

Abstract
The protective effect of ischaemic postconditioning (short cycles of reperfusion and reocclusion of a previously occluded vessel) was identified over a decade ago commanding intense interest as an approach for modifying reperfusion injury which contributes to infarct size in acute myocardial infarction. Elucidation of the major mechanisms of postconditioning has identified potential pharmacological targets for limitation of reperfusion injury. These include ligands for membrane-associated receptors, activators of phosphokinase survival signalling pathways and inhibitors of the mitochondrial permeability transition pore. In experimental models, numerous agents that target these mechanisms have shown promise as postconditioning mimetics. Nevertheless, clinical studies of ischaemic postconditioning and pharmacological postconditioning mimetics are equivocal. The majority of experimental research is conducted in animal models which do not fully portray the complexity of risk factors and comorbidities with which patients present and which we now know modify the signalling pathways recruited in postconditioning. Cohort size and power, patient selection, and deficiencies in clinical infarct size estimation may all represent major obstacles to assessing the therapeutic efficacy of postconditioning. Furthermore, chronic treatment of these patients with drugs like ACE inhibitors, statins and nitrates may modify signalling, inhibiting the protective effect of postconditioning mimetics, or conversely induce a maximally protected state wherein no further benefit can be demonstrated. Arguably, successful translation of postconditioning cannot occur until all of these issues are addressed, that is, experimental investigation requires more complex models that better reflect the clinical setting, while clinical investigation requires bigger trials with appropriate patient selection and standardization of clinical infarct size measurements.
Tables of Links
| LIGANDS | |
|---|---|
| Adenosine | HOE140 |
| AG490 | Hydrogen peroxide |
| ATP | Insulin |
| Atrial natriuretic peptide | Naloxone |
| Bradykinin | Nicorandil |
| Brain natriuretic peptide | Nitric oxide (NO) |
| cGMP | Pravastatin |
| Cyclosporine A | Pro-enkephalin |
| Diazoxide | TNF-α |
| Erythropoietin (EPO) | Wortmannin |
| Exenatide |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,c,dAlexander et al., 2013a,b,c,d,,,).
Development of the postconditioning paradigm for cardioprotection
Death due to acute myocardial infarction (AMI) has declined steadily in the economically developed countries during the last 50 years. Since the 1980s, the development of reperfusion therapies as the ‘standard of care’ for AMI has contributed markedly to the decline in early mortality. However, while case fatality rate has declined, there is evidence of an increasing incidence of chronic heart failure in AMI survivors. It is likely that infarct size is a major determinant of myocardial remodelling processes that predispose patients to the subsequent development of heart failure. Thus, prompt reperfusion is needed to effectively limit the development of ischaemic necrosis during AMI, but it seems plausible that further limitation of infarction is desirable to reduce long-term morbidity and mortality due to heart failure. The identification of potential adjunctive infarct-limiting treatments has been a goal of experimental cardioprotection research for several decades. A vast array of pharmacological and other interventions have been described. A review of all of these is beyond the scope of this paper, but we wish to highlight here three pivotal conceptual developments that have emerged over several decades and converged to provide a new cardioprotection paradigm around 2005.
A mechanism of tissue injury specifically associated with reperfusion, termed ‘lethal reperfusion injury’, was proposed as long ago as the late 1970s (Ashraf et al., 1978; Hearse et al., 1978). This concept implied the rapid irreversible injury, or accelerated death, of cells still viable at the end of an ischaemic period as a result of the sudden reintroduction of oxygen to ischaemic tissue. This reperfusion-associated cell death would be expected to contribute to final infarct size in reperfused AMI. For two decades, the concept of lethal reperfusion injury proved to be controversial. The proposed molecular and cellular mechanisms of lethality were diverse and poorly defined. Most importantly, experimental pharmacological interventions specifically targeted at reperfusion were not consistent in their infarct-limiting ability. However, from the mid-1990s, there was increasing evidence that apoptotic signals are activated during early reperfusion and that reperfused myocardium displays hallmarks of apoptosis. Although the quantitative contribution of apoptosis to infarct size is likely to be small, experimental activation of anti-apoptotic survival signals and inhibition of caspases were found to limit infarct size in experimental models. This work led to the development by Yellon and colleagues of a general hypothesis that attenuation of lethal reperfusion injury and limitation of infarct size could be induced by activating anti-apoptotic survival signals termed the ‘reperfusion injury salvage kinase’ (RISK) pathway (Hausenloy and Yellon, 2004).
In 1986, Murry et al. (1986) made the experimental observation that brief periods of ischaemia preceding AMI led to an acute adaptation of myocardium that limited infarct size. This phenomenon was termed ischaemic preconditioning. Intensive research throughout the 1990s revealed that ischaemic preconditioning is associated with the recruitment of a number of autacoid-stimulated signal transduction mechanisms that enhance the tolerance of myocardium to ischaemia-reperfusion insult, and thereby limit infarct size. The first autacoid to be identified in relation to the preconditioning mechanism was adenosine. This factor had been investigated extensively before the discovery of preconditioning in relation to nucleotide metabolism in ischaemia (Berne and Rubio, 1974; Berne, 1980). Indeed, ATP catabolism had been an area of active investigation in the laboratory of Reimer and Jennings for many years (Reimer et al., 1986) and led directly to the experimental protocol that identified preconditioning (Murry et al., 1986). Subsequently throughout the 1990s, several other autacoid factors and numerous intracellular signal transduction mechanisms were identified, all presumed only to be effective if activated before the onset of AMI. While preconditioning mechanisms induce a marked and very reproducible infarct-limiting effect, the clinical utility of therapies based on these mechanisms is extremely limited since pre-ischaemic treatment is not a possibility for the majority of AMI patients in whom coronary thrombosis is sudden and unexpected.
In 2003, Vinten-Johansen and colleagues reported that intermittent repetitive re-occlusion of the infarct-related coronary artery during the early moments of reperfusion in an experimental model of AMI was able to limit infarct size as effectively as ischaemic preconditioning (Zhao et al., 2003). This reperfusion-specific intervention is termed ischaemic postconditioning. Subsequent early research on postconditioning was remarkable for several reasons. Firstly, postconditioning confirmed that lethal reperfusion injury contributes significantly to final infarct size. Secondly, both ischaemic preconditioning and ischaemic postconditioning were shown to involve the activation, during reperfusion, of the RISK pathway identified a few years previously (Kin et al., 2004). Clearly, the temporal characteristics of postconditioning highlight the relative importance of reperfusion injury in AMI, but have no effect on ischaemic injury even though ischaemia is the sine qua non of AMI.
Introduction of the postconditioning paradigm for cardioprotection has attracted huge interest as a possible therapeutic intervention at reperfusion to limit the injurious combined effect of ischaemia and reperfusion. In this respect, intervention at reperfusion with conditioning protocols or with pharmacological agents that replicate conditioning mechanisms can truly be said to represent a paradigm shift in the field.
Characteristics of postconditioning
Interventions applied in the early reperfusion period to augment tissue salvage, beyond that achieved by reperfusion alone, are now often described as postconditioning treatments. Such interventions may take several forms and it is important to distinguish between them. Here we provide a brief overview of these interventions and their major characteristics: for further discussion, the reader is referred to more detailed reviews elsewhere (Burley and Baxter, 2009; Ovize et al., 2010; Shi and Vinten-Johansen, 2012; Hausenloy, 2013).
Myocardial ischaemic postconditioning
Postconditioning to limit infarct size was first formally described and characterized in the open-chest dog by Zhao et al. (2003). This form of postconditioning is referred to as myocardial ischaemic postconditioning, classical postconditioning or mechanical postconditioning. It has been described in several other experimental species (mouse, rat, rabbit and pig) in vivo (Yang et al., 2004; Schwartz and Lagranha, 2006; Tang et al., 2006; Gomez et al., 2008), in isolated rodent heart preparations (Tsang et al., 2004; Heusch et al., 2006), in humans and in isolated human myocardium (Sivaraman et al., 2007). The major characteristic of the intervention is that brief (typically 10–30 s), repetitive periods (3–10 cycles) of ischaemia, interspersed with brief (10–30 s) periods of reperfusion, are achieved by physical occlusion and reperfusion of the infarct-related coronary artery immediately following the index ischaemic event (see Figure 1). Most studies suggest that the timing of the intervention is critical to the outcome, a reduction in infarct size. A delay of more than 1 min in instituting the first re-occlusion of the coronary artery was associated with a loss of protection (Skyschally et al., 2009). This concurs with the prevailing view that lethal reperfusion injury, associated with opening of the mitochondrial permeability transition pore (MPTP), occurs within the first few minutes of reperfusion (Griffiths and Halestrap, 1995; Di Lisa et al., 2001). However, there is some evidence from the mouse heart suggesting that myocardial ischaemic postconditioning can limit infarct size if instituted even 30 min after reperfusion (Roubille et al., 2011). This effect has been termed ‘delayed’ ischaemic postconditioning. Whether this is a phenomenon that is generally applicable to other species, including humans, is not clear. However, it has been suggested that these observations support the concept of a gradually evolving ‘wavefront of reperfusion injury’, susceptible to later intervention.
Figure 1.

