

Abstract
PKs transfer a phosphate from ATP to the side-chain hydroxyl group of a serine, threonine or tyrosine residue of a substrate protein. This in turn can alter that protein's function; modulating fundamental cellular processes including, metabolism, transcription, growth, division, differentiation, motility and survival. PKs are subdivided into families based on homology. One such group are the stress-activated kinases, which as the name suggests, are activated in response to cellular stresses such as toxins, cytokines, mechanical deformation and osmotic stress. Members include the p38 MAPK family, which is composed of α, β, γ and δ, isoforms which are encoded by separate genes. These kinases transduce extracellular signals and coordinate the cellular responses needed for adaptation and survival. However, in cardiovascular and other disease states, these same systems can trigger maladaptive responses that aggravate, rather than alleviate, the disease. This situation is analogous to adrenergic, angiotensin and aldosterone signalling in heart failure, where inhibition is beneficial despite the importance of these hormones to homeostasis. The question is whether similar benefits could accrue from p38 inhibition? In this review, we will discuss the structure and function of p38, the history of p38 inhibitors and their use in preclinical studies. Finally, we will summarize the results of recent cardiovascular clinical trials with p38 inhibitors.
Tables of Links
| LIGANDS | |
|---|---|
| ACh | NADPH |
| AEA | Nitric oxide (NO) |
| Angiotensin II | SB202190 |
| ATP | SB203580 |
| CD40 ligand | SCH58261 |
| HU210 | TxA2 |
| LPS | U46619 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,c,d,eAlexander et al., 2013a,b,c,d,e,,,,).
p38 isoforms
The four isoforms of p38 are structurally homologous. The α and β isoforms share 74% sequence identity while the γ and δ isoforms are ∼70% similar to each other and ∼60% homologous to the α isoform. Significantly, the isoforms have different sensitivities to p38 inhibitors such as SB203580 (Eyers et al., 1998). These inhibitors compete with ATP and tend to inhibit p38α and p38β, over the p38γ and p38δ isoforms, because of small differences in side-chain length of their gatekeeper residues (Eyers et al., 1998). This feature can be used to create inhibitor resistant p38α and p38β knock-in mouse lines (O'Keefe et al., 2007). This strategy implicated p38α in both lethal myocardial ischaemic injury and cardioprotection following preconditioning (Kumphune et al., 2010; Sicard et al., 2010). Similarly, the p38α knockout mouse dies at embryonal day 10.5–11.5 (Adams et al., 2000; Allen et al., 2000) while ablation of the other isoforms, individually or p38δ and p38γ in combination, results in live mice without major abnormalities (Beardmore et al., 2005; Sabio et al., 2005). In addition, the loss of one isoform does not alter the expression or activity of the other isoforms. While p38α and p38β are ubiquitously expressed, the other isoforms possess a tissue-specific expression pattern, perhaps indicative of their different roles. p38γ has been implicated in myoblast differentiation and is enriched in skeletal muscle (Lechner et al., 1996). p38δ is predominantly expressed in lungs, kidney, testis, spleen, pancreas and small intestine (Kumar et al., 1997). The subcellular distribution of the isoforms may also indicate divergent roles, as p38α and p38β can be found in the cytosol and the nucleus while p38γ isoform is nuclear located in response to chronic pressure overload (Dingar et al., 2010). This theme can be further elaborated on by the substrates identified to be phosphorylated by the different isoforms. While most substrates such as myelin basic protein and activating transcription factor 2 are phosphorylated by multiple isoforms, some substrates appear to be uniquely phosphorylated by a particular isoform. The MAPK activated PK 2 (MAPKAPK2) has been show in vitro to be phosphorylated by α and β isoforms only (Cuenda et al., 1997). Identification of the repertoire of substrates of γ and δ has been more challenging because of the lack of a specific inhibitor for these isoforms. p38γ is unique in possessing a PDZ-binding domain sequence, KETXL, at its C-terminus likely responsible for the observed substrate bias towards α1 syntrophin, synapse-associated protein 90/ postsynaptic density 95 (Hasegawa et al., 1999; Sabio et al., 2004) and synapse-associated protein 97 (Sabio et al., 2005). The δ isoform has been shown to phosphorylate stathmin and Τau in vitro (Parker et al., 1998; Yoshida and Goedert, 2006) indicating the isoform is involved in regulating microtubule dynamics.
All four isoforms have been detected in the murine heart, but with higher levels of expression for p38α and p38γ compared with p38β and p38δ (Dingar et al., 2010). While the relationship between the isoforms is clearly complex and is still being elucidated, p38α appears most relevant to cardiovascular biology.
Activation and structure of p38α MAPK
The structure of p38α is similar to that of other kinases with a smaller N-terminal lobe mainly composed of β-pleated sheets and a C-terminal lobe of α helices. The ATP-binding pocket/catalytic cleft is located at the juncture of these two lobes. In addition, an activation loop, with a number of conserved motifs, lies adjacent to the ATP-binding pocket. The activity of p38 is indicated by the dual phosphorylation of the TGY motif in the activation loop of the kinase (Taylor and Kornev, 2011). Upstream kinases, termed MAPK kinases (MKK), such as MKK3, MKK6, phosphorylate these residues (Figure 1) (Derijard et al., 1995; Raingeaud et al., 1996). The specific activator of the p38 isoform depends on the stimulus and the cell type (Remy et al., 2010). In turn, the MKKs are phosphorylated by upstream MAPK kinase kinases such as the TGF-β activated PK (TAK1). This hierarchical three-tiered level of activation is termed the canonical activation pathway. These components mediate the transduction of an environmental stress signal to elicit a response by the cell.
Figure 1.
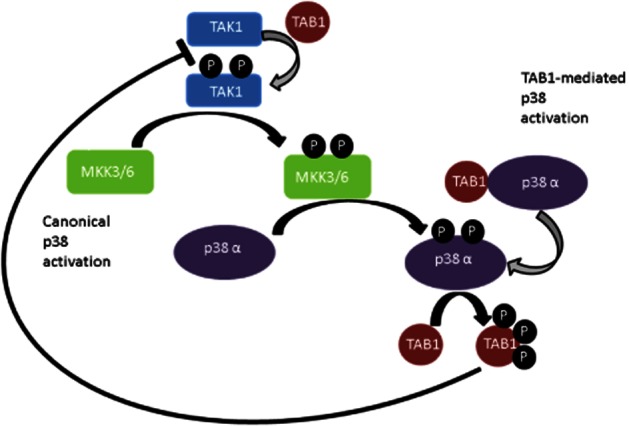
The major pathways of p38 activation. In the canonical activation pathway, TAK1 undergoes autophophorylation (grey arrow) upon binding to TAB1 (left portion of figure). This activated kinase in turn activates MKK3 or MKK6 by a transphosphorylation reaction (black arrow). Subsequently, the activated MKK phosphorylates p38α. p38α can also be activated by a more direct TAB1-mediated mechanism (right portion of figure). Binding of TAB1 to p38α induces autophosphorylation of p38α and activation of the kinase. TAB1 is also a substrate of p38α and phosphorylated TAB1 is a negative regulator of TAK1 autophosphorylation.
The conformation of the non-active kinase differs from that of the active kinase. In the inactive kinase, the activation loop occupies and sterically blocks the peptide-binding channel. The two lobes of the kinase are misaligned and therefore two residues, Lys53 of the N-terminal lobe and Asp168 of the C-terminal lobe, are not in close proximity. An active kinase requires cooperation between these key residues to bind the α phosphate group and ribose group of ATP and the companion Mg2+ ions (Wilson et al., 1996; Gum et al., 1998). Activation of the kinase via phosphorylation occurs in a highly ordered mechanism and results in the movement of the activation loop away from the peptide-binding channel (Humphreys et al., 2013). It is thought this movement exerts a ‘crank-handle effect’ on the tertiary structure of the kinase inducing the movement of the two lobes resulting in the alignment of Lys53 and Asp168 facilitating ATP binding. Overall these conformational changes result in the formation of the catalytic ‘hydrophobic spine’ of the kinase (Taylor and Kornev, 2011). In summary, the transphosphorylation of the TGY motif of the activation loop of the kinase, by upstream kinases, results in significant rearrangement of the kinase and dramatically increases its ability to function. Therefore, the activation of the kinase is a critical factor in the ability of a kinase to phosphorylate its substrates.
In addition to the canonical activation pathway, other additional activation mechanisms of p38 have been identified. Several groups, including ours, have reported a profound effect of binding of p38α isoform to the scaffold protein TAK1 binding protein 1 (TAB1) (Figure 1) (Ge et al., 2002; Tanno et al., 2003; Li et al., 2005). This binding results in activation of the kinase independent of upstream kinases through autophosphorylation. The binding of TAB1 to p38 resulted in significant alterations in the conformation of the kinase to facilitate active intramolecular phosphotransfer (De Nicola et al., 2013). This has been observed with the α isoform only (De Nicola et al., 2013). Interestingly, this scaffold-induced mode of activation was shown to be the mechanism through which p38 is activated during myocardial ischaemia (Kumphune et al., 2010). This indicates a significantly different mechanism than the more usual transactivation by MKK3/6 upstream kinases and may only occur under a limited number of circumstances.
