

Abstract
Background and Purpose
We demonstrated previously that oxycodone has potent antinociceptive effects at supraspinal sites. In this study, we investigated changes in neuronal function and antinociceptive mechanisms of oxycodone at ventrolateral periaqueductal gray (VLPAG) neurons, which are a major site of opioid action, in a femur bone cancer (FBC) model with bone cancer-related pain.
Experimental Approach
We characterized the supraspinal antinociceptive profiles of oxycodone and morphine on mechanical hypersensitivity in the FBC model. Based on the disinhibition mechanism underlying supraspinal opioid antinociception, the effects of oxycodone and morphine on GABAA receptor-mediated inhibitory postsynaptic currents (IPSCs) in VLPAG neurons were evaluated in slices from the FBC model.
Key Results
The supraspinal antinociceptive effects of oxycodone, but not morphine, were abolished by blocking G protein-gated inwardly rectifying potassium1 (Kir3.1) channels. In slices from the FBC model, GABAergic synaptic transmission at VLPAG neurons was enhanced, as indicated by a leftward shift of the input–output relationship curve of evoked IPSCs, the increased paired-pulse facilitation and the enhancement of miniature IPSC frequency. Following treatment with oxycodone and morphine, IPSCs were reduced in the FBC model, and the inhibition of presynaptic GABA release by oxycodone, but not morphine was enhanced and dependent on Kir3.1 channels.
Conclusion and Implications
Our results demonstrate that Kir3.1 channels are important for supraspinal antinociception and presynaptic GABA release inhibition by oxycodone in the FBC model. Enhanced GABAergic synaptic transmission at VLPAG neurons in the FBC model is an important site of supraspinal antinociception by oxycodone via Kir3.1 channel activation.
Tables of Links
| LIGANDS | |
|---|---|
| β-FNA (β-funaltrexamine) | H-89 |
| Bicuculline | Morphine |
| CNQX | Nor-BNI |
| GTPγS | Oxycodone |
| Tertiapin-Q | |
| TTX |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,c,dAlexander et al., 2013a,b,c,d,,,).
Introduction
Bone cancer pain is caused by primary bone tumours or bone metastasis (Banning et al., 1991; Coleman, 1998), and patients suffering from bone cancer often exhibit various types of pain, such as ambulatory pain, ongoing pain, and/or abnormal mechanical or thermal sensitivity, known as allodynia (Mercadante and Arcuri, 1998; Hansen et al., 2012). Evidence of the cellular and neurochemical mechanisms underlying these symptoms is emerging from clinical and non-clinical studies. For example, patients suffering from bone cancer show increased neuronal activity at supraspinal structures, including the prefrontal cortex (Buvanendran et al., 2010). In a bone cancer pain animal model, C fibre-mediated excitatory synaptic transmission was enhanced in the spinal cord (Yanagisawa et al., 2010). To develop effective pain treatments, it is important to understand the underlying mechanisms of bone cancer pain and currently available pain medications.
Clinically used drugs acting on the μ-opioid receptors, such as morphine, oxycodone and fentanyl, exhibit potent analgesic effects against bone cancer pain (Quigley, 2005). These drugs have distinct analgesic and antinociceptive actions, tolerance, and adverse effects in clinical patients and animal models (Quigley, 2005; Raehal and Bohn, 2011; Koyyalagunta et al., 2012). Among the opioids, oxycodone is frequently prescribed for moderate-to-severe bone cancer pain (Heiskanen and Kalso, 1997; Watson and Babul, 1998; Becker et al., 2000; Gimbel et al., 2003; Watson et al., 2003; Kalso, 2005; Bercovitch and Adunsky, 2006; Silvestri et al., 2008; Olkkola et al., 2013). In our previous study using the mouse femur bone cancer (FBC) model, which shows similar pathological symptoms to human bone metastasis, systemically or supraspinally applied oxycodone showed distinct antinociceptive profiles in various types of pain-related behaviours, compared with morphine or fentanyl (Minami et al., 2009; Nakamura et al., 2013; 2014,). Because μ-opioid receptor activation, determined by radiolabelled GTPγS binding, was altered in an opioid-dependent manner at pain-related supraspinal structures, such as the periaqueductal gray (PAG) and mediodorsalthalamus in the FBC model (Nakamura et al., 2013), it is possible that the distinct antinociceptive profile of oxycodone resulted from changes in neuronal functions during bone cancer pain at the supraspinal sites.
The PAG is a major site mediating opioid analgesic effects via the descending pain pathway and is tonically inhibited by GABA release from presynaptic interneurons (Basbaum and Fields, 1984;Reichling et al., 1988). Opioid agonists remove the GABAergic influence, which results in pain relief by excitation of PAG neurons, especially ventrolateral PAG (VLPAG) neurons, projecting to rostral ventromedial medulla-spinal neurons, a process known as disinhibition (Yaksh et al., 1976; Vaughan and Christie, 1997). Previous studies showed that the GABAergic influence on PAG neurons was increased in rats subjected to peripheral nerve ligation (Hahm et al., 2011) and with chronic morphine administration (Ingram et al., 1998; DuPen et al., 2007), which resulted in pain hypersensitivity. Moreover, we found changes in μ-opioid receptor activation by opioid agonists at several pain-related regions including the PAG in the FBC model (Nakamura et al., 2013). Therefore, it is possible that this endogenous inhibitory system at the VLPAG is altered in the bone cancer pain condition.
In the present study, we demonstrated that G protein-gated inwardly rectifying potassium 1 (Kir3.1), also known as GIRK1, channels are important for the supraspinal antinociceptive effects of oxycodone, but not those of morphine, in the FBC model. As the mechanisms underlying the antinociceptive effects of oxycodone related to supraspinal neuronal changes, GABAergic synaptic transmission in VLPAG neurons was enhanced in slices of the FBC model, and the inhibitory effects of oxycodone, but not those of morphine, on evoked inhibitory postsynaptic currents (eIPSCs) were enhanced and its inhibition of presynaptic GABA release by oxycodone was dependent on Kir3.1 channels. Together with the well-established disinhibition mechanisms underlying supraspinal opioid-induced antinociception (Vaughan and Christie, 1997), enhanced GABAergic synaptic transmission at VLPAG neurons in the FBC model is an important site of supraspinal antinociception by oxycodone via Kir3.1 channel activation.
Methods
Experimental animals
The experiments were performed using male C3H/HeN mice (CLEA Japan, Inc., Tokyo, Japan), weighing 18–23 g. In total, 180 mice were used in the behavioural and electrophysiological experiments. The mice were housed under controlled temperature and humidity with a 12/12 h light/dark cycle. All procedures were approved by the Animal Care and Use Committee of Shionogi Research Laboratories, Osaka, Japan. The behavioural assessments of antinociceptive effects were done according to Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC) guidelines. The results of all studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Preparation of the FBC model
The NCTC2472 tumour cells (American Type Culture Collection, Manassas, VA, USA) were maintained in DMEM (Invitrogen, Carlsbad, CA, USA), supplemented with 10% FBS (Invitrogen), 100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin (Invitrogen) and cultured at 37°C in a humidified atmosphere of 5% CO2. To prepare the FBC model, NCTC2472 tumour cells were injected following a protocol described previously (Minami et al., 2009). Briefly, C3H/HeN mice were anaesthetized with 3% isoflurane, and a left knee arthrotomy was performed. Depth of anaesthesia was assessed by applying painful stimuli to tail. If there is no reaction to intense mechanical pressure, mice were considered to undergo sufficient anaesthesia. Tumour cells (1 × 105 in 5 μL HBSS, Invitrogen) were injected directly into the medullary cavity of the distal femur, and the drill hole in the bone was closed with resin cement (ADFA; Shofu, Kyoto, Japan). In the sham group, a left knee arthrotomy was performed and 5 μL HBSS were injected, instead of the tumour cells. We confirmed that the sham operation itself had no influence on behavioural response (Minami et al., 2009), and in a preliminary experiment, we observed identical inhibitory effects of opioid agonists on GABAergic synaptic transmission in slices of sham-operated mice compared with normal mice.