Schematic representation of myocardial postconditioning protocols and reported infarct limitation afforded by these interventions. Index ischaemia, typically of 30 min duration, is followed by intermittent reperfusion-reocclusion of the coronary artery-ischaemic postconditioning. Similarly, when the reocclusion cycles are delayed by as little as 60 s, infarct limitation is no longer afforded. Pharmacological postconditioning typically involves the administration of a postconditioning mimetic during early reperfusion. Remote postconditioning is afforded by occlusion-reperfusion cycles of an artery distal to the myocardium, typically a limb. Modified reperfusion is initiated by gradual reperfusion of the occluded area of the myocardium over several seconds. Temporarily reducing the pH during the first minutes of reperfusion can also limit the infarct. AcR, acidified reperfusion; BAY, cGMP elevating compounds; BNP, brain natriuretic peptide; GrR, gradual reperfusion; I/R, infarct to risk zone size; IPOC, ischaemic postconditioning; RPOC, remote postconditioning.
Although ischaemic postconditioning has been reported in every animal species examined, there is considerable variations in the extent of infarct limitation between species and laboratories. Murine models of postconditioning typically display 30% relative reduction in infarct size whereas in larger models, such as rabbit and canine hearts, infarct size limitation is around 50% (Vinten-Johansen et al., 2011). The postconditioning protocols used and the duration of the period of ischaemia employed in these experimental models vary considerably. Some data suggest that the threshold for ischaemic postconditioning rises as the ischaemic duration increases. There have been some studies showing that ischaemic postconditioning is unable to limit infarct size (Schwartz and Lagranha, 2006; Dow and Kloner, 2007; Hale et al., 2008). Typically, those studies that failed to show infarct limitation following postconditioning used a shorter period of ischaemia, and for larger animals, shorter postconditioning cycles. It is clear that there is not one protocol that suits all models and differences in protocols may account for the varying degrees of protection.
In addition to limiting infarct size, ischaemic postconditioning has been reported to limit the severity of other deleterious consequences of reperfusion. These include the development of arrhythmias in the rat heart (Dow et al., 2008), cardiomyocyte apoptosis and the extent of vascular injury (Schwartz and Kloner, 2012).
Remote ischaemic postconditioning
Numerous experimental and clinical observations suggest that intermittent ischaemia at the onset of myocardial reperfusion of tissues and organs remote from the heart can limit myocardial infarct size (see Figure 1). This phenomenon, called remote ischaemic postconditioning (or inter-organ postconditioning), is the subject of a comprehensive review elsewhere in this issue (Schmidt et al., 2015). The most frequently applied remote ischaemic postconditioning intervention in both experimental and clinical models is intermittent limb ischaemia performed at the onset of myocardial reperfusion (Kharbanda et al., 2001; Loukogeorgakis et al., 2006). The potential utility of such a simple intervention (e.g. repeated inflation of a blood pressure cuff) has attracted considerable interest, further augmented by the recognition that some benefit also accrues if the remote postconditioning intervention is delayed by 30 min after myocardial reperfusion (‘delayed remote ischaemic postconditioning’). The biological mechanisms of remote ischaemic postconditioning are unclear, but there appears to be a dependency on several interacting factors, including neuronal and humoral factors as well as transmission of unknown factors via microvesicles (Giricz et al., 2014).
Pharmacological postconditioning
The administration of pharmacological or other biologically active agents during early reperfusion to effect cardioprotection is frequently termed pharmacological postconditioning. For clarity and precision, we believe that this term should be reserved strictly for approaches that recruit or mimic the established pathways associated with ischaemic postconditioning. These approaches would include pharmacological agonists for receptors that are known to participate in ischaemic postconditioning (e.g. adenosine A2 receptor ligands or kinin B2 receptor ligands) or activators of established signal transduction mechanisms that are invovled in ischaemic postconditioning (e.g. statins and volatile anaesthetics activating the PI3K/Akt pathway or NO donors activating the cGMP/PKG pathway). It is usual for the administration of such agents to be commenced shortly before reperfusion or immediately after reperfusion onset. Over many decades, a wide variety of agents, unrelated directly to the mechanisms of ischaemic postconditioning, have been reported to be adjuncts to reperfusion. These include calcium channel blockers (Kloner and Przyklenk, 1991), magnesium salts (Antman, 1995), caspase inhibitors (Mocanu et al., 2000) and adrenoreceptor antagonists (Broadley and Penson, 2004). Whether or not they are effective at limiting infarct size during reperfusion, such pharmacological treatments should not be described as postconditioning mimetics.
Other modified reperfusion approaches
Several years before the formal description of ischaemic postconditioning, it was recognized that modified forms of reperfusion could limit reperfusion injury. Most notable are staged (gradual) reperfusion and acidic reperfusion (see Figure 1). Several surgical studies in the 1980s showed that gradual, rather than rapid, restoration of coronary blood flow attenuated the development of reperfusion injuries (arrhythmias and stunning) (Casale et al., 1984; Preuss et al., 1987). This manoeuvre was later shown to limit infarct size (Sato et al., 1997). Similarly, mild acidification of the blood or crystalloid perfusate during early reperfusion showed a similarly protective effect (Inserte et al., 2008). Our understanding of the molecular mechanisms of reperfusion injury has led to speculation that both manoeuvres limit the opening of MPTP during early reperfusion, a mechanism shared in common with the various forms of postconditioning and discussed in more detail below.
Overview of mechanisms of ischaemic postconditioning
The prevailing conceptual model (see Figure 2) within which the majority of work on ischaemic postconditioning is currently undertaken postulates opening of MPTP during the early minutes of reperfusion as being a critical event leading to cell death. In the post-conditioned myocardium, a number of complex interlinked signalling pathways are activated by intracellular factors and extracellular autacoids. These signalling pathways ultimately impinge on MPTP, reducing the probability of its opening. This mechanistic framework has been built up through a considerable body of experimental work, including pharmacological and genetic targeting of these pathways, autacoids, and components of the MPTP. We will now describe the key evidence supporting the current model beginning with a discussion of the pivotal role of MPTP.
Figure 2.
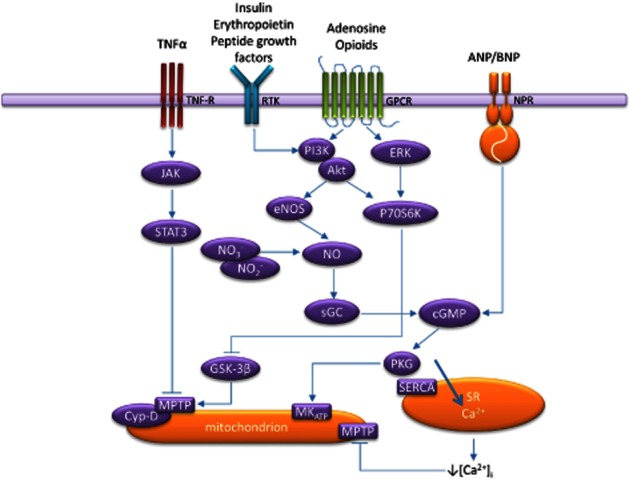
Schematic representation of the identified components of postconditioning signalling in the myocardium. Autacoid factors such as adenosine and opioids along with other extracellular factors initiate cytoprotective signalling through their sarcolemmal receptors. Three distinct signalling pathways have been reported, including RISK, which involves PI3K/Akt and ERK and distal inhibition of GSK-3β. cGMP/PKG signalling through natriuretic peptides and soluble GC activation is identified as an additional pathway distally targeting mitochondrial potassium channels. SAFE, whose major components are TNF-α and JAK/STAT, has also been demonstrated to play a role in postconditioning. Although described as distinct pathways, their cytoprotective actions are demonstrated to culminate on the mitochondria, specifically inhibition of the MPTP. It remains to be fully investigated as to what extent these pathways interact and co-localize. NPR, natriuretic peptide receptor; PKG, cGMP-dependent PK; RTK, receptor tyrosine kinase; SERCA, sarcoplasmic/endoplasmic reticulum calcium ATPase; SR, sarcoplasmic reticulum.
Mitochondrial permeability transition
Hunter and Haworth (1979) and Crompton et al. (1987) identified the MPTP as a non-specific channel of defined diameter spanning the mitochondrial inner and outer membranes. More recent work by Halestrap and colleagues made the association between reperfusion and the formation of this pore in an active state. They observed that the opening of the MPTP is enhanced by adenine nucleotide depletion, as well as elevated phosphate and oxidative stress, which are biochemical anomalies associated with ischaemia-reperfusion injury (Halestrap et al., 1998). Opening of the MPTP permits the passage of molecules up to 1.5 kDa and, with the entry into the mitochondrial matrix of H+, results in the uncoupling of oxidative phosphorylation, ATP depletion and the onset of cell death by necrosis. Work by Crompton and Costi (1988) and Griffiths and Halestrap (1993; 1995,) provided direct evidence of MPTP opening at reperfusion, but not during ischaemia. Particular features of the intracellular environment in reperfusion appear to contribute to this activation of MPTP. They include oxidizing conditions associated with reactive oxygen species (ROS) generation, intracellular Ca2+ overload and the reversal of intracellular acidosis as a result of H+ washout (Buja, 2013). It has been suggested that ischaemic postconditioning and postconditioning mimetic stimuli attenuate the opening of the MPTP by reducing intracellular Ca2+ overload and limiting ROS generation (Leung and Halestrap, 2008).
It remains unclear how Ca2+, ROS and H+ interact with the MPTP, but it has been reported that binding of adenine nucleotide translocase ligands to cyclophilin-D (CYP-D), a subunit of the MPTP, increases sensitivity to Ca2+. Mice deficient in CYP-D could not be protected by an ischaemic postconditioning stimulus (Elrod et al., 2010). On the other hand, cyclosporine-A (Cys-A) that inhibits MPTP opening by binding to CYP-D limits infarct size when administered at reperfusion in most animal models tested and in humans (Gedik et al., 2013).
