

Abstract
This report describes the Pd-catalyzed functionalization of cyclopropanes containing oxazoline, oxime ether, and pyridine directing groups. Three different oxidants were examined in these studies: IOAc, PhI(OAc)2, and benzoquinone. The reactions yielded products derived from 2° sp3 C–H functionalization and/or C–C activation of the cyclopropane ring. The outcome and the product distributions were highly dependent on the structure of the substrate and the nature of the oxidant.
Keywords: cyclopropane, C-H functionalization, palladium, cleavage, homogeneous catalysis
Introduction
Transition metal-catalyzed reactions for the direct functionalization of C–H bonds serve as atom economical methods for the synthesis of diverse organic molecules.1 Research in this area has expanded dramatically over the past decade, and Pd-catalyzed ligand-directed C–H functionalization has become a particularly active sub-field.2 Numerous Pd-catalyzed methods have been developed for transforming C–H bonds into C–O, C–halogen, C–S, C–N, and C–C linkages.2 Furthermore, these reactions have been applied to the functionalization of diverse aromatic, olefinic, and 1° sp3 C–H bonds.2
In marked contrast, the Pd-catalyzed functionalization at 2° sp3 C–H sites remains a significant challenge. Sporadic examples of this type of reactivity have been reported. However, as shown in Scheme 1, the vast majority of these require the presence of an electronically and/or sterically biasing element within the substrate. For example, substrates A and B are activated by the presence of α-heteroatoms and α-aryl groups adjacent to the site of 2° C–H functionalization (Scheme 1, i and ii).3,4 The rigid structure of substrate C provides a conformational bias for 2° sp3 C–H functionalization (Scheme 1, iii).3 Finally, substrate D can participate in two-point binding to the Pd catalyst (through the quinoline and amide nitrogens), and this facilitates cleavage of an unactivated 2° sp3 C–H bond (Scheme 1, iv).5
Scheme 1.
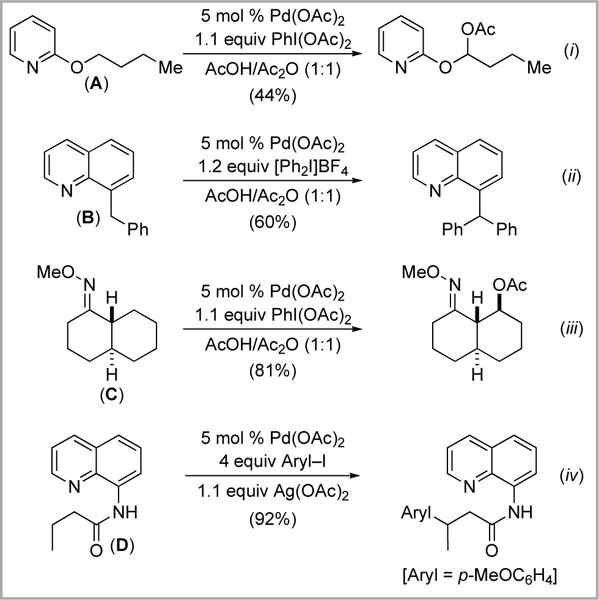
Examples of 2° sp3 C–H activation/acetoxylation
Cyclopropanes have also been successfully utilized as substrates for Pd-catalyzed 2° sp3 C–H functionalization. For example, recent reports have described the Pd-catalyzed ligand-directed C–H halogenation,6 arylation,7 alkylation,8 and olefination8,9 of various substituted cyclopropanes. Interestingly, the C–H bonds of cyclopropanes are not particularly activated towards either homolytic nor heterolytic cleavage (bde = 106 kcal/mol; pKa = 46).11 Nonetheless, in many of these examples, the cyclopropane C–H bonds are cleaved selectively in lieu of adjacent sp2 and/or 1° sp3 C–H bonds.
The reactions in Scheme 2 are of particular interest because substituted cyclopropanes serve as versatile intermediates for further synthetic manipulations.12 In addition, studies of the scope and limitations of such transformations could provide valuable insights for the development of more general Pd-catalyzed reactions for 2° sp3 C–H functionalization. However, to date, these cyclopropane C–H functionalization reactions remain isolated examples, and have not been subject to systematic investigation.
Scheme 2.
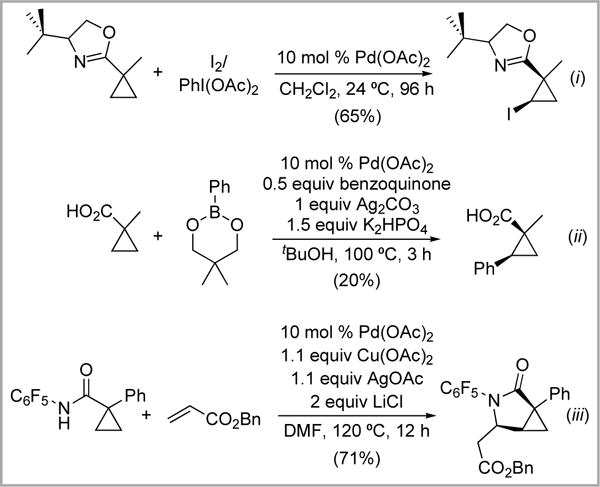
Examples of ligand-directed C–H functionalization of cyclopropane derivatives
To address this gap in knowledge, we have conducted a detailed study of the Pd-catalyzed ligand-directed oxidation of cyclopropane derivatives. This paper describes the Pd-catalyzed reactions of cyclopropyl oxazolines, oxime ethers, and pyridines with three different oxidants: IOAc, PhI(OAc)2, and benzoquinone (BQ). We demonstrate that these reactions are remarkably sensitive to subtle changes in the directing group and reaction conditions. Minor perturbations of both variables often lead to competing C–C bond activation of the cyclopropane to afford ring opened products.
Results
Three different oxidants were utilized for all of the substrates discussed in this manuscript. First, we examined IOAc (generated in situ from PhI(OAc)2 and I2), since this reagent was shown to be effective for the C–H iodination of cyclopropyl oxazoline 1 (Scheme 2, i).6 Second, PhI(OAc)2 was used as an oxidant, since our group13 and others14 have demonstrated that this is a highly effective reagent for the PdII/IV-catalyzed C–H acetoxylation of diverse substrates. Finally, benzoquinone (BQ) was employed in conjunction with AcOH as an external nucleophile. BQ is a much weaker oxidant than PhI(OAc)2, and is frequently used to mediate PdII/0 catalytic cycles. Most relevant to the current manuscript, Yudin has demonstrated the PdCl2-catalyzed oxidative C–C activation of arylcyclopropanes using BQ as a terminal oxidant.15
Oxazoline substrates
Our initial studies focused on the oxazoline substrates 1 and 2. These were selected based on Yu’s report that 1 participates in 2° sp3 C–H iodination at room temperature using IOAc.6 However, interestingly, under analogous conditions compound 2 was completely unreactive towards C–H iodination (Scheme 3). After 96 h at 25 °C, the starting material remained in nearly quantitative yield (99%), as determined by 1H NMR spectroscopic analysis of the crude reaction mixture. This result demonstrates that the C–H iodination reaction is extremely sensitive to the steric/electronic environment around the directing group.
Scheme 3.
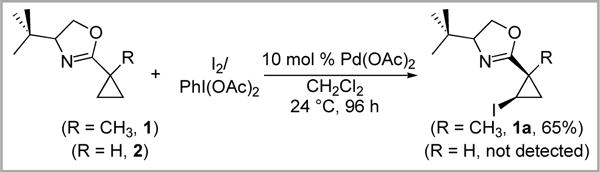
Oxazoline-directed C–H iodination
The Pd-catalyzed oxidation of 1 and 2 was next examined using PhI(OAc)2. Under standard conditions for Pd-catalyzed sp3 C–H acetoxylation (10 mol % Pd(OAc)2, 2 equiv PhI(OAc)2 in AcOH at 100 °C),3,13,14 the major observable products were oxazolium acetate salts 1b (19%) and 2b (29%) (Scheme 4). C–H acetoxylation products were not detected by 1H NMR spectroscopy or by mass spectrometric analysis. The protonated oxazolines 1b and 2b were also formed (in 59% and 62% yield) when 1 and 2 were heated in AcOH at 100 °C for 6 h in the absence of Pd and oxidant.
Scheme 4.
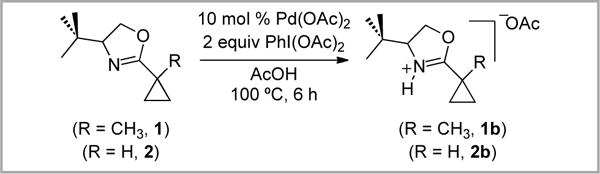
Oxazolium acetate salt formation
In an effort to limit this undesired acid/base chemistry, we switched the solvent for this reaction from AcOH to dichloroethane (DCE). Under these conditions, substrate 1 underwent cyclopropane C–C bond activation to generate olefin 1c in modest 29% yield as a mixture of E and Z isomers (Scheme 5). Notably, similar reactivity was observed at room temperature (with 1c formed in 12% yield). In contrast, substrate 2 did not yield any detectable acetoxylated products under any conditions examined.
Scheme 5.
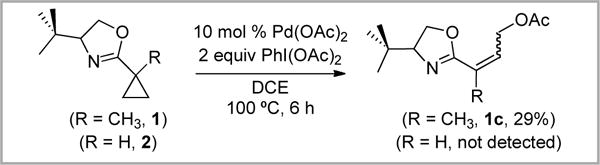
Oxazoline-directed acetoxylation
Finally, oxazolines 1 and 2 were subjected to BQ in AcOH (10 mol % Pd(OAc)2, 2 equiv BQ in AcOH at 100 °C). The major observable products under these conditions were also the oxazolium acetate salts (1b (<10%) and 2b (25%), respectively), and no acetoxylated products were observed upon analysis of the crude reaction mixtures by NMR or electrospray mass spectrometry.