Assessment of guarding behaviour and mechanical pain hypersensitivity as bone cancer-related pain
Pain-related behaviours in the FBC model were evaluated 14–20 days after tumour implantation using a protocol described previously (Minami et al., 2009). The FBC model and sham-operated mice were evaluated for ongoing pain, assessed by guarding behaviour, and mechanical pain hypersensitivity, assessed by paw withdrawal response to von Frey filaments. The mice were placed in a clear plastic observation box (cylinder type: diameter 180 mm, height 250 mm). In the evaluation of the ongoing pain, lifting time of the hind paw on the ipsilateral side was measured for 2 min as a guarding behaviour. The mice exhibiting changes in guarding times of 0–2 s (before tumour implantation) to 8–16 s (14–20 days after tumour implantation) was used in the electrophysiological study. In the assessment of mechanical sensitivity, a series of calibrated von Frey filaments (pressure: 0.008, 0.02, 0.04, 0.07, 0.16, 0.4, 0.6 and 1.0 g, Semmes–Weinstein Monofilamets; North Coast Medical, Inc., Morgan Hill, CA, USA) was used to determine the threshold of a paw withdrawal response by the up-down method (Minami et al., 2009). In behavioural assessment of the antinociceptive effects of oxycodone and morphine on mechanical hypersensitivity in the FBC model, mice exhibiting a threshold of 0.008 g in the von Frey test were used. In the present study, 15/16 mice meeting criteria of changes in guarding time also exhibited mechanical pain hypersensitivity, indicating that almost all mice with guarding behaviour developed mechanical hypersensitivity.
Assessment of the antinociceptive effects of oxycodone and morphine on mechanical pain hypersensitivity in the FBC model
Changes in the paw withdrawal threshold were evaluated 10 min after i.c.v. injection of oxycodone and morphine, according to a previous study (Nakamura et al., 2013, 2014). I.c.v. injection and assessment of antinociceptive effects of oxycodone and morphine were performed according to the AAALAC guideline approved in the Animal Care and Use Committee of Shionogi Research Laboratories, Osaka, Japan. I.c.v. injection was performed in the absence of an anaesthetic as anaesthesia affects the behavioural evaluation in antinociceptive effects of drugs in our experimental paradigm. The well-trained experimenters have advanced animal handling skills and performed i.c.v. injections with a minimum of stress to the mice and carefully observed the condition of the mice before and after injection. The experimenter held the mice by firmly grasping the loose skin behind the head. The head of the mice was held against a V-shaped holder and the skin was pulled taut. On the day before i.c.v. drug administration, a 2 mm double-needle (tip: 27 G, 2 mm; base: 22 G, 10 mm; Natsume Seisakusyo, Tokyo, Japan) attached to a 25 μL Hamilton microsyringe was inserted into a unilateral injection site to make a hole in the skull for injection. The unilateral injection site was ∼2 mm caudal and 2 mm lateral from the bregma, and the needle was inserted perpendicular to the skull. In order to make the hole the mice were restrained for 20 s. If the mice became distressed while being restrained and their condition such as breathing worsened, they were released immediately. When drugs were administered, mice were restrained by the same method described earlier within 10 s and the drug injection was done within 5–10 s. After i.c.v. injection, the mice were returned to the observation box and the experimenter checked that mice exhibited normal behaviours such as moving or rearing. The injection volume was set at 2 μL for each mouse. For the assessment of the effects of the Kir3.1 channel inhibitor tertiapin-Q on the antinociceptive actions of oxycodone or morphine, tertiapin-Q was administered 10 min before the administration of oxycodone and morphine.
Preparation of midbrain slices that included VLPAG neurons
The mice were killed by cervical dislocation. The brains were then quickly removed and placed in ice-coldlow sodium artificial CSF containing the following (in mM): 215.5 sucrose, 3 KCl, 1 NaH2PO4, 25 NaHCO3, 11 D-glucose, 1 CaCl2, and 5 MgCl2 (pH 7.4 after bubbling with 95% O2 and 5% CO2). Coronal slices, 350 μm thick containing VLPAG, were prepared using a vibratome (VT1200S; Leica, Wetzlar, Germany) and then maintained for at least 60 min in standard artificial CSF containing the following (in mM): 113 NaCl, 3 KCl, 1 NaH2PO4, 25 NaHCO3, 11 D-glucose, 2 CaCl2 and 1 MgCl2 (pH 7.4, after bubbling with 95% O2 and 5% CO2 at 30–32°C). Slices were transferred to a recording chamber mounted on the stage of a microscope (BX51-WI; Olympus, Tokyo, Japan) and were superfused with standard artificial CSF (flow rate of 2.5 mL·min−1 at 30–32°C).
Patch-clamp electrophysiology
Whole-cell voltage-clamp recordings were made from visually identified VLPAG neurons in the midbrain using an upright microscope (BX51WI, Olympus) with infrared differential interference contrast optics. Patch electrodes (2.5–3.0 μm tip diameter) were pulled from borosilicate glass capillaries and had resistance of 3–5 MΩ when filled with an internal solution consisting of the following (in mM): 140 CsCl, 10 HEPES, 1.1 EGTA, 2 MgCl2, 3 MgATP, 5 QX-314 and 0.3 Tris-GTP, pH 7.4, adjusted with CsOH. IPSCs were detected as inward deflections (EPC-10; HEKA, Darmstadt, Germany), low-pass filtered at 2.9 kHz, and digitized at 10 kHz for computer analysis with the Pulse software (HEKA). Access resistance was monitored by measuring capacitive transients obtained in response to a hyperpolarizing voltage step (10 mV, 10 ms) from a holding potential of −60 mV. All experiments were performed at 30–32°C.
Assessment of input–output (I–O) relationship curve of evoked IPSCs in VLPAG neurons in the FBC model and in sham-operated mice
Under whole-cell voltage-clamp recording in the presence of 10 μM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) to block glutamatergic excitatory synaptic transmission, GABAergic eIPSCs were induced by stimulating the neighbouring area (within a 40–100 μm radius of the recorded neuron) via a glass pipette filled with 1 M NaCl solution. A voltage pulse 0.2 ms in duration was gradually increased from threshold to maximal response value (the FBC model: 2–20 V, sham-operated mice: 5–25 V). An I–O curve was constructed by plotting the relative amplitude of eIPSCs to the maximal amplitude at each stimulus intensity.
Evaluation of the effects of oxycodone and morphine on action potential-dependent eIPSCs in VLPAG neurons in the FBC model and sham-operated mice
In the presence of 10 μM CNQX to block glutamatergic excitatory synaptic transmission, GABAergic eIPSCs were induced by stimulating the neighbouring area via a glass pipette filled with 1 M NaCl solution. A voltage pulse 0.2 ms in duration at 0.1 Hz was applied at supra-threshold intensity (2× the threshold) via the stimulating electrode. The effects of oxycodone and morphine on eIPSCs were evaluated by comparing averaged eIPSCs before (12 traces for 2 min) and after (last 12 traces for 2 min) application of each drug.
Evaluation of the effects of oxycodone and morphine on the paired-pulse ratio (PPR) of eIPSCs in VLPAG neurons in slices from the FBC model
To evaluate both whether presynaptic GABA release or sensitivity of postsynaptic GABAA receptors were changed in slices from FBC model and the effects of oxycodone, we analysed the PPR as the ratio of the second eIPSCs amplitude divided by the first. In the PPR recording, two successive stimuli of identical strength (2× the threshold) were applied at an interval of 50 ms. The averaged PPR for 2 min in the FBC model slices was compared with that in slices of sham-operated mice. The effects of oxycodone and morphine on the PPR were evaluated by comparing the averaged PPR before (12 traces for 2 min) and after (last 12 traces for 2 min) application of each drug.
Analysis of action potential-independent spontaneous miniature IPSCs in VLPAG neurons of the FBC model
In the presence of 10 μM CNQX and 0.5 μM tetrodotoxin (TTX), action potential-independent spontaneous miniature IPSCs (mIPSCs) were recorded. The frequency and amplitude of mIPSC events were analysed with the Mini Analysis Program (Synaptosoft Inc., Decatur, GA, USA). Changes in the frequency and amplitude of mIPSCs reflect presynaptic changes in GABA release probability and postsynaptic changes, such as the sensitivity of GABAA receptors, respectively. The averaged frequency and amplitude of mIPSCs for 2 min in slices of the FBC model was compared with those in slices of sham-operated mice. The effects of oxycodone and morphine on mIPSCs were evaluated by comparing averaged mIPSCs for 2 min before and after application of each drug.