Mitochondrial KATP (MKATP) channels offer another cytoprotective target through their ability to regulate ROS production and Ca2+ overload. Perfusion with the KATP channel blocker 5-hydroxydecanoate abolished postconditioning protection in the rat, whereas the KATP channel opener diazoxide significantly improved cardiac contractile activity (Jin et al., 2012). It has been also suggested that intermittent targeting of the MKATP channel during reperfusion, mimicking postconditioning, affords cardioprotection by ROS compartmentalization (Penna et al., 2007). Interestingly, postconditioning was blocked by administration of an antioxidant during early reperfusion. It is proposed that the early generation of ROS may trigger MKATP channel opening and PKC activation, which are required for protection; this is supported by the finding that a channel blocker and PKC inhibitor attenuated this protection (Yang et al., 2004). However, the subsequent reduction in ROS may prevent MPTP opening (Clarke et al., 2008).
Receptor-mediated mechanisms
The involvement of a number of extracellular autacoid factors, elaborated or enhanced as a result of ischaemic postconditioning, has been explored extensively. These factors are the subject of a comprehensive discussion elsewhere in this issue (Kleinbongard and Heusch, 2015) and the interested reader is referred there. In brief, the autacoids that have received most attention include adenosine, bradykinin and opioid peptides. Several studies have demonstrated that ischaemic postconditioning delays the washout of endogenous adenosine and the subsequent receptor activation affords protection (Kin et al., 2005). Different receptor subtypes are implicated in different species with A2A and A3 receptors being important in rat (Kin et al., 2005), while A2B receptor signalling is required in the post-conditioned rabbit heart (Philipp et al., 2006). Bradykinin B2 receptors have also been implicated in postconditioning in the rat perfused with the B2 receptor antagonist HOE140, which attenuated the protection (Penna et al., 2007). Interestingly, perfusion of bradykinin for 3 min during early reperfusion was unable to afford protection, yet intermittent perfusion in a protocol that matched mechanical postconditioning demonstrated comparable infarct limitation. This protocol was unsuccessful when using adenosine. The significance of the protection afforded by bradykinin perfusion in this model remains to be elucidated.
Most recently the opioid receptor has been reported to play a part in postconditioning. The opioid receptor antagonist naloxone abolished the protection afforded by postconditioning alone (Zatta et al., 2008). Similar to the observations made with adenosine, postconditioning appears to prevent the washout of pro-enkephalin, suggesting a build-up of endogenous opioid during postconditioning. These observations are supported by recent findings that report that κ opioid receptor activation limits infarct size during early reperfusion, an effect that was blocked by ERK1/2 inhibition (Kim et al., 2011).
Protein kinase mechanisms
The third and most complex element of the postconditioning mechanism is transduction of the extracellular signals described above to the mitochondria, leading to inhibition of MPTP (see above). Signal transduction is via a number of pathways involving protein kinase activation, often sequentially. The discussion below focuses on the major kinases explored to date. While these are grouped discretely for the purposes of this discussion, it needs to be recognized that considerable overlap and crosstalk are likely to exist between these cascades.
RISK pathway (PI3K/Akt and MEK/ERK)
The RISK pathway, initially described by Yellon's group, consists of two related signalling cassettes: PI3K/Akt and MEK/ERK. Both act in a number of biological systems as anti-apoptotic pro-survival signals, classically activated by extracellular ligands including peptide growth factors (Yellon and Baxter, 1999) (see Figure 2). PI3K/Akt and MEK/ERK have been repeatedly demonstrated as major players in mediating the cardioprotective effects of postconditioning in rodent models (Hausenloy, 2009). Tsang et al. (2004) reported that Akt was phosphorylated following six 10 s cycles of reperfusion in the isolated perfused rat heart. Furthermore, endothelial NOS (eNOS) and p70s6K were also phosphorylated more than in hearts that had undergone a standard reperfusion protocol. These findings were corroborated by observations that the classical PI3K inhibitors wortmannin and LY294002 abolished the protective effect of postconditioning. Subsequently, Yang et al. (2004) reported the importance of MEK/ERK signalling in an isolated rabbit heart model where pharmacological inhibition of MEK/ERK activation abolished the protection. Of note, RISK signalling is implicated in the cardioprotective effect of postconditioning in human atrial muscle ex vivo (Sivaraman et al., 2007). Many pharmacological mimetics of postconditioning have been shown to require the participation of either PI3K/Akt or MEK/ERK or both (Hausenloy, 2009).
GSK-3β
Inhibition of glycogen synthase kinase-3β (GSK-3β) is associated with cell survival and may be considered as a downstream component of RISK signalling. Phosphorylation inhibits GSK-3β activity and thereby inhibits MPTP activity (Juhaszova et al., 2009). However, its relative importance has been disputed in different models. Wagner et al. (2008) reported that both GSK-3β and ERK phosphorylation are significantly increased following postconditioning in rats. These observations were subsequently supported by further biochemical analysis demonstrating increased GSK-3β phosphorylation following postconditioning in a rat global ischaemia model. In contrast, GSK-3β double knock-in mice could be protected with a postconditioning stimulus in a global ischaemia model (Nishino et al., 2008). Further evidence is required to ascertain the precise contribution of GSK-3β and how it may vary in different species.
SAFE pathway (JAK/STAT3)
The survivor activating factor enhancement (SAFE) pathway has been identified as an alternative cytoprotective pathway to RISK that is triggered by TNF-α and JAK/STAT signalling. Lecour's laboratory has reported that pharmacological inhibition of the JAK/STAT pathway reverses the infarct limitation afforded by postconditioning (Lacerda et al., 2009). They also demonstrated that TNF-α signalling through TNFR2 also known as TNFRSF1B) and STAT3 is required to afford protection. The protection afforded was independent of PI3K/Akt and MEK/ERK signalling. TNFR2 antibodies abolished protection afforded by postconditioning whereas TNFR1 knockout mice were still conditioned (Lacerda et al., 2009). Protection observed with TNF-α was not present when the JAK/STAT3 inhibitor AG490 was administered at reperfusion (Lecour et al., 2005). The upstream activators of the SAFE pathway have attracted little attention to date, but it is suggested that autacoids such as those found upstream of the RISK cascades could be involved (Hausenloy et al., 2013). Distal to the SAFE pathway, it is suggested that signalling converges on the mitochondria; however, whether the SAFE pathway converges on the same targets as RISK remains to be investigated thoroughly.
cGMP/PKG pathway
Endogenous NO derived from eNOS is implicated in ischaemic postconditioning in several animal models. Pharmacological inhibition of eNOS activity abolished the protective effects of postconditioning (Tsang et al., 2004). Conversely, many studies have demonstrated the cytoprotective effects of administering a NO donor in the first few minutes of reperfusion, although this effect of NO donors is not consistently seen (Bice et al., 2014a). NO activates soluble GC leading to the generation of cGMP and subsequent activation of cGMP-dependent PK (PKG). Several lines of evidence support the effectiveness of this pathway as a cardioprotective cascade (Krieg et al., 2009; Bice et al., 2014b). In addition, cGMP/PKG signalling through particulate GC targeting via natriuretic peptides has also been demonstrated to afford infarct limitation (Burley and Baxter, 2007). However, at present, it is unclear if the PKG pathway is an essential component of ischaemic postconditioning and if it sits alongside the PI3K/Akt pathway or is distal to it (see Figure 2).
Anti-apoptotic mechanisms
The relative contributions that apoptosis and necrosis make in reperfusion injury have long been debated. Specifically, the timing of apoptosis during the development of myocardial ischaemia/reperfusion injury remains unclear. Sun et al. (2009) reported that postconditioning limited myocardial apoptosis in rat neonatal cardiac myocytes. It was reported that TUNEL staining was reduced compared with controls and that ROS generation and intracellular calcium accumulation were reduced. Cytochrome c and caspase-3 have also been implicated in postconditioning signalling associated with a reduction in apoptosis. Penna et al. (2006) reported that these factors were reduced following postconditioning in an ex vivo rat model, while increasing the anti-apoptotic factor Bcl-2. Inflammatory mediators including cytokines have also been associated with apoptotic regulation. Mechanical postconditioning has been shown to decrease TNF-α and limit ROS formation during early reperfusion, resulting in attenuation of apoptosis (Kin et al., 2008). Most recently, the apoptosis repressor with caspase recruitment domain has been shown to decrease caspase-3 activity and subsequent apoptosis in chick embryo myocytes following exposure to hydrogen peroxide (Wu et al., 2013).
Clinical studies of ischaemic and pharmacological postconditioning
From the brief account above, it may be inferred that a number of potential approaches exist for the development of postconditioning as a clinical therapeutic intervention. Indeed, as proof of concept, Staat et al. (2005) demonstrated that a mechanical postconditioning algorithm could be introduced in patients with AMI with significant reduction in a surrogate marker of infarct size [serum creatine kinase (CK) concentration]. Over the last decade, further clinical trials of ischaemic postconditioning have been conducted with mixed outcomes. In those studies that measured CK as an end point, approximately half of them reported positive outcomes (see Table 1). The remaining trials, all with small cohort sizes, reported neutral or negative end points. Most recently, a comparatively large trial reported that four cycles of 60 s reperfusion and re-occlusion failed to reduce peak CK-MB (Hahn et al., 2013) (see Table 1). A number of potential explanations can be posited for the variability of clinical studies of ischaemic postconditioning. These include variations in postconditioning algorithms. These issues are discussed comprehensively in a recent review (Ferdinandy et al., 2014).
