

Abstract
Cardiomyocyte cell death occurring during myocardial reperfusion (reperfusion injury) contributes to final infarct size after transient coronary occlusion. Different interrelated mechanisms of reperfusion injury have been identified, including alterations in cytosolic Ca2+ handling, sarcoplasmic reticulum-mediated Ca2+ oscillations and hypercontracture, proteolysis secondary to calpain activation and mitochondrial permeability transition. All these mechanisms occur during the initial minutes of reperfusion and are inhibited by intracellular acidosis. The cGMP/PKG pathway modulates the rate of recovery of intracellular pH, but has also direct effect on Ca2+ oscillations and mitochondrial permeability transition. The cGMP/PKG pathway is depressed in cardiomyocytes by ischaemia/reperfusion and preserved by ischaemic postconditioning, which importantly contributes to postconditioning protection. The present article reviews the mechanisms and consequences of the effect of ischaemic postconditioning on the cGMP/PKG pathway, the different pharmacological strategies aimed to stimulate it during myocardial reperfusion and the evidence, limitations and promise of translation of these strategies to the clinical practice. Overall, the preclinical and clinical evidence suggests that modulation of the cGMP/PKG pathway may be a therapeutic target in the context of myocardial infarction.
Tables of Links
| LIGANDS | |
|---|---|
| Adenosine | GLP-1, glucagon-like peptide-1 |
| ANP, atrial natriuretic peptide | Insulin |
| Ataciguat | L-arginine |
| BH4, tetrahydrobiopterin | NO, nitric oxide |
| Brain natriuretic peptide | ODQ,[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one |
| cAMP | Riociguat |
| cGMP | Sildenafil |
| Cinaciguat | Vardenafil |
| Exenatide | YC-1 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a,b,).
Ischaemia/reperfusion injury
Cardiomyocyte death is the main cause of the mortality and morbidity in patients with ischaemic heart disease, the leading cause of death in the global world population. It occurs mainly during the acute coronary syndrome, and in particular during myocardial infarction with ST-segment elevation, for which the main treatment is prompt reperfusion. There is solid evidence supporting that part of cell death, secondary to transient coronary occlusion, occurs upon restoration of coronary blood flow, a phenomenon known as reperfusion injury (Piper et al., 1998; Yellon and Hausenloy, 2007).
The mechanisms responsible for cell death during myocardial reperfusion are complex but progressively better known, and have been reviewed recently (Garcia-Dorado et al., 2012; 2014a,). Restoration of coronary blood flow triggers the synthesis of ATP and Ca2+ uptake by the sarcoplasmic reticulum (SR) through the sarcoplasmic reticulum Ca2+-ATPase (SERCA), exceeding its capacity and resulting in the abrupt release of Ca2+(through ryanodine receptors 2), followed by reuptake and resulting in cyclic Ca2+ oscillations. These oscillations can cause arrhythmias, hypercontracture and sarcolemmal rupture (Piper et al., 2006). During reperfusion, Ca2+ influx from the extracellular space occurs mainly via the Na+/Ca2+ exchanger and the increase in cytosolic Ca2+ activates calpain proteolysis, which damages the cytoskeleton and alters the function of the Na+ pump (Inserte et al., 2005), aggravating Na+ and, secondarily, Ca2+ overload. On the other hand, the production of free radicals together with Ca2+ overload induces mitochondrial permeabilization and energy failure leading to cell death (Penna et al., 2013). Due to the preferential communication between mitochondria the and SR, Ca2+ oscillations can cause mitochondrial permeabilization and vice versa (Ruiz-Meana et al., 2007; Abdallah et al., 2011). It is important to note that proteolytic activation, mitochondrial permeabilization and hypercontracture are dependent on intracellular pH (pHi) and do not occur until intracellular acidosis is corrected during the initial minutes of reperfusion (Inserte et al., 2011a).
It is now clear that cAMP/cGMP-dependent PK (cGMP/PKG) signalling modulates not only the rate of pHi normalization, but many other important phenomena determining cell death or survival during myocardial reperfusion. This article will review the effects of ischaemia/reperfusion on the cGMP/PKG pathway, its role in endogenous cardioprotection by postconditioning and its potential as a pharmacological target for the prevention of reperfusion injury.
Effect of ischaemia/reperfusion on the cGMP pathway
cGMP is a ubiquitous intracellular second messenger involved in a large variety of cardiovascular processes (Yuan, 2002; Munzel et al., 2003; Tsai and Kass, 2009). cGMP is produced from the purine nucleotide GTP in an enzymic reaction mediated by two different types of guanylyl cyclase (GC) that differ in their ligands and intracellular distribution (Cerra and Pellegrino, 2007) (Figure 1). One is the soluble GC (sGC), localized in the cytosol and requiring binding of endogenous NO to its haem moiety for cGMP synthesis. The other is the group of particulate GC (pGC) that is the integral proteins of the plasmatic membrane specifically activated by natriuretic peptides (NPs). A natriuretic receptor (natriuretic peptide receptor-C), without GC activity, binds to a Gi protein eliciting activation of endothelial NOS (eNOS) (Costa et al., 2006). The cellular activity of cGMP is controlled through its highly regulated hydrolytic degradation by PDEs (Bender and Beavo, 2006) that differ in their affinity and specificity for cAMP and cGMP, and in their distribution, not only across tissues but also among the different subcellular compartments of cardiomyocytes (Zaccolo and Movsesian, 2007; Rao and Xi, 2009; Francis et al., 2011). The net effect of this system of distribution of PDE activity is that the concentration of cGMP and cAMP varies within different intracellular compartments in response to pGC and sGC stimulation (Castro et al., 2010). The primary cGMP mediator in the cardiovascular system is the cGMP-dependent PKI (PKG), which acts by phosphorylating target proteins and, indirectly, the cAMP-dependent PK (PKA) via cGMP-regulated PDE (Zhang and Kass, 2011).
Figure 1.
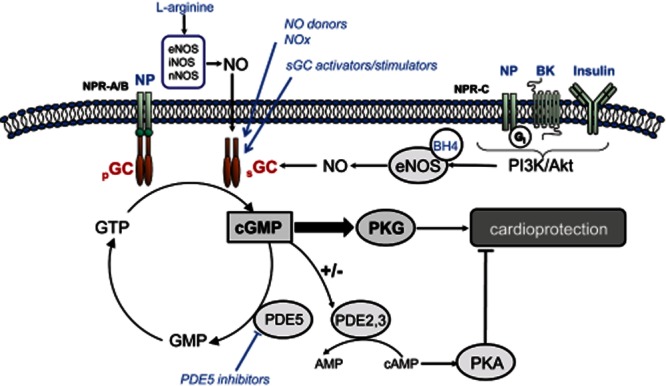
Schematic representation of the cGMP/PKG pathway. cGMP is synthesized by activation of the NO/sGC and the NP/pGC pathways and regulated by compartmentalized PDEs. Agonists of Gi-coupled receptors stimulate the eNOS/sGC/PKG axis via activation of PI3K/Akt. The main cGMP actions are not only mediated by PKG but also by regulation of cAMP-mediated effects through modulation of PDE2 and PDE3 activities. The different pharmacological strategies that have been used to stimulate the cGMP/PKG pathway are shown in blue. BK, bradykinin; NOx, nitrites/nitrates.
Ischaemia/reperfusion has dramatic effects on the basal myocardial content of cGMP and the ability of cardiomyocytes to respond to stimulators of cGMP synthesis. In the isolated heart model, myocardial cGMP levels increase during the first 10–25 min of ischaemia (Depre and Hue, 1994; Lochner et al., 1998) and acutely decrease thereafter (Nesher et al., 1977; Inserte et al., 2000). Reduced myocardial cGMP content has been described in the in situ rat, rabbit and pig heart subjected to transient ischaemia (Yamaguchi et al., 1997; Hoshida et al., 1999; Padilla et al., 2001).
Several factors can modify cGMP synthesis during ischaemia and reperfusion. Energetic depletion could reduce the availability of GTP, the substrate for GC, or affect GC activity by modification in its phosphorylation state (Lucas et al., 2000). However, we have observed no correlation between intracellular ATP content and cGMP synthesis stimulated by NO donors or pGC agonists in isolated cardiomyocytes (Agullo et al., 2003). In agreement with these results, it has been reported that a depletion of GTP to less than 10% of the initial levels did not decrease, but substantially increased cGMP synthesis after stimulation with a NO donor (Geisbuhler and Schwager, 1996). Further studies are needed to understand this absence of effect of ATP concentration on cGMP in cardiomyocytes.
Acidosis has been shown to be critical for cGMP synthesis. Results from our group demonstrated that pHi values close to 6.4 induce a profound depressant effect on pGC in cultures of cardiomyocytes and endothelial cells and on sGC in endothelial cells (Agullo et al., 2003). This study suggests that in situations causing intracellular acidification, as ischaemia, cardiomyocyte cGMP synthesis is largely dependent on NO stimulation of sGC. However, oxidative stress associated with ischaemia/reperfusion may decrease sGC activity by direct oxidation of its haem group (Stasch et al., 2006; Zhou et al., 2008; Derbyshire and Marletta, 2012). In addition, reduced NO bioavailability during reperfusion is a consequence of eNOS uncoupling by oxidation of its cofactor tetrahydrobiopterin (BH4) (Vasquez-Vivar et al., 2002; Chen et al., 2010).
cGMP/PKG pathway as a mediator of endogenous cardioprotection
Repeated non-lethal episodes of ischaemia/reperfusion activate endogenous protective mechanisms that profoundly reduce the infarct caused by a more sustained ischaemic insult. This concept of endogenous cardioprotection was initially described for ischaemic preconditioning (Murry et al., 1986), which is a pretreatment by definition and therefore cannot be used in the setting of acute myocardial infarction (AMI). Subsequently, it has been expanded to the more clinically applicable paradigms of postconditioning (ischaemic postconditioning; PoCo) and remote conditioning (Kharbanda et al., 2002; Zhao et al., 2003; Kerendi et al., 2005).
Postconditioning, consisting in brief episodes of coronary artery reocclusion and reflow applied at the moment of myocardial reperfusion, is by far the cardioprotective strategy that has been tested in more proof-of-concept clinical trials, and the first demonstrating that it is possible to reduce infarct size in patients with ST-segment elevation myocardial infarction (STEMI) using treatments applied at the time of reperfusion. In most of these small clinical trials, cycles ranging from 30 to 90 s of angioplasty balloon inflation and deflation have been shown to improve both, early markers of infarct size and contractile function up to 1 year of infarction (Staat et al., 2005; Thibault et al., 2008; Ovize et al., 2010). There are, however, studies that do not confirm protection by PoCo, questioning the utility of this phenomenon to limit reperfusion injury in clinical practice (Sorensson et al., 2010; Freixa et al., 2012; Tarantini et al., 2012). A recent multicentre, prospective, randomized trial with the largest series reported so far [700 patients with STEMI undergoing primary percutaneous coronary intervention (PCI)] failed to demonstrate a benefit of a PoCo protocol consisting in four cycles of 1 min low-pressure balloon inflation separated by 1 min of reflow (Hahn et al., 2013). The results of a large-scale trial (DANAMI-3, NCT01435408) that is currently underway to determine whether PoCo can reduce the combined end-point of cardiac death, re-infarction and heart failure at 3 years will provide more definitive information about the clinical usefulness of PoCo and the conditions that determine its effectiveness.