Drugs
Drugs were applied by bath application. Oxycodone hydrochloride, morphine hydrochloride, nor-binaltorphimine dihydrochloride (nor-BNI) and β-funaltrexamine hydrochloride (β-FNA) were from Shionogi & Co., Ltd. (Osaka, Japan). TTX and (−)bicuculline methobromide were purchased from Wako (Osaka, Japan). CNQX and H-89 were obtained from Sigma (St. Louis, MO, USA). Tertiapin-Q was purchased from Alomone Labs, Ltd. (Jerusalem, Israel).
Statistical analysis
All data are expressed as means ± SEM. In the assessment of antinociceptive effects of oxycodone and morphine alone and following pretreatment with tertiapin-Q (Figure 1), differences among groups were analysed using a Kruskal–Wallis test and Dunn's multiple-comparison test. In the assessment of I–O relationship curve of evoked IPSCs (Figure 2), the statistical significance of differences among groups was assessed using two-way anova followed by a Bonferroni multiple-comparison post hoc test. In the assessment of the effects of oxycodone and morphine on IPSCs in the FBC model and in sham-operated mice (Figures 9), Student's t-test (two-tailed) was used to compare the two groups between the FBC model and sham-operated mice, and a paired t-test was used to compare the two groups between pre- and post-treatment with each drug. A probability value (P) < 0.05 was considered to indicate statistical significance.
Figure 1.
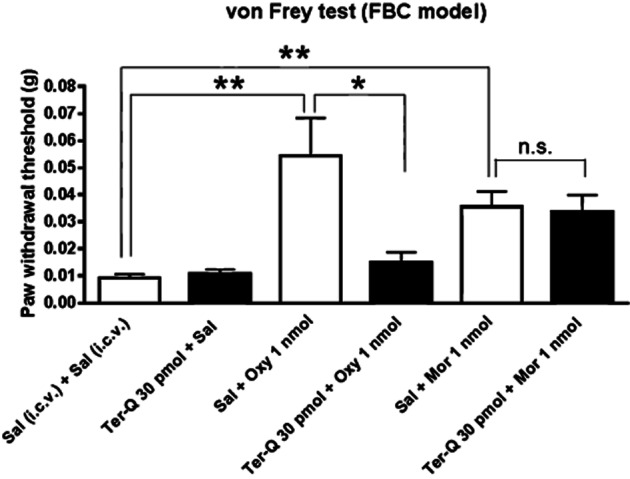
Antinociceptive profiles of oxycodone (Oxy) and morphine (Mor) against mechanical hypersensitivity in the FBC model. The antinociceptive effects of Oxy, but not those of Mor, were abolished by the Kir3.1 channel inhibitor tertiapin-Q (Ter-Q). Each column represents means ± SEM (n = 9–13). A comparison of all groups was performed using a Kruskal–Wallis test and Dunn's multiple-comparison test. In this graph, the antinociceptive effects of Oxy and Mor were followed by asterisks indicating a statistically significant difference between saline (Sal)-treated groups and the Oxy with Sal-treated groups or the Mor with Sal-treated groups; **P < 0.01. The distinct sensitivity to Kir3.1 channel inhibition of the antinociceptive effects of Oxy and Mor were followed by the asterisk indicating the difference betweenOxy with Sal-treated groups and Oxy with trertiapin-Q treated groups; *P < 0.05. No significance (n.s.) indicates a no statistically significant difference between the Mor with Sal-treated groups and the morphine with trertiapin-Q treated group.
Figure 2.
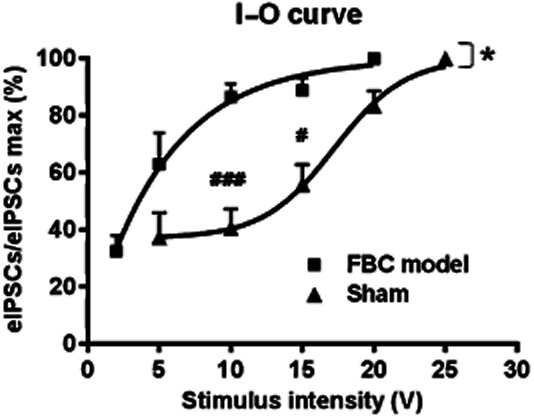
The input-output (I–O) relationship curve of GABAergic eIPSCs in VLPAG neurons was shifted to the left in slices from the FBC model. The asterisk indicates a statistically significant difference in the I–O relationship curve between the FBC model and sham-operated mice, as determined by two-way anova [F(1,32) = 9.098, *P < 0.05]. The sharp indicates a statistically significant difference in the relative amplitude of eIPSCs to maximal (max) eIPSCs at each stimulus intensity between the FBC model and sham-operated mice groups, as determined by a Bonferroni multiple-comparison post hoc test (###P < 0.001 at 10 V, #P < 0.05 at 15 V). Each point represents means ± SEM (n = 6).
Figure 9.
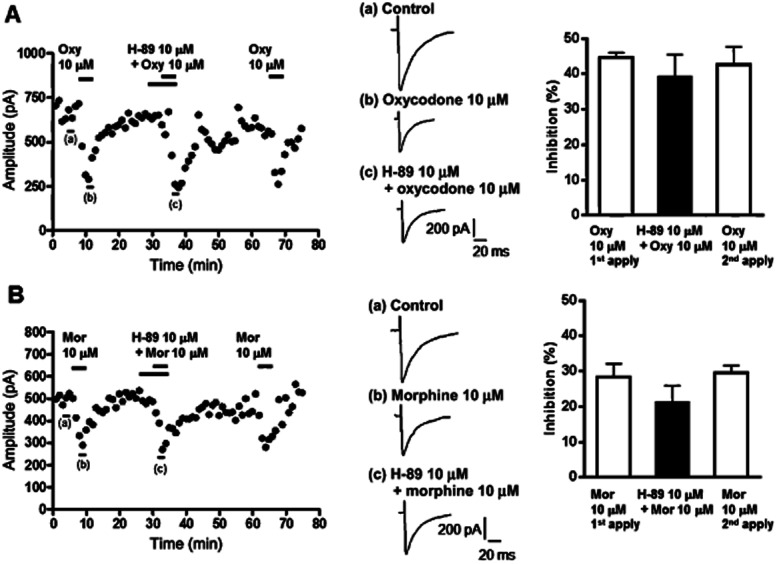
The PKA inhibitor H-89 had negligible effects on the inhibition by oxycodone and morphine of GABAergic eIPSCs in VLPAG neurons of the FBC model. (A) Left panel, a representative time course (plot of average amplitude of six IPSCs traces for 1 min) showing that oxycodone caused potent inhibition of GABAergic eIPSCs in VLPAG neurons from the FBC model, which was not affected by H-89. Middle panel, each trace was recorded during the times indicated on the graph. Right panel, a summary of the effects of H-89 on oxycodone-induced reduction in GABAergic eIPSCs in slices from the FBC model. Each column represents means ± SEM (n = 7), from the oxycodone-only and oxycodone with H-89-treated groups. (B) Left panel, a representative time course showing that morphine caused moderate inhibition of GABAergic eIPSCs in a VLPAG neuron from the FBC model, which was not affected by H-89. Middle panel, each trace was recorded during the times indicated on the graph. Right panel, a summary of the effects of H-89 on morphine-induced reduction in GABAergic eIPSCs in slices from the FBC model. Each column represents means ± SEM (n = 8), from the morphine-only and morphine with H-89-treated groups.
Our drug/molecular target nomenclature conforms to British Journal of Pharmacology's Guide to Receptors and Channels (2013a,b,c,,).
Results
Involvement of Kir3.1 channel in supraspinal oxycodone and morphine-induced antinociceptive actions on mechanical pain hypersensitivity in the FBC model
Kir3.1 channels are activated following binding of opioid agonists to μ-opioid receptors (Spivak and Beglan, 2000; Marker et al., 2004; Virk and Williams, 2008). The goal of the present study was to clarify the distinct mechanism between the antinociceptive effects of oxycodone and morphine in terms of role for Kir3.1 channels using the FBC model. We employed doses of oxycodone and morphine causing the respective maximal antinociceptive effects within the dose range without showing side effects such as sedation (Nakamura et al., 2013), and tested the effects of blockade of Kir3.1 channels on the antinociceptive effects of these two opioids. Injection of oxycodone or morphine at dose of 1 nmol (i.c.v.) ameliorated mechanical hypersensitivity in the FBC model (Figure 1). Oxycodone-induced antinociception was abolished by i.c.v. pretreatment with 30 pmol of the Kir3.1 channel inhibitor tertiapin-Q (oxycodone pretreated with tertiapin-Q, 0.014 ± 0.003 g, n = 11 vs. oxycodone pretreated with saline, 0.054 ± 0.013 g, n = 13, F(13,11) = 24.16; P < 0.05). In contrast, morphine-induced antinociception was not blocked by tertiapin-Q (morphine pretreated with tertiapin-Q, 0.033 ± 0.006 g, n = 12 vs. morphine pretreated with saline, 0.035 ± 0.005 g, n = 12). Thus, Kir3.1 channels are critical for the supraspinal antinociceptive effects of oxycodone, but not those of morphine, on mechanical hypersensitivity in the FBC model. In the following experiments, we evaluated the neuronal changes and supraspinal antinociceptive mechanisms of oxycodone and morphine in the FBC model by focusing on GABAergic synaptic transmission at VLPAG neurons. In the following electrophysiological assessment, we used single concentration of oxycodone and morphine to elicit maximal effects on activation of μ-opioid receptors (Bradaia et al., 2005; Nakamura et al., 2013) and investigated possible alteration of the effects of oxycodone and morphine between the FBC model and sham-operated mice.