Table 1.
Clinical trials utilizing mechanical and pharmacological postconditioning in patients presenting with STEMI
| Study/Reference | Treatment protocol | n number Tx/control | Effect |
|---|---|---|---|
| IPOC | |||
| Laskey, 2005 | 2 cycles of 90 s R/I | 10/7 | Improved ST-segment resolution, no difference in peak CK |
| Staat et al., 2005 | 4 cycles of 60 s R/I | 16/14 | Improved ST-segment resolution, reduction in 72 h CK |
| Ma et al., 2006 | 3 cycles of 30 s R/I | 49/45 | Improved WMSI, endothelial function, reduced CK (NS) |
| Yang et al., 2007 | 3 cycles of 60 s R/I | 23/18 | 27% reduction in 72 h CK |
| Laskey et al., 2008 | 2 cycles of 90 s R/I | 13/11 | Improved ST-segment resolution. Improved coronary flow velocity. |
| Thibault et al., 2008 | 4 cycles of 60 s R/I | 17/21 | 40% reduction in CK. 47% reduction in troponin 1. |
| Zhao et al., 2009 | 3 cycles of 30 or 60 s R/I | 49/26 | Reduced apoptosis at 7 days |
| Lønborg et al., 2012 | 4 cycles of 60 s R/I | 59/59 | 19% relative reduction of MI at 3 months. 31% increase in myocardial salvage. |
| Sörensson et al., 2010 | 4 cycles of 60 s R/I | 38/38 | Neutral outcomes |
| Xue et al., 2010 | 4 cycles of 60 s R/I | 23/20 | Reduction in infarct size (SPECT) |
| Garcia et al., 2011 | 4 cycles of 30 s R/I | 22/21 | Reduction in CK and improved LVEF |
| Freixa et al., 2012 | 4 cycles of 60 s R/I | 39/40 | No improvement in infarct size (CMR) or LVEF |
| Tarantini et al., 2012 | 4 cycles of 60 s R/I | 39/39 | No reduction in infarct size at 30 days (CMR) |
| Thuny et al., 2012 | 4 cycles of 60 s R/I | 25/25 | Reduction in infarct size (CMR) and CK |
| Dwyer et al., 2013 | 4 cycles of 30 s R/I | 51/51 | NS reduction in infarct size (CMR) |
| Hahn et al., 2013 | 4 cycles of 60 s R/I | 350/350 | No improvement in ST-segment resolution |
| PI3K/Akt | |||
| Nikolaidis et al., 2004 | GLP-1 infusion for 72 h post-PCI | 10/11 | Improved LVEF |
| Lønborg et al., 2012 | Exenatide 0.12 μg·min−1 15 min pre-PCI for 6 h | 54/51 | Reduced infarct size at 90 days (CMR) |
| EPO | |||
| Ferrario et al., 2011 | 33000 IU bolus EPO pre-PCI, then 24, 48 h later | 15/15 | 30% reduction in CK-MB |
| Suh et al., 2011 | 50 IU·kg−1 EPO bolus pre-PCI | 29/28 | No improvement in CK, CK-MB or CMR at 96 h |
| The C-ETGI, 2005 | GIK infusion for 24 h | 10 091/10 110 | Neutral outcome |
| Kim et al., 2010 | 80 versus 10 mg oral atorvastatin pre-PCI | 86/85 | No difference in CK-MB or troponin |
| GPCR | |||
| Ross et al., 2005 | 70 μg·kg−1·min−1 adenosine for 3 h within 15 min R | 713/703 | Reduction in infarct size (SPECT) in sub-study (n = 243) |
| Kloner et al., 2006 | 50 or 70 μg·kg−1·min−1 adenosine within 15 min R | 716/350 | Reduction in 1 and 6 month mortality |
| cGMP/PKG | |||
| Kitakaze et al., 2007 | 72 h carperitide infusion post-PCI | 277/292 | 15% reduced total CK. Improved reperfusion. |
| Siddiqi et al., 2014 | 70 μmol IV sodium nitrite over 5 min pre-PCI | 118/111 | No reduction in infarct size (CMR) |
| Mitochondria | |||
| Kitakaze et al., 2007 | Nicorandil IV bolus then 24 h infusion | 276/269 | No difference in total CK or LVEF |
| Piot et al., 2008 | Cys-A 2.5 mg·kg−1 IV 10 min pre-PCI | 30/28 | Reduction in total CK. 27 patients 20% reduction in MI size (CMR). |
| Mewton et al., 2010 | Cys-A 2.5 mg·kg−1 IV 10 min pre-PCI | 28/0 | 24% reduction in MI size (CMR) |
IPOC, ischaemic postconditioning; LVEF, left ventricular ejection fraction; MI, myocardial infarction; NS, not significant; R/I, cycle of reperfusion and ischaemia; SPECT, single-photon emission computer tomography; STEMI, ST-elevated myocardial infarction; WMSI, wall motion score index.
Pharmacological approaches to postconditioning have been assessed in a number of clinical studies. Here we highlight some notable completed studies related to the mechanisms outlined above.
Adenosine
Adenosine was evaluated as an adjunct to clinical reperfusion therapy before the formal identification of postconditioning (Mahaffey et al., 1999). A reduction in infarct size of 33% was demonstrated in patients receiving adenosine before thrombolysis and prompted a larger trial in which the primary end points were development of congestive heart failure or 6 month mortality rates (Ross et al., 2005). The results of this 2118 patient trial were disappointing with no significant improvement in primary outcomes. There was, however, a suggestion that in a subset of patients, infarct size was reduced in patients who received the highest dose of adenosine. Furthermore, post hoc analysis suggested that benefit was only observed in patients who received early adenosine treatment (Kloner et al., 2006). Almost half of the patients in the follow-up trial underwent angioplasty rather than thrombolysis which also needs to be considered.
cGMP/PKG pathway
Most recently, the results of the NIAMI trial have been published in which nitrite was administered before percutaneous coronary intervention (PCI) as a source of exogenous NO (Siddiqi et al., 2014). Extensive experimental studies have demonstrated the protective effects of administering nitrate, nitrite or NO donors before reperfusion. Indeed, nitrite has been shown to have vasorelaxant and anti-platelet properties which may be enhanced under ischaemic conditions, but these are actions unrelated to a postconditioning effect (Rassaf et al., 2014). Post hoc analyses of patients who had been undergoing chronic nitrate therapy were shown to have fewer ST-elevated myocardial infarctions compared with patients who were described as nitrate naïve (Ambrosio et al., 2010). However, in the NIAMI trial, no reduction in infarct size measured by cardiac magnetic resonance imaging was reported in patients receiving sodium nitrite 5 min before PCI.
Targeting the cGMP pathway and the KATP channel has also been explored in the clinical setting. The large multicentre J-WIND trial treated patients with atrial natriuretic peptide after reperfusion treatment which showed approximately 15% reduction in total CK release. Patients treated with the KATP channel opener nicorandil did not show any significant reduction in total CK release (Kitakaze et al., 2007).
MPTP inhibition
In a small pilot study, Cys-A limited infract size by 20% compared with controls when measured by MRI 5 days after treatment (Piot et al., 2008). Furthermore, no adverse effects of Cys-A were reported. An ongoing multicentre trial (CIRCUS) is further investigating the potential of Cys-A as an adjunct to reperfusion, the primary end points being hospitalization for heart failure and left ventricular remodelling at 1 year.
Other pharmacological agents
In addition to exploring pharmacological postconditioning mimetics, other agents that may offer protection in the clinical reperfusion setting have been investigated. Statins, β blockers, erythropoietin (EPO), glucagon-like peptide and glucose-insulin-potassium have all been utilized in clinical trials with varying outcomes. Two small trials in which EPO was administered prior to PCI reported conflicting outcomes (Ferrario et al., 2011; Suh et al., 2011). Similar doses were used; however, a 30% reduction in CK-MB was reported in one and no improvement was reported in the other. Second and third doses were, however, administered at 24 and 48 h in the positive outcome trial. The proposed mechanism of action for EPO protection is said to involve inhibition of the myocardial inflammatory response which may have a delayed component explaining the differences in clinical outcomes.
The challenges and opportunities for successful translation
The picture obtained so far is that myocardial ischaemic postconditioning has the potential to limit infarct size but is of variable efficacy. Clinical studies with pharmacological mimetic approaches (e.g. adjuncts to PCI or thrombolysis for AMI) that target the postconditioning signalling pathways described in experimental studies have not been overwhelmingly positive. There are likely to be many reasons for these inconsistent findings. They include study design features (e.g. patient inclusion criteria, timing of drug administration); technical limitations to accurate end point assessment (e.g. normalized infarct size measurement in humans); and attenuation or the overwhelming of the postconditioning signalling mechanisms in patients. The latter potentially represents the greatest challenge for successful translation of postconditioning into the therapeutic arena.