Although different confounding factors, including co-morbidities, co-medications and interventional procedures, have been proposed to explain the coexistence of positive and negative clinical studies (Heusch, 2012; Hausenloy et al., 2013), it is important to note that not all preclinical studies have consistently shown a benefit of PoCo. The reasons for these discrepancies lie primarily in the PoCo algorithm used, the effectiveness of which varies depending on the species and experimental preparation (Skyschally et al., 2009a). The critical factor determining the threshold for triggering PoCo protection is the ability of the PoCo algorithm to delay pHi normalization at the onset of reperfusion (Cohen et al., 2007; Fujita et al., 2007; Inserte et al., 2009). Measurement of pHi kinetics by NMR spectroscopy in rat hearts showed that only those PoCo protocols that prolong acidosis during reperfusion for at least 3 min are protective and that there is a close correlation between the delay in pHi recovery and the extent of cell death (Inserte et al., 2009). As it is not possible to accurately measure pHi in patients undergoing PCI, we do not know whether the PoCo algorithms used in clinical trials with negative results were adequate to ensure a slow pHi correction at the time of reflow.
The cardioprotective effect of prolongation of intracellular acidosis during the first minutes of reperfusion has been solidly demonstrated in different models (Kaplan et al., 1995; Ohashi et al., 1996; Preckel et al., 1998; Inserte et al., 2008). As detailed in a recent review, acidosis inhibits many mechanisms involved in reperfusion injury including Ca2+ overload and myofibrillar contractility that causes Ca2+-dependent hypercontracture, mitochondrial permeability transition pore (mPTP) formation, calpain activation and propagation of cell death through gap junctions (Inserte et al., 2011a). Preventing cell death during acidosis allows for activation of endogenous mechanisms of protection and/or normalization of Ca2+ homeostasis that would prolong protection once the pHi has been normalized. On the basis of these findings, we propose that the relative timing of the correction of intracellular Ca2+ levels and pHi during the first minutes of reperfusion will determine cell death (recovery of pHi occurs before that of Ca2+) or survival (recovery of Ca2+ control occurs before pHi normalization).
The mechanism by which PoCo delays the normalization of pHi during early reperfusion involves not only reduced lactate washout caused by intermittent reflow but also the activation of the cGMP/PKG pathway (Inserte et al., 2011b). Blockade of cGMP/PKG pathway by the addition of the sGC inhibitor 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) or the PKG inhibitor KT5823 during reperfusion accelerated pHi recovery measured by NMR spectroscopy and abolished PoCo protection in isolated hearts. The effect of PoCo on pHi appears to be mediated by an inhibitory effect of PKG on the Na+/H+ exchanger (NHE) (Inserte et al., 2011b).
Activation of cGMP/PKG pathway by ischaemic conditioning
There is good evidence demonstrating that activation of cGMP/PKG pathway is essential for the protective effects of PoCo (Yang et al., 2004; Penna et al., 2006; Inserte et al., 2011b). As in ischaemic preconditioning (PreC), activation of sGC and PKG by PoCo has been proposed to be dependent on the PI3K/Akt pathway. Hausenloy et al. found that activation of the prosurvival kinases PI3K/Akt and ERK1/2 during the first minutes of reperfusion, which they termed the reperfusion injury salvage kinases (RISK) pathway, is required for PoCo protection (Hausenloy et al., 2005). However, the causal role of RISK in the protection induced by PoCo has been questioned by more recent data. Different studies performed in the in situ pig model and the rat perfused heart demonstrated that PoCo was still effective after pharmacological blockade of RISK and that increased RISK phosphorylation was a consequence but not a cause of the reduction in cell death induced by PoCo (Skyschally et al., 2009b; Inserte et al., 2013). Moreover, the observation that blockade of NOS, sGC and PKG, but not PI3K, abolished the cardioprotective effects of PoCo, confirmed that activation of the cGMP/PKG pathway depends on NOS but is independent of PI3K/Akt signalling (Inserte et al., 2013).
Recently, an alternative mechanism, summarized in Figure 2, for increased cGMP/PKG signalling has been proposed as well. Different studies have observed that PoCo attenuates reactive oxygen species (ROS) generation at reperfusion, probably by limiting the delivery of oxygen during the controlled reperfusion (Halkos et al., 2004; Kin et al., 2004; Fan et al., 2011). Our laboratory reported similar results in an ex vivo rat heart model and demonstrated that the resulted attenuation of oxidative stress by PoCo reduces eNOS uncoupling by preserving cytosolic BH4 levels and results in increased NO-dependent activation of cGMP/PKG pathway (Inserte et al., 2013).
Figure 2.
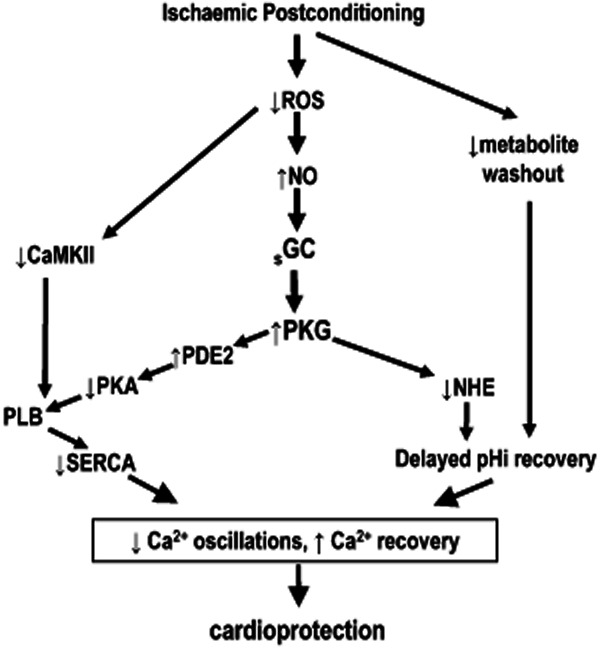
Scheme showing the proposed mechanisms by which cGMP/PKG pathway participates in the cardioprotective effects of ischaemic postconditioning (PoCo). PoCo attenuates the burst of O2− generated at the onset of reperfusion, reducing oxidative stress which limits the oxidation of BH4 and increases eNOS activity. The resulting activation of the cGMP/PKG pathway contributes to PoCo protection at least in part by delaying normalization of pHi during reperfusion via PKG-dependent inhibition of NHE. In addition, PoCo delays PLB phosphorylation at the onset of reperfusion by inhibiting PKA and CaMKII. The resulting transient inhibition of SERCA prevents oscillations Ca2+ and favours Ca2+ extrusion through Na+/Ca2+ exchanger.
Mechanism of cGMP-mediated cardioprotection
Despite the existing evidence supporting that the cGMP/PKG pathway is required for PoCo protection, only few studies have identified target proteins phosphorylated by PKG that are involved in the molecular mechanisms of cardioprotection. Activation of PKG has been suggested to inhibit mPTP by phosphorylation of glycogen synthase kinase-3β (GSK3β) (Juhaszova et al., 2004; Gomez et al., 2008). However, double knockin mice lacking the GSK3β phosphorylation sites that result in kinase inactivation could still be protected by both PreC and PoCo (Nishino et al., 2008), demonstrating that the inhibition of GSK3β is unlikely to serve as a downstream mediator of the cardioprotection mediated by cGMP/PKG activation. More recently, the notion that GSK3β phosphorylation does not play a causal role in PoCo protection has been further supported by data from the in situ pig model and the ex vivo rat heart (Skyschally et al., 2009b; Inserte et al., 2013).
In addition to its contribution to prolong acidosis at the onset of reperfusion by inhibition of NHE, it is proposed that PKG modulates phospholamban (PLB) phosphorylation and, therefore, SR Ca2+ entry through SERCA (Abdallah et al., 2005). At reperfusion, PLB is rapidly phosphorylated by PKA at Ser16 and Ca2+/calmodulin-dependent PK (CaMK) at Thr17 resulting in its dissociation from SERCA and activation of the pump (Vila-Petroff et al., 2009), favouring the formation of SR-driven Ca2+ oscillations that cause reperfusion-induced hypercontracture and mPTP opening (Siegmund et al., 1997). In a recent study, we have demonstrated that PoCo reduces SERCA activity during the first minutes of reperfusion by delaying PLB phosphorylation through activation of PKG and inhibition of PKA and CaMKII. Additionally, our study suggests that cGMP/PKG-dependent activation of PDE2 is responsible for the inhibition of PKA while the reduced activity of CaMKII could be the result of PoCo-dependent attenuation of Ca2+ overload and ROS production during the first minutes of reperfusion (Erickson et al., 2011). The resulting blockade of SERCA activity during the first minutes of reperfusion attenuated the development of hypercontracture and cell death by preventing Ca2+ oscillations and allowed for Ca2+ normalization through the Na+/Ca2+ exchanger and for the activation of endogenous protective signalling pathways that ensure long-term cardioprotection (Inserte et al., 2014).
Prolongation of acidosis by PoCo increases S-nitrosylated protein levels (Penna et al., 2011) and, recently, the selective sGC inhibitor ODQ (Methner et al., 2013; Tong et al., 2014) and the cardiomyocyte-specific ablation of the PKGI gene (Methner et al., 2013) failed to abolish PoCo-induced protection in isolated mouse hearts, suggesting that NO-mediated S-nitrosylation rather than the sGC/PKG pathway plays a key role in the cardioprotective effects of PoCo. However, although ODQ, which inhibits sGC by binding to its haem group in a NO-competitive manner (Schrammel et al., 1996), is commonly used to discriminate cGMP-dependent and cGMP-independent effects of NO, it has been demonstrated that persistence of NO-induced effects in the presence of ODQ does not necessarily prove cGMP independence (Lies et al., 2013). In addition, it cannot be ruled out that the persistence of protection in mice lacking PKGI in cardiomyocytes is a consequence of compensatory mechanisms including the effect of other PKG isoforms or the contribution of PKGI from other cardiac cell types.