Leftward shift of the I–O relation curve of GABAergic eIPSCs and increased inhibitory effect of oxycodone on GABAergic eIPSCs in the FBC model
Based on the disinhibition mechanism underlying supraspinal opioid-induced antinociception in VLPAG area (Vaughan and Christie, 1997; Budai and Fields, 1998; Park et al., 2010), we evaluated the amplitude of GABAergic eIPSCs in slices from the FBC model and sham-operated mice. When the stimulus intensity was gradually increased, electrical stimulation gave rise to larger-amplitude eIPSCs at a lower intensity in the FBC model than the sham-operated mice, indicating a leftward shift of the I–O relationship curve of eIPSCs in slices of the FBC model (Figure 2, F(1,32) = 9.098; P < 0.05. vs. sham-operated mice). At stimulus intensities of 10 and 15 V, the GABAergic synaptic responses in the FBC model were significantly larger than those in sham-operated mice (Figure 2). These results indicate that the GABAergic synaptic influence on VLPAG neurons was enhanced in the FBC model.
When the effects of oxycodone and morphine on GABAergic eIPSCs in VLPAG neurons were evaluated, 10 μM oxycodone elicited potent inhibition of eIPSCs in the FBC model compared with that in sham-operated mice (Figure 3A and C; the FBC model, 38.5 ± 4.6% inhibition vs. sham operation, 24.2 ± 2.5% inhibition, n = 10, t(18) = 2.686; P < 0.05). In contrast, 10 μM morphine inhibited GABAergic eIPSCs in VLPAG neurons of the FBC model to the same extent as those from sham-operated mice (Figure 3B and C; FBC model, 28.8 ± 3.5% inhibition vs. sham operation, 29.2 ± 3.0% inhibition, n = 10). The eIPSCs amplitude before drug application was not significantly different between the FBC model and sham-operated mice (FBC model, 820.1 ± 86.8 pA vs. sham-operated mice, 977.8 ± 127.2 pA) because we evaluated the effects of oxycodone and morphine on eIPSCs induced by electrical stimulation at supra-threshold intensity (2× the threshold). The threshold was different (FBC model, 2.5 ± 0.3 V vs. sham-operated mice, 7.1 ± 1.0 V, n = 10, t(18) = 4.334; P < 0.05). Thus, these results showed that the inhibitory effect of oxycodone on elevated eIPSCs was potentiated in the FBC model.
Figure 3.
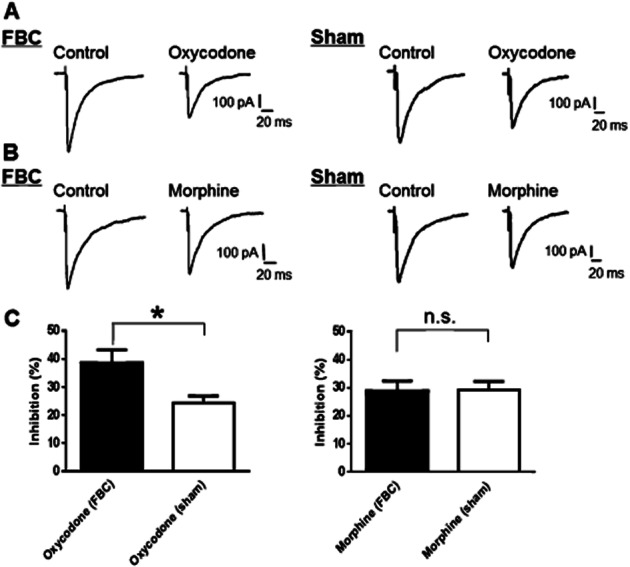
Inhibition of GABAergic eIPSCs by oxycodone in VLPAG neurons was increased in slices from the FBC model. (A) A representative trace showing that oxycodone (10 μM) potently reduced GABAergic eIPSCs in VLPAG neurons in the FBC model, compared with its efficacy in slices of sham-operated mice. (B) A representative trace showing that morphine (10 μM) moderately reduced GABAergic eIPSCs in a VLPAG neuron in the FBC model to a similar extent as in slices of sham-operated mice. (C) Summary graphs showing that oxycodone (10 μM) elicited a statistically significant potent inhibition of GABAergic eIPSCs in the FBC model compared with that in slices of sham-operated mice. In contrast, the efficacy of morphine (10 μM) was unchanged between the FBC model and sham-operated mice.Each column represents means ± SEM (n = 10). The asterisk indicates a significant difference between the FBC model and sham-operated mice, as determined by Student's t-test (two-tailed, *P < 0.05).
Involvement of μ-opioid receptors but not κ-opioid receptors in the inhibitory effects of oxycodone and morphine on GABAergic eIPSCs in the FBC model
We next explored the type of opioid receptors that mediate the inhibitory effects of oxycodone and morphine on GABAergic eIPSCs in VLPAG neurons in slices of the FBC model. It is known that the antinociceptive effects of oxycodone and morphine are mainly mediated by μ-opioid receptors (Lemberg et al., 2006; Peckham and Traynor, 2006), but the involvement of κ-opioid receptors is also reported (Ross and Smith, 1997; Nielsen et al., 2007). Therefore, we evaluated the effects of μ-opioid receptor-selective antagonist and κ-opioid receptor-selective antagonist on inhibition of eIPSCs by oxycodone and morphine in slices of FBC model. In the presence of μ-opioid receptor-selective antagonist β-FNA (1 μM), the inhibitory effect of 10 μM oxycodone on eIPSCs was completely abolished (Figure 4A; oxycodone administered after pretreatment with β-FNA, 2.7 ± 3.8% inhibition vs. oxycodone alone: 45.5 ± 2.3% inhibition, n = 6, t(10) = 9.569; P < 0.001). By contrast, the κ-opioid receptor-selective antagonist nor-BNI (1 μM) did not change the effects of oxycodone (Figure 4B; oxycodone administered after pretreatment with nor-BNI, 37.3 ± 3.2% inhibition vs. oxycodone alone: 37.6 ± 3.5% inhibition, n = 6). The inhibitory effect of 10 μM morphine on eIPSCs was also abolished following pretreatment with β-FNA (Figure 4C; morphine administered after pretreatment with β-FNA, 4.6 ± 3.0% inhibition vs. morphine alone: 32.3 ± 4.2% inhibition, n = 5, t(8) = 5.254; P < 0.01) while it was not blocked by nor-BNI (Figure 4D; morphine administered after pretreatment with nor-BNI, 33.5 ± 4.8% inhibition vs. morphine alone: 31.0 ± 7.4% inhibition, n = 5). Thus, both morphine and oxycodone cause inhibition of eIPSCs via activation of μ-opioid receptors, but not κ-opioid receptors. This result is consistent with our previous studies demonstrating that oxycodone and morphine-induced GTPγS activity in the PAG area is mediated by μ-opioid receptors (Nakamura et al., 2013).
Figure 4.
