhibitory effects in NPM-ALK+ ALCL. Crizotinib appears to effectively downregulate pSTAT3; a potent oncogenic protein in this aggressive lymphoma. The effects of Crizotinib on pSTAT3 are associated with induction of apoptotic cell death, which is attributable to downregulation of members of BCL-2 family of apoptosis promoting molecules that are also known to be targets of STAT3 transcriptional activities.
Acknowledgments
Hesham Amin is supported by the R01CA151533 grant from the National Cancer Institute.
Footnotes
The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Stein H, Mason DY, Gerdes J, O’Connor N, Wainscoat J, Pallesen G, et al. The expression of the Hodgkin’s disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood. 1985;66:848–58. [PubMed] [Google Scholar]
- 2.Stein H, Foss HD, Durkop H, Marafioti T, Delsol G, Pulford K, et al. CD30(+) anaplastic large cell lymphoma: a review of its histopathologic, genetic, and clinical features. Blood. 2000;96:3681–95. [PubMed] [Google Scholar]
- 3.Piccaluga PP, Gazzola A, Mannu C, Agostinelli C, Bacci F, Sabattini E, et al. Pathobiology of anaplastic large cell lymphoma. Adv Hematol. 2010:345053. doi: 10.1155/2010/345053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263:1281–4. doi: 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]
- 5.Duyster J, Bai RY, Morris SW. Translocations involving anaplastic lymphoma kinase (ALK) Oncogene. 2001;20:5623–37. doi: 10.1038/sj.onc.1204594. [DOI] [PubMed] [Google Scholar]
- 6.Morris SW, Naeve C, Mathew P, James PL, Kirstein MN, Cui X, et al. ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin’s lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK) Oncogene. 1997;14:2175–88. doi: 10.1038/sj.onc.1201062. [DOI] [PubMed] [Google Scholar]
- 7.Borer RA, Lehner CF, Eppenberger HM, Nigg EA. Major nucleolar proteins shuttle between nucleus and cytoplasm. Cell. 1989;56:379–90. doi: 10.1016/0092-8674(89)90241-9. [DOI] [PubMed] [Google Scholar]
- 8.Bischof D, Pulford K, Mason DY, Morris SW. Role of the nucleophosmin (NPM) portion of the non-Hodgkin’s lymphoma-associated NPM-anaplastic lymphoma kinase fusion protein in oncogenesis. Mol Cell Biol. 1997;17:2312–25. doi: 10.1128/mcb.17.4.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zamo A, Chiarle R, Piva R, Howes J, Fan Y, Chilosi M, et al. Anaplastic lymphoma kinase (ALK) activates Stat3 and protects hematopoietic cells from cell death. Oncogene. 2002;21:1038–47. doi: 10.1038/sj.onc.1205152. [DOI] [PubMed] [Google Scholar]
- 10.Amin HM, McDonnell TJ, Ma Y, Lin Q, Fujio Y, Kunisada K, et al. Selective inhibition of STAT3 induces apoptosis and G(1) cell cycle arrest in ALK-positive anaplastic large cell lymphoma. Oncogene. 2004;23:5426–34. doi: 10.1038/sj.onc.1207703. [DOI] [PubMed] [Google Scholar]
- 11.Chiarle R, Simmons WJ, Cai H, Dhall G, Zamo A, Raz R, et al. Stat3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat Med. 2005;11:623–9. doi: 10.1038/nm1249. [DOI] [PubMed] [Google Scholar]
- 12.Falini B, Pileri S, Zinzani PL, Carbone A, Zagonel V, Wolf-Peeters C, et al. ALK+ lymphoma: clinico-pathological findings and outcome. Blood. 1999;93:2697–706. [PubMed] [Google Scholar]
- 13.Gascoyne RD, Aoun P, Wu D, Chhanabhai M, Skinnider BF, Greiner TC, et al. Prognostic significance of anaplastic lymphoma kinase (ALK) protein expression in adults with anaplastic large cell lymphoma. Blood. 1999;93:3913–21. [PubMed] [Google Scholar]
- 14.Pulford K, Lamant L, Espinos E, Jiang Q, Xue L, Turturro F, et al. The emerging normal and disease-related roles of anaplastic lymphoma kinase. Cell Mol Life Sci. 2004;61:2939–53. doi: 10.1007/s00018-004-4275-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christensen JG, Zou HY, Arango ME, Li Q, Lee JH, McDonnell SR, et al. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol Cancer Ther. 2007;6:3314–22. doi: 10.1158/1535-7163.MCT-07-0365. [DOI] [PubMed] [Google Scholar]