Activation of cGMP/PKG pathway in remote ischaemic conditioning
A major limitation of PoCo is that it can only be performed during the process of PCI, reducing its target patient population but also increasing the risk of complications during the procedure. In recent years, it has been reported that induction of intermittent non-lethal ischaemia in a remote organ or limb protects the myocardium against the detrimental effects of a sustained lethal episode of ischaemia. This stimulus known as remote ischaemic conditioning (RIC) can be applied before myocardial ischaemia, during myocardial ischaemia or in the first few minutes of reperfusion (Andreka et al., 2007; Schmidt et al., 2007). RIC has multiple advantages over PoCo including the fact that it can be easily applied in the ambulance to all patients with STEMI that receive reperfusion treatment and is extremely safe (Botker et al., 2010). By contrast, the mechanisms by which RIC exerts its protective effect are much less known. The pathway linking the remote organ, on which the stimulus is applied, to the heart remains unclear and has been attributed to neural and/or hormonal pathways. Recently, Donato et al. described that remote preconditioning activates a neural afferent pathway and the cardioprotective signal reaches the heart through the vagus nerve (efferent pathway) and releases ACh that activates the endogenous signal cascade through muscarinic receptors (Donato et al., 2013), which can result in the stimulation of eNOS and increased production of cGMP (Massion and Balligand, 2003; Downey et al., 2007). Other studies provide evidence suggesting the involvement of cGMP/PKG pathway via activation of the PI3K/Akt-dependent signalling by released humoral factors (Breivik et al., 2010; Xin et al., 2010). Hausenloy et al. reported that in the in situ pig model, activation of the PI3K/Akt pathway at the time of reperfusion is required for the cardioprotective effects of remote preconditioning but not of remote perconditioning (Hausenloy et al., 2012).
Myocardial cardioprotection by pharmacological modulation of the cGMP/PKG pathway
There is extensive literature demonstrating that preservation of the cGMP/PKG pathway during myocardial reperfusion by using pharmacological strategies directed to either increase cGMP synthesis or reduce its degradation attenuates cardiomyocyte death occurring with reperfusion (Bice et al., 2013). However, it is important to note that the experimental design used in many of these studies includes the administration of the tested drug before the ischaemic insult, a situation that does not adequately model the clinical context of a patient with ongoing AMI receiving myocardial reperfusion. Unfortunately, several of these pharmacological interventions have provided less conclusive data regarding their effectiveness to increase cGMP production and to limit ultimate infarct size when they are applied at the time of reperfusion.
This section summarizes the pharmacological strategies that have been tested in preclinical studies to stimulate the cGMP/PKG pathway and limit myocardial reperfusion injury, making emphasis in those with potential for their translation to the clinical treatment of AMI.
Stimulation of cGMP synthesis
Most of the preclinical data supporting preservation of cGMP/PKG pathway as a therapeutic strategy for protecting myocardium against reperfusion injury are based in studies that stimulate the synthesis of cGMP. The potential targets that can augment cGMP levels are divided in those acting through the NO/sGC pathway and the NPs/pGC pathway.
NO/sGC pathway
Synthesis of cGMP through sGC is modulated by NO generated by NOS using L-arginine as a substrate, O2 and NADPH. However, in disease states associated with reduced availability of O2 and high-energy phosphates, as occurs during ischaemia, production of NO is severely compromised (Lundberg et al., 2008). L-arginine supplementation has been pursued as a potential approach for restoring NO production in reperfused myocardium. The pre-ischaemic administration of L-arginine has been found to attenuate reperfusion injury in isolated rat and in situ pig hearts by a mechanism dependent on the sGC (Agullo et al., 1999; Padilla et al., 2000). However, its administration at the onset of reperfusion failed to increase cGMP levels and protect myocardium (Agullo et al., 1999). Impaired NOS activity as consequence of the burst of ROS generated during the first minutes of reperfusion may explain the lack of efficacy of L-arginine. Superoxide generated from different sources combines with NO at a very fast rate to form peroxynitrite (ONOO−) ions (Beckman et al., 1990). Peroxynitrite not only reduces the availability of free NO but may also act directly on the NOS haem group producing the inactivation of the enzyme (Chen et al., 2010). In addition, O2− and ONOO− can oxidize BH4, an essential cofactor for NOS, resulting in NOS uncoupling and preferential production of O2− instead of NO (Xia et al., 1998; Vasquez-Vivar et al., 2002).
Because different studies demonstrate that BH4 is markedly reduced during ischaemia (Dumitrescu et al., 2007), the therapeutic potential of BH4 supplementation has been analysed (Moens et al., 2011). Pretreatment with BH4 or the BH4 precursor sepiapterin has been shown to improve endothelium-dependent vasorelaxation in isolated coronary arterioles and ventricular function in isolated rat hearts following ischaemia/reperfusion injury (Tiefenbacher et al., 1996; Verma et al., 2002; Yamashiro et al., 2002). Our laboratory has examined the effect of BH4 supplementation during the first minutes of reperfusion in rat isolated hearts. The results showed that exogenous BH4 was effective in reducing NOS uncoupling and increasing NO production but was associated with no effect on O2− levels, enhanced ONOO− production and failure to protect myocardium (Inserte et al., 2014). This study indicates that NOS uncoupling is not a major source for O2− generated during myocardial reperfusion and support the notion that the effectiveness of those strategies directed to stimulate NOS activity is limited by the excessive O2− production and the subsequent formation of ONOO−, an effect that could counterbalance the potential cardioprotective actions of enhanced NOS activity.
An alternative approach to stimulate the NO/sGC pathway that would bypass NOS involves the use nitrates (nitroglycerin, isosorbide mononitrate) and nitrites (sodium nitrite, amyl nitrite) which are reduced in sequential steps by enzymatic and non-enzymic mechanisms generating NO, and synthetic NO donors which spontaneously release NO (SNAP, NOC9) (Lundberg et al., 2008). The cardioprotective actions of these compounds have been examined by numerous studies and there is solid evidence supporting that their administration before ischaemia diminishes infarct size by NO-dependent, NOS-independent mechanisms (Webb et al., 2004; Duranski et al., 2005; Shiva et al., 2007). However, the literature is inconsistent in reporting protective effects of NO donors when administered at reperfusion in animal models. Bice et al. demonstrated a concentration-dependent effect of NOC9, a rapid release NO donor, limiting infarct size in ex vivo rat hearts (Bice et al., 2014), while in a study by Salloum et al, nitroglycerin given at reperfusion failed to limit infarct size in an in vivo rabbit model (Salloum et al., 2007). The NO donor SNAP in the range of 1–10 μM was not protective in ex vivo hearts when administered for 15 min (Burley and Baxter, 2007), but reduced infarct size when its infusion was extended to 35 min of reperfusion, suggesting that a prolonged time of infusion is necessary to maintain the cardioprotective signalling (Cohen et al., 2010). However, an alternative explanation for this inconsistent effectiveness of NO donors is related to the oxidative stress caused by reperfusion. First, as it occurs with those strategies aimed to stimulate NOS, NO donors can favour the deleterious effects resulting from the formation of ONOO− when they are administered at the time of the burst of ROS generated at reperfusion (Szabo, 1996). Second, sGC is a heterodimeric protein which contains a prosthetic haem group required for stimulation by NO. It is well known that under conditions of oxidative stress, the redox state of this haem group shifts from a reduced (Fe2+) state to a NO-insensitive (Fe3+) state, impairing the NO-dependent activation of sGC (Stasch et al., 2006; Meurer et al., 2009).
To avoid the problems associated to reduced NO bioavailability, formation of ONOO−, a promising new class of drugs has recently been developed which are NO-independent stimulators and activators of sGC. These sGC stimulators (YC-1, riociguat, BAY 41–2272) bind to the haem moiety in its normal Fe2+ state and increase the GTP catalytic activity independently of NO while sGC activators (cinaciguat, ataciguat, BAY 60–2770) activate the oxidized form of sGC in a haem-independent manner (Follmann et al., 2013). In the context of myocardial reperfusion injury, where the oxidized sGC is expected to be the prominent form in cardiomyocytes, sGC activators are, in principle, the most interesting compounds. Administration of the sGC activator cinaciguat during reperfusion improved ventricular function in dog hearts ex vivo (Korkmaz et al., 2009) and decreased infarct size after regional ischaemia in mice (Salloum et al., 2012), rats (Krieg et al., 2009) and rabbits (Krieg et al., 2009; Cohen et al., 2010). More recently, administration of ataciguat during reperfusion reduced infarct size in an ex vivo and in vivo rat model in a concentration-dependent manner (Inserte et al., 2014). In this study, protection was reported at moderate concentrations that were effective in reducing phosphorylation of PLB at Ser16 and Ca2+ oscillations and in favouring Ca2+ normalization in isolated cardiomyocytes, while failing to confer protection at high concentrations lacking effect on PLB. In a recent study, it has been demonstrated that either the sGC stimulator BAY 41–2272 or the activator BAY 60–2770 perfused during early reperfusion limit infarct size in an ex vivo rat model, suggesting that both reduced and oxidized pools of sGC are simultaneously present during reperfusion (Bice et al., 2014). Interestingly, the co-perfusion of both drugs resulted in high cGMP increase and greater infarct size compared with the sGC activator alone, supporting the idea that an excess of cGMP is detrimental.
NP/pGC pathway
There is solid experimental evidence supporting the use of NPs to stimulate pGC as a therapeutic strategy for protecting the myocardium against AMI. Stimulation of pGC by atrial natriuretic peptide (ANP) and brain natriuretic peptide administered at reperfusion has consistently shown to reduce myocardial infarction in isolated cardiomyocytes, ex vivo and in vivo hearts in a concentration-dependent manner (Philipp et al., 2006; Burley and Baxter, 2007; Ren et al., 2007; Gorbe et al., 2010). Our laboratory reported that administration of the NP urodilatin during the first minutes of reperfusion increases cGMP levels, attenuates hypercontracture and reduces infarct size in isolated hearts (Inserte et al., 2000). Furthermore, i.v. doses of urodilatin at the time of reperfusion in the in situ pig model subjected to transient coronary occlusion resulted in a rapid normalization of myocardial cGMP concentration in the absence of significant haemodynamic effects and attenuated infarct size. Interestingly, urodilatin at doses resulting in excessive increase in myocardial cGMP was less cardioprotective (Padilla et al., 2001).
Inhibition of cGMP degradation
Although early research has mostly focused on the stimulation of the cGMP synthesis, it has been more recently demonstrated that cGMP degradation by PDEs plays a critical role in the control of cGMP effects. Preservation of cGMP levels by pharmacological blockade of PDE5 has demonstrated to limit infarct size in different experimental models and species when PDE5 inhibitors are administered prior to ischaemia (Kukreja et al., 2011). The mechanisms proposed involve PKG-dependent phosphorylation of ERK and GSK3β (Das et al., 2008), increased NO generation through activation of eNOS/inducible NOS, activation of PKC and opening of mito-KATP channels (Kukreja et al., 2005). However, the evidence supporting PDE5 inhibition at the time of reperfusion as a cardioprotective strategy is less consistent. Continuous infusion or bolus injection of sildenafil and vardenafil at reperfusion reduced infarct size in in vivo rabbits and rats (Salloum et al., 2007; Ebner et al., 2013), in contrast to the findings of Reffelmann and Kloner who failed to demonstrate a cardioprotective effect with sildenafil in the same model using similar doses (Reffelmann and Kloner, 2003). It is important to note that sildenafil at concentrations that has been reported to limit infarct size did not elevate total cGMP beyond control level (Elrod et al., 2007; Madhani et al., 2010; Ebner et al., 2013). This lack of increase in cGMP observed with sildenafil despite its cardioprotective effects has been explained by the heterogeneous compartmentalization of cGMP within the cell (Castro et al., 2006) and suggests that local elevation of cGMP and no elevation in total cGMP content might be sufficient for protecting the heart. However, as total cGMP levels are severely depleted during prolonged ischaemia, it is possible that PDE inhibition at the onset of reperfusion may not effectively increase cGMP if cGMP synthesis remains depressed because of the oxidative stress and other reasons.