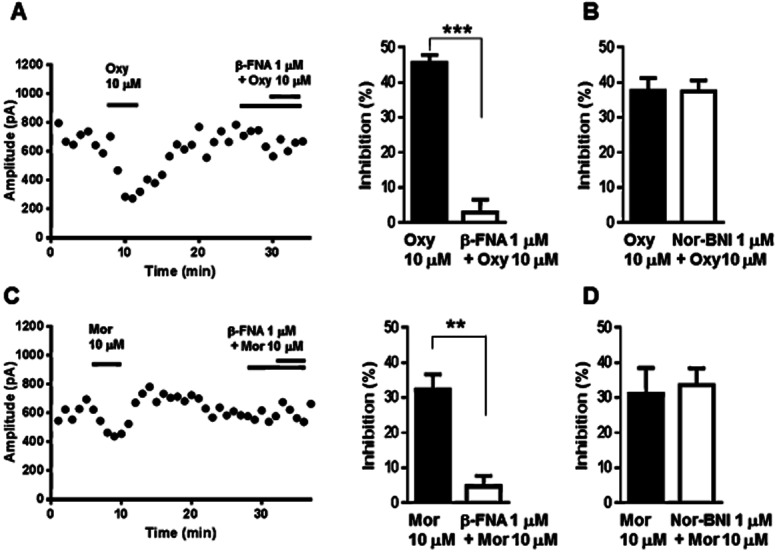
The μ-opioid receptor-selective antagonist β-FNA, but not κ-opioid receptor-selective antagonist nor-BNI abolished the inhibitory effects of oxycodone and morphine, on GABAergic eIPSCs in VLPAG neurons of the FBC model. (A) Left panel, a representative time course, which was shown every 1 min by averaging six IPSCs traces, indicated that oxycodone caused potent inhibition of GABAergic eIPSCs in a VLPAG neuron from the FBC model, which was abolished by β-FNA. On the right panel, a summary of the effects of β-FNA on the oxycodone-induced reduction of GABAergic eIPSCs in slices from the FBC model. Each column represents means ± SEM (n = 6). The asterisk indicates a statistically significant difference between the oxycodone-only and oxycodone with β-FNA-treated groups, as determined by Student's t-test (two-tailed, ***P < 0.001). (B) Summary of the effects of nor-BNI on oxycodone-induced reduction in GABAergic eIPSCs in slices from the FBC model. Each column represents means ± SEM (n = 6) for the oxycodone-only and oxycodone with nor-BNI-treated groups. (C) On the left panel, a representative time course showing that morphine caused inhibition of GABAergic eIPSCs in a VLPAG neuron from the FBC model, which was abolished by β-FNA. On the right panel, a summary of the effects of β-FNA on the morphine-induced reduction of GABAergic eIPSCs in slices from the FBC model. Each column represents means ± SEM (n = 5). The asterisk indicates a statistically significant difference between the morphine-only and morphine with β-FNA-treated groups, as determined by Student's t-test (two-tailed, **P < 0.01). (D) Summary of the effects of nor-BNI on morphine-induced reduction in GABAergic eIPSCs in slices from the FBC model. Each column represents means ± SEM (n = 5) of the morphine-only and morphine with nor-BNI-treated groups.
Changes in the PPR and increased effects of oxycodone on PPR in slices of the FBC model
We then analysed the PPR to evaluate whether presynaptic GABA release or the sensitivity of postsynaptic GABAA receptors was involved in the enhancement of the GABAergic synaptic response and the increased inhibitory effect of oxycodone on GABAergic eIPSCs in slices of the FBC model. A change in the PPR of eIPSCs has been attributed to a presynaptic change in probability of action potential-dependent GABA release (Manabe et al., 1993; Zucker and Regehr, 2002). As seen in Figure 5A–C, the PPR recorded in the FBC model was increased along with an increase in the second eIPSC amplitude (Figure 5C; FBC model, 1.8 ± 0.2, n = 6 vs. sham-operated mice, 1.1 ± 0.1, n = 7, t(11) = 2.431; P < 0.05), indicating enhancement of presynaptic GABA release in the FBC model.
Figure 5.
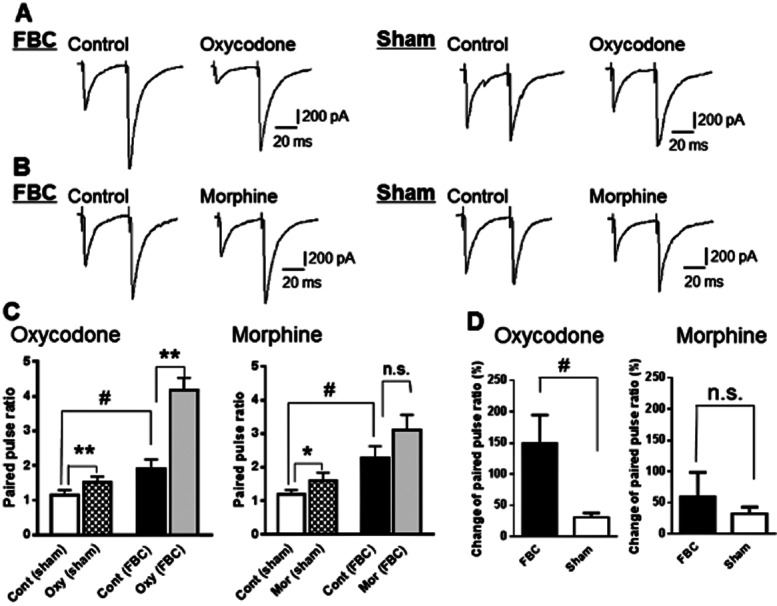
The PPR constructed by two successive GABAergic eIPSCs was increased in the FBC model as compared with sham-operated mice, and oxycodone potently increased the PPR in the FBC model. (A) A representative trace showing that oxycodone (10 μM) potently reduced the first amplitude of GABAergic eIPSCs, resulting in an increase in the PPR in slices of the FBC model. This effect was greater in the FBC model compared with that in sham-operated mice. (B) A representative trace showing that morphine (10 μM) reduced the first amplitude of GABAergic eIPSCs, resulting in a slight increase in the PPR in slices of the FBC model and sham-operated mice. (C) Summary graphs showing a significant increment of the PPR in the FBC model compared with sham-operated mice, indicated by sharp. Each column represents means ± SEM (n = 6 or 7). Oxycodone (10 μM) significantly changed the PPR in the FBC model while morphine(10 μM) induced slight changes in PPR, indicated by asterisk. (D) The increment of PPR by oxycodone (10 μM) was enhanced in the FBC model while that by morphine (10 μM) was not significantly different between the FBC model and sham-operated mice. The sharp indicates a statistically significant difference between the sham-operated mice and the FBC model groups, as determined by Student's t-test (two-tailed, #P < 0.05). The asterisk indicates a statistically significant difference between pre-treatment (control; cont) and post-treatment with each drug in the FBC model and sham-operated mice, as determined by paired t-test (two-tailed, *P < 0.05, **P < 0.01).
In slices from the FBC model, 10 μM oxycodone reduced the initial eIPSC amplitude and significantly increased PPR (Figure 5A and C). The degree of PPR change by oxycodone was significantly greater in the FBC model (Figure 5D; FBC model, 149.5 ± 45.2% increase, n = 6 vs. sham-operated mice, 30.8 ± 6.8% increase, n = 7, t(11) = 2.810; P < 0.05). In contrast, morphine reduced the initial eIPSC amplitude and slightly changed the PPR in the FBC model (Figure 5B and C). The degree of change in PPR caused by morphine did not differ significantly between the FBC model and sham-operated mice (Figure 5D; FBC model, 58.8 ± 39.8% increase, n = 6 vs. sham-operated mice: 32.4 ± 10.6% increase, n = 7). These results showed that inhibition of presynaptic GABA release by oxycodone, but not morphine was increased in the FBC model.
Involvement of Kir3.1 channels in the inhibitory effects of oxycodone on GABAergic eIPSCs in both the FBC model and sham-operated mice
We next explored the involvement of the Kir3.1 channels in the inhibitory effects of oxycodone and morphine on GABAergic eIPSCs in VLPAG neurons in slices of the FBC model. In the presence of the Kir3.1 channel inhibitor tertiapin-Q (100 nM), the inhibitory effect of 10 μM oxycodone on eIPSCs was completely abolished (Figure 6A; oxycodone pretreated with tertiapin-Q, 0.7 ± 4.9% inhibition vs. oxycodone alone: 38.6 ± 5.0% inhibition, n = 7, t(12) = 5.383; P < 0.001). Tertiapin-Q (100 nM) alone did not have significant effects on eIPSCs (3.0 ± 4.9% inhibition, n = 7). The inhibitory effects of tertiapin-Q on the oxycodone-induced reduction of eIPSCs were reversible because a second application of oxycodone alone again reduced eIPSCs. Also, in sham-operated mice, the inhibition of eIPSCs by oxycodone was blocked by pretreatment with tertiapin-Q (Figure 6A; oxycodone pretreated with tertiapin-Q, 3.4 ± 1.5% inhibition vs. oxycodone alone: 23.0 ± 2.1% inhibition, n = 5, t(8) = 7.267; P < 0.001). In contrast, the inhibitory effects of morphine on eIPSCs were not abolished by tertiapin-Q in both the FBC model and sham-operated mice (Figure 6B). The concentration of tertiapin-Q used was sufficient to block Kir3.1 channels as it was reported to effectively block opioid or GABAB receptor agonist baclofen-activated Kir3.1 channels in a brain slice (Nassirpour et al., 2010; Wu et al., 2011). In the present study, Kir3.1 channel activity is important for the inhibitory effects on eIPSCs of oxycodone, but not morphine, in both the FBC model and sham-operated mice.