Oxime ether substrates
Several reports by our group have shown that oxime derivatives are versatile directing groups for Pd-catalyzed sp3 C–H functionalization.3,13c,f As such, we next examined oxime ether derivatives 3–5. These substrates were prepared by oximation of the corresponding ketones and isolated in 68–91% yield as clear oils. Subjecting 3–5 to Yu’s conditions6 for Pd-catalyzed C–H iodination (10 mol % Pd(OAc)2, 1 equiv PhI(OAc)2, 1 equiv I2 in CH2Cl2 at rt) did not produce the desired iodinated products (Scheme 6). In all cases, 1H NMR spectroscopic analysis of the crude reaction mixtures showed that significant quantities of starting material remained after 96 h (22–68%). Complex mixtures of minor products were also observed; however, electrospray mass spectrometry did not show any signals associated with the expected mono-iodinated cyclopropane.
Scheme 6.
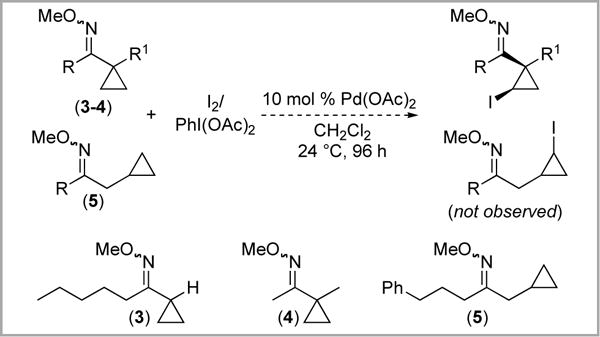
Attempted iodination of oxime ether derivatives
We next studied the Pd-catalyzed reactions of oxime ethers 3–5 with 2 equiv of PhI(OAc)2 in the presence of 10 mol % of Pd(OAc)2 in AcOH at 100 °C. With all three substrates, C–C activation and ring opening of the cyclopropane ring was observed. As shown in Scheme 7, substrate 3 formed the α,β-unsaturated allylic alcohol 3a in low (14%) yield. The mass balance was poor in this reaction; the starting material was completely consumed, and a complex and undecipherable mixture of other by-products was formed. This suggested to us that the product might be unstable under the reaction conditions. Indeed, resubjecting an isolated sample of 3a to Pd(OAc)2/PhI(OAc)2/AcOH showed that only 6% of 3a remained after 6 h at 100 °C.
Scheme 7.
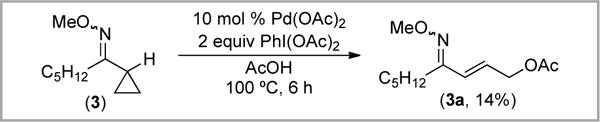
Acetoxylation of 3 with Pd(OAc)2/PhI(OAc)2
Substrate 4 showed somewhat different reactivity with Pd(OAc)2/PhI(OAc)2. Under analogous conditions, it formed a mixture of monoacetoxylated product 4a and trioxygenated 4b in 19% and 24% yield, respectively (Scheme 8,i). We hypothesized that 4b might be generated from 4a via in situ Pd-catalyzed olefin dioxygenation. Consistent with this proposal, subjecting an isolated sample of 4a to the reaction conditions led to formation of 4b in 43% yield (Scheme 8,ii). Notably, similar Pd-catalyzed olefin dioxygenation reactions have been reported in the literature.16
Scheme 8.
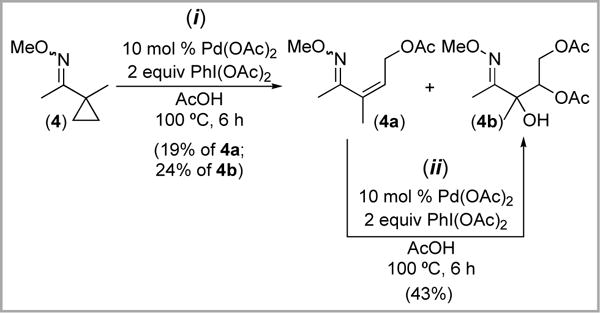
Acetoxylation of 4 with Pd(OAc)2/PhI(OAc)2
Treatment of substrate 5 with Pd(OAc)2/PhI(OAc)2 resulted in cyclopropane ring opening to form diacetoxylated branched products 5a and 5b along with linear product 5c (Scheme 9). The ratio of these three isomers was highly temperature dependent. At 60 °C, 5a was the major product (45% yield) and 5b was not detected (Table 1, entry 1). In contrast, at 120 °C, the yield of 5a was only 9% and 5b was formed in 10% yield (Table 1, entry 4).
Scheme 9.
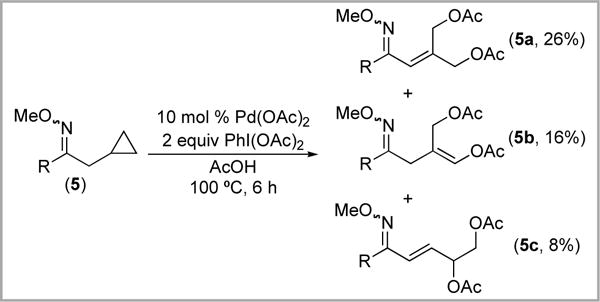
Acetoxylation of 5 with Pd(OAc)2/PhI(OAc)2
Table 1.
Product Distribution as a Function of Temperature
| entry | T (°C) | 5a (%) | 5b (%) | 5c (%) | 5 (%) |
|---|---|---|---|---|---|
| 1 | 60 | 45 | Nd | 7 | 3 |
| 2 | 80 | 44 | Nd | 8 | 6 |
| 3 | 100 | 26 | 12 | 8 | 3 |
| 4 | 120 | 9 | 10 | 9 | 3 |
nd = not detected.
We reasoned that the strong temperature dependence of the 5a/5b ratio might indicate that 5a is kinetically favored, while 5b is the thermodynamic product. Indeed, heating a pure sample of 5a in CD3CO2D for 6 h at 120 °C produced a 1.0: 1.1 mixture of 5a: 5b, implicating isomerization under the reaction conditions (Scheme 10). Collectively these results suggest that isomerization of 5a is likely a major pathway to 5b.
Scheme 10.
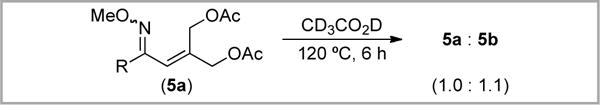
Isomerization of 5a at 120 °C in CD3CO2D
Finally, we examined the use of BQ in AcOH for the Pd-catalyzed oxime ether-directed functionalization of 3–5. As shown in Scheme 11, substrates 3 and 4 underwent ring opening to generate 3a and 4a, respectively. In contrast, no acetoxylated products were detected when substrate 5 was subjected to BQ/AcOH.
Scheme 11.
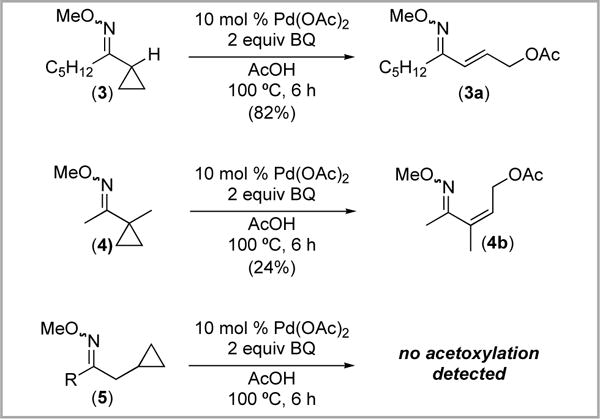
Reaction of substrates 3–5 with BQ/AcOH
Pyridine substrates
Pyridine derivatives have proven to be highly effective directing groups for Pd-catalyzed ligand-directed C–H functionalization reactions.3,4,13d As such, our final set of studies focused on evaluating 2-cyclopropylpyridines 6–10 as substrates for the Pd-catalyzed oxidation of cyclopropanes. Compounds 6–10 were prepared via Suzuki-coupling between cyclopropyl boronic acid and the appropriate 2-bromopyridines.17
In general, the reactivity of substrates 6–10 was extremely sensitive to the substitution pattern on the pyridine ring. For example, the Pd(OAc)2-catalyzed reactions of 6 and 7 with IOAc did not yield any mono-iodinated products, as determined by 1H NMR and electrospray mass spectrometric analysis of the crude reaction mixtures (Table 2, entries 1 and 2). In contrast, when the pyridine methyl substituent was moved to the 3-position (substrate 8), the cis-iodinated product 8a was formed in 19% yield (entry 3). The 3-ethyl and 3-methoxy substituted derivatives showed similar modest reactivity towards C–H iodination (entries 4 and 5). Notably, attempts to further optimize these reactions by varying the reaction time, temperature, and screening iodine salt additives did not lead to significant improvements in yield for any of these transformations.
Table 2.
C–H Iodination of 2-Cyclopropylpyridines with Pd(OAc)2/IOAc
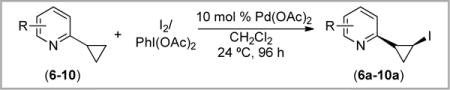
| ||
|---|---|---|
|
| ||
| entry | R | Yield |
| 1 | 4-CH3 (6) | nd |
| 2 | 6-CH3 (7) | nd |
| 3 | 3-CH3 (8) | 19% |
| 4 | 3-CH3CH2 (9) | 21% |
| 5 | 3-CH3O (10) | 19% |
The reactions of 2-cyclopropylpyridines with Pd(OAc)2/PhI(OAc)2 were similarly sensitive to pyridine substitution patterns (Table 3). The 4- and 6-methyl substrates did not yield detectable acetoxylated products (entries 1–2). However, the 3-substituted derivatives 8–10 all underwent ring opening triacetoxylation to afford moderate yields (21–34%) of 8b–10b as mixtures of diastereomers (entries 3–5).
Table 3.
C–C Activation of 2-Cyclopropylpyridines with Pd(OAc)2/PhI(OAc)2
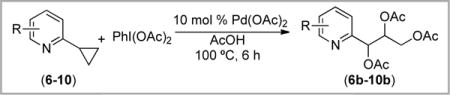
| ||
|---|---|---|
|
| ||
| entry | R | Yield |
| 1 | 4-CH3 (6) | nd |
| 2 | 6-CH3 (7) | nd |
| 3 | 3-CH3 (8) | 29% |
| 4 | 3-CH3CH2 (9) | 21% |
| 5 | 3-CH3O (10) | 34% |
The trioxygenated products 8b–10b are structurally similar to oxime ether 4b. As such, we hypothesized that they likely derive from a similar pathway involving initial cyclopropane ring opening followed by Pd-catalyzed dioxygenation of allylic acetate intermediates 8c–10c (Scheme 12).16 Consistent with this possibility, when an independently synthesized sample of 8c was subjected to Pd(OAc)2/PhI(OAc)2, product 8b was observed, albeit in modest (17%) yield.