Importance of p38 in biology
There are numerous lines of evidence indicating the importance of p38 in biology.
The Saccharomyces cerevisiae homologue of p38α is the pheromone and stress-sensing Hog1 gene (Bell et al., 2001). The high-osmolarity glycerol pathway has numerous functions in yeast including modulation of cell cycle progression at several stages and regulation of the biogenesis and export of mRNAs of stress-responsive genes (Duch et al., 2012; Regot et al., 2013). Similar to the mammalian system, Hog1 is a component in a hierarchical activation cascade and is activated by phosphorylation of its activation loop motif by the upstream kinases, Pbs2 (Brewster et al., 1993). Another conserved feature is the mode of activation through autophosphorylation, in this case by binding to the scaffold protein, Ste5 (Bhattacharyya et al., 2006). The evolutionary conservation of the protein and its mechanisms of activation highlight its biological importance. In addition, the ubiquitous expression of the α isoform and studies indicating this is the predominant isoform mediating the harmful effects during myocardial infarction and the beneficial effects of preconditioning further underline its central function. This is further reflected in the significant interest in p38 MAPK attested by the high number (approaching 25 000) of papers returned on a PubMed search.
Inhibitor design
Designing a specific p38 kinase inhibitor has proven to be challenging for a number of reasons. The 500 known mammalian PKs share a highly homologous ATP-binding site. Therefore, finding unique target areas for enabling the design of selective inhibitors has been difficult. For this reason, many kinase inhibitors have been shown to inhibit multiple kinase in addition to the original target (Bain et al., 2003). Nevertheless, the prominent role of p38α in cytokine production resulted in early success with inhibitors for use in arthritis, inflammatory bowel disease and chronic obstructive pulmonary disease (Lee et al., 1994). A number of crystal structures of p38α and p38γ in the presence or absence of a bound inhibitor are available in the protein databases and they provide a resource for future inhibitor design (Wilson et al., 1996; Wang et al., 1997; Bellon et al., 1999; Chang et al., 2002). The type I inhibitors binds in an ATP competitive manner. As discussed previously, the gatekeeper residue is a key determinant in the binding of the inhibitor and therefore the different isoforms have varied sensitivities to these types of inhibitors (Wilson et al., 1996; Gum et al., 1998). Members of this class also include SB 203580 and losmapimod. A challenge for efficient inhibition with type I inhibitors is that they must compete with physiological concentrations of ATP. To overcome these challenges, second types of inhibitors, which bind to the ATP-binding site and an adjacent region have been designed. Members of these type II inhibitors include BIRB 796 and VX745. BIRB 796 has been shown to inhibit all isoforms of the kinase (Kuma et al., 2005). These inhibitors make use of a pocket vacated by the DFG motif, which lies in the N-terminal portion of the activation loop. When the side chains of Asp and Phe are in the ‘out’ position, the kinase is inactive. BIRB796 binds to the ‘DFG out’ stabilizing it and preventing the kinase changing to an active conformation (Pargellis et al., 2002; Kuma et al., 2005). BIRB796 displays a slow onset of action as the DFG out conformer is rare. Despite the improved characteristics of type II inhibitors over type I inhibitors, translation to clinical studies has not achieved vastly superior results. Therefore, additional scaffolds and modes of inhibition have been sought. One such method has been termed type III inhibitors or allosteric kinase inhibitors. Exploiting less-conserved areas of the kinase distinct from the ATP-binding site offers the opportunity to develop unique inhibitors for a particular kinase with high selectivity because of the divergence of sequence in these less-conserved regions of the kinase.
Alternative sites for inhibitor binding distal to the ATP-binding site have been used by Murga and colleagues (Willemen et al., 2014). As discussed previously, they have shown that phosphorylation of the docking groove of p38 affects partner binding with reduced activation and of the kinase. A candidate inhibitor was designed to target this docking groove with some success. Targeting substrates that depend on this docking groove interaction would enable some modulation of p38 activity without global inhibition. While this approach of targeting p38 is attractive, the benefit would be increased if circumstance-specific inhibition could be achieved. Wang and colleagues have also adopted an alternative design strategy in targeting the TAB1-mediated autophosphorylation (Wang et al., 2013). A cell-permeable peptide was designed to inhibit p38 activity via the non-canonical autophosphorylation activation mechanism. We also had success with this strategy of disrupting p38α and TAB1 interaction, although the sequence targets were different (De Nicola et al., 2013). A detailed structural description of the binding of TAB1 to p38α was used to disrupt the interaction at key contact residues with a cell-permeable peptide preventing binding and therefore p38α autophosphorylation in vitro and in vivo. This strategy indicates that an inhibitor that could also disrupt this interaction would be advantageous in preventing p38α activation during myocardial ischaemia while maintaining its other cellular roles.
Cardioprotection and p38
As summarized earlier, p38 is expressed ubiquitously and implicated in numerous and diverse pathologies. Therefore, perhaps not surprisingly, it plays an important role in many components of the cascade that leads from a healthy vasculature to myocardial infarction and heart failure. Rather than attempt to summarize these studies in the text, we have created a table (see Table 1) that highlights relevant publications addressing the role of p38 in this cascade from endothelial dysfunction → atherosclerosis → platelet activation/thrombosis → myocardial infarction → post-infarction remodelling, contractile dysfunction, arrhythmia and heart failure. As can be seen from the table, generally, p38 is activated by, and aggravates, cardiovascular pathologies. From an evolutionary standpoint, this seems counterintuitive as selection pressure has preserved p38 in yeast and man. However, the benefits of this kinase are apparent under the ‘ischaemic preconditioning’ heading within Table 1. Ischaemic preconditioning describes the paradoxical increase in resistance to lethal ischaemic injury that follows a brief, sublethal, period of ischaemia. Despite the fact that p38 is activated by, and contributes to, lethal ischaemic injury; it also is involved in triggering the protection of ischaemic preconditioning. Some authors have linked these effects; with transient p38 activation during short preconditioning cycles of ischaemia being responsible for diminished p38 activation during subsequent lethal more prolonged ischaemia (Nagarkatti and Sha'afi, 1998; Marais et al., 2001; Sanada et al., 2001) These opposing roles for p38 make it difficult to predict what may happen in a clinical trial of p38 inhibition.
Table 1.
Summary of studies examining the role of p38 in the biological processes associated with cardiovascular disease and the effects of the use of p38 inhibitors on these processes
| Biological process | Study | p38 manipulation | Observed effect |
|---|---|---|---|
| Endothelial dysfunction | Choi et al., 2014 | Mouse mesenteric arteries and isolated endothelial cells, subjected to TNF-α in the presence of SB203580. | TNF-α-activated p38 and impaired endothelium-dependent relaxation to ACh; effect inhibited by SB203580. |
| Kassan et al., 2014 | Isolated coronary arterioles, mesenteric and femoral arteries from type 2 diabetic mice subjected to PEG-SOD and NADPH oxidase p22 (phox) siRNA. | Impaired endothelium-dependent relaxation and elevated p38 phosphorylation was reduced after treatment with ROS scavenger or by p22 (phox) down-regulation. | |
| Thakur et al., 2010 | Mouse thoracic aortic ring preparation and cultured endothelial cells subjected to AngII, SCH58261 A2A R antagonist and A2AR siRNA. | A2AR is involved in the regulation of endothelial ROS production by Nox2 and requires p38 MAPK signalling. Blockade or knockdown of A2AR inhibited p38 activation, inhibiting AngII effects on EC ROS production. | |
| Rajesh et al., 2010 | Cultured primary HCAEC subjected to cannabinoid receptor agonists (AEA and HU210) in the presence of SB203580. | Inhibition of p38 by SB203580 attenuated AEA and HU210-induced cell death. | |
| Bao et al., 2007 | Rat aortic ring preparations and MAPKAPK2 (−/−) mouse hearts subjected to treatment with AngII in the presence of SB239063. | AngII-induced hypertension and p38 phosphorylation was attenuated in MAPKAPK2 null mice. AngII-induced vascular dysfunction, superoxide anion production and cardiac remodelling were attenuated in aortic rings by SB239063. | |