Figure 6.
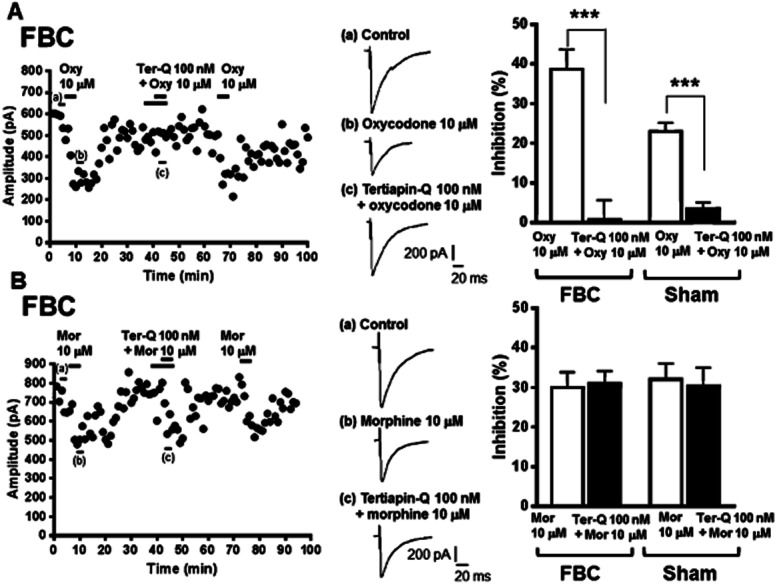
The Kir3.1 channel inhibitor tertiapin-Q abolished the inhibitory effect of oxycodone, but not that of morphine, on GABAergic eIPSCs in VLPAG neurons of the FBC model. (A) Left panel, a representative time course (plot of average amplitude of six IPSCs traces for 1 min) showing that oxycodone caused potent inhibition of GABAergic eIPSCs in a VLPAG neuron from the FBC model, which was abolished by tertiapin-Q. In the middle panel, each trace was recorded during the times indicated on the graph. Right panel, a summary of the effects of tertiapin-Q on the oxycodone-induced reduction of GABAergic eIPSCs in slices from the FBC model and sham-operated mice. Each column represents means ± SEM (n = 5 or 7). The asterisk indicates a statistically significant difference between the oxycodone-only and oxycodone with tertiapin-Q-treated groups, as determined by Student's t-test (two-tailed, ***P < 0.001). (B) Left panel, a representative time course showing that morphine caused inhibition of GABAergic eIPSCs in a VLPAG neuron from the FBC model, which was not abolished by tertiapin-Q. In the middle panel, each trace was recorded during the times indicated on the graph. Right panel, summary of the effects of tertiapin-Q on morphine-induced reduction in GABAergic eIPSCs in slices from the FBC model and sham-operated mice. Each column represents means ± SEM (n = 5 or 6), constituted of the morphine-only and morphine with tertiapin-Q-treated groups.
Enhancement of GABA release in VLPAG neurons in the FBC model and presynaptic inhibition of GABA release by oxycodone and morphine
In this experiment, we evaluated changes in spontaneous GABAergic mIPSC, rather than action potential-dependent GABA release by neuronal firing, following pharmacological elimination of the influence of the neuronal activities of other neurons. In the presence of TTX, which blocks action potential-driven neuronal activity, the frequency of mIPSCs in slices from the FBC model was significantly increased, compared with that in sham-operated mice, whereas the amplitude of mIPSCs in VLPAG neurons was not different between the two groups (Figure 7). This showed that presynaptic, rather than postsynaptic, GABA release in VLPAG neurons was enhanced in the FBC model.
Figure 7.

The frequency, but not the amplitude, of mIPSCs was enhanced in VLPAG neurons from the FBC model. Oxycodone and morphine decreased the frequency, but not the amplitude, of mIPSCs in slices from the FBC model and sham-operated mice. (A and D) A representative trace showing that the frequency of mIPSCs in VLPAG neurons in slices from the FBC model was enhanced compared with that in slices of sham-operated mice. Oxycodone at 10 μM (A–C) and morphine at 10 μM (D–F) reduced the frequency of mIPSCs without altering the amplitude in slices of the FBC model and sham-operated mice. (B and E) Left panel, summary graphs showing that the frequency of mIPSCs in VLPAG neurons was increased in slices of the FBC model compared with sham-operated mice, indicated by sharp. Oxycodone at 10 μM (B) and morphine at 10 μM (E) caused a statistically significant inhibition of the mIPSCs frequency in slices from the FBC model and sham-operated mice, indicated by asterisk. Right panel, the extent of inhibition of mIPSCs frequency by oxycodone at 10 μM (B) and morphine at 10 μM (E) was not different between the FBC model and sham-operated mice. The sharp indicates a significant difference between the FBC model and sham-operated mice, as determined by Student's t-test (two-tailed, #P < 0.05). The asterisk indicates a statistically significant difference between pre- and post-treatment with oxycodone or morphine within the FBC model and sham-operated mice groups, as determined by paired t-test (two-tailed, *P < 0.05, **P < 0.01). Each column represents means ± SEM (n = 5). (C and F) Summary graphs showing that the amplitude of mIPSCs in VLPAG neurons was not changed in slices of the FBC model compared with sham-operated mice, and the mIPSCs' amplitude in both the FBC model and sham-operated mice was not changed by oxycodone and morphine.
When the effects of oxycodone and morphine were tested, 10 μM oxycodone reduced the frequency of mIPSCs in slices of both the FBC model and sham-operated mice without changing the amplitude (Figure 7A–C). As shown in Figure 7B right, the degree of inhibition of mIPSCs frequency by oxycodone did not differ significantly between the FBC model and sham-operated mice (the FBC model, 66.0 ± 4.1% inhibition, n = 5 vs. sham-operated mice, 60.8 ± 7.0% inhibition, n = 5). Morphine also reduced the frequency of mIPSCs in slices of the FBC model and sham-operated mice without changing the amplitude (Figure 7D–F). As shown in Figure 7E right, the degree of inhibition of mIPSCs frequency by morphine did not differ significantly between the FBC model and sham-operated mice (the FBC model, 67.2 ± 5.4% inhibition, n = 5 vs. sham-operated mice, 52.8 ± 11.0% inhibition, n = 5). These results showed that both oxycodone and morphine reduced spontaneous GABA release in VLPAG neurons by acting on presynaptic GABAergic terminals under these experimental conditions.
Involvement of Kir3.1 channels in the inhibitory effects of oxycodone on GABAergic mIPSCs frequency in the FBC model
To further clarify whether presynaptic inhibition of action potential-independent GABA release by oxycodone and morphine was mediated by Kir3.1 channels, we next explored the effects of tertiapin-Q on the inhibitory effects of oxycodone and morphine on GABAergic mIPSCs in VLPAG neurons in slices of the FBC model. In the presence of tertiapin-Q (100 nM), the inhibitory effect of 10 μM oxycodone on mIPSCs was completely abolished (Figure 8A and B; oxycodone pretreated with tertiapin-Q, 2.4 ± 6.7% inhibition vs. oxycodone applied firstly: 54.1 ± 5.5% inhibition, n = 7, t(12) = 5.950; P < 0.001). The inhibitory effects of tertiapin-Q on the oxycodone-induced reduction of mIPSCs were reversible because a second application of oxycodone alone again reduced mIPSCs (Figure 8B). By contrast, the inhibitory effect of morphine on mIPSCs was not abolished by tertiapin-Q (Figure 8D and E; morphine pretreated with tertiapin-Q, 57.0 ± 3.3% inhibition vs. morphine applied firstly, 53.4 ± 4.2% inhibition, n = 6). Both oxycodone and morphine had no effects on mIPSCs amplitude in the FBC model in the absence and the presence of tertiapin-Q (Figure 8C and F). Thus, Kir3.1 channel activity is important for the inhibitory effects on action potential-independent GABA release of oxycodone, but not morphine, in the FBC model.
