

Abstract
Background and Purpose
Acute liver failure (ALF) is a severe and potentially lethal clinical syndrome. 3,3′-Diindolylmethane (DIM) is a natural plant-derived compound with anti-cancer activities. Recently, DIM has also been shown to have anti-inflammatory properties. Here, we tested the hypothesis that DIM would suppress endotoxin-induced ALF.
Experimental Approach
We investigated the therapeutic potential of DIM in a mouse model of D-galactosamine/Lipopolysaccharide (GalN/LPS)-induced ALF. The efficacy of DIM treatment was assessed by survival, liver histopathology, serum levels of alanine transaminase, pro-inflammatory cytokines and number of activated liver macrophages. Effects of DIM on the expression of two miRNAs, 106a and 20b, and their predicted target gene were measured by qRT-PCR and Western blotting. Effects of DIM on the release of TNF-α from RAW264.7 macrophages transfected with mimics of these miRNAs and activated by LPS was assessed by elisa.
Key Results
DIM treatment protected mice from ALF symptoms and reduced the number of activated liver macrophages. DIM increased expression of miR-106a and miR-20b in liver mononuclear cells and decreased expression of their predicted target gene IL-1 receptor-associated kinase 4 (IRAK4), involved in signalling from Toll-like receptor 4 (TLR4). In vitro transfection of RAW264.7 cells using miRNA mimics of miR-106a and 20b decreased expression of IRAK4 and of TNF-α secretion, following LPS stimulation.
Conclusions and Implications
DIM attenuated GalN/LPS-induced ALF by regulating the expression of unique miRNAs that target key molecules in the TLR4 inflammatory pathway. DIM may represent a potential novel hepatoprotective agent.
Tables of Links
| LiGANDS |
|---|
| CCL2 |
| IL-6 |
| LPS |
| TNFα |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guideto PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a,b,).
Introduction
Acute liver failure (ALF) is a severe life-threatening clinical syndrome caused by sudden and severe injury to the liver (Bernal et al., 2010). The syndrome involves rapid loss of liver function and in many cases can lead to multi-organ failure and even death (Bernal et al., 2010). The underlying pathological cause for ALF is the extensive necrosis and apoptosis of hepatocytes (Liang et al., 2013). Common causes for the syndrome include viral hepatitis, drug overdose, idiosyncratic drug reactions and toxins (Zhan et al., 2014). ALF is associated with a very high mortality of up to 80% (Bernal et al., 2010). Currently, there are no effective therapeutic strategies available against ALF and liver transplantation remains the only treatment option. However, non-availability of donors and other associated complications warrant the need to identify novel modalities to treat ALF (Bernal et al., 2010; Sarra et al., 2013).
Increasing evidence suggests that the activation of an acute inflammatory response plays a major role in the pathogenesis of ALF (Antoniades et al., 2008). Among other cell types, the macrophages resident in the liver, Kupffer cells (KCs), have been shown to play an important role in the development of acute liver injury in a number of ALF models (Antoniades et al., 2008). One such experimental model, induced by D-galactosamine and LPS (GalN/LPS), has been widely used to study the mechanisms of the pathogenesis of ALF and identify novel therapeutic drugs (Zhang et al., 2010). The liver injury induced in this model closely resembles clinical ALF (Silverstein, 2004). In GalN/LPS-induced ALF, LPS triggers an innate immune response by stimulating the Toll-like receptor 4 (TLR4) on KCs and infiltrating macrophages to produce pro-inflammatory cytokines such as TNF-α, IL-6 and the chemokine CCL2 (Liang et al., 2013). These cytokines and chemokines then mediate massive infiltration of other immune cells into the liver tissue to cause hepatocyte apoptosis and ultimately ALF (Liang et al., 2013). Among the pro-inflammatory cytokines, TNF-α is an important mediator that induces hepatocyte apoptosis to cause liver injury (Zhan et al., 2014). IL-1 receptor-associated kinases (IRAKs) are a family of proteins involved in signalling downstream of various TLRs (Flannery and Bowie, 2010). TLR4 stimulation by LPS leads to activation of IRAK4 and through a signal transduction pathway to the activation of NF-κB, which then promotes the transcription of various pro-inflammatory genes (Kim et al., 2007; Lu et al., 2008; Flannery and Bowie, 2010).
3,3′-Diindolylmethane (DIM) is a derivative product of its precursor indole-3-carbinol (I3C), a natural indole compound found in cruciferous vegetables, such as broccoli, kale and cauliflower (Cho et al., 2008). Upon exposure to acidic environment of the gastric juices, I3C undergoes condensation to generate several derivatives, including DIM (Cho et al., 2008). Both I3C and DIM have been extensively studied with respect to their anti-cancer properties (Ahmad et al., 2010; Banerjee et al., 2011). However, more recently, a role in immunosuppression has also been identified for these indole compounds in various models of inflammatory diseases (Kim et al., 2009; Dong et al., 2010; Busbee et al., 2013). Previous studies from our laboratory have also shown that both I3C and DIM attenuate disease symptoms in mouse models for experimental multiple sclerosis and staphylococcal enterotoxin B (SEB)-induced toxic shock (Rouse et al., 2013; Busbee et al., 2014). Furthermore, DIM has also been shown to decrease in vitro macrophage (Cho et al., 2008) and microglial (Kim et al., 2014) activation by down-regulating NF-κB activity. However, a mechanistic link between DIM and the decrease in LPS-induced NF-κB transactivation has not been identified. Moreover, the potential of DIM in the treatment of an animal model of ALF has not been studied so far.
miRNAs are a class of evolutionarily conserved short (∼22 nt) non-coding RNAs (Taganov et al., 2006) and they are involved in the regulation of almost all cellular and developmental processes by controlling the expression of a number of genes (Tili et al., 2007). To achieve this, the seed sequence of a miRNA binds the 3′ untranslated region of the target mRNA to cause a translational inhibition or degradation of the mRNA of the target gene. miRNAs have emerged as potential immunomodulators because of their role in a number of clinical disorders such as cancer and inflammatory diseases (O'Neill et al., 2011; Kohanbash and Okada, 2012). In addition, a number of studies have identified miRNAs to be important players in regulating different aspects of macrophage activation and TLR signalling pathways (Chaudhuri et al., 2011; O'Neill et al., 2011). In this study, we investigated if the ability of DIM to ameliorate GalN/LPS-induced ALF involved miRNAs. We found that DIM protected mice from ALF-associated lethality and attenuated disease parameters by suppressing macrophage activation in vivo. Furthermore, our results suggested that DIM protected mice from liver injury, at least in part, through regulation of key miRNAs that target molecules in the TLR4 signalling pathway and thus suppress macrophage activation.
Methods
Animals
All animals were housed and maintained under specific pathogen-free conditions at the AALAC-accredited animal facility of School of Medicine at the University of South Carolina. All experimental procedures complied with NIH guidelines and the protocols were pre-approved by the Institutional Animal Care and Use Committee of the University of South Carolina. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 60 animals were used in the experiments described here. Female C57BL/6 mice (8–10 weeks old) were purchased from the National Cancer Institute (Frederick, MD, USA).
Induction of ALF and treatment with DIM
Mice were randomly allocated to the experimental groups (12 in the group treated with GalN/LPS and 10 in the other three groups, DIM alone, GalN/LPS + DIM and vehicle alone). GalN and LPS (Sigma Aldrich, St. Louis, MO, USA) were dissolved in PBS. To induce ALF, mice were injected i.p. with a combination of GalN (800 mg·kg−1) and LPS (18 μg·kg−1). PBS was used as vehicle for these experiments. DIM (Sigma Aldrich) was dissolved initially in Dimethyl sulfoxide (DMSO) (Sigma Aldrich) and diluted further in corn oil (Sigma Aldrich). For treatment with DIM, mice were pretreated (i.p. injection) with two doses of 40 mg·kg−1 each: the first dose was given 24 h before the GalN/LPS injection and the second 2 h before the GalN/LPS injection. Blood samples were collected 2 and 12 h after GalN/LPS injection, and sera were separated and stored at −20°C until further use. Mice were observed for morbidity each hour after the injection of GalN/LPS and any mice that were very sick (lack of mobility, heavy breathing, tremors and raised fur) were sacrificed (with overdose of isoflurane). None of the mice were found dead during the course of our studies. At 12 h after GalN/LPS treatment, all mice were sacrificed (isoflurane) and liver tissues were collected for histology and isolation of mononuclear cells (MNCs). For some experiments (isolation of liver MNCs and expression of NF-κB; see Figure 6D), mice (groups of 4) were sacrificed at 1.5 h after the GalN/LPS injection. The liver tissues were frozen and stored at −80°C until further use.