Pharmacological stimulation of the cGMP/PKG pathway in clinical trials
Although the cGMP/PKG pathway to date is one of the pharmacological targets for limitation of reperfusion injury with more solid preclinical evidence, only two clinical studies have directly assessed the effectiveness of this strategy in patients with STEMI using different approaches: stimulation of pGC with ANP and of sGC with nitrites respectively.
Consistent with the solid preclinical data previously described, in the J-WIND-ANP trial, stimulation of the pGC with continuous i.v. infusion of ANP during 3 days following reperfusion by either PCI or thrombolytic therapy reduced enzymic release and improved ventricular function at 6 to 12 months compared with controls (Kitakaze et al., 2007).
Recently, the final results of a multicentre trial investigating the effects of i.v. sodium nitrite administered immediately prior to PCI in 118 patients with first STEMI (NIAMI) have been published (Siddiqi et al., 2014). The study showed no significant differences between patients receiving nitrite or placebo neither in the primary end point (infarct size at 6–8 days measured by NMR) nor in the secondary end points (acute enzyme release and ventricular function at 6 months). The failure of nitrites to protect was foreseeable from the contradictory preclinical evidence supporting a protective effect of NO donors when administered at reperfusion in animal models, as it has been discussed earlier, and reinforce the premise that pharmacological therapies should be selected on basis of a robust cardioprotection in different experimental models before being translated to the clinical setting (Hausenloy et al., 2013). The ongoing NITRITE-AMI trial, in the same clinical context of patients with STEMI, analyses whether an intracoronary injection of nitrite, initiated prior to establishment of full reperfusion, reduces infarct size (NITRITE-AMI: ClinicalTrials.gov: NCT01388504).
In addition to these studies, there are a number of small clinical trials using emerging pharmacological treatments for preventing lethal reperfusion injury that, although not specifically addressed at the cGMP/PKG pathway, mediate their effects at least in part through stimulation of this pathway (Figure 3). In general, preclinical studies demonstrate that these therapies have in common the use of agonists for Gi-coupled receptors that stimulate the eNOS/sGC/PKG axis via activation of PI3K/Akt. This is the case of insulin which has been shown to be protective at the onset of reperfusion in different experimental models and is the main component of the glucose-insulin-potassium (GIK) therapy. It has been shown that insulin protects cardiomyocytes against reoxygenation injury by accelerating cytosolic Ca2+ recovery through a mechanism that involves increased eNOS and PKG activities (Abdallah et al., 2006). Although the first attempts to translate the administration of GIK into the clinical practice gave negative results probably because of the late administration of the treatment (Diaz et al., 1998; Ceremuzynski et al., 1999; Mehta et al., 2005), a recent study has assessed the effectiveness of its delivery in the ambulance and in the receiving hospital for 12 h in total to patients with suspected acute coronary syndrome. The results after 30 days and 1 year follow-up demonstrate improved clinical outcomes in those patients with STEMI (reduced composites of cardiac arrest or 1 year mortality, and of cardiac arrest, 1 year mortality or heart failure hospitalization) (Selker et al., 2014).
Figure 3.
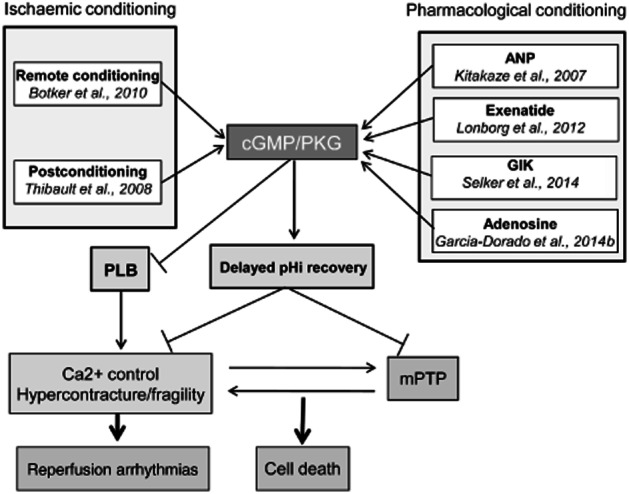
Proposed mechanisms of action of therapeutic interventions that stimulate the cGMP/PKG pathway and have been shown to limit infarct size in clinical trials.
Adenosine is another Gi-coupled receptor agonist that activates Akt and therefore the cGMP/PKG pathway (Yang et al., 2010). Intravenous administration of adenosine or its synthetic agonist AMP579 in the AMISTAD and ADMIRE trials provides inconclusive results with regard to their clinical use (Mahaffey et al., 1999; Kopecky et al., 2003). However, a subsequent reanalysis of the AMISTAD-II gave evidence of protection in those patients receiving early reperfusion (Kloner et al., 2006). More recently, the effect of adenosine delivered in the form of intracoronary injection before reperfusion on the infarct size measured by NMR has been tested in the PROMISE trial (Garcia-Dorado et al., 2014b). Although adenosine failed in its primary end point when considering all patients, the results showed that adenosine limits infarct size and improves ejection fraction in those receiving PCI within the first 3 h after pain onset.
There is solid experimental evidence that stimulation of the glucagon-like peptide-1 (GLP-1) signalling pathway using agonists of GLP-1 or inhibitors of its degradation attenuate reperfusion injury through cGMP/PKG, among other pathways (Ban et al., 2008; Timmers et al., 2009). Two clinical trials have evaluated the effect of the GLP-1 agonist exenatide administered intravenously in patients with STEMI 15 min prior to PCI and for 6 h, and in both studies exenatide increased myocardial salvage (Lonborg et al., 2012; Woo et al., 2013).
Conclusions/limitations
Extensive preclinical evidence supports a pivotal role for the cGMP/PKG pathway as a survival signal and demonstrates that its stimulation at the onset of reperfusion represent a promising therapeutic target to limit infarct size in patients with STEMI. However, although several strategies applied before experimental ischaemia have been shown to enhance cGMP levels and limit cell death, many of these interventions provide less consistent results when initiated at reperfusion mainly because of the negative effects caused by the burst of ROS generated at the onset of reperfusion. This is the case for nitrites, nitrates and synthetic NO donors, stimulators of NOS activity (L-arginine, BH4) and inhibitors of PDE5. In contrast, accumulating evidence suggests that stimulation of pGC with NPs and the new class of NO- and haem-independent sGC activators are the strategies with more potential for being translated into the clinical setting for the benefit of patients with acute coronary diseases.
A potential inherent limitation of interventions aimed to stimulate the cGMP/PKG pathway is that the cardioprotective effects of cGMP are only demonstrated within the physiological concentration range. An excessive stimulation of this pathway resulting in a too large cGMP concentration appears to be detrimental (Padilla et al., 2001; D'Souza et al., 2003; Bice et al., 2013; Inserte et al., 2014). This bell-shaped dose-response curve can be explained at least in part by the complex cross-talk between the effects of cAMP and cGMP. cGMP controls cAMP-mediated signalling in cardiac myocytes by activating or inhibiting PDEs that hydrolyze cAMP. As the binding affinity of myocardial PDEs for cGMP and cAMP is different for each isoform, the global effect is expected to vary depending on the concentration of cGMP (Zaccolo and Movsesian, 2007; Stangherlin et al., 2011; Inserte et al., 2014). In addition, it has been described that high concentrations of cGMP may directly activate PKA and counteract the effects of PKG (Worner et al., 2007).
More proof-of-concept clinical studies with infarct size as main end point are necessary to demonstrate the effectiveness and safety of pharmacological stimulation of the cGMP/PKG pathway during myocardial reperfusion, to determine which of the available drugs are more adequate, to know what is the effect of age and co-morbidities on their efficacy and safety and to establish the potential for combination with other cardioprotective treatments as RIC. However, the definite proof of the utility of these treatments necessary for their inclusion in routine clinical practice will be established only after completion of adequately designed and sized clinical trials with clinical primary end points including mortality and incidence of heart failure.
Acknowledgments
This work was supported by Redes Temáticas de Investigación Cooperativa Sanitaria (RETICS RD12/0042/0021) and Fondo Investigación Sanitaria (FIS-PI121738).
Glossary
- AMI
acute myocardial infarction
- ANP
atrial natriuretic peptide
- BH4
tetrahydrobiopterin
- CaMK
Ca2+/calmodulin-dependent PK
- eNOS
endothelial NOS
- GIK
glucose-insulin-potassium
- GLP-1
glucagon-like peptide-1
- GSK3β
glycogen synthase kinase-3β
- mPTP
mitochondrial permeability transition pore
- NHE
Na+/H + exchanger
- NP
natriuretic peptide
- ODQ
1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one
- PCI
percutaneous coronary intervention
- pGC
particulate GC
- pHi
intracellular pH
- PLB
phospholamban
- PoCo
ischaemic postconditioning
- PreC
ischaemic preconditioning
- RIC
remote ischaemic conditioning
- RISK
reperfusion injury salvage kinases
- ROS
reactive oxygen species
- SERCA
sarcoplasmic reticulum Ca2+-ATPase
- sGC
soluble GC
- SR
sarcoplasmic reticulum
- STEMI
ST-segment elevation myocardial infarction
Conflict of interest
None declared.