Figure 8.

The Kir3.1 channel inhibitor tertiapin-Q abolished the inhibitory effect of oxycodone, but not that of morphine, on GABAergic mIPSCs frequency in VLPAG neurons of the FBC model. (A) A representative trace showing that oxycodone (10 μM) caused inhibition of mIPSCs frequency in a VLPAG neuron from the FBC model, which was completely abolished by tertiapin-Q. (B) Left panel, a summary of inhibition of mIPSCs frequency by oxycodone (10 μM) in the FBC model and its disruption in the presence of tertiapin-Q. Each column represents means ± SEM (n = 7). The asterisk indicates a statistically significant difference between pre- and post-treatment with oxycodone alone groups, as determined by paired t-test (two-tailed, **P < 0.01, ***P < 0.001). Right panel, the extent of inhibition of mIPSCs frequency by oxycodone in the presence of tertiapin-Q and oxycodone alone. The inhibitory effects of tertiapin-Q on the oxycodone-induced reduction of mIPSCs frequency were reversible because a second (2nd) application of oxycodone alone again reduced mIPSCs frequency, to a similar extent as the first (1st) application of oxycodone. (D) A representative trace showing that morphine (10 μM) caused inhibition of mIPSCs frequency in a VLPAG neuron from the FBC model, which was not abolished by tertiapin-Q. (E) Left panel, a summary of inhibition of mIPSCs frequency by morphine (10 μM) and no disruption of morphine-induced inhibition in the presence of tertiapin-Q. Each column represents means ± SEM (n = 6). The asterisk indicates a statistically significant difference between pre- and post-treatment with morphine in the presence of tertiapin-Q and morphine alone, as determined by paired t-test (two-tailed, **P < 0.01, ***P < 0.001). Right panel, extent of inhibition of mIPSCs frequency by morphine in the presence of tertiapin-Q and morphine alone. (C) and (F) Summary graphs showing that the amplitude of mIPSCs in VLPAG neurons was not changed by oxycodone and morphine in the absence and presence of tertiapin-Q.
No involvement of PKA activity in the inhibitory effects of oxycodone on GABAergic eIPSCs in the FBC model
A previous study on β-arrestin2 knock-out mice demonstrated that an elevation of PKA activity in presynaptic terminals causes an enhancement of GABAergic synaptic transmission in PAG neurons and increases the inhibitory effects of morphine on eIPSCs (Bradaia et al., 2005). To investigate whether an elevation of PKA activity underlies the enhancement of GABAergic synaptic transmission in the FBC model, we evaluated the effects of the PKA inhibitor H-89 on GABAergic eIPSC and on the efficacy of oxycodone and morphine. H-89 (10 μM) alone had no significant effect on GABAergic eIPSCs in slices of the FBC model (0.1 ± 2.7% inhibition, n = 15) before application of oxycodone or morphine (Figure 9). In the presence of H-89, the inhibitory effect of 10 μM oxycodone on GABAergic eIPSCs was not altered (Figure 9A; oxycodone pretreated with H-89, 39.1 ± 6.3% inhibition vs. oxycodone applied firstly, 44.6 ± 1.3% inhibition, n = 7). The inhibitory effect of 10 μM morphine on eIPSCs was also unchanged in the presence of H-89 (Figure 9B; morphine pretreated with H-89, 21.2 ± 4.6% inhibition vs. morphine applied firstly, 28.7 ± 3.6% inhibition, n = 8). Our results indicate that an elevation of PKA activity was not involved in either the enhancement of GABAergic synaptic transmission in VLPAGneurons or the effects of oxycodone and morphine in the FBC model.
Discussion
Our study demonstrated that supraspinally administered oxycodone and morphine ameliorated mechanical hypersensitivity in the FBC model, and that oxycodone-induced antinociception was dependent on Kir3.1 channels while that of morphine was not. To clarify these supraspinal antinociceptive effects, we focused on the VLPAG area as a primary site of opioid-induced analgesia, and we evaluated GABAergic synaptic transmission and its modulation by oxycodone and morphine. We found that GABAergic synaptic transmission was enhanced in the FBC model, implying that enhanced GABA release leads to greater inhibition of VLPAG neurons for descending pain control and might be involved in the development of pain hypersensitivity in the FBC model. Furthermore, we found that inhibition of eIPSCs by oxycodone was enhanced in FBC model and inhibitory effects of oxycodone on eIPSCs and mIPSCs frequency were abolished by the Kir3.1 channel inhibitor tertiapin-Q, while the inhibition of eIPSCs by morphine was not altered and inhibitory effects of morphine on eIPSCs and mIPSCs frequency were independent of Kir3.1 channels in the FBC model. This might be responsible for the distinct sensitivity to Kir3.1 channel inhibition of the antinociceptive effects of oxycodone and morphine on mechanical hypersensitivity in the FBC model. Although we cannot rule out the possibility that oxycodone and morphine act on sites other than the VLPAG area because of the methodological limitations of i.c.v administration of drugs, we provided evidence that the enhanced GABAergic synaptic transmission at VLPAG neurons in the FBC model is an important site for supraspinal antinociception by oxycodone via Kir3.1 channel activation.
The present study provided evidence for an increase in GABAergic influence on VLPAG neurons in the FBC model, indicated by a leftward shift of the I–O relationship curve of GABAergic eIPSCs, facilitation of GABAergic eIPSCs by paired stimuli, and the enhanced frequency of spontaneous mIPSCs. These phenomena respectively provided evidence for the enhancement of GABAergic synaptic transmission, as reported by previous studies (Caillard et al., 2000; Park et al., 2010; Kobayashi et al., 2012; Goitia et al., 2013). It was reported that a concurrent leftward shift of the I–O relationship curve and increase in mIPSC frequency reflect an enhancement of GABA release (Herman et al., 2013). By contrast, the observed concurrent increase in PPR and mIPSC frequency contrasts with the general concept that an increase in the initial probability of transmitter release decreases the magnitude of synaptic enhancement (Zucker and Regehr, 2002; Herman et al., 2013). However, it was also reported that synaptic enhancement by electrical stimulation was accompanied by an increase in the neurotransmitter release rate by the mechanism that residual Ca2+ concentration is capable of influencing transmitter release (Felmy et al., 2003; Goitia et al., 2013). The relationship between basal release probability and the direction of paired-pulse synaptic plasticity remains debatable and may vary according to synapse type or pathological state. Based on the relationship between presynaptic release machinery and synaptic facilitation (Wu and Saggau, 1994; Caillard et al., 2000; Xu et al., 2008), our results indicate that the change in presynaptic release machinery might be attributed to the enhancement of GABAergic synaptic transmission in VLPAG neurons in the FBC model. A similar enhanced GABA release mechanism has been reported in rats subjected to peripheral nerve ligation (Hahm et al., 2011) and chronic morphine administration (Ingram et al., 1998; DuPen et al., 2007), both of which resulted in pain hypersensitivity. Together with these studies, several possible mechanisms to explain the enhanced GABAregic synaptic transmission in VLPAG neurons of the FBC model have been suggested. A previous study demonstrated that the GABAA receptor agonist muscimol increased neurotransmitter release in PAG neurons, possibly by mechanisms involving presynaptic or extrasynaptic GABAA receptors, which might cause propagation of presynaptic action potentials (Jang, 2011; Stell, 2011). Thus, GABA release might be triggered by GABAA activation. As another possible mechanism, the enhancement of GABA release in the FBC model might be attributed to diminished inhibition of GABA release by endogenous ligands, such as endocannabinoids or adenosines. These tonically activate VLPAG neurons by modulating GABA release via Gi-coupled receptors with intracellular signal factor and/or ion channels (Hack et al., 2003; Drew et al., 2009). The reduction in tonic inhibition of GABA release by such endogenous ligands might enhance presynaptic GABA release in VLPAG neurons. Thus, an elevation in GABAergic inhibition to VLPAG neurons might elicit strong inhibition of VLPAG neuron activity and weaken descending pain inhibitory control. This might be involved in the development and/or maintenance of chronic pain.