Figure 6.

DIM increases the expression of miR-106a, miR-20b and miR-125b-5p to regulate TLR signalling in liver MNCs. Mice were treated with LPS + Veh or LPS + DIM. (A) miRWalk scores of alignment for the predicted interactions between miRNAs-106a and 20b with their target mRNA IRAK4. (B) Relative expression of miR-106a and miR-20b in liver MNCs as quantified by qRT-PCR at 12 and 2 h after GalN/LPS injection. (C) Immunoblotting of IRAK4 (55 kDa) and β-actin (42 kDa) in liver MNC samples at 12 h after GalN/LPS injection. (D) The expression of NF-κB p65 subunit (65 kDa) in the nuclear protein and cytoplasmic protein fractions of liver MNCs at 1.5 h after LPS administration. (E) Relative expression of miR-125b-5p in liver MNCs as quantified by qRT-PCR at 12 h after GalN/LPS injection. (F) Relative expression of TNF between MNCs from LPS + Veh and LPS + DIM groups at 12 h after GalN/LPS injection. Data are presented as means ± SEM (n = 4 per group). *P < 0.05, **P < 0.01, significantly different as indicated.
Serum cytokine analysis and alanine transaminase (ALT) activity assay
Serum samples collected 2 and 12 h after GalN/LPS injection were analysed for the levels of cytokines using sandwich elisa kits for TNF-α, IL-6 and CCL2 (BioLegend, San Diego, CA, USA). Serum samples collected 12 h after GalN/LPS injections were also analysed to measure levels of the liver enzyme ALT, using the quantitative ALT determination kit (Pointe Scientific, Canton, MI, USA).
Liver histology
Liver tissues isolated at 12 h after GalN/LPS injection were rinsed gently with PBS and fixed in 10% neutral buffered formalin for 24 h, paraffin embedded and sectioned using a microtome to obtain 5 μm thick sections. The sections were stained with haematoxylin-eosin and examined by light microscopy to evaluate tissues for histopathological damage. The sections were number coded before embedding in the paraffin blocks and the stained slides were evaluated without knowledge of the treatment groups.
Isolation of liver MNCs
The cells infiltrating the liver were isolated using a protocol described earlier (Dong et al., 2004). Briefly, livers were perfused with liver perfusion medium, cut into small pieces and digested using liver digestion medium (Gibco Life Technologies, Grand Island, NY, USA). The liver digest was diluted with 8 volumes of PBS, followed by centrifugation (50× g at 4°C for 5 min) to pellet the hepatocytes. MNCs were isolated from the supernatant using centrifugation through a Percoll density gradient (GE Healthcare Life Sciences, Pittsburgh, PA), as described (Dong et al., 2004). MNCs were then filtered using 70 μm nylon mesh filters, counted and stained with fluorochrome conjugated antibodies for the markers: CD11b, CD80 and CD86 (BioLegend) and analysed with the FC 500 flow cytometer (Beckman Coulter, Brea, CA, USA) using the CXP analysis software (Beckman Coulter).
RNA isolation, miRNA microarray, pathway analysis and real-time PCR
Total RNA was isolated from liver MNCs using miRNeasy kit (Qiagen, Valencia, CA, USA). Total RNA isolated from liver MNCs of the (LPS + Veh) group and the (LPS + DIM) group was analysed using Affymetrix microRNA microarray v.1.0, as described previously (Hegde et al., 2013). The miRNA microarray data was analysed using the Affymetrix miRNA QC Tool software, to identify miRNAs that were altered by more than 1.5-fold change in experimental groups, compared with controls. Statistical significance (p values) for ‘detection calls’ was determined by Affymetrix test by using a p value < 0.05. The log-transformed fluorescence intensity values were mean-centered and are shown in the form of the heat map. A scatter plot was developed using Microsoft Excel to show the miRNA that were increased or decreased in LPS + DIM group, compared to the LPS + Veh group.
The TLR pathway regulated by miRNAs was generated through the use of Ingenuity® Systems, Ingenuity Pathway Analysis (IPA) software (IPA®, QIAGEN Redwood City; www.qiagen.com/ingenuity). IPA was also used as described previously (Hegde et al., 2013) to identify the miRNAs in the dataset that were either strongly predicted or have been validated in published papers to target molecules in the TLR signalling pathway. In addition to IPA, we also used the miRWalk database (Dweep et al., 2011) to select miRNAs from the IPA-identified miRNA dataset to further shortlist miRNAs with a minimum predictive score of 4. qRT-PCR was performed to quantify the levels of miRNAs using miScript SYBR Green PCR kits and the following primer assays (Qiagen): mmu-miR-106a, mmu-miR-20b, mmu-miR-125b-5p. Snord61 was used as the reference gene for qRT-PCR.
Immunoblotting
Total protein was isolated from the liver tissues and MNCs collected 12 h after GalN/LPS injection using RIPA buffer (Santa Cruz Biotechnology, Dallas, TX, USA) supplemented with protease inhibitor cocktail (Thermo Fisher Scientific, Rockford, IL, USA). For investigating NF-κB activation and nuclear localization, nuclear and cytosolic protein samples were isolated from liver tissues obtained 1.5 h after GalN/LPS injection using the NE-PER nuclear and cytoplasmic extraction kit (Thermo Fisher Scientific). The protein samples were quantified using a BCA protein estimation kit (Thermo Fisher Scientific). Twenty micrograms of protein was loaded for each sample. The proteins were transferred to the nitrocellulose membrane using the wet transfer method. The membranes were blocked in 5% milk in Tris-buffered saline (pH 7.6), containing Tris base, sodium chloride and 0.1% Tween-20 (TBST) for 1 h and probed with the following antibodies (Cell Signaling, Danvers, MA, USA): β-actin (1:1000), NF-κB p65 subunit (1:500), IRAK4 (1:1000) and caspase-3 (1:1000).
Cell culture and transfections
The mouse macrophage cell line, RAW264.7, was cultured in complete DMEM containing 10% FBS (Life Technologies) and cells were allowed to adhere for 24 h. Thioglycollate-induced macrophages were isolated from the peritoneal cavity of mice, washed in medium, plated for 90 min at 37°C in a 5% CO2 incubator, harvested using a cell scraper, seeded at 0.5 million per well in 24-well plates and allowed to adhere for 24 h. Both cell types were then treated with vehicle (0.05% DMSO) or DIM (10 μM). One hour later, cells were treated with LPS (10 ng·mL−1 for RAW264.7 cells, and 10 and 100 ng·mL−1 for peritoneal macrophages). Twenty-four hours later, cell culture supernatants were obtained for elisas to quantify TNF-α secretion by the RAW264.7 cells and peritoneal macrophages. For transfections, miRNA mimics and negative controls were (Qiagen) used as described (Liu et al., 2009) at 40 nM and the cells were transfected using HiPerFect transfection reagent (Qiagen).
Data analysis
For experiments with mice, groups of 10–12 mice were used. For in vitro assays and qRT-PCRs, all experiments were performed in triplicate. Data have been shown as mean ± SEM wherever applicable. For statistical analysis, one-way anova was used to analyse for significance for each experiment, and Tukey's post hoc test was performed to analyse differences between groups. Survival curves were created using product limit method of Kaplan and Meier and analysed for statistical significance using log-rank (Mantel–Cox) test. A P-value of ≤0.05 was used to determine statistical significance. The graphs were plotted using GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA, USA) and densitometric analysis was performed using ImageJ software (http://rsb.info.nih.gov/ij/).