References
- Abdallah Y, Gkatzoflia A, Pieper H, Zoga E, Walther S, Kasseckert S, et al. Mechanism of cGMP-mediated protection in a cellular model of myocardial reperfusion injury. Cardiovasc Res. 2005;66:123–131. doi: 10.1016/j.cardiores.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Abdallah Y, Gkatzoflia A, Gligorievski D, Kasseckert S, Euler G, Schluter KD, et al. Insulin protects cardiomyocytes against reoxygenation-induced hypercontracture by a survival pathway targeting SR Ca2+ storage. Cardiovasc Res. 2006;70:346–353. doi: 10.1016/j.cardiores.2006.02.020. [DOI] [PubMed] [Google Scholar]
- Abdallah Y, Kasseckert SA, Iraqi W, Said M, Shahzad T, Erdogan A, et al. Interplay between Ca2+ cycling and mitochondrial permeability transition pores promotes reperfusion-induced injury of cardiac myocytes. J Cell Mol Med. 2011;15:2478–2485. doi: 10.1111/j.1582-4934.2010.01249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agullo L, Garcia-Dorado D, Inserte J, Paniagua A, Pyrhonen P, Llevadot J, et al. L-arginine limits myocardial cell death secondary to hypoxia-reoxygenation by a cGMP-dependent mechanism. Am J Physiol. 1999;276:H1574–H1580. doi: 10.1152/ajpheart.1999.276.5.H1574. [DOI] [PubMed] [Google Scholar]
- Agullo L, Garcia-Dorado D, Escalona N, Ruiz-Meana M, Inserte J, Soler-Soler J. Effect of ischemia on soluble and particulate guanylyl cyclase-mediated cGMP synthesis in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2003;284:H2170–H2176. doi: 10.1152/ajpheart.00820.2002. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013a;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Transporters. Br J Pharmacol. 2013b;170:1706–1796. doi: 10.1111/bph.12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreka G, Vertesaljai M, Szantho G, Font G, Piroth Z, Fontos G, et al. Remote ischaemic postconditioning protects the heart during acute myocardial infarction in pigs. Heart. 2007;93:749–752. doi: 10.1136/hrt.2006.114504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban K, Noyan-Ashraf MH, Hoefer J, Bolz SS, Drucker DJ, Husain M. Cardioprotective and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both glucagon-like peptide 1 receptor-dependent and -independent pathways. Circulation. 2008;117:2340–2350. doi: 10.1161/CIRCULATIONAHA.107.739938. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev. 2006;58:488–520. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- Bice JS, Burley DS, Baxter GF. Novel approaches and opportunities for cardioprotective signaling through 3′,5′-cyclic guanosine monophosphate manipulation. J Cardiovasc Pharmacol Ther. 2013;19:269–282. doi: 10.1177/1074248413518971. [DOI] [PubMed] [Google Scholar]
- Bice JS, Keim Y, Stasch JP, Baxter GF. NO-independent stimulation or activation of soluble guanylyl cyclase during early reperfusion limits infarct size. Cardiovasc Res. 2014;101:220–228. doi: 10.1093/cvr/cvt257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botker HE, Kharbanda R, Schmidt MR, Bottcher M, Kaltoft AK, Terkelsen CJ, et al. Remote ischaemic conditioning before hospital admission, as a complement to angioplasty, and effect on myocardial salvage in patients with acute myocardial infarction: a randomised trial. Lancet. 2010;375:727–734. doi: 10.1016/S0140-6736(09)62001-8. [DOI] [PubMed] [Google Scholar]
- Breivik L, Helgeland E, Aarnes EK, Mrdalj J, Jonassen AK. Remote postconditioning by humoral factors in effluent from ischemic preconditioned rat hearts is mediated via PI3K/Akt-dependent cell-survival signaling at reperfusion. Basic Res Cardiol. 2010;106:135–145. doi: 10.1007/s00395-010-0133-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burley DS, Baxter GF. B-type natriuretic peptide at early reperfusion limits infarct size in the rat isolated heart. Basic Res Cardiol. 2007;102:529–541. doi: 10.1007/s00395-007-0672-1. [DOI] [PubMed] [Google Scholar]
- Castro LR, Verde I, Cooper DM, Fischmeister R. Cyclic guanosine monophosphate compartmentation in rat cardiac myocytes. Circulation. 2006;113:2221–2228. doi: 10.1161/CIRCULATIONAHA.105.599241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro LR, Schittl J, Fischmeister R. Feedback control through cGMP-dependent protein kinase contributes to differential regulation and compartmentation of cGMP in rat cardiac myocytes. Circ Res. 2010;107:1232–1240. doi: 10.1161/CIRCRESAHA.110.226712. [DOI] [PubMed] [Google Scholar]
- Ceremuzynski L, Budaj A, Czepiel A, Burzykowski T, Achremczyk P, Smielak-Korombel W, et al. Low-dose glucose-insulin-potassium is ineffective in acute myocardial infarction: results of a randomized multicenter Pol-GIK trial. Cardiovasc Drugs Ther. 1999;13:191–200. doi: 10.1023/a:1007787924085. [DOI] [PubMed] [Google Scholar]
- Cerra MC, Pellegrino D. Cardiovascular cGMP-generating systems in physiological and pathological conditions. Curr Med Chem. 2007;14:585–599. doi: 10.2174/092986707780059715. [DOI] [PubMed] [Google Scholar]
- Chen W, Druhan LJ, Chen CA, Hemann C, Chen YR, Berka V, et al. Peroxynitrite induces destruction of the tetrahydrobiopterin and heme in endothelial nitric oxide synthase: transition from reversible to irreversible enzyme inhibition. Biochemistry. 2010;49:3129–3137. doi: 10.1021/bi9016632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MV, Yang XM, Downey JM. The pH hypothesis of postconditioning: staccato reperfusion reintroduces oxygen and perpetuates myocardial acidosis. Circulation. 2007;115:1895–1903. doi: 10.1161/CIRCULATIONAHA.106.675710. [DOI] [PubMed] [Google Scholar]
- Cohen MV, Yang XM, Liu Y, Solenkova NV, Downey JM. Cardioprotective PKG-independent NO signaling at reperfusion. Am J Physiol Heart Circ Physiol. 2010;299:H2028–H2036. doi: 10.1152/ajpheart.00527.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa MA, Elesgaray R, Balaszczuk AM, Arranz C. Role of NPR-C natriuretic receptor in nitric oxide system activation induced by atrial natriuretic peptide. Regul Pept. 2006;135:63–68. doi: 10.1016/j.regpep.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Das A, Xi L, Kukreja RC. Protein kinase G-dependent cardioprotective mechanism of phosphodiesterase-5 inhibition involves phosphorylation of ERK and GSK3beta. J Biol Chem. 2008;283:29572–29585. doi: 10.1074/jbc.M801547200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depre C, Hue L. Cyclic GMP in the perfused rat heart. Effect of ischaemia, anoxia and nitric oxide synthase inhibitor. FEBS Lett. 1994;345:241–245. doi: 10.1016/0014-5793(94)00459-5. [DOI] [PubMed] [Google Scholar]
- Derbyshire ER, Marletta MA. Structure and regulation of soluble guanylate cyclase. Annu Rev Biochem. 2012;81:533–559. doi: 10.1146/annurev-biochem-050410-100030. [DOI] [PubMed] [Google Scholar]
- Diaz R, Paolasso EA, Piegas LS, Tajer CD, Moreno MG, Corvalan R, et al. Metabolic modulation of acute myocardial infarction. The ECLA (Estudios Cardiologicos Latinoamerica) Collaborative Group. Circulation. 1998;98:2227–2234. doi: 10.1161/01.cir.98.21.2227. [DOI] [PubMed] [Google Scholar]
- Donato M, Buchholz B, Rodriguez M, Perez V, Inserte J, Garcia-Dorado D, et al. Role of the parasympathetic nervous system in cardioprotection by remote hindlimb ischaemic preconditioning. Exp Physiol. 2013;98:425–434. doi: 10.1113/expphysiol.2012.066217. [DOI] [PubMed] [Google Scholar]
- Downey JM, Davis AM, Cohen MV. Signaling pathways in ischemic preconditioning. Heart Fail Rev. 2007;12:181–188. doi: 10.1007/s10741-007-9025-2. [DOI] [PubMed] [Google Scholar]
- D'Souza SP, Yellon DM, Martin C, Schulz R, Heusch G, Onody A, et al. B-type natriuretic peptide limits infarct size in rat isolated hearts via KATP channel opening. Am J Physiol Heart Circ Physiol. 2003;284:H1592–H1600. doi: 10.1152/ajpheart.00902.2002. [DOI] [PubMed] [Google Scholar]
- Dumitrescu C, Biondi R, Xia Y, Cardounel AJ, Druhan LJ, Ambrosio G, et al. Myocardial ischemia results in tetrahydrobiopterin (BH4) oxidation with impaired endothelial function ameliorated by BH4. Proc Natl Acad Sci U S A. 2007;104:15081–15086. doi: 10.1073/pnas.0702986104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duranski MR, Greer JJ, Dejam A, Jaganmohan S, Hogg N, Langston W, et al. Cytoprotective effects of nitrite during in vivo ischemia-reperfusion of the heart and liver. J Clin Invest. 2005;115:1232–1240. doi: 10.1172/JCI22493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebner B, Ebner A, Reetz A, Bohme S, Schauer A, Strasser RH, et al. Pharmacological postconditioning by bolus injection of phosphodiesterase-5 inhibitors vardenafil and sildenafil. Mol Cell Biochem. 2013;379:43–49. doi: 10.1007/s11010-013-1625-7. [DOI] [PubMed] [Google Scholar]
- Elrod JW, Greer JJ, Lefer DJ. Sildenafil-mediated acute cardioprotection is independent of the NO/cGMP pathway. Am J Physiol Heart Circ Physiol. 2007;292:H342–H347. doi: 10.1152/ajpheart.00306.2006. [DOI] [PubMed] [Google Scholar]
- Erickson JR, He BJ, Grumbach IM, Anderson ME. CaMKII in the cardiovascular system: sensing redox states. Physiol Rev. 2011;91:889–915. doi: 10.1152/physrev.00018.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Q, Yang XC, Liu Y, Wang LF, Liu SH, Ge YG, et al. Postconditioning attenuates myocardial injury by reducing nitro-oxidative stress in vivo in rats and in humans. Clin Sci (Lond) 2011;120:251–261. doi: 10.1042/CS20100369. [DOI] [PubMed] [Google Scholar]
- Follmann M, Griebenow N, Hahn MG, Hartung I, Mais FJ, Mittendorf J, et al. The chemistry and biology of soluble guanylate cyclase stimulators and activators. Angew Chem Int Ed Engl. 2013;52:9442–9462. doi: 10.1002/anie.201302588. [DOI] [PubMed] [Google Scholar]