| Riad et al., 2007 | In vivo rat model of diabetes by administration of streptozotocin in the presence of SB239063. | SB239063 attenuated diabetes-induced p38 phosphorylation and improved impairments in LV and endothelial function. | |
| Widder et al., 2004 | Rat aortic ring preparation of hearts subjected to ischaemia by LAD ligation in the presence of SB239063. | SB239063 preserved endothelium-dependent vasodilation, reduced vascular superoxide anion production and p38 and MAPKAPK2 phosphorylation. | |
| Ju et al., 2003 | Spontaneously hypertensive rat and cultured HUVECs exposed to TNF-α and LPS in the presence of SB239063. | TNF-α and LPS-induced p38 activation and ICAM expression in HUVECSs and in aortas from hypertensive rats. All inhibited by SB239063 and endothelial dysfunction restored in aortic rings. | |
| Atherosclerosis | Cheriyan et al., 2011 | Randomized controlled trial of losmapimod (p38α/β MAPK inhibitor) on untreated hypercholesterolaemic patients. Endothelial function assessed by venous occlusion plethysmography. | Losmapimod attenuated inflammation by inhibiting p38 activity and improved NO-mediated vasodilation. |
| Seeger et al., 2010 | MicroCT and planimetry of mouse ApoE−/− aortas and isolated bone marrow-derived mononuclear cells. | Proangiogenic activation of p38 inhibited by SB and resulted in reduction of atherosclerotic lesion size, inflammation and increased vasculogenic cells. | |
| Sun et al., 2009 | Isolated cultured macrophages from ApoE−/−, Npc1−/− mice exposed to free cholesterol. | Lipid accumulation in atherosclerotic plaques activates p38 and its targets Ctsk, S100a8, MMP8 and MMP14 via TLR signalling. | |
| Proctor et al., 2008 | Transgenic mice expressing inducible SMC-specific DN-p38α subjected to carotid injury and ectopic expression of DN-p38α in cultured rat SMC, in presence of SB202190 or siRNA. | Mice expressing DN-p38α resistant to carotid injury and reduced p38 activity. | |
| SB202190 or siRNA blocked PDGF-induced p38 activation and cell proliferation, in A10 SMC. | |||
| Morris et al., 2008 | In vivo MRI assessment of ApoE−/− mouse aortas with AngII and SB239063. | SB239063 inhibited p38 activity, inflammation in atherosclerotic plaques and phagocytic activity of macrophages and diminished aortic root lesion size. | |
| Platelet activation | Alrehani et al., 2013 | HEK293 cells overexpressing αIIbβ3, and protein phosphate 1 knockdown by siRNA. | p38 negatively regulated PP1cα-mediated adhesion to immobilized fibrinogen and fibrin clot retraction, which were abrogated in the presence of p38 inhibitor, SB203580. |
| Yacoub et al., 2010 | Isolated human and CD40−/− mouse platelets treated with soluble CD40 ligand (sCD40L). | Pro-thrombotic inflammatory CD40L enhanced platelet activation and aggregation via Rac1 and p38. SB203580, prevented p38 phosphorylation and impaired the effects of sCD40L on platelet P-selectin expression and aggregation. | |
| Rauch et al., 2007 | Human vascular smooth muscle cell migration in Boyden–Chamber assay by fibrinogen in the presence of SB203580. | Fibrinogen-induced migration and mediated the activation of p38 in an ICAM-1-dependent manner. SB203580 inhibited p38 activity and cell migration. | |
| Shen et al., 2007 | Isolated human platelets activated by exposure to collagen in the presence of resveratrol. | Resveratrol inhibited collagen-induced platelet aggregation, p38 MAPK phosphorylation and thromboxane A2 formation. | |
| Vega-Ostertag et al., 2007 | Pressure-perfused CD1 mouse aorta, isolated platelets and cultured endothelial cells exposed to human aPL. | aPL-induced thrombosis and EC activation in vivo and aPL-induced monocyte adherence to HUVEC in vitro, were all attenuated by SB203580. | |
| Brooks et al., 2007 | Isolated adult horse platelets and leukocytes following exposure to LPS in vivo and in vitro respectively. | LPS infusion enhanced p38 phosphorylation and TxA2 production in platelets and leukocytes. SB203580 attenuated LPS-induced TxA2 release in platelets. | |
| Sakurai et al., 2004 | p38α+/− mice subjected to FeCl3-induced carotid injury model of thrombus formation. | Time to thrombotic occlusion was prolonged in p38α+/− mice and U46619-induced aggregatory response in platelets was impaired with poor binding to fibrinogen. | |
| MI | Gray et al., 2011 | Administration of GlcNAc-coated SB239063 in rat hearts subjected to regional ischaemia-reperfusion. | GlcNAc-decorated particles loaded with the p38 inhibitor SB239063 reduced apoptotic events and infarct size and improved acute cardiac function. |
| Kumphune et al., 2010 | Drug-resistant-p38α mouse hearts subjected to global ischaemia in the presence of SB203580. | p38α is the dominant active isoform during myocardial ischaemia that contributes to infarction | |
| Sy et al., 2008 | PCADK-mediated delivery of SB239063 in rat hearts subjected to regional ischaemia. | Inhibition of p38 by SB239063 improved cardiac function following MI. | |
| Kaiser et al., 2004 | Transgenic mice expressing DN-p38 and DN-MKK6, subjected to regional ischaemia by LAD ligation and ectopic expression in cultured rat neonatal cardiac myocytes and transgenic mice. | p38 functions as a pro-death signalling effector in both cultured myocytes as well as the intact heart. | |
| Tanno et al., 2003 | MKK3-knockout mouse hearts and H9c2 expressing drug-resistant-p38α subjected to global ischaemia-reperfusion or SI, respectively, in the presence of SB203580. | Absence of MKK3 had no significant effect on post-ischaemic infarction or p38 activation. p38 activation is SB203580-sensitive and TAB1-associated and contributes to myocardial injury. | |
| Otsu et al., 2003 | Regional ischaemia-reperfusion by coronary occlusion in p38α−/+ mouse hearts. | Ischaemia-reperfusion induces activation of p38 leading to injury. Reduction of p38α expression results in protection. | |
| Martin et al., 2001 | Ectopic expression of drug-resistant-p38α in cultured rat adult cardiac myocytes and H9c2 myoblasts, subjected to simulated ischaemia. | Cardioprotective effect of SB203580 is through specific inhibition of p38α | |
| Ma et al., 1999 | Langendorff-perfused rabbit hearts subjected to global ischaemia-reperfusion in presence of SB203580. | Inhibition of p38 by SB203580 is cardioprotective. SB203580 decreased myocardial apoptosis by ischaemia-reperfusion injury and improved cardiac function. This effect is most evident when SB203580 exposure precedes reperfusion. | |
| MacKay et al., 1999 | Cultured rat neonatal cardiac myocytes subjected to simulated ischaemia in the presence of SB203580. | Inhibition of p38 activation during SI by SB203580 and reduced activation of caspase-3, affording protection against cardiomyocyte apoptosis. | |
| Yada et al., 2004 | CD-1 mice, regional myocardial IR with or without FR167653 | Pretreatment with FR167653 before IR reduced inflammatory cytokine expression and infarct size. | |
| LV remodelling | Koivisto et al., 2011 | Ectopic co-expression of p38α or p38β with CA-MKK3 or MKK6 in cultured rat neonatal cardiac myocytes. | p38β increased mRNA levels of pro-hypertrophic BNP and ANF, whereas p38α augmented the expression of the pro-fibrotic genes, CTGF, bFGF and MMP9. |
| Lau et al., 2007 | 14-3-3(+/−) mouse coronary artery occlusion in the presence of SB202190 and cultured adult mouse cardiac myocytes. | SB202190 inhibited p38 activation and increased survival in 14-3-3(+/−mice), which developed pathological ventricular remodelling with increased cardiomyocyte apoptosis post MI. | |
| Engel et al., 2006 | Rat hearts subjected to regional ischaemia by LAD Ligation in the presence of FGF-2 and SB203580. | Administration of SB203580 and FGF-2, post infarction resulted in cardiomyocyte mitosis, reduction of scarring and rescued heart function. | |
| Tenhunen et al., 2006 | Rat hearts subjected to LAD ligation and adenoviral co-transfection of p38α and MKK3b. | p38 activation reduced infarct size, improved ejection fraction, fractional shortening, decreased LV diameter and increased capillary density and reduced apoptosis and fibrosis. | |
| Ren et al., 2005 | Transgenic mice expressing cardiac-specific DN-p38α, subjected to regional ischaemia by LAD ligation. | DN-p38α mice had increased ventricular systolic function 7 days post MI and reduced infarct size, cardiomyocye apoptosis and Bcl-XL deamination. | |
| Nishida et al., 2004 | Cardiac-specific p38α conditional KO mice subjected to TAC and cultured isolated neonatal cardiac myocytes. | p38α plays a critical role in cardiomyocyte survival in response to pressure with no effect on hypertrophic growth, despite down-regulation of p38α. | |
| See et al., 2004 | Rat hearts subjected to regional ischaemia by LAD ligation in the presence of RWJ-67657. | RWJ-67657 treatment following myocardial ischaemia had beneficial effects on LV remodelling and dysfunction. | |
| Andrews et al., 2003 | Ectopic expression of CA-MKK6 in cultured rat neonatal cardiac myocytes. | MKK6-mediated activation of p38 prolonged contractile calcium transient, reduced SERCA2 expression, resulting in increased diastolic [Ca2+]i and enhanced NFAT activity. | |