Results
Attenuation of ALF in DIM-treated mice
ALF induced by GalN/LPS is a well-established animal model for screening hepatoprotective compounds to identify new therapeutic agents. The model involves severe mortality and an increase in serum ALT levels of mice with ALF (Zhang et al., 2010). In this study, we investigated the possible use of DIM in the treatment of ALF induced by GalN/LPS (henceforth referred to as LPS-induced ALF). We used a dose of 40 mg·kg−1 of DIM in our studies based on our previous findings where DIM was able to attenuate other inflammatory disorders (Rouse et al., 2013; Busbee et al., 2014). All mice treated with vehicle or DIM alone survived. However, mice injected with LPS had a survival rate of 70% at 10 h and 50% at 12 h (Figure 1A). In contrast, mice pretreated with DIM and injected with LPS had 100% survival rate (Figure 1A). Injection with LPS increased the serum ALT levels 12 h later, and these raised levels were decreased by DIM pretreatment (Figure 1B). Also, mice injected with DIM alone did not show any increase in serum ALT levels (Figure 1B). Histological analysis of the liver tissues 12 h after injection with LPS showed severe tissue damage and infiltration of inflammatory cells (Figure 1C). On the other hand, liver tissues from the DIM pretreated group showed less tissue damage and a much reduced infiltration of inflammatory cells (Figure 1C). Mice injected with DIM alone did not show any mortality, increase in serum ALT levels or liver tissue damage over the 12h experimental period (Figure 1A–C).
Figure 1.
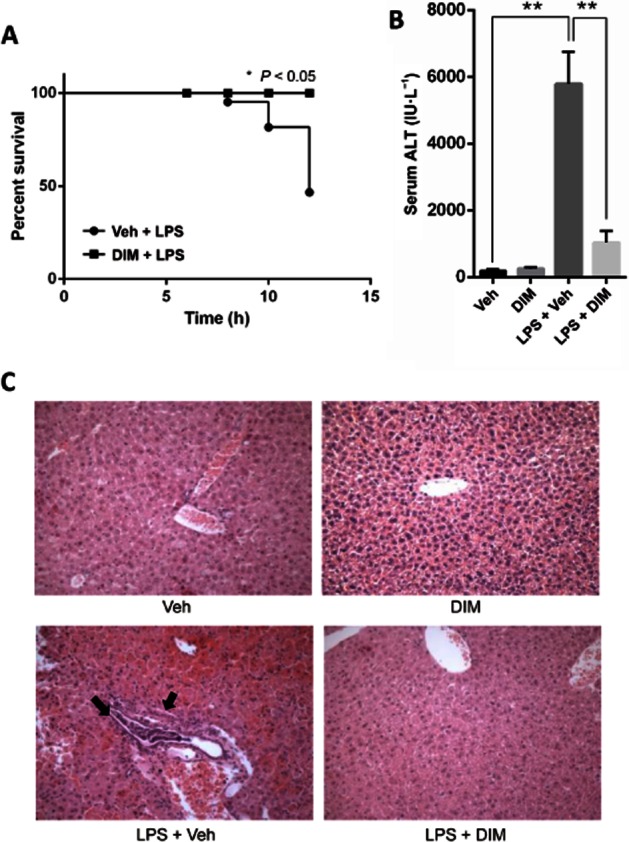
DIM treatment attenuates LPS-induced ALF. (A) Survival of mice (n = 10–12 per group) at different times after co-administration of GalN (800 mg·kg−1) + LPS (18 μg·kg−1) (shown as LPS) along with vehicle or DIM (40 mg·kg−1). (B) Sera collected at 12 h after LPS treatment were analysed for ALT levels. Data presented as mean ± SEM (n = 6 for the LPS + Veh group; n = 10–12 for the other groups). (C) Representative images of histological examination of livers (10×) from mice injected with vehicle alone, DIM alone, LPS or LPS + DIM following haematoxylin-eosin staining. *P < 0.05, **P < 0.01, significantly different as indicated.
DIM treatment decreases production of pro-inflammatory cytokines and liver apoptosis upon LPS injection
Pro-inflammatory cytokines have been shown to play a major role in the development and progression of ALF in the GalN/LPS model (Liang et al., 2013). Therefore we investigated the serum levels of pro-inflammatory cytokines, TNF-α, CCL2 and IL-6, 2 and 12 h after injecting mice with LPS and observed an increase in all these cytokines in LPS-injected mice at both 2 and 12 h (Figure 2A and B). On the other hand, in mice pretreated with DIM, the levels of these pro-inflammatory cytokines after LPS, were decreased (Figure 2A and B). Because hepatocyte apoptosis is a significant event (Liang et al., 2013) that leads to ALF and caspase-3 is a major mediator of apoptosis, we investigated the effect of DIM on LPS-induced apoptosis in liver tissues by performing immunoblotting on 12 h liver tissue protein samples to determine caspase-3 cleavage. LPS treatment triggered liver tissue apoptosis as shown by caspase-3 cleavage, while DIM pretreatment significantly reduced the caspase-3 cleavage (Figure 2C), showing an attenuation of apoptotic damage in the liver tissues.
Figure 2.
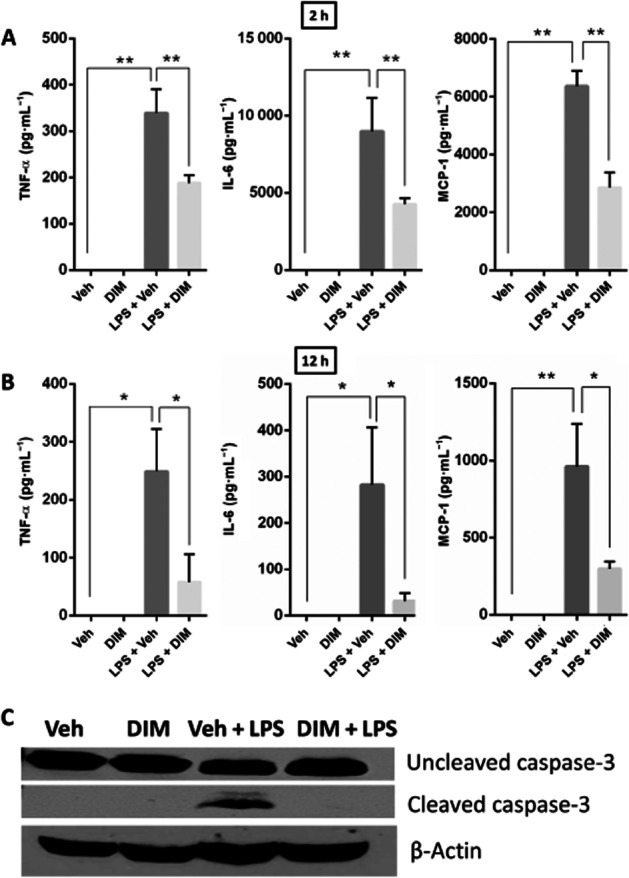
DIM treatment suppresses the production of systemic pro-inflammatory cytokines and apoptotic damage in livers of mice with LPS-induced ALF. Blood was collected from mice at (A) 2 and (B) 12 h after GalN + LPS (shown as LPS) injection. Sera were analysed for cytokines using sandwich elisa for TNF-α, CCL2 and IL-6. Data presented as mean ± SEM (n = 6 for the LPS + Veh group); n = 8 for the other groups). (C) Protein samples were isolated from liver tissues harvested 12 h after LPS injection and run on SDS-PAGE for immunoblotting to probe for caspase-3 (uncleaved: 35 kDa and cleaved: 17 kDa) and β-actin (42 kDa). *P < 0.05, **P < 0.01, significantly different as indicated.