Scheme 12.
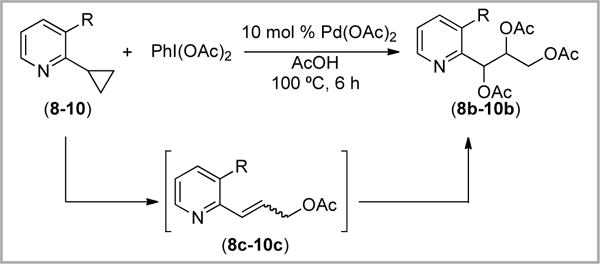
Plausible pathway to triacetoxylated product
Finally, cyclopropyl pyridines 8–10 were subjected to 10 mol % Pd(OAc)2 and 2 equiv of BQ at 100 °C in AcOH. Interestingly, these substrates did not undergo ring opening or C–H acetoxylation under these conditions. In all cases, the major organic compound at the end of the reaction was the starting material (55–81%). Furthermore, 1H NMR spectroscopic analysis and electrospray mass spectrometric analysis of the crude reaction mixtures did not show detectable quantities of monoacetoxylated products.
Discussion
The results reported herein show that cyclopropanes are not very general substrates for ligand-directed 2° sp3 C–H activation/functionalization. Under certain conditions, cyclopropane C–H functionalization was observed. For example, iodinated cyclopropyl oxazolines and pyridines could be isolated using IOAc as the oxidant. However, the yields of these transformations were typically modest, and the reactions were extremely sensitive to the substitution patterns of the substrate.
Under most conditions that are commonly used for Pd-catalyzed C–H functionalization, C–C activation of the cyclopropane was observed. These ring-opening reactions proceeded with concomitant incorporation of an acetate nucleophile derived from the solvent and/or the oxidant. While the detailed mechanism of cyclopropane ring opening has not been elucidated in the current systems, it is likely to be initiated by nucleopalladation of the cyclopropane. Such reactions have significant precedent in the organopalladium(II) literature. For example, Backvall has shown the stoichiometric acetoxypalladation and methoxypalladation of vinylcyclopropanes in AcOH and MeOH solvent, respectively.18 A similar mechanism has also been proposed by Yudin in the catalytic conversion of substituted cyclopropanes to heterocycles.15
Summary and Conclusions
In summary, this paper describes studies of the ligand-directed oxidative functionalization of cyclopropane derivatives. In certain cases, these substrates undergo 2° sp3 C–H oxidation reactions. However, relatively minor perturbations of the substrate structure, oxidant, and/or reaction conditions can lead to oxidative ring opening of the cyclopropane moiety. As a result, it is likely to be hard to extrapolate the reactivity of cyclopropanes to the functionalization of other substrates containing 2° sp3 C–H sites. Furthermore, future efforts to achieve Pd-catalyzed C–H and/or C– C activation of cyclopropane derivatives should take into account the possibility of generating products derived from these two different pathways.
Experimental Section
NMR spectra were obtained on a Varian Inova 500 (499.90 MHz for 1H; 125.70 MHz for 13C), a Varian Inova 400 (399.96 MHz for 1H; 100.57 MHz for 13C), or a MR400 (400.53 MHz for 1H; 100.71 MHz for 13C) spectrometer. 1H and 13C NMR chemical shifts are reported in parts per million (ppm) relative to TMS, with the residual solvent peak used as an internal reference. Multiplicities are reported as follows: singlet (s), doublet (d), doublet of doublets (dd), doublet of doublets of doublets (ddd), doublet of doublets of doublets of doublets (dddd), doublet of triplets (dt), triplet (t), quartet (q), quintet (quin), septet (sept), multiplet (m), and broad resonance (br). Flash chromatography was performed on EM Science silica gel 60 (0.040–0.063 mm particle size, 230–400 mesh) and thin layer chromatography was performed on Merck TLC plates pre-coated with silica gel 60 F254. HPLC was performed on a Varian ProStar 210 HPLC using Waters SunFire Prep Silica 5μm (19 × 150 mm) column. IR spectra were obtained on a Perkin-Elmer Spectrum BX FT-IR spectrometer. HRMS were obtained on a Micromass AutoSpec Ultima Magnetic sector mass spectrometer.
All reagents mentioned below were obtained from commercial sources and used as received unless noted otherwise. Benzoquinone (BQ) was obtained from Acros and sublimed prior to use. The parent ketone of 3 was synthesized from 1-octen-3-ol via Simmons-Smith cyclopropanation followed by oxidation of the alcohol.19 The parent ketone of 5 was synthesized by the addition of allymagnesium bromide to 4-phenylbutanal, Simmons-Smith cyclopropanation, and oxidation of the alcohol.19,20
Procedure for synthesis of substrates 1 and 2
Substrates 1 and 2 were synthesized according to the reported procedure6 by converting cyclopropanecarboxylic acid to its acid chloride and coupling with (S)-tert-leucinol. The coupled product was then cyclized using PPh3.
4-(tert-butyl)-2-(1-methylcyclopropyl)-4,5-dihydrooxazole (1)
The three-step synthesis6 from 1-methylcyclopropanecarboxylic acid (320 mg, 3.2 mmol, 1.0 equiv) afforded substrate 1 as a clear oil after purification by chromatography on silica gel using 80% petroleum ether/20% Et2O (349 mg, 60% yield over 3 steps).
Rf = 0.31 (85% hexanes/15% EtOAc).
IR (neat film): 3008, 1663 cm−1.
1H NMR (400 MHz, CDCl3) δ: 4.06 (dd, J = 9.6, 8.8 Hz, 1H), 3.99 (dd, J = 8.8, 6.8 Hz, 1H), 3.78 (dd, J = 9.6, 6.8 Hz, 1H), 1.34 (s, 3H), 1.12 (m, 1H), 1.06 (m, 1H), 0.86 (s, 9H), 0.59 (m, 2H).
13C NMR (100 MHz, CDCl3) δ: 170.00, 75.68, 68.52, 33.82, 25.64, 20.94, 14.80, 14.58, 14.01.
HRMS electrospray (m/z): [M+H]+ calcd for C11H20NO, 182.1539; found, 182.1536.
4-(tert-butyl)-2-cyclopropyl-4,5-dihydrooxazole (2)
The three-step synthesis6 from cyclopropanecarboxylic acid (2.0 g, 23.2 mmol, 1.0 equiv) afforded substrate 2 as a clear oil after purification by chromatography on silica gel using 80% petroleum ether/20% Et2O (1.7 g, 43% yield over 3 steps).
Rf = 0.24 (80% hexanes/20% EtOAc).
IR (neat film): 3015, 1669 cm−1.
1H NMR (400 MHz, CDCl3) δ: 4.06 (dd, J = 8.0, 6.8 Hz, 1H), 3.94 (dd, J = 6.8, 6.0 Hz, 1H), 3.75 (dd, J = 8.0, 6.0 Hz, 1H), 1.62 (dddd, J = 6.8, 6.8, 4.0, 4.0 Hz, 1H), 0.90 (m, 1H), 0.87 (m, 1H), 0.87 (s, 9H), 0.79 (m, 1H), 0.78 (m, 1H).
13C NMR (100 MHz, CDCl3) δ: 168.01, 75.57, 68.28, 33.60, 25.71, 8.43, 6.75, 6.34.
HRMS electrospray (m/z): [M+H]+ calcd for C10H18NO, 168.1383; found, 168.1377.
General procedure for synthesis of oxime substrates 3–5
The oxime substrates were prepared by combining the corresponding ketones (1.0 equiv) and NH2OMe•HCl (1.35 equiv) in pyridine (2.7 M). The resulting solution was stirred at 80 °C for 15 min and then at rt overnight. The reaction mixture was diluted with Et2O and washed with H2O containing a few drops of concentrated AcOH, H2O, saturated aqueous NaHCO3, and brine. The organic layer was dried over MgSO4, filtered, and concentrated under vacuum. The products were then purified by flash chromatography if necessary.
1-cyclopropylhexan-1-one O-methyl oxime (3)
1-Cyclopropylhexan-1-one (1.07 g, 7.6 mmol) reacted to form 3 as a 2.3:1 mixture of oxime isomers as a clear oil (1.18 g, 91% yield). Purification by column chromatography was not necessary.
IR (neat film, mixture of oxime isomers): 3088, 1621 cm−1.
HRMS obtained for mixture of oxime isomers, electron impact (m/z): [M]+ calcd for C10H19NO, 169.1467; found, 169.1467.
Major oxime isomer
Rf = 0.53 (90% hexanes/10% EtOAc).
1H NMR (500 MHz, CDCl3) δ: 3.76 (s, 3H), 2.13 (dd, J = 8.0, 7.5 Hz, 2H), 1.53-1.44 (multiple peaks, 3H), 1.36-1.25 (multiple peaks, 4H), 0.94-0.87 (multiple peaks, 5H), 0.70 (m, 2H).
13C NMR (100 MHz, CDCl3) δ: 161.99, 61.07, 32.05, 27.35, 25.96, 22.39, 13.98, 13.96, 5.02.
Minor oxime isomer
Rf = 0.28 (90% hexanes/10% EtOAc).
1H NMR (500 MHz, CDCl3) δ: 3.85 (s, 3H), 2.19 (dddd, J = 11.0, 11.0, 5.5, 5.5 Hz, 1H), 1.76 (dd, J = 8.0, 8.0 Hz, 2H), 1.48 (m, 2H), 1.36-1.25 (multiple peaks, 4H), 0.82 (m, 2H), 0.71-0.66 (multiple peaks, 5H).
13C NMR (100 MHz, CDCl3) δ: 161.12, 61.26, 31.71, 28.98, 27.30, 22.42, 14.03, 8.64, 5.06.