| Braz et al., 2003 | Transgenic mice expressing cardiac- specific DN mutants of p38α, MKK3 or MKK6, subjected to abdominal aortic banding or AngII, ISO or PE. | p38 signalling antagonizes the hypertrophic growth response of the adult heart through a dominant mechanism involving crosstalk with the calcineurin–NFAT signalling pathway. | |
| Myocardial contractile dysfunction | Wang et al., 2012 | Langendorff-perfused HHcy−/+ mouse hearts subjected to global ischaemia-reperfusion and cultured rat adult cardiac myocytes subjected to Hcy in the presence of SB203580. | Ischaemia-reperfusion resulted in activation of p38 and in impaired cardiac relaxation, contractile function and increased apoptosis that was markedly exaggerated in HHcy mice and cardiomyocytes. SB203580 prevented the Hcy-induced changes. |
| Yin et al., 2008 | Rat hearts subjected to ischaemia by LAD ligation in the presence of SB203580. | SB203580 suppressed myocardial fibrosis and LV remodelling, attenuated p38 activation and expression of TNF-α, α-SMA and collagen I. | |
| Vahebi et al., 2007 | Isolated skinned cardiac muscle fibre bundles from transgenic mice expressing DN-p38α and CA-MKK6. | Activation of p38α directly depresses sarcomeric function by decreased phosphorylation of α-tropomyosin, which is reversed by overexpression of DN-p38α. | |
| Bellahcene et al., 2006 | Langendorff-perfused MKK3-knockout mouse hearts and isolated cardiac myocytes subjected to TNF-α. | TNF-α induces contractile depression by activating p38 in the intact heart and in isolated cardiac myocytes in an MKK3-dependent and SB203580-sensitive manner. | |
| Arrhythmia | Lu et al., 2013 | Whole cell patch-clamp analysis on isolated rabbit PV cardiomyocytes, treated with collagen. | Collagen modulated PV electrical activity with changes in AP morphology, increased triggered and spontaneous activity. SB203580 ameliorated collagen-induced p38 phosphorylation and arrythmogenesis. |
| De Jong et al., 2013 | Cultured isolated rat neonatal cardiac myocytes, subjected to mechanical stretch. | Stretch induced p38 phosphorylation and hypertrophy-related changes including increased cell diameter, reinduction of the foetal gene programme and cell death. | |
| Surinkaew et al., 2013 | Rat hearts subjected to LAD ligation in the presence of SB203580. | SB203580 decreased ischaemia-induced ventricular tachycardia/ventricular fibrillation incidence and heat shock protein 27 phosphorylation, and increased connexin 43 phosphorylation. | |
| Tang et al., 2011 | Rat hearts subjected to LAD ligation, Langendorff perfusion and cultured, isolated adult ventricular myocytes in the presence of SB203580 or auranofin. | Expression of ventricular K(+) channels is redox regulated and impairment of the Trx system post-MI heart contributes to I(to) remodelling through sustained activation of apoptosis signal-regulating kinase-1-JNK-p38 signalling. | |
| Ischaemic preconditioning | Sicard et al., 2010 | Drug-resistant-p38α or p38β mouse hearts subjected to ischaemic preconditioning in the presence of SB203580. | SB203580 attenuated activity of p38 and its substrates and abolished infarct size reduction by IP in WT and drug-resistant-p38β hearts but not in drug-resistant-p38α hearts. p38α is necessary for ischaemic preconditioning. |
| Schulz et al., 2003 | In vivo pig model of ischaemic preconditioning by LAD ligation (regional ischaemia). | IP increases co-localization of p38 with Cx43 and preserves phosphorylation of Cx43 during ischaemia. Inhibition of p38MAPK by SB203580 attenuated IP-induced IS-reduction and led to dephosphorylation of Cx43 that correlates with the propagation of I/R injury. | |
| Sanada et al., 2001 | In vivo canine model of ischaemic preconditioning by coronary occlusion (regional ischaemia). | p38 MAPK activation during IP mainly mediates the cardioprotection followed by HSP27 phosphorylation/translocation. SB203580 treatment during IP blunted the infarct size limitation by IP and attenuated phosphorylation/translocation of HSP27. | |
| Marais et al., 2001 | Langendorff-perfused rat hearts subjected to global ischaemia- reperfusion and cultured rat neonatal cardiac myocytes | p38 was activated during preconditioning and attenuated during subsequent ischaemia. Non-preconditioned hearts had elevated p38 activation in comparison. p38 inhibition by SB203580 during ischaemia and reperfusion is cardioprotective. | |
| Saurin et al., 2000 | Ectopic expression p38α or p38β isoforms in cultured rat neonatal cardiac myocytes subjected to simulated ischaemia in the presence of SB203580. | Inhibition of p38 during prolonged ischaemia reduced injury and contributed to preconditioning-induced cardioprotection. | |
| p38α and p38β differentially activated or deactivated respectively, during ischaemia. | |||
| Nagarkatti et al., 1998 | Simulated ischaemia in rat myoblast cell line H9c2. | Inhibition of p38 before the onset of SI blocks preconditioning, but is protective during prolonged ischaemia. | |
| Weinbrenner et al., 1997 | Langendorff-perfused rabbit hearts subjected to global ischaemia-reperfusion in presence of SB203580 | Inhibition of p38 activation abolished protection in preconditioned hearts and cardiomyocytes. | |
| Tong et al., 2000 | Langendorff-perfused rat hearts, preconditioned with or without SB202190 | Preconditioning induced uptake of glucose was abrogated by the presence of SB202190 |
AEA, anandamide; ANF, atrial natriuretic peptide; AP, action potentials; aPL, antiphospholipid antibodies; ApoE, apolipoprotein E; AngII, angiotensin II; bFGF, basic fibroblast growth factor; CTGF, connective tissue growth factor; Ctsk, cathepsin K; Cx43, gap junction protein connexin43; DN, dominant negative; FGF, fibroblast growth factor; GlcNAc, N-acetylglucosamine; H9c2, rat myoblast cell line; Hcy, homocysteine; HHcy, hyperhomocysteinaemia; ICAM, intercellular cell adhesion molecule; IP, ischaemic preconditioning; I/R, ischaemia/reperfusion; IS, infarct size; ISO, isoproterenol; LAD, left anterior descending coronary artery; LV, left ventricle; MAPKAPK2, MAPK activated PK 2; MI, myocardial infarction; MMP, matrix metalloproteinase; NADPH, nicotinamide adenine dinucleotide phosphate; NFAT, nuclear factor of activated T-cell; Npc1, Niemann–Pick disease type C1; PCADK, poly(cyclohexane-1,4-diyl acetone dimethylene ketal); PE, phenylephrine; PEG-SOD, polyethylene glycol superoxide dismutase; PV, pulmonary vein; ROS, reactive oxygen species; SB, SB203580; SERCA2, sarcoplasmic reticulum Ca2+ ATPase; SI, simulated ischaemia; SMA, smooth muscle actin; SMC, smooth muscle cell; TAC, transverse aortic constriction; TLR, Toll-like receptor; TxA2, thromboxane A2; WT, wild type.
The findings that p38 activation aggravates many components of atherothrombosis and myocardial infarction, have laid the foundation for recent and relevant clinical trial activity. The companies with agents under investigation include GlaxoSmithKline (losmapimod, various trials), ArrayBioPharma (ARRY-371797, NCT02057341) and Bristol-Myers Squibb (BMS-582949, NCT00570752), although the latter programme seems inactive. GlaxoSmithKline has the most active programme with a number of phase 1 trials suggesting a potential benefit in patients with early (Cheriyan et al., 2011) and late (Sarov-Blat et al., 2010; Elkhawad et al., 2012) atherosclerosis. We have summarized these trials previously (Martin et al., 2012), and here will concentrate on the recently published SOLSTICE trial (Newby et al., 2014). SOLSTICE was a GlaxoSmithKline-sponsored phase 2 study performed by the Duke Clinical Research Institute (NCT00910962) (Newby et al., 2014). This study enrolled 535 patients with non-ST elevation myocardial infarction who were randomized in a 3:3:2 ratio to oral losmapimod (two regimes, both of 7.5 mg twice daily with, or without, a 15 mg initial dose) or placebo. The trial medication was initiated within 18 h of hospital presentation and continued for 12 weeks. The first dose had to be administered at least 2 h before planned percutaneous coronary intervention (PCI). This design attempted to ensure p38 was inhibited during any procedure-related exacerbation of myocardial injury. The primary end points were of safety and efficacy. The safety end point was mainly designed to detect hepatotoxicity. The efficacy end points were high-sensitivity C-reactive protein (hsCRP) at 12 weeks and biomarker measures of extent of myocardial infarction.
In terms of safety, there was no statistically significant increase in liver enzymes, but alanine transaminase elevations three times above the upper limit of normal were more common in the losmapimod groups. In addition, there was a statistically significant, but very small (∼2 μmol·L−1), increase in serum creatinine at 12 weeks in the losmapimod groups. Study drug discontinuation was twice as frequent among patients receiving losmapimod than among those receiving placebo. This observation was of borderline significance and the cause was unknown. Serious adverse events were very similar across groups.