DIM decreases liver infiltration of MNCs and decreases macrophage activation in the infiltrating MNCs
ALF induced by GalN/LPS is accompanied by an extensive infiltration of inflammatory cells in the liver tissues (Liang et al., 2013). Next, we determined the absolute cell numbers of MNCs that had infiltrated the liver tissues in different groups of mice. LPS-treated mice had a significant increase in the number of liver-infiltrating MNCs (Figure 3A), compared with the control mice injected with saline. The mice pretreated with DIM and then given LPS, in contrast, showed significantly decreased infiltration of MNCs in the liver tissues (Figure 3A). A number of studies have identified macrophages as playing a major role in ALF induced by GalN/LPS (Stuart et al., 2011). CD11b+ macrophages are the main phenotype that infiltrates liver during inflammatory responses (Holt et al., 2008; Ramachandran et al., 2012; Ikarashi et al., 2013; Nakashima et al., 2013). In our study, we found that LPS injection increased both the percentage and absolute numbers of CD11b+ macrophages in the liver (Figure 3B and D) and that pretreatment with DIM reduced the infiltration of these macrophages in the liver tissue, following LPS (Figure 3B and D). CD80 is an activation marker that has been shown to be up-regulated upon stimulation of macrophages with LPS (Xia et al., 2009). Furthermore, previous studies have shown that CD80+ macrophages are significantly increased in liver tissues of both ALF patients and mice with GalN/LPS-induced ALF and may contribute to the pathogenesis of liver failure (Leifeld et al., 1999). Therefore, we double-stained the CD11b+ macrophages for the marker CD80 and determined the proportion of macrophages expressing this activation marker CD80. Mice with LPS-induced ALF exhibited both significantly increased percentages and absolute cell numbers of macrophages expressing CD80, while DIM pretreatment decreased these effects (Figure 3C and D). These results together suggested that pretreatment with DIM suppressed macrophage activation in liver after GalN/LPS injection in vivo.
Figure 3.

DIM treatment reduces infiltration of MNCs and suppresses activation of infiltrating macrophages in vivo. Mice were treated with LPS + Veh or LPS + DIM as described earlier and 12h later, infiltrating MNCs were isolated. (A) The cells were counted to obtain absolute numbers of infiltrating cells in liver. Liver MNCs were stained using monoclonal antibodies for the macrophage marker CD11b and the activation marker CD80 and were analysed by flow cytometry to identify (B) CD11b+ cells and (C) CD11b+ CD80+ cells. Representative histogram plots for these cell populations are shown (B and C). (D) These data were used to calculate the absolute numbers of infiltrating CD11b+ macrophages and CD11b+ CD80+ macrophages in the liver using the formula: Total number of MNCs × Percentage of CD11b+ CD80+ macrophages/100. Data are presented as means ± SEM (n = 4 per group). *P < 0.05, **P < 0.01, significantly different as indicated.
DIM treatment suppresses the LPS-induced activation of RAW264.7 cells in vitro
We next investigated the effects of DIM on in vitro macrophage activation using the mouse macrophage cell line, RAW264.7. In our pilot studies, we performed a dose response of DIM and studied apoptosis of RAW264.7 cells and peritoneal macrophages, and found that a concentration of 10 μM was non-cytotoxic, and therefore that dose of DIM was used to study changes in macrophage activation and responses. RAW264.7 cells were treated with DIM (10 μM), and 1 h later, LPS (10 ng·mL−1) was added to the cultures to stimulate the cells. Flow cytometric analysis showed that RAW264.7 cells activated with LPS exhibited significant increase in the expression of activation markers CD80 (Figure 4A) and CD86 (Figure 4B), and the co-stimulatory molecule CD40 (Figure 4C). Treatment with DIM attenuated these increases in expression of these three markers in response to LPS (Figure 4A–C). LPS stimulation also triggered a robust increase in TNF-α, which was also decreased by pretreatment of the RAW264.7 cells with DIM (Figure 4D). DIM also decreased TNF-α secretion from LPS-stimulated peritoneal macrophages (Figure 4E).
Figure 4.

DIM treatment reduces LPS-induced activation of macrophages in vitro. RAW264.7 cells were treated with DIM (10 μM) or Veh. One hour later, the cells were stimulated with 10 ng·mL−1 of LPS. Twenty-four hours later, the cells were harvested to perform flow cytometry after staining with monoclonal antibodies for the activation markers: (A) CD80, (B) CD86 and the co-stimulatory molecule (C) CD40. Representative histogram plots for RAW264.7 cells stained with these markers are shown. (D) Levels of TNF-α in the culture supernatants were measured by elisa. (E) Peritoneal macrophages elicited with thioglycollate were treated with DIM (10 μM) and 1 h later LPS was added at a concentration of 100 or 10 ng·mL−1. Culture supernatants were obtained 24 h after LPS stimulation and analysed for TNF-α by elisa. Data are presented as means ± SEM (n = 3 per group). *P < 0.05, **P < 0.01, significantly different as indicated.
DIM treatment of LPS-injected mice significantly alters the miRNA profile of liver MNCs
miRNAs have been implicated in a number of disease pathologies and inflammatory disorders (Kohanbash and Okada, 2012). Thus, we tested the role of miRNAs in the protection by DIM against LPS-induced ALF, by performing a miRNA microarray analysis that screened for 609 miRNAs. In mice treated with DIM and LPS, there was a significant change in the miRNA expression profile of liver MNCs as seen in the heat map, compared with those from mice that received LPS alone (Figure 5A). Further analysis led us to identify 42 miRNAs that were up-regulated and 21 miRNAs that were down-regulated in DIM + LPS groups when compared with LPS + Veh groups (Figure 5B).
Figure 5.
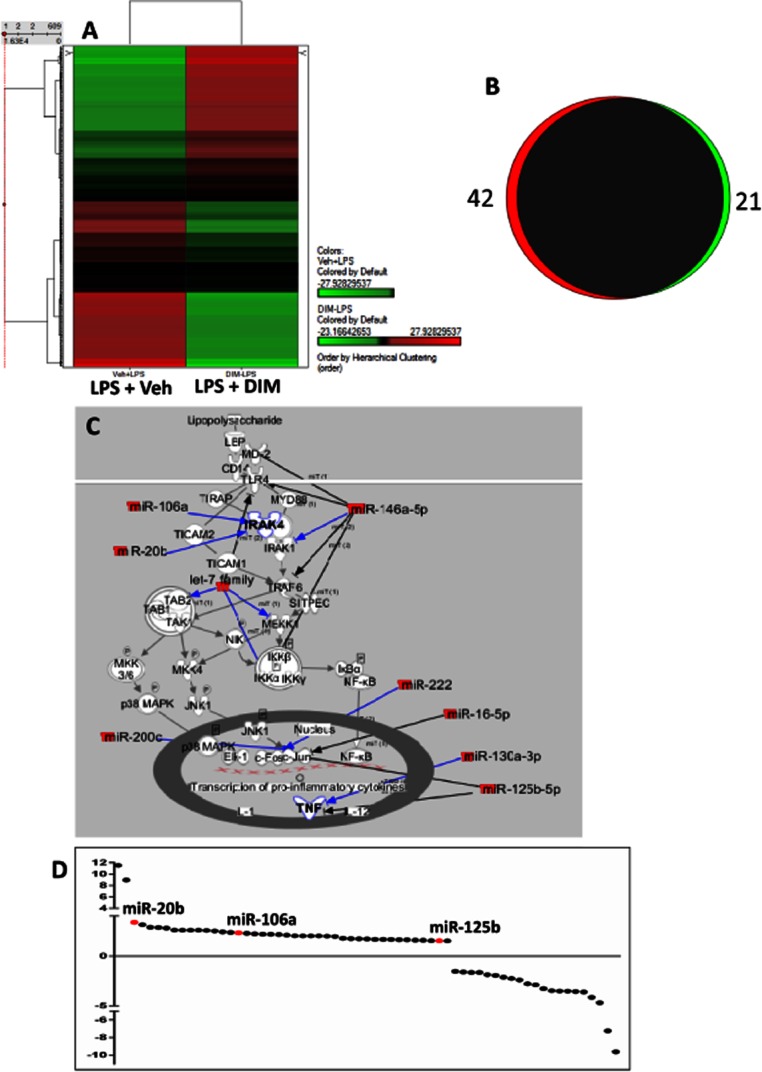
DIM treatment up-regulates a number of miRNAs in liver MNCs that can regulate TLR signalling. Mice were treated with LPS + Veh or LPS + DIM. (A) Heat map of 602 miRNAs differentially altered between liver MNCs of Veh + LPS and DIM + LPS groups. (B) A Venn diagram to show the number of miRNAs up-regulated (in red) and down-regulated (in green) by more than 1.5-fold in the DIM + LPS group, compared with controls. (C) An IPA pathway analysis of TLR pathway based on the predicted (blue arrows) and confirmed (black arrows) regulation by several miRNAs identified. (D) Scatter plot of miRNAs obtained from the microarray dataset that were altered by more than 1.5-fold in the DIM + LPS group. The miRNAs: 106a, 20b and 125b-5p have been coloured in red.