1-(1-methylcyclopropyl)ethanone O-methyl oxime (4)
Methyl 1-methylcyclopropyl ketone (1.0 g, 10.2 mmol) reacted to form 4 as a single detectable oxime isomer as a clear oil (0.88 g, 68% yield). Purification by column chromatography was not necessary.
IR (neat film): 3084, 1613 cm−1.
1H NMR (400 MHz, CDCl3) δ: 3.82 (s, 3H), 1.72 (s, 3H), 1.24 (s, 3H), 0.85 (m, 2H), 0.51 (m, 2H).
13C NMR (100 MHz, CDCl3) δ: 160.06, 61.13, 21.78, 20.13, 12.51, 11.29.
HRMS electron impact (m/z): [M]+calcd for C7H13NO, 127.0997; found, 127.0999.
1-cyclopropyl-5-phenylpentan-2-one O-methyl oxime (5)
Reaction with 1-cyclopropyl–5-phenylpentan-2-one (1.27 g, 6.3 mmol) afforded 5 as 1:1 mixture of oxime isomers as a clear oil after purification by chromatography on silica gel using 95% hexanes/5% EtOAc (1.41 g, 97% yield).
IR (neat film, mixture of oxime isomers): 3079 cm−1.
HRMS obtained for mixture of oxime isomers, electron impact (m/z): [M]+ calcd for C15H21NO, 231.1631; found, 231.1625.
Oxime isomer 1
Rf = 0.52 (90% hexanes/10% EtOAc).
1H NMR (400 MHz, CDCl3) δ: 7.28 (m, 2H), 7.20-7.16 (multiple peaks, 3H), 3.81 (s, 3H), 2.65 (dd, J = 7.6, 7.6 Hz, 2H), 2.30 (dd, J = 8.0, 7.6 Hz, 2H), 2.20 (d, J = 6.8 Hz, 2H), 1.86 (m, 2H), 0.84 (m, 1H), 0.45 (m, 2H), 0.10 (m, 2H).
13C NMR (100 MHz, CDCl3) δ: 160.58, 142.01, 128.43, 128.28, 125.77, 61.04, 35.53, 33.53, 32.27, 28.39, 7.68, 4.70.
Oxime isomer 2
Rf = 0.48 (90% hexanes/10% EtOAc).
1H NMR (400 MHz, CDCl3) δ: 7.29 (m, 2H), 7.20-7.17 (multiple peaks, 3H), 3.81 (s, 3H), 2.64 (dd, J = 7.6, 7.6 Hz, 2H), 2.41 (dd, J = 8.0, 8.0 Hz, 2H), 2.04 (d, J = 7.2 Hz, 2H), 1.83 (m, 2H), 0.81 (m, 1H), 0.46 (m, 2H), 0.11 (m, 2H).
13C NMR (100 MHz, CDCl3) δ: 161.01, 141.90, 128.36, 128.28, 125.82, 61.01, 38.84, 36.03, 27.56, 27.51, 8.55, 4.63.
General procedure for synthesis of pyridine substrates 6–8 and 10
The pyridine substrates were prepared from the corresponding 2-bromopyridine and cyclopropylboronic acid via a modification of a literature procedure.17 The reactions were run overnight and then cooled to room temperature. A 3 M aqueous solution of HCl was added, and the aqueous layer was extracted with EtOAc. The EtOAc extracts were discarded, the aqueous layer was basicified with 3 M aqueous NaOH, and the product was extracted with Et2O. The ether extracts were dried over MgSO4, filtered, and concentrated under vacuum. The products were then purified by flash chromatography.
2-cyclopropyl-4-methylpyridine (6)
2-Bromo-4-methylpyridine (1.0 g, 5.8 mmol) reacted to afford substrate 6 as a clear oil after purification by chromatography on silica gel using 90% hexanes/10% EtOAc (310 mg, 40% yield).
Rf = 0.22 (90% hexanes/10% EtOAc).
IR (neat film): 3089 cm−1.
1H NMR (400 MHz, CDCl3) δ: 8.28 (d, J = 5.2 Hz, 1H), 6.94 (m, 1H), 6.84 (m, 1H), 2.29 (s, 3H), 1.98 (dddd, J = 8.0, 8.0, 5.2, 5.2 Hz, 1H), 0.98-0.96 (multiple peaks, 3H), 0.95 (m, 1H).
13C NMR (100 MHz, CDCl3) δ: 162.56, 148.97, 146.75, 122.03, 121.46, 20.90, 16.94, 9.49.
HRMS electrospray (m/z): [M+H]+ calcd for C9H12N, 134.0964; found, 134.0964.
2-cyclopropyl-6-methylpyridine (7)
2-Bromo-6-methylpyridine (344 mg, 2.0 mmol) reacted to afford substrate 7 as a clear oil after purification by chromatography on silica gel using 90% petroleum ether/10% Et2O (168 mg, 63% yield).
Rf = 0.41 (90% hexanes/10% EtOAc).
IR (neat film): 3064 cm−1.
1H NMR (400 MHz, CDCl3) δ: 7.40 (t, J = 7.6 Hz, 1H), 6.88 (d, J = 7.6 Hz, 1H), 6.83 (d, J = 7.6 Hz, 1H), 2.47 (s, 3H), 2.02 (m, 1H), 0.96 (m, 2H), 0.94 (m, 2H).
13C NMR (100 MHz, CDCl3) δ: 162.18, 157.66, 136.06, 119.79, 117.09, 24.57, 17.25, 9.45.
HRMS electrospray (m/z): [M+H]+ calcd for C9H12N, 132.0964; found, 132.0965.
2-cyclopropyl-3-methylpyridine (8)
2-Bromo-3-methylpyridine (5.0 g, 29.1 mmol) reacted to afford substrate 8 as a clear oil after purification by chromatography on silica gel using 90% petroleum ether/10% Et2O (3.1 g, 80% yield).
Rf = 0.28 (90% hexanes/10% EtOAc).
IR (neat film): 3087 cm−1.
1H NMR (400 MHz, CDCl3) δ: 8.28 (d, J = 4.8 Hz, 1H), 7.36 (m, 1H), 6.93 (dd, J = 7.6, 4.8 Hz, 1H), 2.41 (s, 3H), 2.08 (dddd, J = 9.6, 9.6, 5.2, 5.2 Hz, 1H), 1.06 (m, 2H), 0.95 (m, 2H).
13C NMR (100 MHz, CDCl3) δ: 160.45, 146.56, 136.75, 130.98, 120.07, 18.83, 13.54, 8.76.
HRMS electron impact (m/z): [M-H]+ calcd for C9H10N, 132.0813; found, 132.0816.
2-cyclopropyl-3-methoypyridine (10)
2-Bromo-3-methoxypyridine (1.0 g, 5.3 mmol) afforded substrate 10 as a clear oil after purification by chromatography on silica gel using 90% petroleum ether/10% Et2O (590 mg, 74% yield).
Rf = 0.36 (85% hexanes/15% EtOAc).
IR (neat film): 3063, 1430 cm−1.
1H NMR (400 MHz, CDCl3) δ: 8.03 (dd, J = 4.8, 1.2 Hz, 1H), 7.05 (dd, J = 8.4, 1.2 Hz, 1H), 6.93 (dd, J = 8.4, 4.8 Hz, 1H), 3.86 (s, 3H), 2.47 (dddd, J = 10.0, 10.0, 4.8, 4.8 Hz, 1H), 1.05 (m, 2H), 0.95 (m, 2H).
13C NMR (100 MHz, CDCl3) δ: 153.68, 152.55, 140.57, 120.28, 116.05, 55.37, 10.24, 9.01.
HRMS electron impact (m/z): [M-H]+ calcd for C9H10NO, 148.0762; found, 148.0766.
Procedure for synthesis of substrate 9
A solution of 8 (1.0 g, 7.5 mmol, 1.0 equiv) in THF (1.5 mL) was added to a solution of LDA (1.5 equiv) in THF (23 mL) at −40 °C under N2. The reaction was stirred for 30 min and then cooled to −78 °C. MeI (5.3 g, 37.5 mmol, 5.0 equiv) was added, and resulting solution was stirred for 2 h at −78 °C. The reaction was quenched with H2O (15 mL) at −78 °C and then slowly warmed to rt. The product was obtained using the same work-up as that for the synthesis of substrates 6, 7, and 10.
2-cyclopropyl-3-ethylpyridine (9)
Substrate 9 was obtained as a clear oil after purification by chromatography on silica gel using 97.5% petroleum ether/2.5% Et2O (1.1 g, 98% yield).
Rf = 0.23 (95% hexanes/5% EtOAc).
IR (neat film): 3051, 1586, 1572 cm−1.
1H NMR (400 MHz, CDCl3) δ: 8.29 (d, J = 4.8 Hz, 1H), 7.37 (d, J = 7.6 Hz, 1H), 6.97 (dd, J = 7.6, 4.8 Hz, 1H), 2.79 (q, J = 7.6 Hz, 2H), 2.13 (dddd, J = 9.6, 9.6, 4.8, 4.8 Hz, 1H), 1.27 (t, J = 7.6 Hz, 3H), 1.08 (m, 2H), 0.95 (m, 2H).
13C NMR (100 MHz, CDCl3) δ: 159.90, 146.53, 136.76, 135.17, 120.22, 25.49, 14.55, 13.09, 9.03.
HRMS electron impact (m/z): [M-H]+calcd for C10H12N, 146.0970; found, 146.0971.
Procedure for synthesis of oxazolium acetate salt
The appropriate substrate (100 mg, 0.5 mmol, 1.0 equiv) was weighed into a scintillation vial containing a stir bar. Acetic acid (4.6 mL) was added, and the vial was sealed with Teflon lined cap. The resulting solution was stirred at 100 °C for 6–12 h. The reaction mixture was diluted with H2O (5 mL) and neutralized with a 3M aqueous solution of NaOH (to get to pH 7). The product was extracted into CH2Cl2 (5 mL × 3), and the organic extracts were dried over MgSO4, filtered, and concentrated under vacuum. The product was then purified by flash chromatography. The isolated products were then dissolved in CHCl3 and the resulting solution was filtered through K2CO3 to remove residual AcOH.