In terms of efficacy, the principal end point, hsCRP concentration at 12 weeks, did not differ between groups. However, by 12 weeks, CRP had returned to baseline, so a discernable difference was unlikely. However, at earlier timepoints, both hsCRP and IL-6 concentrations were significantly lower in the losmapimod groups. Biomarkers of necrosis (creatine kinase and troponin I) did not differ by treatment group. However, in an MRI substudy (92 patients with paired MRI scans) losmapimod was associated with significant improvements in ejection fraction and left ventricular end-diastolic and end-systolic volumes, and also a significant reduction in discharge brain natriuretic peptide in the main cohort. Collectively, these signals hint of a benefit with losmapimod and p38 inhibition in the setting of myocardial infarction despite the fact that SOLSTICE failed to meet its primary efficacy end points. Moreover, this suggestion of benefit was observed despite a losmapimod oral dosing regime that probably only achieved a maximum of 50% inhibition of p38 at the time of peak effect which occurred 4 h after oral dosing (Barbour et al., 2013). This time–concentration profile matched the timing of PCI in SOLSTICE, where approximately 60% of patients in losmapimod and placebo groups underwent PCI at a median of about 5 h after randomization.
The potential efficacy signal in SOLSTICE has encouraged a much larger phase 3 trial with clinical end points. LATITUDE-TIMI 60 (Losmapimod to inhibit p38 MAPK as a therapeutic target and modify outcomes after an acute coronary syndrome–thrombolysis in myocardial infarction 60; NCT02145468) will involve approximately 26 000 patients worldwide with myocardial infarction and have a study design similar to SOLSTICE, but without the 15 mg loading group (i.e. losmapimod 7.5 mg twice per day vs. placebo). The primary efficacy end point is a composite of major adverse cardiovascular events comprising cardiovascular death, reinfarction or recurrent myocardial ischaemia requiring urgent revascularization at 12 weeks. The main inclusion criterion is type 1 myocardial infarction (an atherosclerotic plaque rupture event) and will include presentation electrocardiograms with both ST and no ST elevation. Study medication will be continued for 12 weeks and follow-up for 24 weeks. It is anticipated that the study will be completed in December 2018.
Interaction with other signalling pathways
The p38 MAPK pathway is integrated with multiple other signalling pathways at different levels. For example, p38α has been shown to phosphorylate glycogen synthase kinase 3 and the Wnt signalling pathways (Bikkavilli et al., 2008; Thornton et al., 2008). p38α is part of numerous feedback mechanisms. A recent paper has also indicated p38α may participate in multiprotein complex hypertrophy mediated by a PKA anchoring protein, in combination with MKK3. In addition, p38α is located within a number of feedback loops. These studies reflect p38α as a signalling nexus of multiple signalling pathways the specifics of which are continuing to be elucidated. The multiple pathways also illustrate the complexity in targeting a single PK. It is clear that multiple related pathways may be affected if a single protein is inhibited. Therefore, the ideal inhibition of a kinase requires selective, temporally discrete and circumstance-specific inhibition.
Global inhibition of p38α can lead to detrimental effects to the homeostasis of the organism. Pharmacological inhibition of p38 has been achieved, but the results of the preclinical trials, although encouraging, have not translated to clinical trials with a similar level of success. Therefore, a more selective targeted inhibition of the kinase that could ameliorate the detrimental consequence of activation of the kinase while maintaining the essential functions of the kinase would be highly desirable. The recent studies indicating the TAB1-induced activation of the kinase that mediates myocardial damage would be an eligible target for inhibition and that would allow the circumstance-specific mode of activation to be exploited.
Acknowledgments
M. S. M. is supported by the UK Department of Health through the National Institute for Health Research Biomedical Research Centre award to Guy's and St Thomas' National Health Service Foundation Trust. M. S. M. through King's College London, has acted as a consultant for GlaxoSmithKline and was a member of the SOLSTICE steering committee. E. D. M. is funded by a project grant from the Medical Research Council (MR/J007501/1).
Glossary
- hsCRP
high-sensitivity CRP
- PCI
percutaneous coronary intervention
- TAB1
TGF-β-activated binding protein 1
- TAK1
TGF-β-activated kinase 1
References
- Adams RH, Porras A, Alonso G, Jones M, Vintersten K, Panelli S, et al. Essential role of p38alpha MAP kinase in placental but not embryonic cardiovascular development. Mol Cell. 2000;6:109–116. [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, McGrath JC, et al. The Concise Guide to PHARMACOLOGY 2013/14: Overview. Br J Pharmacol. 2013a;170:1449–1458. [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013b;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ion channels. Br J Pharmacol. 2013c;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14:Transporters. Br J Pharmacol. 2013d;170:1706–1796. doi: 10.1111/bph.12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013e;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen M, Svensson L, Roach M, Hambor J, McNeish J, Gabel CA. Deficiency of the stress kinase p38alpha results in embryonic lethality: characterization of the kinase dependence of stress responses of enzyme-deficient embryonic stem cells. J Exp Med. 2000;191:859–870. doi: 10.1084/jem.191.5.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alrehani N, Pradhan S, Khatlani T, Kailasam L, Vijayan KV. Distinct roles for the alpha, beta and gamma1 isoforms of protein phosphatase 1 in the outside-in alphaIIbbeta3 integrin signalling-dependent functions. Thromb Haemost. 2013;109:118–126. doi: 10.1160/TH12-04-0237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews C, Ho PD, Dillmann WH, Glembotski CC, McDonough PM. The MKK6-p38 MAPK pathway prolongs the cardiac contractile calcium transient, downregulates SERCA2, and activates NF-AT. Cardiovasc Res. 2003;59:46–56. doi: 10.1016/s0008-6363(03)00329-8. [DOI] [PubMed] [Google Scholar]
- Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371(Pt 1):199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao W, Behm DJ, Nerurkar SS, Ao Z, Bentley R, Mirabile RC, et al. Effects of p38 MAPK inhibitor on angiotensin II-dependent hypertension, organ damage, and superoxide anion production. J Cardiovasc Pharmacol. 2007;49:362–368. doi: 10.1097/FJC.0b013e318046f34a. [DOI] [PubMed] [Google Scholar]
- Barbour AM, Sarov-Blat L, Cai G, Fossler MJ, Sprecher DL, Graggaber J, et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of losmapimod following a single intravenous or oral dose in healthy volunteers. Br J Clin Pharmacol. 2013;76:99–106. doi: 10.1111/bcp.12063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beardmore VA, Hinton HJ, Eftychi C, Apostolaki M, Armaka M, Darragh J, et al. Generation and characterization of p38beta (MAPK11) gene-targeted mice. Mol Cell Biol. 2005;25:10454–10464. doi: 10.1128/MCB.25.23.10454-10464.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell M, Capone R, Pashtan I, Levitzki A, Engelberg D. Isolation of hyperactive mutants of the MAPK p38/Hog1 that are independent of MAPK kinase activation. J Biol Chem. 2001;276:25351–25358. doi: 10.1074/jbc.M101818200. [DOI] [PubMed] [Google Scholar]
- Bellahcene M, Jacquet S, Cao XB, Tanno M, Haworth RS, Layland J, et al. Activation of p38 mitogen-activated protein kinase contributes to the early cardiodepressant action of tumor necrosis factor. J Am Coll Cardiol. 2006;48:545–555. doi: 10.1016/j.jacc.2006.02.072. [DOI] [PubMed] [Google Scholar]
- Bellon S, Fitzgibbon MJ, Fox T, Hsiao H-M, Wilson KP. The structure of phosphorylated P38γ is monomeric and reveals a conserved activation-loop conformation. Structure. 1999;7:1057–1065. doi: 10.1016/s0969-2126(99)80173-7. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya RP, Remenyi A, Good MC, Bashor CJ, Falick AM, Lim WA. The Ste5 scaffold allosterically modulates signaling output of the yeast mating pathway. Science. 2006;311:822–826. doi: 10.1126/science.1120941. [DOI] [PubMed] [Google Scholar]