We then performed pathway analysis using IPA to identify the miRNAs in the dataset that could potentially regulate the TLR signalling pathway or those that have been already shown to regulate this pathway, by published results (Figure 5C, black arrows). In addition to IPA, we also used the miRWalk database (Dweep et al., 2011) to select miRNAs from the IPA miRNA dataset to further shortlist miRNAs with a minimum predictive score of 4. These steps provided a list of miRNAs that were significantly altered in the LPS + DIM group and were strongly predicted to regulate the expression of various target molecules in TLR signalling pathway (shown in blue arrows in Figure 5C). From these analyses, we identified three miRNAs that were up-regulated following DIM treatment of GalN/LPS-injected mice and potentially targeted the TLR signalling pathway: miR-106a, miR-20b and miR-125b-5p (Figure 5D).
DIM treatment increases the expression of miRs-106a, 20b and 125b to regulate IRAK4 and TNF-α expression
miRNAs-106a and 20b that were up-regulated by DIM treatment belong to the miR-17 family. These miRNAs were predicted to target IRAK4 with a miRWalk analysis score of 4 for each miR-mRNA alignment (Figure 6A). IRAK4 is a crucial mediator of TLR4 signalling located upstream in the TLR signalling pathway involved in the transcription of pro-inflammatory genes (Li and Qin, 2005). miR-125b-5p has been shown previously to regulate the responses to LPS-activated TLRs and to down-regulate TNF-α (Tili et al., 2007). We confirmed the expression of miR-106a and miR-20b using qRT-PCR in liver MNCs isolated at 2 and 12 h after LPS injection and demonstrated that they were indeed up-regulated in DIM + LPS treated groups, compared with levels after LPS treatment alone (Figure 6B). We then investigated the expression of IRAK4 in liver MNCs by immunoblotting and found it to be down-regulated in liver MNCs from DIM + LPS mice, compared with that in LPS-treated groups (Figure 6C). Because NF-κB activation and nuclear translocation is an event downstream of the IRAK4 activation in the TLR signalling pathway, we also investigated the nuclear translocation of NF-κB in liver MNCs. The data suggested that NF-κB translocation was increased in LPS-treated mice, while DIM treatment decreased this translocation (Figure 6D). We also confirmed the expression of miR-125b-5p and found it to be increased in liver MNCs isolated at 12 h after GalN/LPS injection (Figure 6E). The expression of TNF-α was also investigated in the liver MNCs and we noted that DIM pretreatment of LPS-injected mice decreased its expression, compared with that in the mice treated with LPS + Veh (Figure 6F).
miR-106a and miR-20b mimics inhibit LPS-induced increase in TNF-α secretion by regulating IRAK4 expression
We next investigated the effects of miRNA mimics on LPS-mediated activation of RAW264.7 cells. Mimics for miRNAs-106a and 20b suppressed the secretion of TNF-α upon stimulation by LPS, compared with the transfection control (Figure 7A and B). In RAW264.7 cells stimulated with LPS, these mimics also decreased expression of IRAK4 (Figure 7C and D). Together, these results suggested that, in RAW264.7 cells, DIM may attenuate LPS-mediated induction of TNF-α through up-regulation of miRs-106a and miR-20b that target IRAK4.
Figure 7.
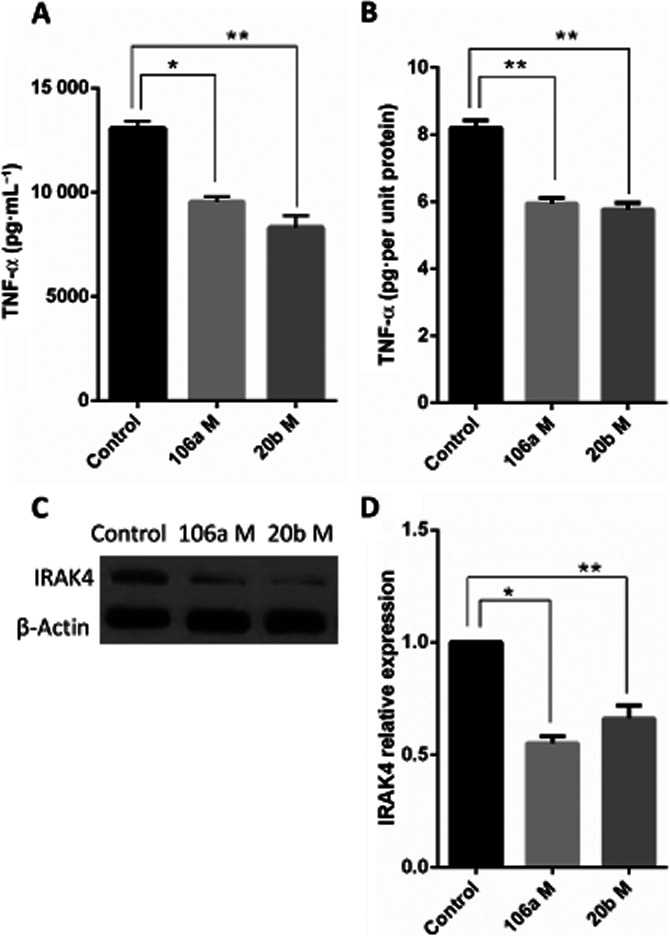
miR-106a and miR-20b regulate IRAK4 expression to suppress LPS-induced TLR responses. RAW264.7 cells were transfected with transfection control (mock) or miRNA mimics for miRNAs-106a and 20b. Twenty-four hours later, LPS was added at 10 ng·mL−1. (A) Cells and culture supernatants were obtained 24 h after LPS stimulation and the supernatants were analysed for TNF-α by elisa. (B) Total protein was isolated from the harvested cells to normalize the cytokine production to total protein content for each group. (C) Immunoblotting was performed for IRAK4 and the obtained data were quantified using (D) densitometric analysis by ImageJ software. Data are presented as means ± SEM (n = 3 per group). *P < 0.05, **P < 0.01, significantly different as indicated.
Discussion and conclusions
ALF is a severe clinical disorder caused by massive injury to the liver (Bernal et al., 2010). Over the past few decades, the role of LPS in the initiation and progression of a number of acute and chronic inflammatory liver pathologies has been established (Nolan, 2010; Zhan et al., 2014). This evidence and the similarity of the pathology of GalN/LPS-induced model of ALF to clinical ALF (Silverstein, 2004) make this model highly likely to identify new hepatoprotective drugs. Inasmuch as there is considerable evidence to point out the contribution of a systemic inflammatory response syndrome (SIRS) in ALF (Antoniades et al., 2008), a therapeutic agent that ameliorates ALF could be a promising drug against other inflammatory disorders based on SIRS, such as sepsis as well. In the current study, we demonstrated that pretreatment with DIM protected mice from GalN/LPS-induced ALF by increasing their survival, decreasing serum ALT levels and decreasing histological damage to the liver tissues. In addition, our results here suggest that the changes in the expression of miRNAs, specifically 106a, 20b and 125b-5p, induced by DIM in vivo could be involved in the protective effects of DIM. Treatment with DIM increased the expression of these miRNAs in the liver-infiltrating MNCs with a concurrent decrease in the expression of IRAK4, suppressed nuclear translocation of NF-κB and decreased TNF-α expression.