The confounding effect of comorbidities
The majority of experimental studies of ischaemic postconditioning or pharmacological postconditioning mimetics have been performed in healthy, juvenile male animals. These models are devoid of associated risk factors for cardiovascular disease and do not represent the comorbidities often present in the clinical setting. It is now clear that many of the risk factors and comorbid conditions that contribute to or are present in coronary artery disease (senescence, gender-related hormonal background, dyslipidaemia, hypertension, diabetes, etc.) modify the signalling pathways underpinning postconditioning (Downey and Cohen, 2009; Przyklenk, 2013; Vander Heide and Steenbergen, 2013; Ferdinandy et al., 2014). In experimental models that address these factors, both ischaemic and pharmacological postconditioning effects may be abolished or severely attenuated because of biochemical perturbations brought about by these conditions. The worrying possibility that the majority of experimental models have not predicted or do not resemble clinical reality might be regarded by some as the killer blow for successful development of clinical postconditioning and may go some way to explaining the massive variability in clinical trials to date. However, we are not so pessimistic. It seems plausible that at least with some of these comorbidities, postconditioning is not completely abolished, but rather the threshold for activation of the pathways is raised. For example, in experimental studies where protection by either ischaemic postconditioning or Cys-A was abolished in diabetic hearts, the combination of both interventions restored protection suggesting that the diabetic heart could be protected if an increased cardioprotective threshold could be met (Badalzadeh et al., 2012). Moreover, in some cases, treatment or resolution of the comorbidity restores the postconditioning effect. For example, in a rabbit model, postconditioning alone could not limit infarct size in high-cholesterol-fed animals. However, administration of pravastatin was able to afford protection in these resistant animals, an effect that was blocked by eNOS inhibition (Andreadou et al., 2012).
The confounding effect of current drug therapies
Another intriguing possible explanation for variability in clinical postconditioning studies is that many patients are in fact already in a maximally conditioned state as a result of their existing drug therapy. Bell and Yellon (2014) have recently proposed a ‘success hypothesis’ suggesting that many, perhaps the majority of, patients presenting with acute coronary syndromes are already conditioned by the polypharmaceutical regimen of drugs that they are already taking. Statins, ACE inhibitors, β blockers and opioid analgesics are all commonly prescribed to these patients and indeed have all been shown to be cardioprotective or to have conditioning-like properties in the experimental setting. On the other hand, these drugs have been found to inhibit conditioning mechanisms in some studies. The term ‘hidden cardiotoxicity’ has been proposed which suggests that some of the adjunct therapies used may increase the threshold for cardioprotection (Ferdinandy et al., 2014).
Clinical trial design and translating postconditioning
The disparity between the experimental studies and the clinical trial data obtained so far suggests that translation – both from bench to bedside and vice versa – needs to be improved. As identified above, experimental study design needs to be refined for further mechanistic studies to represent better the clinical setting. At the very least, experimental models in which comorbidities can be simulated should be used following initial mechanistic studies. It is clear that we need to focus on building on the well-documented signalling cascades and the spatial and temporal modifications to signalling in diseased states.
To date, the majority of clinical trials assessing pharmacological postconditioning mimetics have been unsuccessful or of only modest benefit (see Table 1). But their limited success may be explained in two ways. Firstly, the design of the preclinical animal experiments may fail to resemble the complexities of the clinical situation and this leads to inappropriate target selection. Secondly, the design of a clinical trial needs to account for the massive heterogeneity of the patient population and recognise the currently limited ability to quantify tissue salvage or measure infarct size standardized to risk zone size accurately and reliably. Unlike laboratory species, the clinical population presenting with AMI is a heterogeneous mix of high-risk and low-risk patients, those with large infarcts and those with small infarcts. Unlike the laboratory experiment, the ischaemic risk zone size, the duration of the ischaemic episode and the speed of successful reperfusion are highly variable in human AMI. Perhaps most importantly, the high degree of standardization of infarct size measurement required in the experimental laboratory is effectively unachievable in the clinical setting with presently available methods.
Thus, it seems unlikely that we will achieve a postconditioning intervention that guarantees benefit for all. Much more likely is that an agent that is safe and easy to administer as a single dose – probably a repurposed drug such as Cys-A – could be given to all AMI patients undergoing reperfusion with the expectation that a proportion might benefit. Given the very large number of patients undergoing reperfusion therapy, the global benefit of such an approach could be large.
Conflict of interest
There is no conflict of interest to disclose.
Glossary
- AMI
acute myocardial infarction
- CK
creatine kinase
- CK-MB
creatine kinase assay
- CYP-D
cyclophilin-D
- Cys-A
cyclosporine-A
- eNOS
endothelial NOS
- EPO
erythropoietin
- GSK-3β
glycogen synthase-3β
- MKATP
ATP-sensitive mitochondrial potassium channel
- MPTP
mitochondrial permeability transition pore
- PCI
percutaneous coronary intervention
- RISK
reperfusion injury salvage kinase
- ROS
reactive oxygen species
- SAFE
survivor activating factor enhancement
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ion channels. Br J Pharmacol. 2013b;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Catalytic receptors. Br J Pharmacol. 2013c;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013d;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosio G, Del Pinto M, Tritto I, Agnelli G, Bentivoglio M, Zuchi C, et al. Chronic nitrate therapy is associated with different presentation and evolution of acute coronary syndromes: insights from 52 693 patients in the Global Registry of Acute Coronary Events. Eur Heart J. 2010;31:430–438. doi: 10.1093/eurheartj/ehp457. [DOI] [PubMed] [Google Scholar]
- Andreadou I, Farmakis D, Prokovas E, Sigala F, Zoga A, Spyridaki K, et al. Short-term statin administration in hypercholesterolaemic rabbits resistant to postconditioning: effects on infarct size, endothelial nitric oxide synthase, and nitro-oxidative stress. Cardiovasc Res. 2012;94:501–509. doi: 10.1093/cvr/cvs121. [DOI] [PubMed] [Google Scholar]
- Antman EM. Magnesium in acute MI: timing is critical. Circulation. 1995;92:2367–2372. doi: 10.1161/01.cir.92.9.2367. [DOI] [PubMed] [Google Scholar]
- Ashraf M, White F, Bloor CM. Ultrastructural influence of reperfusing dog myocardium with calcium-free blood after coronary artery occlusion. Am J Pathol. 1978;90:423–434. [PMC free article] [PubMed] [Google Scholar]
- Badalzadeh R, Mohammadi M, Najafi M, Ahmadiasl N, Farajnia S, Ebrahimi H. The additive effects of ischemic postconditioning and cyclosporine-a on nitric oxide activity and functions of diabetic myocardium injured by ischemia/reperfusion. J Cardiovasc Pharmacol Ther. 2012;17:181–189. doi: 10.1177/1074248411416118. [DOI] [PubMed] [Google Scholar]
- Bell RM, Yellon DM. Conditioning the Heart by stealth: the Success hypothesis. Bull Br Soc Cardiovasc Res. 2014;27:25–29. [Google Scholar]
- Berne RM. The role of adenosine in the regulation of coronary blood flow. Circ Res. 1980;47:807–813. doi: 10.1161/01.res.47.6.807. [DOI] [PubMed] [Google Scholar]
- Berne RM, Rubio R. Adenine nucleotide metabolism in the heart. Circ Res. 1974;35(Suppl. 3):109–120. [PubMed] [Google Scholar]
- Bice JS, Burley DS, Baxter GF. Novel approaches and opportunities for cardioprotective signaling through 3′,5′-cyclic guanosine monophosphate manipulation. J Cardiovasc Pharmacol Ther. 2014a;19:269–282. doi: 10.1177/1074248413518971. [DOI] [PubMed] [Google Scholar]
- Bice JS, Keim Y, Stasch J-P, Baxter GF. NO-independent stimulation or activation of soluble guanylyl cyclase during early reperfusion limits infarct size. Cardiovasc Res. 2014b;101:220–228. doi: 10.1093/cvr/cvt257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadley KJ, Penson PE. The roles of alpha- and beta-adrenoceptor stimulation in myocardial ischaemia. Auton Autacoid Pharmacol. 2004;24:87–93. doi: 10.1111/j.1474-8673.2004.00324.x. [DOI] [PubMed] [Google Scholar]