4-(tert-butyl)-2-(1-methylcyclopropyl)-4,5-dihydrooxazol-3-ium acetate salt (1b)
Substrate 1 (100 mg, 0.55 mmol, 1.0 equiv) yielded 1b as a white solid after purification by chromatography on silica gel using 65% hexanes/35% EtOAc (82 mg, 62% yield).
Mp 92.8–93.7 °C; Rf = 0.48 (50% hexanes/50% EtOAc).
IR (thin film with CH2Cl2): 3310, 3002, 1739, 1632 cm−1.
1H NMR (400 MHz, CDCl3) δ: 5.70 (d, J = 9.2 Hz, 1H), 4.28 (m, 1H), 4.11-4.05 (multiple peaks, 2H), 2.03 (s, 3H), 1.31 (s, 3H), 1.18 (dd, J = 3.2, 9.6 Hz, 1H), 1.12 (dd, J = 3.2, 9.6 Hz, 1H), 0.95 (s, 9H), 0.56 (m, 2H).
13C NMR (100 MHz, CDCl3) δ: 174.79, 171.42, 63.54, 55.85, 33.94, 26.69, 20.91, 19.59, 19.10, 15.82, 15.74.
HRMS electrospray (m/z): [M+Na]+ calcd for NaC13H23NO, 264.1570; found, 264.1565.
4-(tert-butyl)-2-cyclopropyl-4,5-dihydrooxazol-3-ium acetate salt (2b)
Substrate 2 (100 mg, 0.60 mmol, 1.0 equiv) yielded 2b as a white solid after purification by chromatography on silica gel using 60% hexanes/40% EtOAc (80 mg, 59% yield).
Mp 53.8–54.9 °C; Rf = 0.31 (50% hexanes/50% EtOAc).
IR (thin film with CH2Cl2): 3301, 3010, 1739, 1645 cm−1.
1H NMR (400 MHz, CDCl3) δ: 5.64 (d, J = 9.6 Hz, 1H), 4.22 (dd, J = 11.6, 8.8 Hz, 1H), 4.13-4.05 (multiple peaks, 2H), 2.02 (s, 3H), 1.62 (dddd, J = 8.0, 8.0, 4.4, 4.4 Hz, 1H), 0.95 (s, 9H), 0.93 (m, 2H), 0.71 (dd, J = 8.0, 2.4 Hz, 2H).
13C NMR (100 MHz, CDCl3) δ: 173.54, 171.38, 63.68, 55.80, 33.85, 26.67, 20.89, 14.79, 7.01, 6.97.
HRMS electrospray (m/z): [M+Na]+ calcd for NaC12H21NO3, 250.1414; found, 250.1412.
General iodination procedure
The appropriate pyridine substrate (1.0 equiv), Pd(OAc)2 (10 mol %), PhI(OAc)2 (1.0 equiv), and I2 (1.0 equiv) were weighed into a scintillation vial containing a stir bar. Methylene chloride was added to make a 0.2 M solution (in substrate), and the vial was sealed with Teflon lined cap. The reaction was stirred at rt overnight. The reaction mixture was washed with saturated sodium thiosulfate solution. The organic layer was collected, the solvent was removed under vacuum, NO2Ph (0.25 equiv, 1H NMR resonance at 8.2 ppm) or 1,3-dinitrobenzene (0.25 equiv, 1H NMR resonance at 9.1 ppm) was added as an internal standard, and the crude mixture was analyzed by 1H NMR spectroscopy. The products were then purified by flash chromatography.
2-(cis-2-iodocyclopropyl)-3-methylpyridine (8a)
1H NMR spectroscopic analysis of the crude reaction mixture showed that substrate 8 (60 mg, 0.45 mmol) reacted to form 8a in 19% yield. Product 8a was purified by chromatography on silica gel using 90% CH2Cl2/10% EtOAc and was isolated as a white solid (19 mg, 16% yield).
Rf = 0.42 (90% CH2Cl2/10% EtOAc).
IR (thin film with CH2Cl2): 3051 cm−1.
1H NMR (400 MHz, CDCl3) δ: 8.40 (m, 1H), 7.48 (m, 1H), 7.12 (dd, J = 7.6, 4.8 Hz, 1H), 3.02 (td, J = 7.6, 5.6 Hz, 1H), 2.38 (s, 3H), 2.25 (td, J = 8.0, 7.6 Hz, 1H), 1.98 (td, J = 6.4, 5.6 Hz, 1H), 1.65 (ddd, J = 8.8, 7.6, 6.0 Hz, 1H).
13C NMR (100 MHz, CDCl3) δ: 156.67, 146.11, 137.31, 133.27, 121.94, 21.07, 18.70, 13.54, −7.97.
HRMS electrospray (m/z): [M+H]+calcd for C9H11IN, 259.9936; found, 259.9936.
2-(cis-2-iodocyclopropyl)-3-ethylpyridine (9a)
1H NMR spectroscopic analysis of the crude reaction mixture showed that substrate 9 (50 mg, 0.34 mmol) reacted to form 9a in 30% yield. Product 9a was purified by chromatography on silica gel using 85% hexanes/15% EtOAc and was isolated as a yellow oil (15 mg, 16% yield).
Rf = 0.32 (85% hexanes/15% EtOAc).
IR (neat film): 3049 cm−1.
1H NMR (400 MHz, CDCl3) δ: 8.41 (dd, J = 4.8,1.2 Hz, 1H), 7.52 (dd, J = 7.6, 1.6 Hz, 1H), 7.15 (dd, J = 7.6, 5.2 Hz, 1H), 3.02 (td, J = 7.6, 5.6 Hz, 1H), 2.76 (q, J = 3.6 Hz, 2H), 2.08 (td, J = 7.6, 7.6 Hz, 1H), 1.98 (td, J = 6.4, 6.4 Hz, 1H), 1.64 (td, J = 8.0, 6.4 Hz, 1H), 1.31 (t, J = 3.6 Hz, 3H).
13C NMR (100 MHz, CDCl3) δ: 155.94, 145.93, 138.75, 135.32, 122.05, 24.76, 20.29, 13.70, 13.52, −7.04.
HRMS electron impact (m/z): [M+H]+ calcd for C10H13IN, 274.0087; found, 274.0085.
2-(cis-2-iodocyclopropyl)-3-methoxypyridine (10a)
1H NMR spectroscopic analysis of the crude reaction mixture showed that substrate 10 (50 mg, 0.34 mmol) reacted to form 10a in 13% yield. Product 10a was purified by chromatography on silica gel using 85% hexanes/15% EtOAc and was isolated as a yellow oil (14 mg, 15% yield).
Rf = 0.21 (85% hexanes/15% EtOAc).
IR (neat film): 3058, 1435 cm−1
1H NMR (400 MHz, CDCl3) δ: 8.16 (dd, J = 4.0, 2.0 Hz, 1H), 7.19-7.13 (multiple peaks, 2H), 3.89 (s, 3H), 3.01 (td, J = 8.0, 5.6 Hz, 1H), 2.48 (td, J = 8.0, 7.2 Hz, 1H), 1.90 (td, J = 7.2, 5.6 Hz, 1H), 1.59 (td, J = 8.0, 6.4 Hz, 1H).
13C NMR (100 MHz, CDCl3) δ: 154.97, 148.38, 139.98, 122.34, 116.68, 55.45, 17.93, 13.29, −6.87.
HRMS electron impact (m/z): [M+H]+ calcd for C9H11INO, 275.9880; found, 275.9881.
General acetoxylation procedure with PhI(OAc)2 or benzoquinone
Substrate (1.0 equiv), Pd(OAc)2 (10 mol %), and PhI(OAc)2 (2.0 equiv) or benzoquinone (2.0 equiv) were weighed into a scintillation vial containing a stir bar. Solvent was added to make a 0.12 M solution in substrate, and the vial was sealed with Teflon lined cap. The reaction was stirred at 100 °C for 6–12 h. The reaction mixture was filtered through Celite, and the Celite was washed with Et2O. The solvent was removed under vacuum, NO2Ph (0.25–0.5 equiv, 1H NMR resonance at 8.2 ppm) or 1,3-dinitrobenzene (0.25 equiv, 1H NMR resonance at 9.1 ppm) was added as an internal standard, and the crude mixture was analyzed by 1H NMR spectroscopy. The products were then purified by flash chromatography.
3-(4-(tert-butyl)-4,5-dihydrooxazol-2-yl)but-2-en-1-yl acetate (1c)
1H NMR spectroscopic analysis of the crude reaction mixture after 6 h at 100 °C showed that substrate 1 (50 mg, 0.28 mmol) reacted with PhI(OAc)2 in DCE to form 1c in 29% yield as a 2.6:1 mixture of E:Z isomers. The products were purified by chromatography on silica gel using 90% CH2Cl2/10% EtOAc and were isolated a clear oils (12 mg (1c–E), 18% yield; 5 mg (1c–Z), 8% yield).
(E)-isomer
Rf = 0.16 (85% hexanes/15% EtOAc).
IR (neat film): 1741 cm−1.
1H NMR (400 MHz, CDCl3) δ: 6.41 (t, J = 6.4 Hz, 1H), 4.74 (d, J = 6.4 Hz, 2H), 4.20 (dd, J = 9.6, 8.8 Hz, 1H), 4.10 (dd, J = 8.8, 8.0 Hz, 1H), 3.93 (dd, J = 9.6, 8.0 Hz, 1H), 2.08 (s, 3H), 1.99 (br s, 3H), 0.89 (s, 9H).
13C NMR (125 MHz, CDCl3) δ: 170.79, 164.03, 129.95, 128.09, 76.13, 68.47, 60.96, 33.93, 25.79, 20.86, 13.70.
HRMS electrospray (m/z): [M+H]+ calcd for C13H22NO3, 240.1594; found, 240.1586.
(Z)-isomer
Rf = 0.25 (85% hexanes/15% EtOAc).
IR (neat film): 1744 cm−1.
1H NMR (400 MHz, CDCl3) δ: 5.85 (t, J = 6.0 Hz, 1H), 5.03 (dd, J = 15.2, 6.0 Hz, 1H), 4.96 (dd, J = 15.2, 6.0 Hz, 1H), 4.20 (dd, J = 10.0, 8.4 Hz, 1H), 4.08 (dd, J = 8.4, 8.0 Hz, 1H), 3.93 (dd, J = 10.0, 8.0 Hz, 1H), 2.07 (s, 3H), 1.98 (br s, 3H), 0.90 (s, 9H).