Upon stimulation of macrophage TLR4 by LPS, NF-κB is activated and translocated to the nucleus leading to increased expression of pro-inflammatory cytokines and chemokines such as TNF-α, CCL2 and IL-6 (Lu et al., 2008). The release of these cytokines and chemokines from macrophages plays an important role in the pathogenesis of ALF (Ogushi et al., 2003; Antoniades et al., 2008; Stuart et al., 2011). The initial release of pro-inflammatory cytokines and chemokines in the liver tissues triggers an extensive infiltration of immune cells in the liver (Antoniades et al., 2008). In our study, we observed that DIM treatment decreased the serum levels of these pro-inflammatory cytokines and chemokines that were increased by LPS. In addition, DIM treatment caused a decrease in the infiltration of MNCs. KCs and liver-infiltrating macrophages have been identified to play an important role in the pathogenesis of a number of liver diseases such acute liver injury (Brown et al., 1997; Luckey and Petersen, 2001), viral hepatitis (Adams and Hubscher, 2006), steatohepatitis (Malaguarnera et al., 2006) and alcoholic liver injury (Adachi et al., 1994; Enomoto et al., 1998; Kolios et al., 2006). The activation of KCs and liver-infiltrating macrophages by LPS has been established as the first event that ultimately leads to the development of ALF (Antoniades et al., 2008). One of the possible mechanisms underlying the DIM-mediated attenuation of ALF in our studies could be the suppression of the LPS-induced activation of liver resident macrophages. For instance, DIM decreased LPS-induced activation of macrophages and microglia in vitro, by suppressing NF-κB trans-activation induced by TLR signalling (Cho et al., 2008; Kim et al., 2014). In addition, DIM suppressed microglial and macrophage infiltration in an animal model of LPS-induced microglial hyperactivation (Kim et al., 2014). In the present study, DIM treatment suppressed the infiltration of immune cells, including macrophages. More interestingly, DIM decreased the numbers of activated macrophages that infiltrated the liver tissues. Our results from in vitro studies also corroborate earlier findings, where DIM has been shown to suppress LPS-induced activation of macrophages in vitro (Cho et al., 2008).
Several miRNAs regulate immune responses by altering the function of a number of cell types, such as B cells, T cells, neutrophils and macrophages (Lindsay, 2008; O'Connell et al., 2010). In addition, activation of TLRs has been shown to increase miR-146a, miR-155 and miR-21 and decrease miR-125b-5p (O'Neill et al., 2011). Also, miRNAs regulate the expression of various genes involved in TLR4 signalling in response to LPS (O'Neill et al., 2011).
The role of IRAK4 as an important component of TLR signalling is well established (Suzuki and Saito, 2006; Kim et al., 2011). The absence of IRAK4 (Suzuki et al., 2002) or inhibition of its kinase activity (Kim et al., 2007) impaired TLR signalling and induced resistance to LPS-induced sepsis through a decreased production of cytokines and chemokines. These findings imply that decreased IRAK4 expression or function in diseases mediated by TLR activation may be beneficial as such changes should down-regulate all the events downstream of IRAK4. In our present study, DIM treatment increased the expression of miR-106a and miR-20b in liver MNCs. Interestingly enough, our in silico analyses suggested that both miR-106a and miR-20b target the mRNA of IRAK4. We confirmed these findings and found decreased IRAK4 expression in liver MNCs of DIM-treated mice. These observations suggested that DIM treatment may suppress TLR responses by decreasing expression of IRAK4 through increased expression of miRNAs-106a and 20b. The significance of NF-κB in the culmination of TLR signalling pathways (Kawai and Akira, 2007) leading to the increased expression of pro-inflammatory cytokines that ultimately lead to ALF (Lu et al., 2008) is well described. To further provide evidence for the regulation of IRAK4 by miRNAs 106a and 20b, we transfected RAW264.7 macrophages with mimics for both miRNAs. This procedure decreased expression of IRAK4, with a concomitant decrease in TNF-α secretion after LPS stimulation of the transfected macrophages. In accordance with a previous report of in vitro suppression of NF-κB activity by DIM (Cho et al., 2008), DIM treatment caused a decrease in the nuclear translocation of NF-κB in liver MNCs, which could be a downstream effect of the decreased IRAK4 expression mediated by DIM.
Among the pro-inflammatory cytokines released during the initial stages, TNF-α is the main mediator of apoptosis in hepatocytes leading to ALF (Hishinuma et al., 1990; Leist et al., 1995; Josephs et al., 2000). This suggests that a therapeutic approach based on suppression of TNF-α in response to TLR stimulation could be effective in an ALF model (Zhan et al., 2014). In addition, expression of miR-125b-5p decreased after stimulation of macrophages with LPS and this miRNA down-regulated expression of TNF-α by targeting its mRNA directly (Tili et al., 2007). In the current study, DIM treatment increased expression of several miRNAs including miR-125b-5p along with decreased expression of TNF-α in MNCs infiltrating the liver. Our findings are, therefore, consistent with the direct decrease of TNF-α by miR-125b-5p (Tili et al., 2007), and further strengthen the role of miR-125b-5p in mitigating responses to TLR activation.
DIM is a ligand for the aryl hydrocarbon receptor (AhR), one of the transcription factors for cytochrome P450 enzymes (Chen et al., 1996; Banerjee et al., 2011). Modulation of AhR signalling by DIM itself attenuated colonic inflammation and experimental arthritis (Kim et al., 2009; Dong et al., 2010), which suggests the use of such AhR ligands as anti-inflammatory agents to treat inflammatory diseases (Busbee et al., 2013). In addition, research findings from our laboratory have shown that indoles including I3C and DIM suppress disease symptoms in experimental autoimmune encephalomyelitis, a mouse model for multiple sclerosis (Rouse et al., 2013) and in an animal model of SEB-induced acute inflammation (Busbee et al., 2014). Signalling through the AhR modulated and altered miRNA expression profiles in immune cells (Singh et al., 2012; Nakahama et al., 2013). A possible mechanism for DIM-mediated regulation of miRNAs observed in our studies could be the formation of a transcriptional activation complex between STAT1 protein and AhR. Indeed, previous studies have shown that AhR forms a complex with STAT1 to inhibit NF-κB triggered transcription of pro-inflammatory genes and thus suppress LPS-induced inflammatory responses in macrophages (Kimura et al., 2008; 2009,). Furthermore, STAT proteins are known to regulate miRNAs and vice versa (Kohanbash and Okada, 2012). It would be interesting to see if such a STAT1–AhR complex or AhR by itself also binds to miRNA gene promoters to regulate their transcription as well. Our findings here provide support for the anti-inflammatory potential of indole compounds, in general, and serve to identify a possible therapeutic agent in the GalN/LPS-induced ALF model that targets IRAK4 as its mode of action.
In summary, we have demonstrated here for the first time that DIM was protective in an animal model of GalN/LPS-induced ALF and that this protective effect was mediated by the up-regulation of miRNAs 106a, 20b, and 125b-5p that decreased expression of IRAK4 and TNF-α to limit responses to TLRs activated by LPS. Our studies suggest that DIM could represent a novel therapeutic approach to the treatment of ALF.
Author contributions
S. T. performed the research. S. T., M. N. and P. S. N. designed the research study. M. N. and P. S. N. provided reagents/materials/analysis tools. S. T., M. N. and P. S. N. analysed the data and wrote the manuscript.
Conflict of interest
None.