- Bikkavilli RK, Feigin ME, Malbon CC. p38 mitogen-activated protein kinase regulates canonical Wnt–β-catenin signaling by inactivation of GSK3β. J Cell Sci. 2008;121:3598–3607. doi: 10.1242/jcs.032854. [DOI] [PubMed] [Google Scholar]
- Braz JC, Bueno OF, Liang Q, Wilkins BJ, Dai YS, Parsons S, et al. Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin-NFAT signaling. J Clin Invest. 2003;111:1475–1486. doi: 10.1172/JCI17295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewster JL, de Valoir T, Dwyer ND, Winter E, Gustin MC. An osmosensing signal transduction pathway in yeast. Science. 1993;259:1760–1763. doi: 10.1126/science.7681220. [DOI] [PubMed] [Google Scholar]
- Brooks AC, Menzies-Gow NJ, Wheeler-Jones C, Bailey SR, Cunningham FM, Elliott J. Endotoxin-induced activation of equine platelets: evidence for direct activation of p38 MAPK pathways and vasoactive mediator production. Inflamm Res. 2007;56:154–161. doi: 10.1007/s00011-006-6151-6. [DOI] [PubMed] [Google Scholar]
- Chang CI, Xu BE, Akella R, Cobb MH, Goldsmith EJ. Crystal structures of MAP kinase p38 complexed to the docking sites on its nuclear substrate MEF2A and activator MKK3b. Mol Cell. 2002;9:1241–1249. doi: 10.1016/s1097-2765(02)00525-7. [DOI] [PubMed] [Google Scholar]
- Cheriyan J, Webb AJ, Sarov-Blat L, Elkhawad M, Wallace SML, Maki-Petaja KM, et al. Inhibition of p38 mitogen-activated protein kinase improves nitric oxide-mediated vasodilatation and reduces inflammation in hypercholesterolemia. Circulation. 2011;123:515–523. doi: 10.1161/CIRCULATIONAHA.110.971986. [DOI] [PubMed] [Google Scholar]
- Choi H, Nguyen HN, Lamb FS. Inhibition of endocytosis exacerbates TNF-alpha-induced endothelial dysfunction via enhanced JNK and p38 activation. Am J Physiol Heart Circ Physiol. 2014;306:H1154–H1163. doi: 10.1152/ajpheart.00885.2013. [DOI] [PubMed] [Google Scholar]
- Cuenda A, Cohen P, Buee-Scherrer V, Goedert M. Activation of stress-activated protein kinase-3 (SAPK3) by cytokines and cellular stresses is mediated via SAPKK3 (MKK6); comparison of the specificities of SAPK3 and SAPK2 (RK/p38) EMBO J. 1997;16:295–305. doi: 10.1093/emboj/16.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jong AM, Maass AH, Oberdorf-Maass SU, De Boer RA, Van Gilst WH, Van Gelder IC. Cyclical stretch induces structural changes in atrial myocytes. J Cell Mol Med. 2013;17:743–753. doi: 10.1111/jcmm.12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Nicola GF, Martin ED, Chaikuad A, Bassi R, Clark J, Martino L, et al. Mechanism and consequence of the autoactivation of p38alpha mitogen-activated protein kinase promoted by TAB1. Nat Struct Mol Biol. 2013;20:1182–1190. doi: 10.1038/nsmb.2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derijard B, Raingeaud J, Barrett T, Wu IH, Han J, Ulevitch RJ, et al. Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science. 1995;267:682–685. doi: 10.1126/science.7839144. [DOI] [PubMed] [Google Scholar]
- Dingar D, Merlen C, Grandy S, Gillis MA, Villeneuve LR, Mamarbachi AM, et al. Effect of pressure overload-induced hypertrophy on the expression and localization of p38 MAP kinase isoforms in the mouse heart. Cell Signal. 2010;22:1634–1644. doi: 10.1016/j.cellsig.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duch A, de Nadal E, Posas F. The p38 and Hog1 SAPKs control cell cycle progression in response to environmental stresses. FEBS Lett. 2012;586:2925–2931. doi: 10.1016/j.febslet.2012.07.034. [DOI] [PubMed] [Google Scholar]
- Elkhawad M, Rudd JH, Sarov-Blat L, Cai G, Wells R, Davies LC, et al. Effects of p38 mitogen-activated protein kinase inhibition on vascular and systemic inflammation in patients with atherosclerosis. JACC Cardiovasc Imaging. 2012;5:911–922. doi: 10.1016/j.jcmg.2012.02.016. [DOI] [PubMed] [Google Scholar]
- Engel FB, Hsieh PC, Lee RT, Keating MT. FGF1/p38 MAP kinase inhibitor therapy induces cardiomyocyte mitosis, reduces scarring, and rescues function after myocardial infarction. Proc Natl Acad Sci U S A. 2006;103:15546–15551. doi: 10.1073/pnas.0607382103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyers PA, Craxton M, Morrice N, Cohen P, Goedert M. Conversion of SB 203580-insensitive MAP kinase family members to drug-sensitive forms by a single amino-acid substitution. Chem Biol. 1998;5:321–328. doi: 10.1016/s1074-5521(98)90170-3. [DOI] [PubMed] [Google Scholar]
- Ge B, Gram H, Di Padova F, Huang B, New L, Ulevitch RJ, et al. MAPKK-independent activation of p38alpha mediated by TAB1-dependent autophosphorylation of p38alpha. Science. 2002;295:1291–1294. doi: 10.1126/science.1067289. [DOI] [PubMed] [Google Scholar]
- Gray WD, Che P, Brown M, Ning X, Murthy N, Davis ME. N-acetylglucosamine conjugated to nanoparticles enhances myocyte uptake and improves delivery of a small molecule p38 inhibitor for post-infarct healing. J Cardiovasc Transl Res. 2011;4:631–643. doi: 10.1007/s12265-011-9292-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gum RJ, McLaughlin MM, Kumar S, Wang Z, Bower MJ, Lee JC, et al. Acquisition of sensitivity of stress-activated protein kinases to the p38 inhibitor, SB 203580, by alteration of one or more amino acids within the ATP binding pocket. J Biol Chem. 1998;273:15605–15610. doi: 10.1074/jbc.273.25.15605. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Cuenda A, Spillantini MG, Thomas GM, Buée-Scherrer V, Cohen P, et al. Stress-activated protein kinase-3 interacts with the PDZ domain of α1-syntrophin: a mechanism for specific substrate recognition. J Biol Chem. 1999;274:12626–12631. doi: 10.1074/jbc.274.18.12626. [DOI] [PubMed] [Google Scholar]
- Humphreys JM, Piala AT, Akella R, He H, Goldsmith EJ. Precisely ordered phosphorylation reactions in the p38 MAP kinase cascade. J Biol Chem. 2013;288:23322–23330. doi: 10.1074/jbc.M113.462101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju H, Behm DJ, Nerurkar S, Eybye ME, Haimbach RE, Olzinski AR, et al. p38 MAPK inhibitors ameliorate target organ damage in hypertension: part 1. p38 MAPK-dependent endothelial dysfunction and hypertension. J Pharmacol Exp Ther. 2003;307:932–938. doi: 10.1124/jpet.103.057422. [DOI] [PubMed] [Google Scholar]
- Kaiser RA, Bueno OF, Lips DJ, Doevendans PA, Jones F, Kimball TF, et al. Targeted inhibition of p38 mitogen-activated protein kinase antagonizes cardiac injury and cell death following ischemia-reperfusion in vivo. J Biol Chem. 2004;279:15524–15530. doi: 10.1074/jbc.M313717200. [DOI] [PubMed] [Google Scholar]
- Kassan M, Choi SK, Galan M, Lee YH, Trebak M, Matrougui K. Enhanced p22phox expression impairs vascular function through p38 and ERK1/2 MAP kinase-dependent mechanisms in type 2 diabetic mice. Am J Physiol Heart Circ Physiol. 2014;306:H972–H980. doi: 10.1152/ajpheart.00872.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivisto E, Kaikkonen L, Tokola H, Pikkarainen S, Aro J, Pennanen H, et al. Distinct regulation of B-type natriuretic peptide transcription by p38 MAPK isoforms. Mol Cell Endocrinol. 2011;338:18–27. doi: 10.1016/j.mce.2011.02.015. [DOI] [PubMed] [Google Scholar]
- Kuma Y, Sabio G, Bain J, Shpiro N, Marquez R, Cuenda A. BIRB796 inhibits all p38 MAPK isoforms in vitro and in vivo. J Biol Chem. 2005;280:19472–19479. doi: 10.1074/jbc.M414221200. [DOI] [PubMed] [Google Scholar]
- Kumar S, McDonnell PC, Gum RJ, Hand AT, Lee JC, Young PR. Novel homologues of CSBP/p38 MAP kinase: activation, substrate specificity and sensitivity to inhibition by pyridinyl imidazoles. Biochem Biophys Res Commun. 1997;235:533–538. doi: 10.1006/bbrc.1997.6849. [DOI] [PubMed] [Google Scholar]