- Francis SH, Blount MA, Corbin JD. Mammalian cyclic nucleotide phosphodiesterases: molecular mechanisms and physiological functions. Physiol Rev. 2011;91:651–690. doi: 10.1152/physrev.00030.2010. [DOI] [PubMed] [Google Scholar]
- Freixa X, Bellera N, Ortiz-Perez JT, Jimenez M, Pare C, Bosch X, et al. Ischaemic postconditioning revisited: lack of effects on infarct size following primary percutaneous coronary intervention. Eur Heart J. 2012;33:103–112. doi: 10.1093/eurheartj/ehr297. [DOI] [PubMed] [Google Scholar]
- Fujita M, Asanuma H, Hirata A, Wakeno M, Takahama H, Sasaki H, et al. Prolonged transient acidosis during early reperfusion contributes to the cardioprotective effects of postconditioning. Am J Physiol Heart Circ Physiol. 2007;292:H2004–H2008. doi: 10.1152/ajpheart.01051.2006. [DOI] [PubMed] [Google Scholar]
- Garcia-Dorado D, Ruiz-Meana M, Inserte J, Rodriguez-Sinovas A, Piper HM. Calcium-mediated cell death during myocardial reperfusion. Cardiovasc Res. 2012;94:168–180. doi: 10.1093/cvr/cvs116. [DOI] [PubMed] [Google Scholar]
- Garcia-Dorado D, Rodriguez-Sinovas A, Ruiz-Meana M, Inserte J. Protection against myocardial ischemia-reperfusion injury in clinical practice. Rev Esp Cardiol. 2014a;67:394–404. doi: 10.1016/j.rec.2014.01.010. [DOI] [PubMed] [Google Scholar]
- Garcia-Dorado D, García-del-Blanco B, Otaegui I, Rodríguez-Palomares J, Pineda V, Gimeno F, et al. Intracoronary injection of adenosine before reperfusion in patients with ST-segment elevation myocardial infarction: a randomized controlled clinical trial. Int J Cardiol. 2014b;34:669. doi: 10.1016/j.ijcard.2014.09.203. http://dx.doi.org/10.1016/j.ijcard.2014.09.203. [DOI] [PubMed] [Google Scholar]
- Geisbuhler TP, Schwager TL. Effect of anoxia on cyclic nucleotides and inositol phosphate turnover in cardiac myocytes. J Mol Cell Cardiol. 1996;28:1857–1866. doi: 10.1006/jmcc.1996.0178. [DOI] [PubMed] [Google Scholar]
- Gomez L, Paillard M, Thibault H, Derumeaux G, Ovize M. Inhibition of GSK3beta by postconditioning is required to prevent opening of the mitochondrial permeability transition pore during reperfusion. Circulation. 2008;117:2761–2768. doi: 10.1161/CIRCULATIONAHA.107.755066. [DOI] [PubMed] [Google Scholar]
- Gorbe A, Giricz Z, Szunyog A, Csont T, Burley DS, Baxter GF, et al. Role of cGMP-PKG signaling in the protection of neonatal rat cardiac myocytes subjected to simulated ischemia/reoxygenation. Basic Res Cardiol. 2010;105:643–650. doi: 10.1007/s00395-010-0097-0. [DOI] [PubMed] [Google Scholar]
- Hahn JY, Song YB, Kim EK, Yu CW, Bae JW, Chung WY, et al. Ischemic postconditioning during primary percutaneous coronary intervention: the effects of postconditioning on myocardial reperfusion in patients with ST-segment elevation myocardial infarction (POST) randomized trial. Circulation. 2013;128:1889–1896. doi: 10.1161/CIRCULATIONAHA.113.001690. [DOI] [PubMed] [Google Scholar]
- Halkos ME, Kerendi F, Corvera JS, Wang NP, Kin H, Payne CS, et al. Myocardial protection with postconditioning is not enhanced by ischemic preconditioning. Ann Thorac Surg. 2004;78:961–969. doi: 10.1016/j.athoracsur.2004.03.033. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ, Tsang A, Yellon DM. The reperfusion injury salvage kinase pathway: a common target for both ischemic preconditioning and postconditioning. Trends Cardiovasc Med. 2005;15:69–75. doi: 10.1016/j.tcm.2005.03.001. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ, Iliodromitis EK, Andreadou I, Papalois A, Gritsopoulos G, Anastasiou-Nana M, et al. Investigating the signal transduction pathways underlying remote ischemic conditioning in the porcine heart. Cardiovasc Drugs Ther. 2012;26:87–93. doi: 10.1007/s10557-011-6364-y. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ, Erik Botker H, Condorelli G, Ferdinandy P, Garcia-Dorado D, Heusch G, et al. Translating cardioprotection for patient benefit: position paper from the Working Group of Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc Res. 2013;98:7–27. doi: 10.1093/cvr/cvt004. [DOI] [PubMed] [Google Scholar]
- Heusch G. Reduction of infarct size by ischaemic post-conditioning in humans: fact or fiction? Eur Heart J. 2012;33:13–15. doi: 10.1093/eurheartj/ehr341. [DOI] [PubMed] [Google Scholar]
- Hoshida S, Yamashita N, Kawahara K, Kuzuya T, Hori M. Amelioration by quinapril of myocardial infarction induced by coronary occlusion/reperfusion in a rabbit model of atherosclerosis: possible mechanisms. Circulation. 1999;99:434–440. doi: 10.1161/01.cir.99.3.434. [DOI] [PubMed] [Google Scholar]
- Inserte J, Garcia-Dorado D, Agullo L, Paniagua A, Soler-Soler J. Urodilatin limits acute reperfusion injury in the isolated rat heart. Cardiovasc Res. 2000;45:351–359. doi: 10.1016/s0008-6363(99)00371-5. [DOI] [PubMed] [Google Scholar]
- Inserte J, Garcia-Dorado D, Hernando V, Soler-Soler J. Calpain-mediated impairment of Na+/K+-ATPase activity during early reperfusion contributes to cell death after myocardial ischemia. Circ Res. 2005;97:465–473. doi: 10.1161/01.RES.0000181170.87738.f3. [DOI] [PubMed] [Google Scholar]
- Inserte J, Barba I, Hernando V, Abellan A, Ruiz-Meana M, Rodriguez-Sinovas A, et al. Effect of acidic reperfusion on prolongation of intracellular acidosis and myocardial salvage. Cardiovasc Res. 2008;77:782–790. doi: 10.1093/cvr/cvm082. [DOI] [PubMed] [Google Scholar]
- Inserte J, Barba I, Hernando V, Garcia-Dorado D. Delayed recovery of intracellular acidosis during reperfusion prevents calpain activation and determines protection in postconditioned myocardium. Cardiovasc Res. 2009;81:116–122. doi: 10.1093/cvr/cvn260. [DOI] [PubMed] [Google Scholar]
- Inserte J, Ruiz-Meana M, Rodriguez-Sinovas A, Barba I, Garcia-Dorado D. Contribution of delayed intracellular pH recovery to ischemic postconditioning protection. Antioxid Redox Signal. 2011a;14:923–939. doi: 10.1089/ars.2010.3312. [DOI] [PubMed] [Google Scholar]
- Inserte J, Barba I, Poncelas-Nozal M, Hernando V, Agullo L, Ruiz-Meana M, et al. cGMP/PKG pathway mediates myocardial postconditioning protection in rat hearts by delaying normalization of intracellular acidosis during reperfusion. J Mol Cell Cardiol. 2011b;50:903–909. doi: 10.1016/j.yjmcc.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Inserte J, Hernando V, Vilardosa U, Abad E, Poncelas-Nozal M, Garcia-Dorado D. Activation of cGMP/protein kinase G pathway in postconditioned myocardium depends on reduced oxidative stress and preserved endothelial nitric oxide synthase coupling. J Am Heart Assoc. 2013;2:e005975. doi: 10.1161/JAHA.112.005975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inserte J, Hernando V, Ruiz-Meana M, Poncelas-Nozal M, Fernández C, Agulló L, et al. Delayed phospholamban phosphorylation in postconditioned heart favors Ca2+ normalization and contributes to protection. Cardiovasc Res. 2014;103:542–553. doi: 10.1093/cvr/cvu163. [DOI] [PubMed] [Google Scholar]
- Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, et al. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113:1535–1549. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan SH, Yang H, Gilliam DE, Shen J, Lemasters JJ, Cascio WE. Hypercapnic acidosis and dimethyl amiloride reduce reperfusion induced cell death in ischaemic ventricular myocardium. Cardiovasc Res. 1995;29:231–238. [PubMed] [Google Scholar]
- Kerendi F, Kin H, Halkos ME, Jiang R, Zatta AJ, Zhao ZQ, et al. Remote postconditioning. Brief renal ischemia and reperfusion applied before coronary artery reperfusion reduces myocardial infarct size via endogenous activation of adenosine receptors. Basic Res Cardiol. 2005;100:404–412. doi: 10.1007/s00395-005-0539-2. [DOI] [PubMed] [Google Scholar]
- Kharbanda RK, Mortensen UM, White PA, Kristiansen SB, Schmidt MR, Hoschtitzky JA, et al. Transient limb ischemia induces remote ischemic preconditioning in vivo. Circulation. 2002;106:2881–2883. doi: 10.1161/01.cir.0000043806.51912.9b. [DOI] [PubMed] [Google Scholar]
- Kin H, Zhao ZQ, Sun HY, Wang NP, Corvera JS, Halkos ME, et al. Postconditioning attenuates myocardial ischemia-reperfusion injury by inhibiting events in the early minutes of reperfusion. Cardiovasc Res. 2004;62:74–85. doi: 10.1016/j.cardiores.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Kitakaze M, Asakura M, Kim J, Shintani Y, Asanuma H, Hamasaki T, et al. Human atrial natriuretic peptide and nicorandil as adjuncts to reperfusion treatment for acute myocardial infarction (J-WIND): two randomised trials. Lancet. 2007;370:1483–1493. doi: 10.1016/S0140-6736(07)61634-1. [DOI] [PubMed] [Google Scholar]
- Kloner RA, Forman MB, Gibbons RJ, Ross AM, Alexander RW, Stone GW. Impact of time to therapy and reperfusion modality on the efficacy of adenosine in acute myocardial infarction: the AMISTAD-2 trial. Eur Heart J. 2006;27:2400–2405. doi: 10.1093/eurheartj/ehl094. [DOI] [PubMed] [Google Scholar]
- Kopecky SL, Aviles RJ, Bell MR, Lobl JK, Tipping D, Frommell G, et al. A randomized, double-blinded, placebo-controlled, dose-ranging study measuring the effect of an adenosine agonist on infarct size reduction in patients undergoing primary percutaneous transluminal coronary angioplasty: the ADMIRE (AmP579 Delivery for Myocardial Infarction REduction) study. Am Heart J. 2003;146:146–152. doi: 10.1016/S0002-8703(03)00172-8. [DOI] [PubMed] [Google Scholar]
- Korkmaz S, Radovits T, Barnucz E, Hirschberg K, Neugebauer P, Loganathan S, et al. Pharmacological activation of soluble guanylate cyclase protects the heart against ischemic injury. Circulation. 2009;120:677–686. doi: 10.1161/CIRCULATIONAHA.109.870774. [DOI] [PubMed] [Google Scholar]