Glossary
- ALF
Acute liver failure
- ALT
Alanine transaminase
- DIM
3,3′-Diindolylmethane
- GalN/LPS
D-galactosamine and LPS
- IRAK4
IL-1 receptor-associated kinase 4
- KCs
Kupffer cells
- MNCs
Mononuclear cells
References
- Adachi Y, Bradford BU, Gao W, Bojes HK, Thurman RG. Inactivation of Kupffer cells prevents early alcohol-induced liver injury. Hepatology. 1994;20:453–460. [PubMed] [Google Scholar]
- Adams DH, Hubscher SG. Systemic viral infections and collateral damage in the liver. Am J Pathol. 2006;168:1057–1059. doi: 10.2353/ajpath.2006.051296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad A, Sakr WA, Rahman KM. Anticancer properties of indole compounds: mechanism of apoptosis induction and role in chemotherapy. Curr Drug Targets. 2010;11:652–666. doi: 10.2174/138945010791170923. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Catalytic Receptors. Br J Pharmacol. 2013a;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniades CG, Berry PA, Wendon JA, Vergani D. The importance of immune dysfunction in determining outcome in acute liver failure. J Hepatol. 2008;49:845–861. doi: 10.1016/j.jhep.2008.08.009. [DOI] [PubMed] [Google Scholar]
- Banerjee S, Kong D, Wang Z, Bao B, Hillman GG, Sarkar FH. Attenuation of multi-targeted proliferation-linked signaling by 3,3′-diindolylmethane (DIM): from bench to clinic. Mutat Res. 2011;728:47–66. doi: 10.1016/j.mrrev.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal W, Auzinger G, Dhawan A, Wendon J. Acute liver failure. Lancet. 2010;376:190–201. doi: 10.1016/S0140-6736(10)60274-7. [DOI] [PubMed] [Google Scholar]
- Brown AP, Harkema JR, Schultze AE, Roth RA, Ganey PE. Gadolinium chloride pretreatment protects against hepatic injury but predisposes the lungs to alveolitis after lipopolysaccharide administration. Shock. 1997;7:186–192. doi: 10.1097/00024382-199703000-00006. [DOI] [PubMed] [Google Scholar]
- Busbee PB, Rouse M, Nagarkatti M, Nagarkatti PS. Use of natural AhR ligands as potential therapeutic modalities against inflammatory disorders. Nutr Rev. 2013;71:353–369. doi: 10.1111/nure.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busbee PB, Nagarkatti M, Nagarkatti PS. Natural indoles, indole-3-carbinol and 3,3′-diindolymethane, inhibit T cell activation by staphylococcal enterotoxin B through epigenetic regulation involving HDAC expression. Toxicol Appl Pharmacol. 2014;274:7–16. doi: 10.1016/j.taap.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri AA, So AY, Sinha N, Gibson WS, Taganov KD, O'Connell RM, et al. MicroRNA-125b potentiates macrophage activation. J Immunol. 2011;187:5062–5068. doi: 10.4049/jimmunol.1102001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I, Safe S, Bjeldanes L. Indole-3-carbinol and diindolylmethane as aryl hydrocarbon (Ah) receptor agonists and antagonists in T47D human breast cancer cells. Biochem Pharmacol. 1996;51:1069–1076. doi: 10.1016/0006-2952(96)00060-3. [DOI] [PubMed] [Google Scholar]
- Cho HJ, Seon MR, Lee YM, Kim J, Kim JK, Kim SG, et al. 3,3′-Diindolylmethane suppresses the inflammatory response to lipopolysaccharide in murine macrophages. J Nutr. 2008;138:17–23. doi: 10.1093/jn/138.1.17. [DOI] [PubMed] [Google Scholar]
- Dong L, Xia S, Gao F, Zhang D, Chen J, Zhang J. 3,3′-Diindolylmethane attenuates experimental arthritis and osteoclastogenesis. Biochem Pharmacol. 2010;79:715–721. doi: 10.1016/j.bcp.2009.10.010. [DOI] [PubMed] [Google Scholar]
- Dong ZJ, Wei HM, Sun R, Tian ZG, Gao B. Isolation of murine hepatic lymphocytes using mechanical dissection for phenotypic and functional analysis of NK1.1+ cells. World J Gastroenterol. 2004;10:1928–1933. doi: 10.3748/wjg.v10.i13.1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dweep H, Sticht C, Pandey P, Gretz N. miRWalk – database: prediction of possible miRNA binding sites by ‘walking’ the genes of three genomes. J Biomed Inform. 2011;44:839–847. doi: 10.1016/j.jbi.2011.05.002. [DOI] [PubMed] [Google Scholar]
- Enomoto N, Ikejima K, Bradford B, Rivera C, Kono H, Brenner DA, et al. Alcohol causes both tolerance and sensitization of rat Kupffer cells via mechanisms dependent on endotoxin. Gastroenterology. 1998;115:443–451. doi: 10.1016/s0016-5085(98)70211-2. [DOI] [PubMed] [Google Scholar]
- Flannery S, Bowie AG. The interleukin-1 receptor-associated kinases: critical regulators of innate immune signalling. Biochem Pharmacol. 2010;80:1981–1991. doi: 10.1016/j.bcp.2010.06.020. [DOI] [PubMed] [Google Scholar]
- Hegde VL, Tomar S, Jackson A, Rao R, Yang X, Singh UP, et al. Distinct microRNA expression profile and targeted biological pathways in functional myeloid-derived suppressor cells induced by delta9-tetrahydrocannabinol in vivo: regulation of CCAAT/enhancer-binding protein alpha by microRNA-690. J Biol Chem. 2013;288:36810–36826. doi: 10.1074/jbc.M113.503037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hishinuma I, Nagakawa J, Hirota K, Miyamoto K, Tsukidate K, Yamanaka T, et al. Involvement of tumor necrosis factor-alpha in development of hepatic injury in galactosamine-sensitized mice. Hepatology. 1990;12:1187–1191. doi: 10.1002/hep.1840120518. [DOI] [PubMed] [Google Scholar]
- Holt MP, Cheng L, Ju C. Identification and characterization of infiltrating macrophages in acetaminophen-induced liver injury. J Leukoc Biol. 2008;84:1410–1421. doi: 10.1189/jlb.0308173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikarashi M, Nakashima H, Kinoshita M, Sato A, Nakashima M, Miyazaki H, et al. Distinct development and functions of resident and recruited liver Kupffer cells/macrophages. J Leukoc Biol. 2013;94:1325–1336. doi: 10.1189/jlb.0313144. [DOI] [PubMed] [Google Scholar]
- Josephs MD, Bahjat FR, Fukuzuka K, Ksontini R, Solorzano CC, Edwards CK, 3rd, et al. Lipopolysaccharide and D-galactosamine-induced hepatic injury is mediated by TNF-alpha and not by Fas ligand. Am J Physiol Regul Integr Comp Physiol. 2000;278:R1196–R1201. doi: 10.1152/ajpregu.2000.278.5.R1196. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. Signaling to NF-kappaB by toll-like receptors. Trends Mol Med. 2007;13:460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HW, Kim J, Kim J, Lee S, Choi BR, Han JS, et al. 3,3′-Diindolylmethane inhibits lipopolysaccharide-induced microglial hyperactivation and attenuates brain inflammation. Toxicol Sci. 2014;137:158–167. doi: 10.1093/toxsci/kft240. [DOI] [PubMed] [Google Scholar]
- Kim TW, Staschke K, Bulek K, Yao J, Peters K, Oh KH, et al. A critical role for IRAK4 kinase activity in toll-like receptor-mediated innate immunity. J Exp Med. 2007;204:1025–1036. doi: 10.1084/jem.20061825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TW, Febbraio M, Robinet P, Dugar B, Greene D, Cerny A, et al. The critical role of IL-1 receptor-associated kinase 4-mediated NF-kappaB activation in modified low-density lipoprotein-induced inflammatory gene expression and atherosclerosis. J Immunol. 2011;186:2871–2880. doi: 10.4049/jimmunol.1002242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YH, Kwon HS, Kim DH, Shin EK, Kang YH, Park JH, et al. 3,3′-diindolylmethane attenuates colonic inflammation and tumorigenesis in mice. Inflamm Bowel Dis. 2009;15:1164–1173. doi: 10.1002/ibd.20917. [DOI] [PubMed] [Google Scholar]