- Buja LM. The pathobiology of acute coronary syndromes: clinical implications and central role of the mitochondria. Tex Heart Inst J. 2013;40:221–228. [PMC free article] [PubMed] [Google Scholar]
- Burley DS, Baxter GF. B-type natriuretic peptide at early reperfusion limits infarct size in the rat isolated heart. Basic Res Cardiol. 2007;102:529–541. doi: 10.1007/s00395-007-0672-1. [DOI] [PubMed] [Google Scholar]
- Burley DS, Baxter GF. Pharmacological targets revealed by myocardial postconditioning. Curr Opin Pharmacol. 2009;9:177–188. doi: 10.1016/j.coph.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Casale AS, Bolling SF, Rui J-A, Flaherty JT, Bulkley BH, Jacobus WE, et al. Progression and resolution of myocardial reflow injury. J Surg Res. 1984;37:94–99. doi: 10.1016/0022-4804(84)90167-7. [DOI] [PubMed] [Google Scholar]
- Clarke SJ, Khaliulin I, Das M, Parker JE, Heesom KJ, Halestrap AP. Inhibition of mitochondrial permeability transition pore opening by ischemic preconditioning is probably mediated by reduction of oxidative stress rather than mitochondrial protein phosphorylation. Circ Res. 2008;102:1082–1090. doi: 10.1161/CIRCRESAHA.107.167072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M, Costi A. Kinetic evidence for a heart mitochondrial pore activated by Ca2+, inorganic phosphate and oxidative stress. A potential mechanism for mitochondrial dysfunction during cellular Ca2+ overload. Eur J Biochem. 1988;178:489–501. doi: 10.1111/j.1432-1033.1988.tb14475.x. [DOI] [PubMed] [Google Scholar]
- Crompton M, Costi A, Hayat L. Evidence for the presence of a reversible Ca2+-dependent pore activated by oxidative stress in heart mitochondria. Biochem J. 1987;245:915–918. doi: 10.1042/bj2450915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lisa F, Menabo R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem. 2001;276:2571–2575. doi: 10.1074/jbc.M006825200. [DOI] [PubMed] [Google Scholar]
- Dow J, Kloner RA. Postconditioning does not reduce myocardial infarct size in an in vivo regional ischemia rodent model. J Cardiovasc Pharmacol Ther. 2007;12:153–163. doi: 10.1177/1074248407300897. [DOI] [PubMed] [Google Scholar]
- Dow J, Bhandari A, Kloner RA. Ischemic postconditioning's benefit on reperfusion ventricular arrhythmias is maintained in the senescent heart. J Cardiovasc Pharmacol Ther. 2008;13:141–148. doi: 10.1177/1074248408317705. [DOI] [PubMed] [Google Scholar]
- Downey JM, Cohen MV. Why do we still not have cardioprotective drugs? Circ J. 2009;73:1171–1177. doi: 10.1253/circj.cj-09-0338. [DOI] [PubMed] [Google Scholar]
- Dwyer NB, Mikami Y, Hilland D, Aljizeeri A, Friedrich MG, Traboulsi M, et al. No cardioprotective benefit of ischemic postconditioning in patients with ST-segment elevation myocardial infarction. J Interv Cardiol. 2013;26:482–490. doi: 10.1111/joic.12064. [DOI] [PubMed] [Google Scholar]
- Elrod JW, Wong R, Mishra S, Vagnozzi RJ, Sakthievel B, Goonasekera SA, et al. Cyclophilin D controls mitochondrial pore-dependent Ca2+ exchange, metabolic flexibility, and propensity for heart failure in mice. J Clin Invest. 2010;120:3680–3687. doi: 10.1172/JCI43171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferdinandy P, Hausenloy DJ, Heusch G, Baxter GF, Schulz R. Interaction of cardiovascular risk factors, comorbidities and comedications with ischaemia/reperfusion injury and cardioprotection by preconditioning, postconditioning and remote conditioning: an update. Pharmacol Rev. 2014;66:1142–1174. doi: 10.1124/pr.113.008300. [DOI] [PubMed] [Google Scholar]
- Ferrario M, Arbustini E, Massa M, Rosti V, Marziliano N, Raineri C, et al. High-dose erythropoietin in patients with acute myocardial infarction: a pilot, randomised, placebo-controlled study. Int J Cardiol. 2011;147:124–131. doi: 10.1016/j.ijcard.2009.10.028. [DOI] [PubMed] [Google Scholar]
- Freixa X, Bellera N, Ortiz-Perez JT, Jimenez M, Pare C, Bosch X, et al. Ischaemic postconditioning revisited: lack of effects on infarct size following primary percutaneous coronary intervention. Eur Heart J. 2012;33:103–112. doi: 10.1093/eurheartj/ehr297. [DOI] [PubMed] [Google Scholar]
- Garcia S, Henry T, Wang Y, Chavez I, Pedersen W, Lesser J, et al. Long-term Follow-up of Patients Undergoing Postconditioning During ST-Elevation Myocardial Infarction. J Cardiovasc Transl Res. 2011;4:92–98. doi: 10.1007/s12265-010-9252-0. [DOI] [PubMed] [Google Scholar]
- Gedik N, Heusch G, Skyschally A. Infarct size reduction by cyclosporine A at reperfusion involves inhibition of the mitochondrial permeability transition pore but does not improve mitochondrial respiration. Arch Med Sci. 2013;9:968–975. doi: 10.5114/aoms.2013.38175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giricz Z, Varga ZV, Baranyai T, Sipos P, Paloczi K, Kittel A, et al. Cardioprotection by remote ischemic preconditioning of the rat heart is mediated by extracellular vesicles. J Mol Cell Cardiol. 2014;68:75–78. doi: 10.1016/j.yjmcc.2014.01.004. [DOI] [PubMed] [Google Scholar]
- Gomez L, Paillard M, Thibault H, Derumeaux G, Ovize M. Inhibition of GSK3β by postconditioning is required to prevent opening of the mitochondrial permeability transition pore during reperfusion. Circulation. 2008;117:2761–2768. doi: 10.1161/CIRCULATIONAHA.107.755066. [DOI] [PubMed] [Google Scholar]
- Griffiths EJ, Halestrap AP. Pyrophosphate metabolism in the perfused heart and isolated heart mitochondria and its role in regulation of mitochondrial function by calcium. Biochem J. 1993;290(Pt 2):489–495. doi: 10.1042/bj2900489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths EJ, Halestrap AP. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J. 1995;307(Pt 1):93–98. doi: 10.1042/bj3070093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn J-Y, Song YB, Kim EK, Yu CW, Bae J-W, Chung W-Y, et al. Ischemic postconditioning during primary percutaneous coronary intervention: the effects of postconditioning on myocardial reperfusion in patients with ST-segment elevation myocardial infarction (POST) randomized trial. Circulation. 2013;128:1889–1896. doi: 10.1161/CIRCULATIONAHA.113.001690. [DOI] [PubMed] [Google Scholar]
- Hale SL, Mehra A, Leeka J, Kloner RA. Postconditioning fails to improve no reflow or alter infarct size in an open-chest rabbit model of myocardial ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2008;294:H421–H425. doi: 10.1152/ajpheart.00962.2007. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Kerr PM, Javadov S, Woodfield KY. Elucidating the molecular mechanism of the permeability transition pore and its role in reperfusion injury of the heart. Biochim Biophys Acta. 1998;1366:79–94. doi: 10.1016/s0005-2728(98)00122-4. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ. Signalling pathways in ischaemic postconditioning. Thromb Haemost. 2009;101:626–634. [PubMed] [Google Scholar]
- Hausenloy DJ. Cardioprotection techniques: preconditioning, postconditioning and remote conditioning (basic science) Curr Pharm Des. 2013;19:4544–4563. doi: 10.2174/1381612811319250004. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ, Yellon DM. New directions for protecting the heart against ischaemia-reperfusion injury: targeting the Reperfusion Injury Salvage Kinase (RISK)-pathway. Cardiovasc Res. 2004;61:448–460. doi: 10.1016/j.cardiores.2003.09.024. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ, Erik Botker H, Condorelli G, Ferdinandy P, Garcia-Dorado D, Heusch G, et al. Translating cardioprotection for patient benefit: position paper from the Working Group of Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc Res. 2013;98:7–27. doi: 10.1093/cvr/cvt004. [DOI] [PubMed] [Google Scholar]
- Hearse DJ, Humphrey SM, Boink AB, Ruigrok TJ. The calcium paradox: metabolic, electrophysiological, contractile and ultrastructural characteristics in four species. Eur J Cardiol. 1978;7:241–256. [PubMed] [Google Scholar]
- Heusch G, Buchert A, Feldhaus S, Schulz R. No loss of cardioprotection by postconditioning in connexin 43-deficient mice. Basic Res Cardiol. 2006;101:354–356. doi: 10.1007/s00395-006-0589-0. [DOI] [PubMed] [Google Scholar]
- Hunter DR, Haworth RA. The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch Biochem Biophys. 1979;195:453–459. doi: 10.1016/0003-9861(79)90371-0. [DOI] [PubMed] [Google Scholar]
- Inserte J, Barba I, Hernando V, Abellan A, Ruiz-Meana M, Rodriguez-Sinovas A, et al. Effect of acidic reperfusion on prolongation of intracellular acidosis and myocardial salvage. Cardiovasc Res. 2008;77:782–790. doi: 10.1093/cvr/cvm082. [DOI] [PubMed] [Google Scholar]