- Kumphune S, Bassi R, Jacquet S, Sicard P, Clark JE, Verma S, et al. A chemical genetic approach reveals that p38alpha MAPK activation by diphosphorylation aggravates myocardial infarction and is prevented by the direct binding of SB203580. J Biol Chem. 2010;285:2968–2975. doi: 10.1074/jbc.M109.079228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau JM, Jin X, Ren J, Avery J, DeBosch BJ, Treskov I, et al. The 14-3-3tau phosphoserine-binding protein is required for cardiomyocyte survival. Mol Cell Biol. 2007;27:1455–1466. doi: 10.1128/MCB.01369-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner C, Zahalka MA, Giot JF, Møller NP, Ullrich A. ERK6, a mitogen-activated protein kinase involved in C2C12 myoblast differentiation. PNAS. 1996;93:4355–4359. doi: 10.1073/pnas.93.9.4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- Li J, Miller EJ, Ninomiya-Tsuji J, Russell RR, Young LH. Amp-activated protein kinase activates p38 mitogen-activated protein kinase by increasing recruitment of p38 MAPK to TAB1 in the ischemic heart. Circ Res. 2005;97:872–879. doi: 10.1161/01.RES.0000187458.77026.10. [DOI] [PubMed] [Google Scholar]
- Lu YY, Chen YC, Kao YH, Chen SA, Chen YJ. Extracellular matrix of collagen modulates arrhythmogenic activity of pulmonary veins through p38 MAPK activation. J Mol Cell Cardiol. 2013;59:159–166. doi: 10.1016/j.yjmcc.2013.03.008. [DOI] [PubMed] [Google Scholar]
- Ma XL, Kumar S, Gao F, Louden CS, Lopez BL, Christopher TA, et al. Inhibition of p38 mitogen-activated protein kinase decreases cardiomyocyte apoptosis and improves cardiac function after myocardial ischemia and reperfusion. Circulation. 1999;99:1685–1691. doi: 10.1161/01.cir.99.13.1685. [DOI] [PubMed] [Google Scholar]
- MacKay K, Mochly-Rosen D. An inhibitor of p38 mitogen-activated protein kinase protects neonatal cardiac myocytes from ischemia. J Biol Chem. 1999;274:6272–6279. doi: 10.1074/jbc.274.10.6272. [DOI] [PubMed] [Google Scholar]
- Marais E, Genade S, Huisamen B, Strijdom JG, Moolman JA, Lochner A. Activation of p38 MAPK induced by a multi-cycle ischaemic preconditioning protocol is associated with attenuated p38 MAPK activity during sustained ischaemia and reperfusion. J Mol Cell Cardiol. 2001;33:769–778. doi: 10.1006/jmcc.2001.1347. [DOI] [PubMed] [Google Scholar]
- Martin ED, De Nicola GF, Marber MS. New therapeutic targets in cardiology. Circulation. 2012;126:357–368. doi: 10.1161/CIRCULATIONAHA.111.071886. [DOI] [PubMed] [Google Scholar]
- Martin JL, Avkiran M, Quinlan RA, Cohen P, Marber MS. Antiischemic effects of SB203580 are mediated through the inhibition of p38alpha mitogen-activated protein kinase: evidence from ectopic expression of an inhibition-resistant kinase. Circ Res. 2001;89:750–752. doi: 10.1161/hh2101.099504. [DOI] [PubMed] [Google Scholar]
- Morris JB, Olzinski AR, Bernard RE, Aravindhan K, Mirabile RC, Boyce R, et al. p38 MAPK inhibition reduces aortic ultrasmall superparamagnetic iron oxide uptake in a mouse model of atherosclerosis: MRI assessment. Arterioscler Thromb Vasc Biol. 2008;28:265–271. doi: 10.1161/ATVBAHA.107.151175. [DOI] [PubMed] [Google Scholar]
- Nagarkatti DS, Sha'afi RI. Role of p38 MAP kinase in myocardial stress. J Mol Cell Cardiol. 1998;30:1651–1664. doi: 10.1006/jmcc.1998.0733. [DOI] [PubMed] [Google Scholar]
- Newby LK, Marber MS, Melloni C, Sarov-Blat L, Aberle LH, Aylward PE, et al. Losmapimod, a novel p38 mitogen-activated protein kinase inhibitor, in non-ST-segment elevation myocardial infarction: a randomised phase 2 trial. Lancet. 2014;384:1187–1195. doi: 10.1016/S0140-6736(14)60417-7. [DOI] [PubMed] [Google Scholar]
- Nishida K, Yamaguchi O, Hirotani S, Hikoso S, Higuchi Y, Watanabe T, et al. p38alpha mitogen-activated protein kinase plays a critical role in cardiomyocyte survival but not in cardiac hypertrophic growth in response to pressure overload. Mol Cell Biol. 2004;24:10611–10620. doi: 10.1128/MCB.24.24.10611-10620.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Keefe SJ, Mudgett JS, Cupo S, Parsons JN, Chartrain NA, Fitzgerald C, et al. Chemical genetics define the roles of p38alpha and p38beta in acute and chronic inflammation. J Biol Chem. 2007;282:34663–34671. doi: 10.1074/jbc.M704236200. [DOI] [PubMed] [Google Scholar]
- Otsu K, Yamashita N, Nishida K, Hirotani S, Yamaguchi O, Watanabe T, et al. Disruption of a single copy of the p38alpha MAP kinase gene leads to cardioprotection against ischemia-reperfusion. Biochem Biophys Res Commun. 2003;302:56–60. doi: 10.1016/s0006-291x(03)00096-2. [DOI] [PubMed] [Google Scholar]
- Pargellis C, Tong L, Churchill L, Cirillo PF, Gilmore T, Graham AG, et al. Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat Struct Biol. 2002;9:268–272. doi: 10.1038/nsb770. [DOI] [PubMed] [Google Scholar]
- Parker CG, Hunt J, Diener K, McGinley M, Soriano B, Keesler GA, et al. Identification of stathmin as a novel substrate for p38 delta. Biochem Biophys Res Commun. 1998;249:791–796. doi: 10.1006/bbrc.1998.9250. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098-106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor BM, Jin X, Lupu TS, Muglia LJ, Semenkovich CF, Muslin AJ. Requirement for p38 mitogen-activated protein kinase activity in neointima formation after vascular injury. Circulation. 2008;118:658–666. doi: 10.1161/CIRCULATIONAHA.107.734848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raingeaud J, Whitmarsh AJ, Barrett T, Derijard B, Davis RJ. MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol Cell Biol. 1996;16:1247–1255. doi: 10.1128/mcb.16.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajesh M, Mukhopadhyay P, Hasko G, Liaudet L, Mackie K, Pacher P. Cannabinoid-1 receptor activation induces reactive oxygen species-dependent and -independent mitogen-activated protein kinase activation and cell death in human coronary artery endothelial cells. Br J Pharmacol. 2010;160:688–700. doi: 10.1111/j.1476-5381.2010.00712.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch BH, Muschenborn B, Braun M, Weber AA, Schror K. ICAM-1 and p38 MAPK mediate fibrinogen-induced migration of human vascular smooth muscle cells. Eur J Pharmacol. 2007;577:54–57. doi: 10.1016/j.ejphar.2007.08.041. [DOI] [PubMed] [Google Scholar]
- Regot S, de Nadal E, Rodríguez-Navarro S, González-Novo A, Pérez-Fernandez J, Gadal O, et al. The Hog1 stress-activated protein kinase targets nucleoporins to control mRNA export upon stress. J Biol Chem. 2013;288:17384–17398. doi: 10.1074/jbc.M112.444042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remy G, Risco AM, Inesta-Vaquera FA, Gonzalez-Teran B, Sabio G, Davis RJ, et al. Differential activation of p38MAPK isoforms by MKK6 and MKK3. Cell Signal. 2010;22:660–667. doi: 10.1016/j.cellsig.2009.11.020. [DOI] [PubMed] [Google Scholar]
- Ren J, Zhang S, Kovacs A, Wang Y, Muslin AJ. Role of p38alpha MAPK in cardiac apoptosis and remodeling after myocardial infarction. J Mol Cell Cardiol. 2005;38:617–623. doi: 10.1016/j.yjmcc.2005.01.012. [DOI] [PubMed] [Google Scholar]
- Riad A, Unger D, Du J, Westermann D, Mohr Z, Sobirey M, et al. Chronic inhibition of p38MAPK improves cardiac and endothelial function in experimental diabetes mellitus. Eur J Pharmacol. 2007;554:40–45. doi: 10.1016/j.ejphar.2006.08.065. [DOI] [PubMed] [Google Scholar]
- Sabio G, Reuver S, Feijoo C, Hasegawa M, Thomas GM, Centeno F, et al. Stress- and mitogen-induced phosphorylation of the synapse-associated protein SAP90/PSD-95 by activation of SAPK3/p38gamma and ERK1/ERK2. Biochem J. 2004;380(Pt 1):19–30. doi: 10.1042/BJ20031628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabio G, Arthur JS, Kuma Y, Peggie M, Carr J, Murray-Tait V, et al. p38gamma regulates the localisation of SAP97 in the cytoskeleton by modulating its interaction with GKAP. EMBO J. 2005;24:1134–1145. doi: 10.1038/sj.emboj.7600578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai K, Matsuo Y, Sudo T, Takuwa Y, Kimura S, Kasuya Y. Role of p38 mitogen-activated protein kinase in thrombus formation. J Recept Signal Transduct Res. 2004;24:283–296. doi: 10.1081/rrs-200040324. [DOI] [PubMed] [Google Scholar]
- Sanada S, Kitakaze M, Papst PJ, Hatanaka K, Asanuma H, Aki T, et al. Role of phasic dynamism of p38 mitogen-activated protein kinase activation in ischemic preconditioning of the canine heart. Circ Res. 2001;88:175–180. doi: 10.1161/01.res.88.2.175. [DOI] [PubMed] [Google Scholar]
- Sarov-Blat L, Morgan JM, Fernandez P, James R, Fang Z, Hurle MR, et al. Inhibition of p38 mitogen-activated protein kinase reduces inflammation after coronary vascular injury in humans. Arterioscler Thromb Vasc Biol. 2010;30:2256–2263. doi: 10.1161/ATVBAHA.110.209205. [DOI] [PubMed] [Google Scholar]