- Krieg T, Liu Y, Rutz T, Methner C, Yang XM, Dost T, et al. BAY 58–2667, a nitric oxide-independent guanylyl cyclase activator, pharmacologically post-conditions rabbit and rat hearts. Eur Heart J. 2009;30:1607–1613. doi: 10.1093/eurheartj/ehp143. [DOI] [PubMed] [Google Scholar]
- Kukreja RC, Salloum F, Das A, Ockaili R, Yin C, Bremer YA, et al. Pharmacological preconditioning with sildenafil: basic mechanisms and clinical implications. Vascul Pharmacol. 2005;42:219–232. doi: 10.1016/j.vph.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Kukreja RC, Salloum FN, Das A, Koka S, Ockaili RA, Xi L. Emerging new uses of phosphodiesterase-5 inhibitors in cardiovascular diseases. Exp Clin Cardiol. 2011;16:e30–e35. [PMC free article] [PubMed] [Google Scholar]
- Lies B, Groneberg D, Gambaryan S, Friebe A. Lack of effect of ODQ does not exclude cGMP signalling via NO-sensitive guanylyl cyclase. Br J Pharmacol. 2013;170:317–327. doi: 10.1111/bph.12275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochner A, Genade S, Tromp E, Opie L, Moolman J, Thomas S, et al. Role of cyclic nucleotide phosphodiesterases in ischemic preconditioning. Mol Cell Biochem. 1998;186:169–175. [PubMed] [Google Scholar]
- Lonborg J, Vejlstrup N, Kelbaek H, Botker HE, Kim WY, Mathiasen AB, et al. Exenatide reduces reperfusion injury in patients with ST-segment elevation myocardial infarction. Eur Heart J. 2012;33:1491–1499. doi: 10.1093/eurheartj/ehr309. [DOI] [PubMed] [Google Scholar]
- Lucas KA, Pitari GM, Kazerounian S, Ruiz-Stewart I, Park J, Schulz S, et al. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol Rev. 2000;52:375–414. [PubMed] [Google Scholar]
- Lundberg JO, Weitzberg E, Gladwin MT. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov. 2008;7:156–167. doi: 10.1038/nrd2466. [DOI] [PubMed] [Google Scholar]
- Madhani M, Hall AR, Cuello F, Charles RL, Burgoyne JR, Fuller W, et al. Phospholemman Ser69 phosphorylation contributes to sildenafil-induced cardioprotection against reperfusion injury. Am J Physiol Heart Circ Physiol. 2010;299:H827–H836. doi: 10.1152/ajpheart.00129.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahaffey KW, Puma JA, Barbagelata NA, DiCarli MF, Leesar MA, Browne KF, et al. Adenosine as an adjunct to thrombolytic therapy for acute myocardial infarction: results of a multicenter, randomized, placebo-controlled trial: the Acute Myocardial Infarction STudy of ADenosine (AMISTAD) trial. J Am Coll Cardiol. 1999;34:1711–1720. doi: 10.1016/s0735-1097(99)00418-0. [DOI] [PubMed] [Google Scholar]
- Massion PB, Balligand JL. Modulation of cardiac contraction, relaxation and rate by the endothelial nitric oxide synthase (eNOS): lessons from genetically modified mice. J Physiol. 2003;546:63–75. doi: 10.1113/jphysiol.2002.025973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta SR, Yusuf S, Diaz R, Zhu J, Pais P, Xavier D, et al. Effect of glucose-insulin-potassium infusion on mortality in patients with acute ST-segment elevation myocardial infarction: the CREATE-ECLA randomized controlled trial. JAMA. 2005;293:437–446. doi: 10.1001/jama.293.4.437. [DOI] [PubMed] [Google Scholar]
- Methner C, Lukowski R, Grube K, Loga F, Smith RA, Murphy MP, et al. Protection through postconditioning or a mitochondria-targeted S-nitrosothiol is unaffected by cardiomyocyte-selective ablation of protein kinase G. Basic Res Cardiol. 2013;108:337. doi: 10.1007/s00395-013-0337-1. [DOI] [PubMed] [Google Scholar]
- Meurer S, Pioch S, Pabst T, Opitz N, Schmidt PM, Beckhaus T, et al. Nitric oxide-independent vasodilator rescues heme-oxidized soluble guanylate cyclase from proteasomal degradation. Circ Res. 2009;105:33–41. doi: 10.1161/CIRCRESAHA.109.198234. [DOI] [PubMed] [Google Scholar]
- Moens AL, Kietadisorn R, Lin JY, Kass D. Targeting endothelial and myocardial dysfunction with tetrahydrobiopterin. J Mol Cell Cardiol. 2011;51:559–563. doi: 10.1016/j.yjmcc.2011.03.009. [DOI] [PubMed] [Google Scholar]
- Munzel T, Feil R, Mulsch A, Lohmann SM, Hofmann F, Walter U. Physiology and pathophysiology of vascular signaling controlled by guanosine 3′,5′-cyclic monophosphate-dependent protein kinase. Circulation. 2003;108:2172–2183. doi: 10.1161/01.CIR.0000094403.78467.C3. [DOI] [PubMed] [Google Scholar]
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- Nesher R, Robinson WF, Gibb L, Bishop SP, Kruger FA. Cyclic nucleotide levels in the perfused rat heart subjected to hypoxia. Experientia. 1977;33:215–217. doi: 10.1007/BF02124074. [DOI] [PubMed] [Google Scholar]
- Nishino Y, Webb IG, Davidson SM, Ahmed AI, Clark JE, Jacquet S, et al. Glycogen synthase kinase-3 inactivation is not required for ischemic preconditioning or postconditioning in the mouse. Circ Res. 2008;103:307–314. doi: 10.1161/CIRCRESAHA.107.169953. [DOI] [PubMed] [Google Scholar]
- Ohashi T, Yamamoto F, Yamamoto H, Ichikawa H, Shibata T, Kawashima Y. Transient reperfusion with acidic solution affects postischemic functional recovery: studies in the isolated working rat heart. J Thorac Cardiovasc Surg. 1996;111:613–620. doi: 10.1016/s0022-5223(96)70313-6. [DOI] [PubMed] [Google Scholar]
- Ovize M, Baxter G, Di Lisa F, Ferdinandy P, Garcia-Dorado D, Hausenloy DJ, et al. Postconditioning and protection from reperfusion injury: where do we stand? Cardiovasc Res. 2010;87:406–423. doi: 10.1093/cvr/cvq129. [DOI] [PubMed] [Google Scholar]
- Padilla F, Garcia-Dorado D, Agullo L, Inserte J, Paniagua A, Mirabet S, et al. L-Arginine administration prevents reperfusion-induced cardiomyocyte hypercontracture and reduces infarct size in the pig. Cardiovasc Res. 2000;46:412–420. doi: 10.1016/s0008-6363(00)00048-1. [DOI] [PubMed] [Google Scholar]
- Padilla F, Garcia-Dorado D, Agullo L, Barrabes JA, Inserte J, Escalona N, et al. Intravenous administration of the natriuretic peptide urodilatin at low doses during coronary reperfusion limits infarct size in anesthetized pigs. Cardiovasc Res. 2001;51:592–600. doi: 10.1016/s0008-6363(01)00242-5. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 2014;42(Database Issue):D1098–1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penna C, Rastaldo R, Mancardi D, Raimondo S, Cappello S, Gattullo D, et al. Post-conditioning induced cardioprotection requires signaling through a redox-sensitive mechanism, mitochondrial ATP-sensitive K + channel and protein kinase C activation. Basic Res Cardiol. 2006;101:180–189. doi: 10.1007/s00395-006-0584-5. [DOI] [PubMed] [Google Scholar]
- Penna C, Perrelli MG, Tullio F, Moro F, Parisella ML, Merlino A, et al. Post-ischemic early acidosis in cardiac postconditioning modifies the activity of antioxidant enzymes, reduces nitration, and favors protein S-nitrosylation. Pflugers Arch. 2011;462:219–233. doi: 10.1007/s00424-011-0970-1. [DOI] [PubMed] [Google Scholar]
- Penna C, Perrelli MG, Pagliaro P. Mitochondrial pathways, permeability transition pore, and redox signaling in cardioprotection: therapeutic implications. Antioxid Redox Signal. 2013;18:556–599. doi: 10.1089/ars.2011.4459. [DOI] [PubMed] [Google Scholar]
- Philipp S, Yang XM, Cui L, Davis AM, Downey JM, Cohen MV. Postconditioning protects rabbit hearts through a protein kinase C-adenosine A2b receptor cascade. Cardiovasc Res. 2006;70:308–314. doi: 10.1016/j.cardiores.2006.02.014. [DOI] [PubMed] [Google Scholar]
- Piper HM, Garcia-Dorado D, Ovize M. A fresh look at reperfusion injury. Cardiovasc Res. 1998;38:291–300. doi: 10.1016/s0008-6363(98)00033-9. [DOI] [PubMed] [Google Scholar]
- Piper HM, Kasseckert S, Abdallah Y. The sarcoplasmic reticulum as the primary target of reperfusion protection. Cardiovasc Res. 2006;70:170–173. doi: 10.1016/j.cardiores.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Preckel B, Schlack W, Obal D, Barthel H, Ebel D, Grunert S, et al. Effect of acidotic blood reperfusion on reperfusion injury after coronary artery occlusion in the dog heart. J Cardiovasc Pharmacol. 1998;31:179–186. doi: 10.1097/00005344-199802000-00002. [DOI] [PubMed] [Google Scholar]
- Rao YJ, Xi L. Pivotal effects of phosphodiesterase inhibitors on myocyte contractility and viability in normal and ischemic hearts. Acta Pharmacol Sin. 2009;30:1–24. doi: 10.1038/aps.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reffelmann T, Kloner RA. Effects of sildenafil on myocardial infarct size, microvascular function, and acute ischemic left ventricular dilation. Cardiovasc Res. 2003;59:441–449. doi: 10.1016/s0008-6363(03)00435-8. [DOI] [PubMed] [Google Scholar]
- Ren B, Shen Y, Shao H, Qian J, Wu H, Jing H. Brain natriuretic peptide limits myocardial infarct size dependent of nitric oxide synthase in rats. Clin Chim Acta. 2007;377:83–87. doi: 10.1016/j.cca.2006.08.027. [DOI] [PubMed] [Google Scholar]
- Ruiz-Meana M, Abellan A, Miro-Casas E, Garcia-Dorado D. Opening of mitochondrial permeability transition pore induces hypercontracture in Ca2+ overloaded cardiac myocytes. Basic Res Cardiol. 2007;102:542–552. doi: 10.1007/s00395-007-0675-y. [DOI] [PubMed] [Google Scholar]
- Salloum FN, Takenoshita Y, Ockaili RA, Daoud VP, Chou E, Yoshida K, et al. Sildenafil and vardenafil but not nitroglycerin limit myocardial infarction through opening of mitochondrial K(ATP) channels when administered at reperfusion following ischemia in rabbits. J Mol Cell Cardiol. 2007;42:453–458. doi: 10.1016/j.yjmcc.2006.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salloum FN, Das A, Samidurai A, Hoke NN, Chau VQ, Ockaili RA, et al. Cinaciguat, a novel activator of soluble guanylate cyclase, protects against ischemia/reperfusion injury: role of hydrogen sulfide. Am J Physiol Heart Circ Physiol. 2012;302:H1347–H1354. doi: 10.1152/ajpheart.00544.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt MR, Smerup M, Konstantinov IE, Shimizu M, Li J, Cheung M, et al. Intermittent peripheral tissue ischemia during coronary ischemia reduces myocardial infarction through a KATP-dependent mechanism: first demonstration of remote ischemic perconditioning. Am J Physiol Heart Circ Physiol. 2007;292:H1883–H1890. doi: 10.1152/ajpheart.00617.2006. [DOI] [PubMed] [Google Scholar]