13C NMR (100 MHz, CDCl3) δ: 170.87, 163.17, 132.60, 126.33, 75.88, 68.27, 62.91, 33.81, 25.82, 21.08, 20.99.
HRMS electrospray (m/z): [M+H]+ calcd for C13H22NO3, 240.1594; found, 240.1586.
(2E)-4-(methoxyimino)non-2-en-1-yl acetate (3a)
1H NMR spectroscopic analysis of the crude reaction mixture after 12 h at 100 °C showed that substrate 3 (50 mg, 0.30 mmol) reacted with benzoquinone to afford 3a in 76% yield as a 2.2:1 mixture of oxime isomers. Product 3a was purified by chromatography on silica gel using 95% hexanes/5% EtOAc and was isolated as a 3.0:1.0 mixture of oxime isomers as a clear oil (44 mg, 66% yield).
IR (neat film, mixture of isomers): 1741 cm−1.
HRMS obtained for mixture of isomers, electrospray (m/z): [M+Na]+calcd for NaC12H21NO3, 250.1419; found, 250.1417.
Major oxime isomer
Rf = 0.40 (85% hexanes/15% EtOAc).
1H NMR (400 MHz, CDCl3) δ: 6.26 (dt, J = 16.0, 1.6 Hz, 1H), 6.06 (dt, J = 16.0, 6.0 Hz, 1H), 4.68 (dd, J = 6.0, 1.6 Hz, 2H), 3.89 (s, 3H), 2.42 (m, 2H), 2.09 (s, 3H), 1.46 (m, 2H), 1.35-1.29 (multiple peaks, 4H), 0.89 (t, J = 7.2 Hz, 3H).
13C NMR (100 MHz, CDCl3) δ: 170.66, 158.51, 129.93, 127.48, 64.29, 61.81, 31.96, 26.22, 24.64, 22.38, 20.89, 13.94.
Minor oxime isomer
Rf = 0.47 (85% hexanes/15% EtOAc).
1H NMR (400 MHz, CDCl3) δ: 6.87 (dt, J = 16.4, 1.6 Hz, 1H), 6.15 (dt, J = 16.4, 5.6 Hz, 1H), 4.69 (dd, J = 5.6, 1.6 Hz, 2H), 3.87 (s, 3H), 2.34 (m, 2H), 2.10 (s, 3H), 1.54 (m, 2H), 1.34-1.29 (multiple peaks, 4H), 0.89 (t, J = 7.2 Hz, 3H).
13C NMR (100 MHz, CDCl3) δ: 170.61, 154.67, 131.08, 121.41, 64.30, 61.60, 31.63, 30.95, 27.42, 22.38, 20.88, 13.99.
4-(methoxyimino)-3-methylpent-2-en-1-yl acetate (4a)
1H NMR spectroscopic analysis of the crude reaction mixture after 12 h at 100 °C showed that substrate 4 (60 mg, 0.47 mmol) reacted with benzoquinone to afford 4a in 35% yield as a 3.4:1 mixture of oxime isomers. Product 4a was purified by chromatography on silica gel using 90% hexanes/10% EtOAc and was isolated as a 7.0:1.0 mixture of oxime isomers as a clear oil (29 mg, 33% yield).
Major oxime isomer
Rf = 0.42 (85% hexanes/15% EtOAc).
IR (neat film, major oxime isomer): 1589, 1739 cm−1.
1H NMR (400 MHz, CDCl3) δ: 5.90 (t, J = 6.8 Hz, 1H), 4.76 (d, J = 6.8 Hz, 2H), 3.92 (s, 3H), 2.08 (s, 3H), 1.96 (s, 3H), 1.91 (s, 3H).
13C NMR (100 MHz, CDCl3) δ: 170.93, 155.64, 137.14, 124.61, 61.83, 61.37, 20.94, 12.86, 10.55.
HRMS obtained for E isomer, electrospray (m/z): [M+Na]+ calcd for NaC9H15NO3, 208.0950; found, 208.0947.
Minor oxime isomer
Rf = 0.35 (85% hexanes/15%/EtOAc).
1H NMR (400 MHz, CDCl3) δ: 5.56 (t, J = 6.4 Hz, 1H), 4.70 (d, J = 6.4 Hz, 2H), 3.89 (s, 3H), 2.05 (s, 3H), 1.92 (s, 3H), 1.90 (s, 3H).
13C NMR (100 MHz, CDCl3) δ: 170.83, 155.43, 136.22, 124.92, 62.23, 61.74, 21.37, 20.98, 13.44.
3-hydroxy-4-(methoxyimino)-3-methylpentane-1,2-diyl diacetate (4b)
1H NMR spectroscopic analysis of the crude reaction mixture after 12 h at 100 °C showed that substrate 4 (100 mg, 0.79 mmol) reacted with PhI(OAc)2 to form 4b in 37% yield as a 1.6:1 mixture of diastereomers. Product 4b was purified by chromatography on silica gel using 80% hexanes/20% EtOAc and was isolated as a 1.7:1.0 mixture of diastereomers as a yellow oil (83 mg, 40% yield).
Major diastereomer
Rf = 0.17 (80% hexanes/20% EtOAc).
IR (neat film): 3481, 1744 cm−1.
1H NMR (400 MHz, CDCl3) δ: 5.21 (dd, J = 8.0, 2.8 Hz, 1H), 4.51 (dd, J = 12.0, 2.8 Hz, 1H), 4.16 (dd, J = 12.0, 8.0 Hz, 1H), 4.03 (s, 1H), 3.87 (s, 3H), 2.07 (s, 3H), 2.02 (s, 3H), 1.82 (s, 3H), 1.38 (s, 3H).
13C NMR (100 MHz, CDCl3) δ: 170.75, 170.03, 156.93, 74.64, 74.18, 62.41, 62.08, 23.10, 20.78, 20.74, 10.93.
HRMS electrospray (m/z): [M+Na]+calcd for NaC11H19NO6, 284.1105; found, 284.1100.
Minor diastereomer
Rf = 0.12 (80% hexanes/20% EtOAc).
IR (neat film): 3475, 1744 cm−1.
1H NMR (400 MHz, CDCl3) δ: 5.27 (dd, J = 8.4, 3.6 Hz, 1H), 4.49 (dd, J = 12.0, 3.6 Hz, 1H), 4.02 (s, 1H), 4.01 (dd, J = 12.0, 8.4 Hz, 1H), 3.86 (s, 3H), 2.14 (s, 3H), 1.98 (s, 3H), 1.90 (s, 3H), 1.29 (s, 3H).
13C NMR (100 MHz, CDCl3) δ: 170.71, 170.65, 156.90, 74.89, 73.80, 62.71, 62.18, 23.32, 20.88, 20.69, 11.13.
HRMS electrospray (m/z): [M+Na]+calcd for NaC11H19NO6, 284.1105; found, 284.1099.
Acetoxylation of substrate 5
1H NMR spectroscopic analysis of the crude reaction mixture after 6 h at 100 °C showed that substrate 5 (200 mg, 0.87 mmol) reacted with PhI(OAc)2 to form 5a (26% yield as a 2.3:1.0 mixture of oxime isomers), 5b (16% yield as a 3.0:1.0 mixture of oxime isomers), and 5c (8% yield as a single detectable oxime isomer). The products were purified by chromatography on silica gel using 90% hexanes/10% EtOAc gradient to 80% hexanes/20% EtOAc. The mixture of isomers 5a–c was isolated as a yellow oil (145 mg, 48% total yield). Each isomers was separated and isolated using HPLC (93% hexanes/7% EtOAc, 22 mL/min, Waters SunFire Prep Silica 5μm).
2-(2-(methoxyimino)-5-phenylpentylidene)propane-1,3-diyl diacetate (5a)
Major oxime isomer
Rf = 0.47 (70% hexanes/30% EtOAc).
IR (neat film): 1742 cm−1.
1H NMR (400 MHz, CDCl3) δ: 7.28 (m, 2H), 7.20-7.16 (multiple peaks, 3H), 6.00 (s, 1H), 4.96 (s, 2H), 4.67 (s, 2H), 3.90 (s, 3H), 2.62 (t, J = 7.6 Hz, 2H), 2.42 (t, J = 7.6 Hz, 2H), 2.08 (s, 3H), 2.06 (s, 3H), 1.78 (m, 2H).
13C NMR (100 MHz, CDCl3) δ: 170.52, 170.43, 155.77, 141.64, 135.75, 128.35, 128.30, 126.21, 125.88, 65.22, 62.99, 61.45, 35.66, 28.66, 27.51, 20.85, 20.79.
HRMS, electrospray (m/z): [M+Na]+ calcd for NaC19H25NO5, 370.1630; found, 370.1621.
Minor oxime isomer
Rf = 0.42 (70% hexanes/30% EtOAc).
1H NMR (400 MHz, CDCl3) δ: 7.26 (m, 2H), 7.18-7.16 (multiple peaks, 3H), 5.94 (s, 1H), 4.64 (s, 2H), 4.50 (s, 2H), 3.84 (s, 3H), 2.62 (t, J = 7.6 Hz, 2H), 2.29 (t, J = 7.6 Hz, 2H), 2.10 (s, 3H), 2.03 (s, 3H), 1.81 (m, 2H).
13C NMR (100 MHz, CDCl3) δ: 170.46, 170.39, 154.08, 141.69, 135.36, 128.42, 128.33, 125.88, 122.73, 64.51, 62.01, 61.63, 35.35, 33.97, 28.39, 20.84, 20.68.
(Z)-2-(2-(methoxyimino)-5-phenylpentyl)prop-1-ene-1,3-diyl diacetate (5b)
Major oxime isomer
Rf = 0.47 (70% hexanes/30% EtOAc).
IR (neat film): 1761, 1740 cm−1.
1H NMR (400 MHz, CDCl3) δ: 7.27 (m, 2H), 7.20-7.16 (multiple peaks, 3H), 7.08 (s, 1H), 4.65 (s, 2H), 3.81 (s, 3H), 3.06 (s, 2H), 2.63 (t, J = 7.6 Hz, 2H), 2.19 (t, J = 7.6 Hz, 2H), 2.16 (s, 3H), 2.04 (s, 3H), 1.84 (quin, J = 7.6 Hz, 2H).