- Jin C, Wu J, Watanabe M, Okada T, Iesaki T. Mitochondrial K+ channels are involved in ischemic postconditioning in rat hearts. J Physiol Sci. 2012;62:325–332. doi: 10.1007/s12576-012-0206-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhaszova M, Zorov DB, Yaniv Y, Nuss HB, Wang S, Sollott SJ. Role of glycogen synthase kinase-3beta in cardioprotection. Circ Res. 2009;104:1240–1252. doi: 10.1161/CIRCRESAHA.109.197996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharbanda RK, Peters M, Walton B, Kattenhorn M, Mullen M, Klein N, et al. Ischemic preconditioning prevents endothelial injury and systemic neutrophil activation during ischemia-reperfusion in humans in vivo. Circulation. 2001;103:1624–1630. doi: 10.1161/01.cir.103.12.1624. [DOI] [PubMed] [Google Scholar]
- Kim J-S, Kim J, Choi D, Lee CJ, Lee SH, Ko Y-G, et al. Efficacy of high-dose atorvastatin loading before primary percutaneous coronary intervention in st-segment elevation myocardial infarction: the STATIN STEMI trial. JACC Cardiovasc Interv. 2010;3:332–339. doi: 10.1016/j.jcin.2009.11.021. [DOI] [PubMed] [Google Scholar]
- Kim JH, Jang YH, Chun KJ, Kim J, Park YH, Kim JS, et al. Kappa-opioid receptor activation during reperfusion limits myocardial infarction via ERK1/2 activation in isolated rat hearts. Korean J Anesthesiol. 2011;60:351–356. doi: 10.4097/kjae.2011.60.5.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kin H, Zhao ZQ, Sun HY, Wang NP, Corvera JS, Halkos ME, et al. Postconditioning attenuates myocardial ischemia-reperfusion injury by inhibiting events in the early minutes of reperfusion. Cardiovasc Res. 2004;62:74–85. doi: 10.1016/j.cardiores.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Kin H, Zatta AJ, Lofye MT, Amerson BS, Halkos ME, Kerendi F, et al. Postconditioning reduces infarct size via adenosine receptor activation by endogenous adenosine. Cardiovasc Res. 2005;67:124–133. doi: 10.1016/j.cardiores.2005.02.015. [DOI] [PubMed] [Google Scholar]
- Kin H, Wang N-P, Mykytenko J, Reeves J, Deneve J, Jiang R, et al. Inhibition of myocardial apoptosis by postconditioning is associated with attenuation of oxidative stress-mediated nuclear factor-kappa b translocation and TNF alpha release. Shock. 2008;29:761–768. doi: 10.1097/SHK.0b013e31815cfd5a. [DOI] [PubMed] [Google Scholar]
- Kitakaze M, Asakura M, Kim J, Shintani Y, Asanuma H, Hamasaki T, et al. Human atrial natriuretic peptide and nicorandil as adjuncts to reperfusion treatment for acute myocardial infarction (J-WIND): two randomised trials. Lancet. 2007;370:1483–1493. doi: 10.1016/S0140-6736(07)61634-1. [DOI] [PubMed] [Google Scholar]
- Kleinbongard P, Heusch G. Extracellular signalling molecules in the ischaemic/reperfused heart – druggable and translatable for cardioprotection. Br J Pharmacol. 2015;172:2010–2025. doi: 10.1111/bph.12902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloner R, Przyklenk K. Experimental infarct size reduction with calcium channel blockers. J Am Coll Cardiol. 1991;18:876–878. doi: 10.1016/0735-1097(91)90815-q. [DOI] [PubMed] [Google Scholar]
- Kloner RA, Forman MB, Gibbons RJ, Ross AM, Alexander RW, Stone GW. Impact of time to therapy and reperfusion modality on the efficacy of adenosine in acute myocardial infarction: the AMISTAD-2 trial. Eur Heart J. 2006;27:2400–2405. doi: 10.1093/eurheartj/ehl094. [DOI] [PubMed] [Google Scholar]
- Krieg T, Liu Y, Rutz T, Methner C, Yang XM, Dost T, et al. BAY 58–2667, a nitric oxide-independent guanylyl cyclase activator, pharmacologically post-conditions rabbit and rat hearts. Eur Heart J. 2009;30:1607–1613. doi: 10.1093/eurheartj/ehp143. [DOI] [PubMed] [Google Scholar]
- Lacerda L, Somers S, Opie LH, Lecour S. Ischaemic postconditioning protects against reperfusion injury via the SAFE pathway. Cardiovasc Res. 2009;84:201–208. doi: 10.1093/cvr/cvp274. [DOI] [PubMed] [Google Scholar]
- Laskey WK. Brief repetitive balloon occlusions enhance reperfusion during percutaneous coronary intervention for acute myocardial infarction: a pilot study. Catheter Cardiovasc Interv. 2005;65:361–367. doi: 10.1002/ccd.20397. [DOI] [PubMed] [Google Scholar]
- Laskey WK, Yoon S, Calzada N, Ricciardi MJ. Concordant improvements in coronary flow reserve and ST-segment resolution during percutaneous coronary intervention for acute myocardial infarction: a benefit of postconditioning. Catheter Cardiovasc Interv. 2008;72:212–220. doi: 10.1002/ccd.21583. [DOI] [PubMed] [Google Scholar]
- Lecour S, Suleman N, Deuchar GA, Somers S, Lacerda L, Huisamen B, et al. Pharmacological preconditioning with tumor necrosis factor-alpha activates signal transducer and activator of transcription-3 at reperfusion without involving classic prosurvival kinases (Akt and extracellular signal-regulated kinase) Circulation. 2005;112:3911–3918. doi: 10.1161/CIRCULATIONAHA.105.581058. [DOI] [PubMed] [Google Scholar]
- Leung AWC, Halestrap AP. Recent progress in elucidating the molecular mechanism of the mitochondrial permeability transition pore. Biochim Biophys Acta. 2008;1777:946–952. doi: 10.1016/j.bbabio.2008.03.009. [DOI] [PubMed] [Google Scholar]
- Loukogeorgakis SP, Panagiotidou AT, Yellon DM, Deanfield JE, MacAllister RJ. Postconditioning protects against endothelial ischemia-reperfusion injury in the human forearm. Circulation. 2006;113:1015–1019. doi: 10.1161/CIRCULATIONAHA.105.590398. [DOI] [PubMed] [Google Scholar]
- Lønborg J, Vejlstrup N, Kelbæk H, Bøtker HE, Kim WY, Mathiasen AB, et al. Exenatide reduces reperfusion injury in patients with ST-segment elevation myocardial infarction. Eur Heart J. 2012;33:1491–1499. doi: 10.1093/eurheartj/ehr309. [DOI] [PubMed] [Google Scholar]
- Ma X, Zhang X, Li C, Luo MAN. Effect of postconditioning on coronary blood flow velocity and endothelial function and LV recovery after myocardial infarction. J Interv Cardiol. 2006;19:367–375. doi: 10.1111/j.1540-8183.2006.00191.x. [DOI] [PubMed] [Google Scholar]
- Mahaffey KW, Puma JA, Barbagelata NA, DiCarli MF, Leesar MA, Browne KF, et al. Adenosine as an adjunct to thrombolytic therapy for acute myocardial infarction: results of a multicenter, randomized, placebo-controlled trial: the Acute Myocardial Infarction STudy of ADenosine (AMISTAD) Trial. J Am Coll Cardiol. 1999;34:1711–1720. doi: 10.1016/s0735-1097(99)00418-0. [DOI] [PubMed] [Google Scholar]
- Mewton N, Croisille P, Gahide G, Rioufol G, Bonnefoy E, Sanchez I, et al. Effect of cyclosporine on left ventricular remodeling after reperfused myocardial infarction. J Am Coll Cardiol. 2010;55:1200–1205. doi: 10.1016/j.jacc.2009.10.052. [DOI] [PubMed] [Google Scholar]
- Mocanu MM, Baxter GF, Yellon DM. Caspase inhibition and limitation of myocardial infarct size: protection against lethal reperfusion injury. Br J Pharmacol. 2000;130:197–200. doi: 10.1038/sj.bjp.0703336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- Nikolaidis LA, Mankad S, Sokos GG, Miske G, Shah A, Elahi D, et al. Effects of glucagon-like peptide-1 in patients with acute myocardial infarction and left ventricular dysfunction after successful reperfusion. Circulation. 2004;109:962–965. doi: 10.1161/01.CIR.0000120505.91348.58. [DOI] [PubMed] [Google Scholar]
- Nishino Y, Webb IG, Davidson SM, Ahmed AI, Clark JE, Jacquet S, et al. Glycogen synthase kinase-3 inactivation is not required for ischemic preconditioning or postconditioning in the mouse. Circ Res. 2008;103:307–314. doi: 10.1161/CIRCRESAHA.107.169953. [DOI] [PubMed] [Google Scholar]
- Ovize M, Baxter GF, Di Lisa F, Ferdinandy PT, Garcia-Dorado D, Hausenloy DJ, et al. Postconditioning and protection from reperfusion injury: where do we stand? Position paper from the working group of cellular biology of the heart of the European society of cardiology. Cardiovasc Res. 2010;87:406–423. doi: 10.1093/cvr/cvq129. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penna C, Cappello S, Mancardi D, Raimondo S, Rastaldo R, Gattullo D, et al. Post–conditioning reduces infarct size in the isolated rat heart: role of coronary flow and pressure and the nitric oxide/cGMP pathway. Basic Res Cardiol. 2006;101:168–179. doi: 10.1007/s00395-005-0543-6. [DOI] [PubMed] [Google Scholar]
- Penna C, Mancardi D, Rastaldo R, Losano G, Pagliaro P. Intermittent activation of bradykinin B2 receptors and mitochondrial KATP channels trigger cardiac postconditioning through redox signaling. Cardiovasc Res. 2007;75:168–177. doi: 10.1016/j.cardiores.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Philipp S, Yang XM, Cui L, Davis AM, Downey JM, Cohen MV. Postconditioning protects rabbit hearts through a protein kinase C-adenosine A2b receptor cascade. Cardiovasc Res. 2006;70:308–314. doi: 10.1016/j.cardiores.2006.02.014. [DOI] [PubMed] [Google Scholar]
- Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N, et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. NEJM. 2008;359:473–481. doi: 10.1056/NEJMoa071142. [DOI] [PubMed] [Google Scholar]