- Schrammel A, Behrends S, Schmidt K, Koesling D, Mayer B. Characterization of 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one as a heme-site inhibitor of nitric oxide-sensitive guanylyl cyclase. Mol Pharmacol. 1996;50:1–5. [PubMed] [Google Scholar]
- Selker HP, Udelson JE, Massaro JM, Ruthazer R, D'Agostino RB, Griffith JL, et al. One-year outcomes of out-of-hospital administration of intravenous glucose, insulin, and potassium (GIK) in patients with suspected acute coronary syndromes (from the IMMEDIATE [Immediate Myocardial Metabolic Enhancement During Initial Assessment and Treatment in Emergency Care] Trial) Am J Cardiol. 2014;113:1599–1605. doi: 10.1016/j.amjcard.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiva S, Sack MN, Greer JJ, Duranski M, Ringwood LA, Burwell L, et al. Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J Exp Med. 2007;204:2089–2102. doi: 10.1084/jem.20070198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqi N, Neil C, Bruce M, MacLennan G, Cotton S, Papadopoulou S, et al. Intravenous sodium nitrite in acute ST-elevation myocardial infarction: a randomized controlled trial (NIAMI) Eur Heart J. 2014;35:1255–1262. doi: 10.1093/eurheartj/ehu096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegmund B, Schlack W, Ladilov YV, Balser C, Piper HM. Halothane protects cardiomyocytes against reoxygenation-induced hypercontracture. Circulation. 1997;96:4372–4379. doi: 10.1161/01.cir.96.12.4372. [DOI] [PubMed] [Google Scholar]
- Skyschally A, van Caster P, Iliodromitis EK, Schulz R, Kremastinos DT, Heusch G. Ischemic postconditioning: experimental models and protocol algorithms. Basic Res Cardiol. 2009a;104:469–483. doi: 10.1007/s00395-009-0040-4. [DOI] [PubMed] [Google Scholar]
- Skyschally A, van Caster P, Boengler K, Gres P, Musiolik J, Schilawa D, et al. Ischemic postconditioning in pigs: no causal role for RISK activation. Circ Res. 2009b;104:15–18. doi: 10.1161/CIRCRESAHA.108.186429. [DOI] [PubMed] [Google Scholar]
- Sorensson P, Saleh N, Bouvier F, Bohm F, Settergren M, Caidahl K, et al. Effect of postconditioning on infarct size in patients with ST elevation myocardial infarction. Heart. 2010;96:1710–1715. doi: 10.1136/hrt.2010.199430. [DOI] [PubMed] [Google Scholar]
- Staat P, Rioufol G, Piot C, Cottin Y, Cung TT, L'Huillier I, et al. Postconditioning the human heart. Circulation. 2005;112:2143–2148. doi: 10.1161/CIRCULATIONAHA.105.558122. [DOI] [PubMed] [Google Scholar]
- Stangherlin A, Gesellchen F, Zoccarato A, Terrin A, Fields LA, Berrera M, et al. cGMP signals modulate cAMP levels in a compartment-specific manner to regulate catecholamine-dependent signaling in cardiac myocytes. Circ Res. 2011;108:929–939. doi: 10.1161/CIRCRESAHA.110.230698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasch JP, Schmidt PM, Nedvetsky PI, Nedvetskaya TY, Arun Kumar HS, Meurer S, et al. Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J Clin Invest. 2006;116:2552–2561. doi: 10.1172/JCI28371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C. The pathophysiological role of peroxynitrite in shock, inflammation, and ischemia-reperfusion injury. Shock. 1996;6:79–88. doi: 10.1097/00024382-199608000-00001. [DOI] [PubMed] [Google Scholar]
- Tarantini G, Favaretto E, Marra MP, Frigo AC, Napodano M, Cacciavillani L, et al. Postconditioning during coronary angioplasty in acute myocardial infarction: the POST-AMI trial. Int J Cardiol. 2012;162:33–38. doi: 10.1016/j.ijcard.2012.03.136. [DOI] [PubMed] [Google Scholar]
- Thibault H, Piot C, Staat P, Bontemps L, Sportouch C, Rioufol G, et al. Long-term benefit of postconditioning. Circulation. 2008;117:1037–1044. doi: 10.1161/CIRCULATIONAHA.107.729780. [DOI] [PubMed] [Google Scholar]
- Tiefenbacher CP, Chilian WM, Mitchell M, DeFily DV. Restoration of endothelium-dependent vasodilation after reperfusion injury by tetrahydrobiopterin. Circulation. 1996;94:1423–1429. doi: 10.1161/01.cir.94.6.1423. [DOI] [PubMed] [Google Scholar]
- Timmers L, Henriques JP, de Kleijn DP, Devries JH, Kemperman H, Steendijk P, et al. Exenatide reduces infarct size and improves cardiac function in a porcine model of ischemia and reperfusion injury. J Am Coll Cardiol. 2009;53:501–510. doi: 10.1016/j.jacc.2008.10.033. [DOI] [PubMed] [Google Scholar]
- Tong G, Aponte AM, Kohr MJ, Steenbergen C, Murphy E, Sun J. Postconditioning leads to an increase in protein S-nitrosylation. Am J Physiol Heart Circ Physiol. 2014;306:H825–H832. doi: 10.1152/ajpheart.00660.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai EJ, Kass DA. Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol Ther. 2009;122:216–238. doi: 10.1016/j.pharmthera.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez-Vivar J, Martasek P, Whitsett J, Joseph J, Kalyanaraman B. The ratio between tetrahydrobiopterin and oxidized tetrahydrobiopterin analogues controls superoxide release from endothelial nitric oxide synthase: an EPR spin trapping study. Biochem J. 2002;362:733–739. doi: 10.1042/0264-6021:3620733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma S, Maitland A, Weisel RD, Fedak PW, Pomroy NC, Li SH, et al. Novel cardioprotective effects of tetrahydrobiopterin after anoxia and reoxygenation: identifying cellular targets for pharmacologic manipulation. J Thorac Cardiovasc Surg. 2002;123:1074–1083. doi: 10.1067/mtc.2002.121687. [DOI] [PubMed] [Google Scholar]
- Vila-Petroff M, Mundina-Weilenmann C, Lezcano N, Snabaitis AK, Huergo MA, Valverde CA, et al. Ca(2+)/calmodulin-dependent protein kinase II contributes to intracellular pH recovery from acidosis via Na(+)/H(+) exchanger activation. J Mol Cell Cardiol. 2009;49:106–112. doi: 10.1016/j.yjmcc.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb A, Bond R, McLean P, Uppal R, Benjamin N, Ahluwalia A. Reduction of nitrite to nitric oxide during ischemia protects against myocardial ischemia-reperfusion damage. Proc Natl Acad Sci U S A. 2004;101:13683–13688. doi: 10.1073/pnas.0402927101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo JS, Kim W, Ha SJ, Kim JB, Kim SJ, Kim WS, et al. Cardioprotective effects of exenatide in patients with ST-segment-elevation myocardial infarction undergoing primary percutaneous coronary intervention: results of exenatide myocardial protection in revascularization study. Arterioscler Thromb Vasc Biol. 2013;33:2252–2260. doi: 10.1161/ATVBAHA.113.301586. [DOI] [PubMed] [Google Scholar]
- Worner R, Lukowski R, Hofmann F, Wegener JW. cGMP signals mainly through cAMP kinase in permeabilized murine aorta. Am J Physiol Heart Circ Physiol. 2007;292:H237–H244. doi: 10.1152/ajpheart.00079.2006. [DOI] [PubMed] [Google Scholar]
- Xia Y, Tsai AL, Berka V, Zweier JL. Superoxide generation from endothelial nitric-oxide synthase. A Ca2+/calmodulin-dependent and tetrahydrobiopterin regulatory process. J Biol Chem. 1998;273:25804–25808. doi: 10.1074/jbc.273.40.25804. [DOI] [PubMed] [Google Scholar]
- Xin P, Zhu W, Li J, Ma S, Wang L, Liu M, et al. Combined local ischemic postconditioning and remote perconditioning recapitulate cardioprotective effects of local ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2010;298:H1819–H1831. doi: 10.1152/ajpheart.01102.2009. [DOI] [PubMed] [Google Scholar]
- Yamaguchi F, Nasa Y, Yabe K, Ohba S, Hashizume Y, Ohaku H, et al. Activation of cardiac muscarinic receptor and ischemic preconditioning effects in in situ rat heart. Heart Vessels. 1997;12:74–83. doi: 10.1007/BF02820870. [DOI] [PubMed] [Google Scholar]
- Yamashiro S, Noguchi K, Matsuzaki T, Miyagi K, Nakasone J, Sakanashi M, et al. Beneficial effect of tetrahydrobiopterin on ischemia-reperfusion injury in isolated perfused rat hearts. J Thorac Cardiovasc Surg. 2002;124:775–784. doi: 10.1067/mtc.2002.124393. [DOI] [PubMed] [Google Scholar]
- Yang X, Cohen MV, Downey JM. Mechanism of cardioprotection by early ischemic preconditioning. Cardiovasc Drugs Ther. 2010;24:225–234. doi: 10.1007/s10557-010-6236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XM, Proctor JB, Cui L, Krieg T, Downey JM, Cohen MV. Multiple, brief coronary occlusions during early reperfusion protect rabbit hearts by targeting cell signaling pathways. J Am Coll Cardiol. 2004;44:1103–1110. doi: 10.1016/j.jacc.2004.05.060. [DOI] [PubMed] [Google Scholar]
- Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- Yuan SY. Protein kinase signaling in the modulation of microvascular permeability. Vascul Pharmacol. 2002;39:213–223. doi: 10.1016/s1537-1891(03)00010-7. [DOI] [PubMed] [Google Scholar]
- Zaccolo M, Movsesian MA. cAMP and cGMP signaling cross-talk: role of phosphodiesterases and implications for cardiac pathophysiology. Circ Res. 2007;100:1569–1578. doi: 10.1161/CIRCRESAHA.106.144501. [DOI] [PubMed] [Google Scholar]
- Zhang M, Kass DA. Phosphodiesterases and cardiac cGMP: evolving roles and controversies. Trends Pharmacol Sci. 2011;32:360–365. doi: 10.1016/j.tips.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, et al. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2003;285:H579–H588. doi: 10.1152/ajpheart.01064.2002. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Pyriochou A, Kotanidou A, Dalkas G, van Eickels M, Spyroulias G, et al. Soluble guanylyl cyclase activation by HMR-1766 (ataciguat) in cells exposed to oxidative stress. Am J Physiol Heart Circ Physiol. 2008;295:H1763–H1771. doi: 10.1152/ajpheart.51.2008. [DOI] [PubMed] [Google Scholar]