- Kimura A, Naka T, Nohara K, Fujii-Kuriyama Y, Kishimoto T. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc Natl Acad Sci U S A. 2008;105:9721–9726. doi: 10.1073/pnas.0804231105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura A, Naka T, Nakahama T, Chinen I, Masuda K, Nohara K, et al. Aryl hydrocarbon receptor in combination with Stat1 regulates LPS-induced inflammatory responses. J Exp Med. 2009;206:2027–2035. doi: 10.1084/jem.20090560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohanbash G, Okada H. MicroRNAs and STAT interplay. Semin Cancer Biol. 2012;22:70–75. doi: 10.1016/j.semcancer.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolios G, Valatas V, Kouroumalis E. Role of Kupffer cells in the pathogenesis of liver disease. World J Gastroenterol. 2006;12:7413–7420. doi: 10.3748/wjg.v12.i46.7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leifeld L, Trautwein C, Dumoulin FL, Manns MP, Sauerbruch T, Spengler U. Enhanced expression of CD80 (B7-1), CD86 (B7-2), and CD40 and their ligands CD28 and CD154 in fulminant hepatic failure. Am J Pathol. 1999;154:1711–1720. doi: 10.1016/S0002-9440(10)65427-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leist M, Gantner F, Bohlinger I, Tiegs G, Germann PG, Wendel A. Tumor necrosis factor-induced hepatocyte apoptosis precedes liver failure in experimental murine shock models. Am J Pathol. 1995;146:1220–1234. [PMC free article] [PubMed] [Google Scholar]
- Li X, Qin J. Modulation of toll-interleukin 1 receptor mediated signaling. J Mol Med (Berl) 2005;83:258–266. doi: 10.1007/s00109-004-0622-4. [DOI] [PubMed] [Google Scholar]
- Liang DY, Liu LM, Ye CG, Zhao L, Yu FP, Gao DY, et al. Inhibition of UII/UTR system relieves acute inflammation of liver through preventing activation of NF-kappaB pathway in ALF mice. PLoS ONE. 2013;8:e64895. doi: 10.1371/journal.pone.0064895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay MA. microRNAs and the immune response. Trends Immunol. 2008;29:343–351. doi: 10.1016/j.it.2008.04.004. [DOI] [PubMed] [Google Scholar]
- Liu G, Friggeri A, Yang Y, Park YJ, Tsuruta Y, Abraham E. miR-147, a microRNA that is induced upon toll-like receptor stimulation, regulates murine macrophage inflammatory responses. Proc Natl Acad Sci U S A. 2009;106:15819–15824. doi: 10.1073/pnas.0901216106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42:145–151. doi: 10.1016/j.cyto.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Luckey SW, Petersen DR. Activation of Kupffer cells during the course of carbon tetrachloride-induced liver injury and fibrosis in rats. Exp Mol Pathol. 2001;71:226–240. doi: 10.1006/exmp.2001.2399. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaguarnera L, Di Rosa M, Zambito AM, Dell'ombra N, Di Marco R, Malaguarnera M. Potential role of chitotriosidase gene in nonalcoholic fatty liver disease evolution. Am J Gastroenterol. 2006;101:2060–2069. doi: 10.1111/j.1572-0241.2006.00680.x. [DOI] [PubMed] [Google Scholar]
- Nakahama T, Hanieh H, Nguyen NT, Chinen I, Ripley B, Millrine D, et al. Aryl hydrocarbon receptor-mediated induction of the microRNA-132/212 cluster promotes interleukin-17-producing T-helper cell differentiation. Proc Natl Acad Sci U S A. 2013;110:11964–11969. doi: 10.1073/pnas.1311087110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima H, Ogawa Y, Shono S, Kinoshita M, Nakashima M, Sato A, et al. Activation of CD11b+ Kupffer cells/macrophages as a common cause for exacerbation of TNF/Fas-ligand-dependent hepatitis in hypercholesterolemic mice. PLoS ONE. 2013;8:e49339. doi: 10.1371/journal.pone.0049339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan JP. The role of intestinal endotoxin in liver injury: a long and evolving history. Hepatology. 2010;52:1829–1835. doi: 10.1002/hep.23917. [DOI] [PubMed] [Google Scholar]
- O'Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol. 2010;10:111–122. doi: 10.1038/nri2708. [DOI] [PubMed] [Google Scholar]
- Ogushi I, Iimuro Y, Seki E, Son G, Hirano T, Hada T, et al. Nuclear factor kappa B decoy oligodeoxynucleotides prevent endotoxin-induced fatal liver failure in a murine model. Hepatology. 2003;38:335–344. doi: 10.1053/jhep.2003.50298. [DOI] [PubMed] [Google Scholar]
- O'Neill LA, Sheedy FJ, McCoy CE. MicroRNAs: the fine-tuners of toll-like receptor signalling. Nat Rev Immunol. 2011;11:163–175. doi: 10.1038/nri2957. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucleic Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci U S A. 2012;109:E3186–E3195. doi: 10.1073/pnas.1119964109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouse M, Singh NP, Nagarkatti PS, Nagarkatti M. Indoles mitigate the development of experimental autoimmune encephalomyelitis by induction of reciprocal differentiation of regulatory T cells and Th17 cells. Br J Pharmacol. 2013;169:1305–1321. doi: 10.1111/bph.12205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarra M, Cupi ML, Bernardini R, Ronchetti G, Monteleone I, Ranalli M, et al. IL-25 prevents and cures fulminant hepatitis in mice through a myeloid-derived suppressor cell-dependent mechanism. Hepatology. 2013;58:1436–1450. doi: 10.1002/hep.26446. [DOI] [PubMed] [Google Scholar]
- Silverstein R. D-galactosamine lethality model: scope and limitations. J Endotoxin Res. 2004;10:147–162. doi: 10.1179/096805104225004879. [DOI] [PubMed] [Google Scholar]
- Singh NP, Singh UP, Guan H, Nagarkatti P, Nagarkatti M. Prenatal exposure to TCDD triggers significant modulation of microRNA expression profile in the thymus that affects consequent gene expression. PLoS ONE. 2012;7:e45054. doi: 10.1371/journal.pone.0045054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart WD, Kulkarni RM, Gray JK, Vasiliauskas J, Leonis MA, Waltz SE. Ron receptor regulates Kupffer cell-dependent cytokine production and hepatocyte survival following endotoxin exposure in mice. Hepatology. 2011;53:1618–1628. doi: 10.1002/hep.24239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N, Saito T. IRAK-4 – a shared NF-kappaB activator in innate and acquired immunity. Trends Immunol. 2006;27:566–572. doi: 10.1016/j.it.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Suzuki N, Suzuki S, Duncan GS, Millar DG, Wada T, Mirtsos C, et al. Severe impairment of interleukin-1 and toll-like receptor signalling in mice lacking IRAK-4. Nature. 2002;416:750–756. doi: 10.1038/nature736. [DOI] [PubMed] [Google Scholar]
- Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tili E, Michaille JJ, Cimino A, Costinean S, Dumitru CD, Adair B, et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol. 2007;179:5082–5089. doi: 10.4049/jimmunol.179.8.5082. [DOI] [PubMed] [Google Scholar]
- Xia W, Hilgenbrink AR, Matteson EL, Lockwood MB, Cheng JX, Low PS. A functional folate receptor is induced during macrophage activation and can be used to target drugs to activated macrophages. Blood. 2009;113:438–446. doi: 10.1182/blood-2008-04-150789. [DOI] [PubMed] [Google Scholar]
- Zhan Y, Wang Z, Yang P, Wang T, Xia L, Zhou M, et al. Adenosine 5′-monophosphate ameliorates D-galactosamine/lipopolysaccharide-induced liver injury through an adenosine receptor-independent mechanism in mice. Cell Death Dis. 2014;5:e985. doi: 10.1038/cddis.2013.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Li HZ, Gong X, Luo FL, Wang B, Hu N, et al. Protective effects of Asiaticoside on acute liver injury induced by lipopolysaccharide/D-galactosamine in mice. Phytomedicine. 2010;17:811–819. doi: 10.1016/j.phymed.2010.01.008. [DOI] [PubMed] [Google Scholar]