- 16.Zou HY, Li Q, Lee JH, Arango ME, McDonnell SR, Yamazaki S, et al. An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007;67:4408–17. doi: 10.1158/0008-5472.CAN-06-4443. [DOI] [PubMed] [Google Scholar]
- 17.Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wood GS, Hardman DL, Boni R, Dummer R, Kim YH, Smoller BR, et al. Lack of the t(2;5) or other mutations resulting in expression of anaplastic lymphoma kinase catalytic domain in CD30+ primary cutaneous lymphoproliferative disorders and Hodgkin’s disease. Blood. 1996;88:1765–70. [PubMed] [Google Scholar]
- 19.Lowe EJ, Sposto R, Perkins SL, Gross TG, Finlay J, Zwick D, et al. Intensive chemotherapy for systemic anaplastic large cell lymphoma in children and adolescents: final results of Children’s Cancer Group Study 5941. Pediatr Blood Cancer. 2009;52:335–9. doi: 10.1002/pbc.21817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woessmann W, Zimmermann M, Lenhard M, Burkhardt B, Rossig C, Kremens B, et al. Relapsed or refractory anaplastic large-cell lymphoma in children and adolescents after Berlin-Frankfurt-Muenster (BFM)-type first-line therapy: a BFM-group study. J Clin Oncol. 2011;29:3065–71. doi: 10.1200/JCO.2011.34.8417. [DOI] [PubMed] [Google Scholar]
- 21.Kasprzycka M, Marzec M, Liu X, Zhang Q, Wasik MA. Nucleophosmin/anaplastic lymphoma kinase (NPM/ALK) oncoprotein induces the T regulatory cell phenotype by activating STAT3. Proc Natl Acad Sci U S A. 2006;103:9964–9. doi: 10.1073/pnas.0603507103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nasr MR, Laver JH, Chang M, Hutchison RE. Expression of anaplastic lymphoma kinase, tyrosine-phosphorylated STAT3, and associated factors in pediatric anaplastic large cell lymphoma: A report from the children’s oncology group. Am J Clin Pathol. 2007;127:770–8. doi: 10.1309/FNY8Y4H6PK1V2MGE. [DOI] [PubMed] [Google Scholar]
- 23.Khoury JD, Medeiros LJ, Rassidakis GZ, Yared MA, Tsioli P, Leventaki V, et al. Differential expression and clinical significance of tyrosine-phosphorylated STAT3 in ALK+ and ALK- anaplastic large cell lymphoma. Clin Cancer Res. 2003;9:3692–9. [PubMed] [Google Scholar]
- 24.Piva R, Pellegrino E, Mattioli M, Agnelli L, Lombardi L, Boccalatte F, et al. Functional validation of the anaplastic lymphoma kinase signature identifies CEBPB and BCL2A1 as critical target genes. J Clin Invest. 2006;116:3171–82. doi: 10.1172/JCI29401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Desjobert C, Renalier MH, Bergalet J, Dejean E, Joseph N, Kruczynski A, et al. MiR-29a down-regulation in ALK-positive anaplastic large cell lymphomas contributes to apoptosis blockade through MCL-1 overexpression. Blood. 2011;117:6627–37. doi: 10.1182/blood-2010-09-301994. [DOI] [PubMed] [Google Scholar]
- 26.Rust R, Harms G, Blokzijl T, Boot M, Diepstra A, Kluiver J, et al. High expression of Mcl-1 in ALK positive and negative anaplastic large cell lymphoma. J Clin Pathol. 2005;58:520–4. doi: 10.1136/jcp.2004.022335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rassidakis GZ, Sarris AH, Herling M, Ford RJ, Cabanillas F, McDonnell TJ, et al. Differential expression of BCL-2 family proteins in ALK-positive and ALK-negative anaplastic large cell lymphoma of T/null-cell lineage. Am J Pathol. 2001;159:527–35. doi: 10.1016/S0002-9440(10)61724-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rassidakis GZ, Jones D, Lai R, Ramalingam P, Sarris AH, McDonnell TJ, et al. BCL-2 family proteins in peripheral T-cell lymphomas: correlation with tumour apoptosis and proliferation. J Pathol. 2003;200:240–8. doi: 10.1002/path.1346. [DOI] [PubMed] [Google Scholar]