- Preuss KC, Gross GJ, Brooks HL, Warltier DC. Time course of recovery of ‘stunned’ myocardium following variable periods of ischemia in conscious and anesthetized dogs. Am Heart J. 1987;114:696–703. doi: 10.1016/0002-8703(87)90777-0. [DOI] [PubMed] [Google Scholar]
- Przyklenk K. Reduction of myocardial infarct size with ischemic ‘conditioning’: physiologic and technical considerations. Anesth Analg. 2013;117:891–901. doi: 10.1213/ANE.0b013e318294fc63. [DOI] [PubMed] [Google Scholar]
- Rassaf T, Ferdinandy P, Schulz R. Nitrite in organ protection. Br J Pharmacol. 2014;171:1–11. doi: 10.1111/bph.12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimer KA, Murry CE, Yamasawa I, Hill ML, Jennings RB. Four brief periods of myocardial ischemia cause no cumulative ATP loss or necrosis. Am J Physiol. 1986;251:H1306–H1315. doi: 10.1152/ajpheart.1986.251.6.H1306. [DOI] [PubMed] [Google Scholar]
- Ross AM, Gibbons RJ, Stone GW, Kloner RA, Alexander RW. A randomized, double-blinded, placebo-controlled multicenter trial of adenosine as an adjunct to reperfusion in the treatment of acute myocardial infarction (AMISTAD-II) J Am Coll Cardiol. 2005;45:1775–1780. doi: 10.1016/j.jacc.2005.02.061. [DOI] [PubMed] [Google Scholar]
- Roubille F, Franck-Miclo A, Covinhes A, Lafont C, Cransac F, Combes S, et al. Delayed postconditioning in the mouse heart in vivo. Circulation. 2011;124:1330–1336. doi: 10.1161/CIRCULATIONAHA.111.031864. [DOI] [PubMed] [Google Scholar]
- Sato H, Jordan JE, Zhao ZQ, Sarvotham SS, Vinten-Johansen J. Gradual reperfusion reduces infarct size and endothelial injury but augments neutrophil accumulation. Ann Thorac Surg. 1997;64:1099–1107. doi: 10.1016/s0003-4975(97)00734-0. [DOI] [PubMed] [Google Scholar]
- Schmidt MR, Redington A, Bøtker E. Remote conditioning the heart overview – translatability and mechanism. Br J Pharmacol. 2015;172:1947–1960. doi: 10.1111/bph.12933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz BG, Kloner RA. Coronary no reflow. J Mol Cell Cardiol. 2012;52:873–882. doi: 10.1016/j.yjmcc.2011.06.009. [DOI] [PubMed] [Google Scholar]
- Schwartz LM, Lagranha CJ. Ischemic postconditioning during reperfusion activates Akt and ERK without protecting against lethal myocardial ischemia-reperfusion injury in pigs. Am J Physiol Heart Circ Physiol. 2006;290:H1011–H1018. doi: 10.1152/ajpheart.00864.2005. [DOI] [PubMed] [Google Scholar]
- Shi W, Vinten-Johansen J. Endogenous cardioprotection by ischaemic postconditioning and remote conditioning. Cardiovasc Res. 2012;94:206–216. doi: 10.1093/cvr/cvs088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqi N, Neil C, Bruce M, MacLennan G, Cotton S, Papadopoulou S, et al. Intravenous sodium nitrite in acute ST-elevation myocardial infarction: a randomized controlled trial (NIAMI) Eur Heart J. 2014;35:1255–1262. doi: 10.1093/eurheartj/ehu096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivaraman V, Mudalagiri NR, Di Salvo C, Kolvekar S, Hayward M, Yap J, et al. Postconditioning protects human atrial muscle through the activation of the RISK pathway. Basic Res Cardiol. 2007;102:453–459. doi: 10.1007/s00395-007-0664-1. [DOI] [PubMed] [Google Scholar]
- Skyschally A, van Caster P, Iliodromitis EK, Schulz R, Kremastinos DT, Heusch G. Ischemic postconditioning: experimental models and protocol algorithms. Basic Res Cardiol. 2009;104:469–483. doi: 10.1007/s00395-009-0040-4. [DOI] [PubMed] [Google Scholar]
- Sörensson P, Saleh N, Bouvier F, Böhm F, Settergren M, Caidahl K, et al. Effect of postconditioning on infarct size in patients with ST elevation myocardial infarction. Heart. 2010;96:1710–1715. doi: 10.1136/hrt.2010.199430. [DOI] [PubMed] [Google Scholar]
- Staat P, Rioufol G, Piot C, Cottin Y, Cung TT, L'Huillier I, et al. Postconditioning the human heart. Circulation. 2005;112:2143–2148. doi: 10.1161/CIRCULATIONAHA.105.558122. [DOI] [PubMed] [Google Scholar]
- Suh J-W, Chung W-Y, Kim Y-S, Kim K-I, Jeon E-J, Cho Y-S, et al. The effect of intravenous administration of erythropoietin on the infarct size in primary percutaneous coronary intervention. Int J Cardiol. 2011;149:216–220. doi: 10.1016/j.ijcard.2010.02.002. [DOI] [PubMed] [Google Scholar]
- Sun Y, Zhang Y, Yan M, Wu Y, Zheng X. B-type natriuretic peptide-induced cardioprotection against reperfusion is associated with attenuation of mitochondrial permeability transition. Biol Pharm Bull. 2009;32:1545–1551. doi: 10.1248/bpb.32.1545. [DOI] [PubMed] [Google Scholar]
- Tang XL, Sato H, Tiwari S, Dawn B, Bi Q, Li Q, et al. Cardioprotection by postconditioning in conscious rats is limited to coronary occlusions <45 min. Am J Physiol Heart Circ Physiol. 2006;291:H2308–H2317. doi: 10.1152/ajpheart.00479.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarantini G, Favaretto E, Marra MP, Frigo AC, Napodano M, Cacciavillani L, et al. Postconditioning during coronary angioplasty in acute myocardial infarction: the POST-AMI trial. Int J Cardiol. 2012;162:33–38. doi: 10.1016/j.ijcard.2012.03.136. [DOI] [PubMed] [Google Scholar]
- The C-ETGI. Effect of glucose-insulin-potassium infusion on mortality in patients with acute st-segment elevation myocardial infarction: the CREATE-ECLA randomized controlled trial. JAMA. 2005;293:437–446. doi: 10.1001/jama.293.4.437. [DOI] [PubMed] [Google Scholar]
- Thibault H, Piot C, Staat P, Bontemps L, Sportouch C, Rioufol G, et al. Long-term benefit of postconditioning. Circulation. 2008;117:1037–1044. doi: 10.1161/CIRCULATIONAHA.107.729780. [DOI] [PubMed] [Google Scholar]
- Thuny F, Lairez O, Roubille F, Mewton N, Rioufol G, Sportouch C, et al. Post-conditioning reduces infarct size and edema in patients with ST-segment elevation myocardial infarction. J Am Coll Cardiol. 2012;59:2175–2181. doi: 10.1016/j.jacc.2012.03.026. [DOI] [PubMed] [Google Scholar]
- Tsang A, Hausenloy DJ, Mocanu MM, Yellon DM. Postconditioning: a form of ‘modified reperfusion’ protects the myocardium by activating the phosphatidylinositol 3-kinase-Akt pathway. Circ Res. 2004;95:230–232. doi: 10.1161/01.RES.0000138303.76488.fe. [DOI] [PubMed] [Google Scholar]
- Vander Heide RS, Steenbergen C. Cardioprotection and myocardial reperfusion: pitfalls to clinical application. Circ Res. 2013;113:464–477. doi: 10.1161/CIRCRESAHA.113.300765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinten-Johansen J, Granfeldt A, Mykytenko J, Undyala VV, Dong Y, Przyklenk K. The multidimensional physiological responses to postconditioning. Antioxid Redox Signal. 2011;14:791–810. doi: 10.1089/ars.2010.3396. [DOI] [PubMed] [Google Scholar]
- Wagner C, Kloeting I, Strasser RH, Weinbrenner C. Cardioprotection by postconditioning is lost in WOKW rats with metabolic syndrome: robe of glycogen synthase kanase 3P. J Cardiovasc Pharmacol. 2008;52:430–437. doi: 10.1097/FJC.0b013e31818c12a7. [DOI] [PubMed] [Google Scholar]
- Wu L, Xi Z, Guo R, Liu S, Yang S, Liu D, et al. Exogenous ARC down-regulates caspase-3 expression and inhibits apoptosis of broiler chicken cardiomyocytes exposed to hydrogen peroxide. Avian Pathol. 2013;42:32–37. doi: 10.1080/03079457.2012.757289. [DOI] [PubMed] [Google Scholar]
- Xue F, Yang X, Zhang B, Zhao C, Song J, Jiang T, et al. Postconditioning the human heart in percutaneous coronary intervention. Clin Cardiol. 2010;33:439–444. doi: 10.1002/clc.20796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X-M, Proctor JB, Cui L, Krieg T, Downey JM, Cohen MV. Multiple, brief coronary occlusions during early reperfusion protect rabbit hearts by targeting cell signaling pathways. J Am Coll Cardiol. 2004;44:1103–1110. doi: 10.1016/j.jacc.2004.05.060. [DOI] [PubMed] [Google Scholar]
- Yang XC, Liu Y, Wang LF, Cui L, Wang T, Ge YG, et al. Reduction in myocardial infarct size by postconditioning in patients after percutaneous coronary intervention. J Invasive Cardiol. 2007;19:424–430. [PubMed] [Google Scholar]
- Yellon DM, Baxter GF. Reperfusion injury revisited: is there a role for growth factor signaling in limiting lethal reperfusion injury? Trends Cardiovasc Med. 1999;9:245–249. doi: 10.1016/s1050-1738(00)00029-3. [DOI] [PubMed] [Google Scholar]
- Zatta AJ, Kin H, Yoshishige D, Jiang R, Wang N, Reeves JG, et al. Evidence that cardioprotection by postconditioning involves preservation of myocardial opioid content and selective opioid receptor activation. Am J Physiol Heart Circ Physiol. 2008;294:H1444–H1451. doi: 10.1152/ajpheart.01279.2006. [DOI] [PubMed] [Google Scholar]
- Zhao W-S, Xu L, Wang L-F, Zhang L, Zhang Z-Y, Liu Y, et al. A 60-s postconditioning protocol by percutaneous coronary intervention inhibits myocardial apoptosis in patients with acute myocardial infarction. Apoptosis. 2009;14:1204–1211. doi: 10.1007/s10495-009-0387-x. [DOI] [PubMed] [Google Scholar]
- Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, et al. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2003;285:H579–H588. doi: 10.1152/ajpheart.01064.2002. [DOI] [PubMed] [Google Scholar]