13C NMR (100 MHz, CDCl3) δ: 170.80, 167.30, 156.51, 141.82, 135.25, 128.44, 128.34, 125.85, 115.03, 61.30, 59.80, 35.41, 33.10, 29.69, 28.14, 20.77, 20.65.
HRMS, electrospray (m/z): [M+Na]+ calcd for NaC19H25NO5, 370.1630; found, 370.1640.
Minor oxime isomer
Rf = 0.41 (70% hexanes/30% EtOAc).
1H NMR (400 MHz, CDCl3) δ: 7.27 (m, 2H), 7.20-7.16 (multiple peaks, 3H), 7.11 (s, 1H), 4.70 (s, 2H), 3.81 (s, 3H), 2.89 (s, 2H), 2.61 (t, J = 7.6 Hz, 2H), 2.29 (t, J = 7.6 Hz, 2H), 2.17 (s, 3H), 2.03 (s, 3H), 1.77 (m, 2H).
13C NMR (100 MHz, CDCl3) δ: 170.80, 167.36, 157.57, 135.26, 128.45, 128.33, 126.23, 125.89, 115.78, 61.33, 59.27, 36.38, 35.83, 34.88, 27.27, 20.82, 20.67.
(3E)-5-(methoxyimino)-8-phenyloct-3-ene-1,2-diyl diacetate (5c)
1H NMR analysis of the crude reaction mixture showed a single oxime isomer of 5c. However, this compound underwent isomerization to a mixture of oxime isomers during chromatographic purification on silica gel.
Major oxime isomer
Rf = 0.43 (70% hexanes/30% EtOAc).
IR (neat film): 1744 cm−1.
1H NMR (400 MHz, CDCl3) δ: 7.29 (m, 2H), 7.21-7.17 (multiple peaks, 3H), 6.27 (dd, J = 16.4, 1.6 Hz, 1H), 5.75 (dd, J = 16.4, 6.0 Hz, 1H), 5.57 (m, 1H), 4.24 (dd, J = 12.0, 4.0 Hz, 1H), 4.08 (dd, J = 12.0, 6.8 Hz, 1H), 3.90 (s, 3H), 2.64 (t, J = 7.6 Hz, 2H), 2.45 (t, J = 7.6 Hz, 2H), 2.10 (s, 3H), 2.05 (s, 3H), 1.77 (m, 2H).
13C NMR (100 MHz, CDCl3) δ: 170.59, 169.89, 157.81, 141.67, 129.94, 128.40, 128.34, 127.56, 125.94, 71.15, 64.58, 61.92, 35.75, 27.87, 24.10, 21.02, 20.74.
HRMS, electrospray (m/z): [M+Na]+ calcd for NaC19H25NO5, 370.1625; found, 370.1622.
Minor oxime isomer
Rf = 0.45 (70% hexanes/30% EtOAc).
1H NMR (400 MHz, CDCl3) δ: 7.29 (m, 2H), 7.21-7.17 (multiple peaks, 3H), 6.89 (dd, J = 16.4, 1.6 Hz, 1H), 5.90 (dd, J = 16.4, 6.0 Hz, 1H), 5.57 (m, 1H), 4.27 (dd, J = 12.0, 4.0 Hz, 1H), 4.10 (dd, J = 11.6, 6.8 Hz, 1H), 3.88 (s, 3H), 2.64 (t, J = 7.6 Hz, 2H), 2.36 (t, J = 7.6 Hz, 2H), 2.11 (s, 3H), 2.05 (s, 3H), 1.77 (m, 2H).
13C NMR (100 MHz, CDCl3) δ: 170.57, 169.88, 153.68, 141.82, 130.95, 128.48, 128.35, 125.89, 121.72, 71.33, 64.49, 61.72, 35.42, 30.20, 29.06, 21.01, 20.72.
1-(3-methylpyridin-2-yl)propane-1,2,3-triyl triacetate (8b)
1H NMR spectroscopic analysis of the crude reaction mixture after 12 h at 100 °C showed that substrate 8 (20 mg, 0.15 mmol) reacted with PhI(OAc)2 to form 8b in 41% yield as a 2.4:1 mixture of diastereomers. Product 8b was purified by chromatography on silica gel using 50% hexanes/50% EtOAc and was isolated as a 2.6:1.0 mixture of diastereomers as a yellow oil (17 mg, 37% yield). Pure samples of each diastereomer were obtained from individual column fractions.
IR (neat film, mixture of diastereomers): 1739 cm−1.
Major diastereomer
Rf = 0.30 in 60% EtOAc/40% hexanes.
1H NMR (400 MHz, CDCl3) δ: 8.45 (m, 1H), 7.46 (m, 1H), 7.13 (m, 1H), 6.15 (d, J = 6.4 Hz, 1H), 5.52 (td, J = 6.4, 2.4 Hz, 1H), 4.60 (dd, J = 12.0, 2.4 Hz, 1H), 4.46 (dd, J = 12.0, 6.4 Hz, 1H), 2.48 (s, 3H), 2.13 (s, 3H), 2.05 (s, 3H), 1.93 (s, 3H).
13C NMR (100 MHz, CDCl3) δ: 170.63, 170.03, 169.65, 153.25, 147.20, 138.25, 132.40, 123.29, 72.23, 70.24, 62.02, 20.84, 20.74, 20.68, 18.08.
HRMS, electrospray (m/z): [M+Na]+ calcd for NaC15H19NO6, 332.1110; found, 332.1106.
Minor diastereomer
Rf = 0.27 in 60% EtOAc/40% hexanes.
1H NMR (400 MHz, CDCl3) δ: 8.48 (m, 1H), 7.48 (m, 1H), 7.16 (m, 1H), 6.25 (d, J = 8.0 Hz, 1H), 5.82 (ddd, J = 8.0, 4.8, 3.2 Hz, 1H), 4.38 (dd, J = 12.0, 3.2 Hz, 1H), 3.74 (dd, J = 12.0, 4.8 Hz, 1H), 2.49 (s, 3H), 2.08 (s, 3H), 2.07 (s, 3H), 2.05 (s, 3H).
13C NMR (100 MHz, CDCl3) δ: 170.35, 170.26, 169.98, 152.78, 147.44, 138.70, 132.57, 123.63, 71.85, 70.82, 62.43, 20.82, 20.73, 20.67, 18.11.
1-(3-ethylpyridin-2-yl)propane-1,2,3-triyl triacetate (6b)
1H NMR spectroscopic analysis of the crude reaction mixture after 12 h at 100 °C showed that substrate 9 (50 mg, 0.34 mmol) reacted with PhI(OAc)2 to form 9b in 81% yield as a 13:5 mixture of diastereomers. The product was purified by chromatography on silica gel using 50% EtOAc/50% hexanes and was isolated as a 3.2:1.0 mixture of diastereomers as a yellow oil (23 mg, 21% yield). Pure samples of each diastereomer were obtained from individual column fractions.
Rf = 0.18 in 50% EtOAc/50% hexanes.
Major diastereomer
IR (neat film): 1741 cm−1.
1H NMR (400 MHz, CDCl3) δ: 8.47 (m, 1H), 7.51 (m, 1H), 7.18 (m, 1H), 6.24 (d, J = 6.8 Hz, 1H), 5.55 (ddd, J = 6.8, 6.0, 2.4 Hz, 1H), 4.61 (dd, J = 12.4, 2.4 Hz, 1H), 4.46 (dd, J = 12.4, 6.0 Hz, 1H), 2.86 (m, 2H), 2.12 (s, 3H), 2.06 (s, 3H), 1.91 (s, 3H), 1.23 (t, J = 7.2 Hz, 3H).
13C NMR (100 MHz, CDCl3) δ: 170.65, 170.04, 169.61, 152.55, 147.18, 138.26, 136.62, 123.56, 72.34, 69.64, 62.03, 24.27, 20.92, 20.76, 20.68, 14.68.
HRMS, electron impact (m/z): [M+H]+ calcd for C16H22NO6, 324.1442; found, 324.1443.
Minor diastereomer
1H NMR (400 MHz, CDCl3) δ: 8.49 (m, 1H), 7.54 (m, 1H), 7.22(m, 1H), 6.37 (d, J = 7.6 Hz, 1H), 5.85 (ddd, J = 7.6, 4.8, 3.2 Hz, 1H), 4.36 (dd, J = 12.4, 3.2 Hz, 1H), 3.79 (dd, J = 12.4, 4.8 Hz, 1H), 2.86 (m, 2H), 2.08 (s, 3H), 2.06 (s, 3H), 2.06 (s, 3H), 1.26 (t, J = 7.2 Hz, 3H).
13C NMR (100 MHz, CDCl3) δ: 170.39, 170.16, 169.99, 151.99, 147.29, 138.22, 136.84, 123.87, 72.06, 70.13, 62.44, 23.99, 20.90, 20.84, 20.71, 14.59.
1-(3-methoxypyridin-2-yl)propane-1,2,3-triyl triacetate (10b)
1H NMR spectroscopic analysis of the crude reaction mixture after 12 h at 100 °C showed that substrate 10 (50 mg, 0.34 mmol) reacted with PhI(OAc)2 to form 10b in 28% yield as a 1.3:1.0 mixture of diastereomers. The product was purified by chromatography on silica gel using 60% EtOAc/50% hexanes and was isolated as a 1.4:1.0 mixture of diastereomers as a yellow oil (37 mg, 34% yield). Pure samples of each diastereomer were obtained from individual column fractions.
IR (neat film, mixture of diastereomers): 1734 cm−1.
HRMS obtained for mixture of diastereomers, electrospray (m/z): [M+Na]+ calcd for NaC15H19NO7, 348.1059; found, 348.1051.
Major diastereomer
Rf = 0.16 (60% EtOAc/40% hexanes).
1H NMR (400 MHz, CDCl3) δ: 8.19 (m, 1H), 7.22 (m, 1H), 7.17 (m, 1H), 6.40 (d, J = 5.6 Hz, 1H), 5.74 (ddd, J = 6.4, 5.6, 4.0 Hz, 1H), 4.29 (dd, J = 12.0, 4.0 Hz, 1H), 4.09 (dd, J = 12.0, 6.4 Hz, 1H), 3.88 (s, 3H), 2.14 (s, 3H), 2.03 (s, 3H), 1.99 (s, 3H).