- Saurin AT, Martin JL, Heads RJ, Foley C, Mockridge JW, Wright MJ, et al. The role of differential activation of p38-mitogen-activated protein kinase in preconditioned ventricular myocytes. FASEB J. 2000;14:2237–2246. doi: 10.1096/fj.99-0671com. [DOI] [PubMed] [Google Scholar]
- Schulz R, Gres P, Skyschally A, Duschin A, Belosjorow S, Konietzka I, et al. Ischemic preconditioning preserves connexin 43 phosphorylation during sustained ischemia in pig hearts in vivo. FASEB J. 2003;17:1355–1357. doi: 10.1096/fj.02-0975fje. [DOI] [PubMed] [Google Scholar]
- See F, Thomas W, Way K, Tzanidis A, Kompa A, Lewis D, et al. p38 mitogen-activated protein kinase inhibition improves cardiac function and attenuates left ventricular remodeling following myocardial infarction in the rat. J Am Coll Cardiol. 2004;44:1679–1689. doi: 10.1016/j.jacc.2004.07.038. [DOI] [PubMed] [Google Scholar]
- Seeger F, Sedding D, Langheinrich A, Haendeler J, Zeiher A, Dimmeler S. Inhibition of the p38 MAP kinase in vivo improves number and functional activity of vasculogenic cells and reduces atherosclerotic disease progression. Basic Res Cardiol. 2010;105:389–397. doi: 10.1007/s00395-009-0072-9. [DOI] [PubMed] [Google Scholar]
- Shen MY, Hsiao G, Liu CL, Fong TH, Lin KH, Chou DS, et al. Inhibitory mechanisms of resveratrol in platelet activation: pivotal roles of p38 MAPK and NO/cyclic GMP. Br J Haematol. 2007;139:475–485. doi: 10.1111/j.1365-2141.2007.06788.x. [DOI] [PubMed] [Google Scholar]
- Sicard P, Clark JE, Jacquet S, Mohammadi S, Arthur JS, O'Keefe SJ, et al. The activation of p38 alpha, and not p38 beta, mitogen-activated protein kinase is required for ischemic preconditioning. J Mol Cell Cardiol. 2010;48:1324–1328. doi: 10.1016/j.yjmcc.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Ishibashi M, Seimon T, Lee M, Sharma SM, Fitzgerald KA, et al. Free cholesterol accumulation in macrophage membranes activates Toll-like receptors and p38 mitogen-activated protein kinase and induces cathepsin K. Circ Res. 2009;104:455–465. doi: 10.1161/CIRCRESAHA.108.182568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surinkaew S, Kumphune S, Chattipakorn S, Chattipakorn N. Inhibition of p38 MAPK during ischemia, but not reperfusion, effectively attenuates fatal arrhythmia in ischemia/reperfusion heart. J Cardiovasc Pharmacol. 2013;61:133–141. doi: 10.1097/FJC.0b013e318279b7b1. [DOI] [PubMed] [Google Scholar]
- Sy JC, Seshadri G, Yang SC, Brown M, Oh T, Dikalov S, et al. Sustained release of a p38 inhibitor from non-inflammatory microspheres inhibits cardiac dysfunction. Nat Mater. 2008;7:863–868. doi: 10.1038/nmat2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang K, Li X, Zheng MQ, Rozanski GJ. Role of apoptosis signal-regulating kinase-1-c-Jun NH2-terminal kinase-p38 signaling in voltage-gated K+ channel remodeling of the failing heart: regulation by thioredoxin. Antioxid Redox Signal. 2011;14:25–35. doi: 10.1089/ars.2010.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanno M, Bassi R, Gorog DA, Saurin AT, Jiang J, Heads RJ, et al. Diverse mechanisms of myocardial p38 mitogen-activated protein kinase activation: evidence for MKK-independent activation by a TAB1-associated mechanism contributing to injury during myocardial ischemia. Circ Res. 2003;93:254–261. doi: 10.1161/01.RES.0000083490.43943.85. [DOI] [PubMed] [Google Scholar]
- Taylor SS, Kornev AP. Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem Sci. 2011;36:65–77. doi: 10.1016/j.tibs.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenhunen O, Soini Y, Ilves M, Rysa J, Tuukkanen J, Serpi R, et al. p38 kinase rescues failing myocardium after myocardial infarction: evidence for angiogenic and anti-apoptotic mechanisms. FASEB J. 2006;20:1907–1909. doi: 10.1096/fj.05-5618fje. [DOI] [PubMed] [Google Scholar]
- Thakur S, Du J, Hourani S, Ledent C, Li JM. Inactivation of adenosine A2A receptor attenuates basal and angiotensin II-induced ROS production by Nox2 in endothelial cells. J Biol Chem. 2010;285:40104–40113. doi: 10.1074/jbc.M110.184606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton TM, Pedraza-Alva G, Deng B, Wood CD, Aronshtam A, Clements JL, et al. Phosphorylation by p38 MAPK as an alternative pathway for GSK3β inactivation. Science. 2008;320:667–670. doi: 10.1126/science.1156037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong H, Chen W, London RE, Murphy E, Steenbergen C. Preconditioning enhanced glucose uptake is mediated by p38 MAP kinase not by phosphatidylinositol 3-kinase. J Biol Chem. 2000;275:11981–11986. doi: 10.1074/jbc.275.16.11981. [DOI] [PubMed] [Google Scholar]
- Vahebi S, Ota A, Li M, Warren CM, de Tombe PP, Wang Y, et al. p38-MAPK induced dephosphorylation of alpha-tropomyosin is associated with depression of myocardial sarcomeric tension and ATPase activity. Circ Res. 2007;100:408–415. doi: 10.1161/01.RES.0000258116.60404.ad. [DOI] [PubMed] [Google Scholar]
- Vega-Ostertag ME, Ferrara DE, Romay-Penabad Z, Liu X, Taylor WR, Colden-Stanfield M, et al. Role of p38 mitogen-activated protein kinase in antiphospholipid antibody-mediated thrombosis and endothelial cell activation. J Thromb Haemost. 2007;5:1828–1834. doi: 10.1111/j.1538-7836.2007.02680.x. [DOI] [PubMed] [Google Scholar]
- Wang Q, Feng J, Wang J, Zhang X, Zhang D, Zhu T, et al. Disruption of TAB1/p38alpha interaction using a cell-permeable peptide limits myocardial ischemia/reperfusion injury. Mol Ther. 2013;21:1668–1677. doi: 10.1038/mt.2013.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Cui L, Joseph J, Jiang B, Pimental D, Handy DE, et al. Homocysteine induces cardiomyocyte dysfunction and apoptosis through p38 MAPK-mediated increase in oxidant stress. J Mol Cell Cardiol. 2012;52:753–760. doi: 10.1016/j.yjmcc.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Harkins PC, Ulevitch RJ, Han J, Cobb MH, Goldsmith EJ. The structure of mitogen-activated protein kinase p38 at 2.1-Ã resolution. Proc Natl Acad Sci U S A. 1997;94:2327–2332. doi: 10.1073/pnas.94.6.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinbrenner C, Liu GS, Cohen MV, Downey JM. Phosphorylation of tyrosine 182 of p38 mitogen-activated protein kinase correlates with the protection of preconditioning in the rabbit heart. J Mol Cell Cardiol. 1997;29:2383–2391. doi: 10.1006/jmcc.1997.0473. [DOI] [PubMed] [Google Scholar]
- Widder J, Behr T, Fraccarollo D, Hu K, Galuppo P, Tas P, et al. Vascular endothelial dysfunction and superoxide anion production in heart failure are p38 MAP kinase-dependent. Cardiovasc Res. 2004;63:161–167. doi: 10.1016/j.cardiores.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Willemen HLDM, Campos PM, Lucas E, Morreale A, Gil-Redondo R, Agut J, et al. A novel p38 MAPK docking-groove-targeted compound is a potent inhibitor of inflammatory hyperalgesia. Biochem J. 2014;459:427–439. doi: 10.1042/BJ20130172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson KP, Fitzgibbon MJ, Caron PR, Griffith JP, Chen W, McCaffrey PG, et al. Crystal structure of p38 mitogen-activated protein kinase. J Biol Chem. 1996;271:27696–27700. doi: 10.1074/jbc.271.44.27696. [DOI] [PubMed] [Google Scholar]
- Yacoub D, Hachem A, Theoret JF, Gillis MA, Mourad W, Merhi Y. Enhanced levels of soluble CD40 ligand exacerbate platelet aggregation and thrombus formation through a CD40-dependent tumor necrosis factor receptor-associated factor-2/Rac1/p38 mitogen-activated protein kinase signaling pathway. Arterioscler Thromb Vasc Biol. 2010;30:2424–2433. doi: 10.1161/ATVBAHA.110.216143. [DOI] [PubMed] [Google Scholar]
- Yada M, Shimamoto A, Hampton CR, Chong AJ, Takayama H, Rothnie CL, et al. FR167653 diminishes infarct size in a murine model of myocardial ischemia-reperfusion injury. J Thorac Cardiovasc Surg. 2004;128:588–594. doi: 10.1016/j.jtcvs.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Yin H, Zhang J, Lin H, Wang R, Qiao Y, Wang B, et al. p38 mitogen-activated protein kinase inhibition decreases TNFalpha secretion and protects against left ventricular remodeling in rats with myocardial ischemia. Inflammation. 2008;31:65–73. doi: 10.1007/s10753-007-9050-2. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Goedert M. Sequential phosphorylation of tau protein by cAMP-dependent protein kinase and SAPK4/p38δ or JNK2 in the presence of heparin generates the AT100 epitope. J Neurochem. 2006;99:154–164. doi: 10.1111/j.1471-4159.2006.04052.x. [DOI] [PubMed] [Google Scholar]