13C NMR (100 MHz, CDCl3) δ: 170.52, 170.05, 170.05, 153.29, 144.06, 141.03, 124.12, 117.83, 70.80, 68.72, 62.47, 55.63, 20.88, 20.72, 20.70.
Minor diastereomer
Rf = 0.22 (60% EtOAc/40% hexanes).
1H NMR (400 MHz, CDCl3) δ: 8.19 (m, 1H), 7.22 (m, 1H), 7.18 (m, 1H), 6.37 (d, J = 4.8 Hz, 1H), 5.62 (m, 1H), 4.47 (dd, J = 12.0, 2.0 Hz, 1H), 4.36 (dd, J = 12.0, 7.2 Hz, 1H), 3.88 (s, 3H), 2.15 (s, 3H), 2.00 (s, 6H).
13C NMR (100 MHz, CDCl3) δ: 170.68, 169.95, 169.87, 153.48, 144.15, 140.99, 124.09, 117.83, 71.14, 69.46, 62.23, 55.65, 20.91, 20.84, 20.74.
Procedure for synthesis of 8c
Substrate 8 (200 mg, 1.5 mmol, 1.0 equiv), Pd(OAc)2 (33.7 mg, 0.15 mmol, 10 mol %), and PhI(OAc)2 (484 mg, 1.5 mmol, 1.0 equiv) were weighed into a scintillation vial containing a stir bar. Methylene chloride (7.5 mL) was added, and the vial was sealed with a Teflon lined cap. The reaction was stirred at rt for 48 h. The solvent was then removed under vacuum.
3-(3-methylpyridin-2-yl)allyl acetate (8c)
The product 8c was obtained as a yellow oil after purification by flash chromatography on silica gel using 80% petroleum ether/20% Et2O (5 mg 8c–E, 14 mg 8c–Z, 7% total yield).
(E)-isomer
Rf = 0.35 (70% EtOAc/30% hexanes).
IR (neat film, isomer 1): 1736 cm−1.
1H NMR (400 MHz, CDCl3) δ: 8.45 (d, J = 4.8 Hz, 1H), 7.43 (d, J = 7.6 Hz, 1H), 7.05 (dd, J = 7.6, 4.8 Hz, 1H), 6.68 (d, J = 11.6 Hz, 1H), 5.97 (ddd, J = 11.6, 5.6, 5.6 Hz, 1H), 5.22 (dd, J = 5.6, 1.6 Hz, 2H), 2.33 (s, 3H), 2.08 (s, 3H).
13C NMR (100 MHz, CDCl3) δ: 170.98, 154.00, 146.61, 137.57, 131.58, 131.52, 126.98, 121.85, 63.48, 21.07, 18.95.
HRMS electron impact (m/z): [M+H]+ calcd for C11H14NO2, 192.1019; found, 192.1017.
(Z)-isomer
Rf = 0.17 (70% EtOAc/30% hexanes).
IR (neat film, isomer 1): 1736 cm−1.
1H NMR (400 MHz, CDCl3) δ: 8.41 (m, 1H), 7.43 (m, 1H), 7.07 (dd, J = 7.6, 4.8 Hz, 1H), 6.90-6.89 (multiple peaks, 2H), 4.81 (dd, J = 2.8, 1.2 Hz, 2H), 2.36 (s, 3H), 2.10 (s, 3H).
13C NMR (100 MHz, CDCl3) δ: 170.71, 152.47, 147.06, 138.19, 130.62, 129.04, 128.81, 122.51, 64.60, 20.95, 18.65.
HRMS electron impact (m/z): [M+H]+ calcd for C11H14NO2, 192.1019; found, 192.1019.
Acknowledgments
This work was supported by the NIH (R01-GM073836). We also thank Dr. Lopa Desai for preliminary studies of Pd-catalyzed oxidative functionalization of cyclopropanes.
References
- 1.For some recent reviews on C–H functionalization, see; (a) Kakiuchi F, Chatani N. Adv Synth Catal. 2003;345:1077. [Google Scholar]; (b) Diaz-Requejo MM, Perez P. J Chem Rev. 2008;108:3379. doi: 10.1021/cr078364y. [DOI] [PubMed] [Google Scholar]; (c) Jazzar R, Hitce J, Renaudat A, Sofack-Kreutzer J, Baudoin O. Chem Eur J. 2010;16:2654. doi: 10.1002/chem.200902374. [DOI] [PubMed] [Google Scholar]; (d) Colby DA, Bergman RG, Ellman JA. Chem Rev. 2010;110:624. doi: 10.1021/cr900005n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Doyle MP, Duffy R, Ratnikov M, Zhou L. Chem Rev. 2010;110:704. doi: 10.1021/cr900239n. [DOI] [PubMed] [Google Scholar]; (f) Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. Chem Rev. 2010;110:890. doi: 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]; (g) Yu JQ, Shi Z, editors. C-H Activation. Springer; Berlin, Germany: 2010. (Topics in Current Chemistry 292). [PubMed] [Google Scholar]
- 2.Lyons TW, Sanford MS. Chem Rev. 2010;110:1147. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Desai LV, Hull KL, Sanford MS. J Am Chem Soc. 2004;126:9542. doi: 10.1021/ja046831c. [DOI] [PubMed] [Google Scholar]
- 4.Kalyani D, Deprez NR, Desai LV, Sanford MS. J Am Chem Soc. 2005;127:7330. doi: 10.1021/ja051402f. [DOI] [PubMed] [Google Scholar]
- 5.(a) Zaitsev VG, Shabashov D, Daugulis O. J Am Chem Soc. 2005;127:13154. doi: 10.1021/ja054549f. [DOI] [PubMed] [Google Scholar]; (b) Shabashov D, Daugulis O. J Am Chem Soc. 2010;132:3965. doi: 10.1021/ja910900p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Giri R, Chen X, Yu JQ. Angew Chem Int Ed. 2005;44:2112. doi: 10.1002/anie.200462884. [DOI] [PubMed] [Google Scholar]
- 7.Giri R, Maugel N, Li JJ, Wang DH, Breazzano SP, Saunders LB, Yu JQ. J Am Chem Soc. 2007;129:3510. doi: 10.1021/ja0701614. [DOI] [PubMed] [Google Scholar]
- 8.Wang DH, Wasa M, Giri R, Yu JQ. J Am Chem Soc. 2008;130:7190. doi: 10.1021/ja801355s. [DOI] [PubMed] [Google Scholar]
- 9.Wasa M, Engle KM, Yu JQ. J Am Chem Soc. 2010;132:3680. doi: 10.1021/ja1010866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stowers KJ, Fortner KC, Sanford MS. 2011 doi: 10.1021/ja2015586. manuscript submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rappoport Z, editor. The Chemistry of the Cyclopropyl Group. John Wiley & Sons Ltd; Chichester, UK: 1987. [Google Scholar]
- 12.For selected reviews on synthetic applications of cyclopropanes, see:; (a) Reissig HU, Zimmer R. Chem Rev. 2003;103:1151. doi: 10.1021/cr010016n. [DOI] [PubMed] [Google Scholar]; (b) Yu M, Pagenkopf BL. Tetrahedron. 2005;61:321. [Google Scholar]; (c) Carson CA, Kerr MA. Chem Soc Rev. 2009;38:3051. doi: 10.1039/b901245c. [DOI] [PubMed] [Google Scholar]
- 13.(a) Dick AR, Hull KL, Sanford MS. J Am Chem Soc. 2004;126:2300. doi: 10.1021/ja031543m. [DOI] [PubMed] [Google Scholar]; (b) Kalyani D, Sanford MS. Org Lett. 2005;7:4149. doi: 10.1021/ol051486x. [DOI] [PubMed] [Google Scholar]; (c) Desai LV, Malik HA, Sanford MS. Org Lett. 2006;8:1141. doi: 10.1021/ol0530272. [DOI] [PubMed] [Google Scholar]; (d) Desai LV, Stowers KJ, Sanford MS. J Am Chem Soc. 2008;130:13285. doi: 10.1021/ja8045519. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Stowers KJ, Sanford MS. Org Lett. 2009;11:4584. doi: 10.1021/ol901820w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Neufeldt SR, Sanford MS. Org Lett. 2010;12:532. doi: 10.1021/ol902720d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Yoneyama T, Crabtree RH. J Mol Cat A. 1996;108:35. [Google Scholar]; (b) Gou FR, Wang XC, Huo PF, Bi HP, Guan ZH, Liang YM. Org Lett. 2009;11:5726. doi: 10.1021/ol902497k. [DOI] [PubMed] [Google Scholar]; (c) Mutule I, Suna E, Olofsson K, Pelcman B. J Org Chem. 2009;74:7195. doi: 10.1021/jo901321b. [DOI] [PubMed] [Google Scholar]
- 15.He Z, Yudin AK. Org Lett. 2006;8:5829. doi: 10.1021/ol062476e. [DOI] [PubMed] [Google Scholar]
- 16.(a) Li Y, Song D, Dong VM. J Am Chem Soc. 2008;130:2962. doi: 10.1021/ja711029u. [DOI] [PubMed] [Google Scholar]; (b) Wang W, Wang F, Shi M. Organometallics. 2010;29:928. [Google Scholar]
- 17.Wallace DJ, Chen CY. Tetrahedron Lett. 2002;43:6987. [Google Scholar]
- 18.Wilhelm D, Backvall JE, Nordberg RE, Norin T. Organometallics. 1985;4:1296. [Google Scholar]
- 19.(a) Charette AB, Juteau H, Lebel H, Molinaro C. J Am Chem Soc. 1998;120:11943. [Google Scholar]; (b) Cryle MJ, Ortiz de Montellano PR, De Voss JJ. J Org Chem. 2005;70:2455. doi: 10.1021/jo047985d. [DOI] [PubMed] [Google Scholar]
- 20.Denmark SE, Yang SM. J Am Chem Soc. 2002;124:2102. doi: 10.1021/ja0178158. [DOI] [PubMed] [Google Scholar]