We observed a distinct inhibition of eIPSCs by oxycodone and morphine in slices from the FBC model; the inhibitory effect of oxycodone on eIPSCs was enhanced and dependent on Kir3.1 channels whereas that of morphine was neither changed nor sensitive to these channels. This finding might account for the different sensitivity to Kir3.1 channels inhibition in the antinociceptive effects of oxycodone and morphine in the FBC model. To date, the mechanisms mediating the potent antinociceptive effects of oxycodone, despite its weaker affinity for the μ-opioid receptors compared with morphine, have not yet been clarified. In the present study using the FBC model, we have demonstrated the importance of Kir3.1 channel activity in the supraspinal antinociceptive effects and inhibition of GABAergic transmission in VLPAG neurons by oxycodone. As the inhibitory effects of oxycodone and morphine on eIPSCs were completely abolished by the selective μ-opioid receptor antagonist β-FNA, but not the κ-opioid receptor antagonist nor-BNI, these two opioids act on μ-opioid receptors. The distinct sensitivity to Kir3.1 channel inhibition in the inhibitory effects of oxycodone and morphine on IPSCs might be due to the distinct receptor conformationsand intracellular signalling imposed by binding of different ligands. A previous study of ligand-directed signalling demonstrated that oxycodone, compared with morphine, induced less μ-opioid receptor desensitization and elicited prolonged activation of Kir3.1 channels in locus coeruleus neurons of midbrain slices (Virk and Williams, 2008). The inhibitory effects of oxycodone on eIPSCs, in slices of both the FBC model and sham-operated mice, were dependent on Kir3.1 channels while those of morphine were not. Thus, the Kir3.1 channels dependency of oxycodone was observed not only in the FBC model, but also in the sham-operated mice, and oxycodone might act primarily on a homogeneous population of GABAergic synapses where high-density oxycodone-sensitive μ-opioid receptors coupled with Kir3.1 channels could modulate GABA release. These action sites of oxycodone might be mainly presynaptic μ-opioid receptors coupled with Kir3.1 channels in GABAergic neurons because the inhibitory effect of oxycodone on mIPSCs frequency was completely abolished by tertiapin-Q. Thus, Kir3.1 channels at presynapitc GABAergic neurons, which alter GABA release, are an important target for oxycodone-induced reduction of GABA release and the potent antinociceptive effects in the FBC model.
In the present study, nevertheless, the inhibitory effects of oxycodone on the frequency of both eIPSCs and mIPSCs were mediated by Kir3.1 channels, it was demonstrated that the enhanced efficacy of oxycodone was observed in only the frequency of eIPSCs, but not mIPSC in the FBC model. This appears to be due to the fact that different mechanisms mediate spontaneous and action potential-dependent GABA release, and it is suggested that oxycodone mainly activates Kir3.1 channels located at presynaptic sites. In central synapses, GABAergic eIPSCs induced by electrical stimulation, are dependent on activation of voltage-dependent Ca2+ channels via depolarization followed by propagation of action potentials in cells near the recording sites, while the frequency of mIPSCs reflects the quantal release machinery in presynaptic terminals via PK or the exocytosis step itself, independent of propagation of action potentials by neuronal activity in cells near the recording sites (Scanziani et al., 1992; Sillar and Simmers, 1994; Trudeau et al., 1996, but see Patel et al., 2000). Activation of Kir3.1 channels causes hyperpolarization and sequentially reduces action potential-dependent and -independent GABA release by reducing the release probability and neuronal spike firing in GABAergic neurons (Virk and Williams, 2008; Yum et al., 2008; Liu et al., 2012). The influence of hyperpolarization induced by Kir3.1 channels on GABA release might be greater when these channels are activated in more sites or in cells with higher excitability, as indicated by a previous study (Kohno et al., 2005; Xu et al., 2008). In the present study, oxycodone, but not morphine, elicited Kir3.1 channel-dependent inhibition of eIPSCs and mIPSCs frequency in VLPAG neurons. The inhibitory effects of oxycodone on eIPSCs were enhanced in the FBC model while those on mIPSCs frequency were not changed. Thus, oxycodone more effectively acts on μ-opioid receptors coupled with Kir3.1 channels at presynaptic terminals and/or near the site of generation of the action potentials in the enhanced GABAergic neurons of VLPAG area and reduces the enhanced GABA release in the FBC model. As other possible mechanism, we could not exclude the possibility that oxycodone acts on μ-opioid receptors coupled with Kir3.1 channels at postsynaptic GABAergic neuron and reduces its neuronal firing, because eIPSCs were dependent on postsynaptic neuronal activity and Kir3.1 channels are expressed in post-synaptic neurons (Liu et al., 2012). The possible mechanisms remain to be clarified in future studies.
It was demonstrated that GABAergic synaptic transmission in VLPAG neurons was enhanced in the FBC model and the inhibitory effects of oxycodone on eIPSCs were increased; a previous study with β-arrestin2 knock-out mice demonstrated that an elevation of PKA activity in presynaptic terminals caused an enhancement of GABAergic synaptic transmission in PAG neurons and increased the inhibitory effects of morphine on eIPSCs (Bradaia et al., 2005). To investigate whether the elevation of PKA activity might underlie the enhancement of GABAergic synaptic transmission in the FBC model, we evaluated the effects of PKA inhibitor H-89. However, H-89 had negligible effects on both GABAergic eIPSCs and the efficacy of morphine and oxycodone in the FBC model. Our results indicate that elevation of PKA activity was not involved in either the enhancement of GABAergic synaptic transmission in the FBC model or the effects of the two opioids. Furthermore, in contrast to enhanced inhibitory effects of morphine on GABAergic eIPSCs in the β-arrestin2 knock-out mice, the present study demonstrated that the inhibition of GABAergic eIPSCs by morphine was not increased in the FBC model. This might be explained by a distinct mechanism of enhancement of GABAergic synaptic transmission between the FBC model and the β-arrestin2 knock-out mice in terms of elevation of PKA activity.
In the previous study, we demonstrated that the effects of oxycodone on [3H]-DAMGO binding and [35S]-GTPγS activation were attenuated in the FBC model (Nakamura et al., 2013). This seems to be opposite to the enhanced inhibition of eIPSCs by oxycodone in the FBC model. In the intracellular signalling, μ-opioid receptors coupled to Kir3.1 activation could be modified by GPCR kinase 2, PKC and PKA (Chen and Yu, 1994; Johnson et al., 2006). The effects of oxycodone on GABA release might be enhanced in the intracellular signalling pathway after the activation of μ-opioid receptors. The intracellular mechanisms in the FBC model underlying the modulation of GABA release by μ-opioid receptors coupled with Kir3.1 channels remain to be clarified in future studies.
In conclusion, we demonstrated that Kir3.1 channels are important for the supraspinal antinociceptive effects of oxycodone, but not those of morphine, in the FBC model. As supraspinal mechanisms of oxycodone associated with supraspinal neuronal functional changes, we found that GABAergic synaptic transmission in VLPAG neurons was enhanced in the FBC model and that the inhibitory effects of oxycodone, but not those of morphine, on eIPSCs, were enhanced and dependent on Kir3.1 channels. This is consistent with data that the supraspinal antinociceptive effects of oxycodone were dependent on Kir3.1 channels while those of morphine were not. Considered together with the well-established disinhibition mechanisms underlying supraspinal opioid-induced antinociception (Vaughan and Christie, 1997), the enhanced GABAergic synaptic transmission at VLPAG neurons in the FBC model is an important site of supraspinal antinociception by oxycodone via Kir3.1 channel activation.
Author contributions
K. T. carried out all electrophysiological studies and wrote the paper. K. O. participated in study design and helped to prepare the paper. A. N. and M. H. helped to prepare the paper. T. K. and T. T. carried out the preparation of the FBC model. H. O. carried out behavioural tests in the FBC model. Y. M. conceived of the study and participated in its design. M. S., T. M. and T. S. conceived of the study and helped to prepare the paper. G. S. performed study design and is a corresponding author of this study. All authors read and approved the final paper.
Conflict of interest
K. T., K. O., A. N., T. K., H. O., T. T., Y. M., M. H. and G. S. are employees of Shionogi Co., Ltd., the manufacturer of oxycodone and morphine.
Glossary
- β-FNA
β-funaltrexamine hydrochloride
- CNQX
6-cyano-7-nitroquinoxaline-2,3-dione
- FBC
femur bone cancer
- IPSCs
inhibitory postsynaptic currents
- Kir3.1
G protein-gated inwardly rectifying potassium 1
- nor-BNI
nor-binaltorphimine dihydrochloride
- PPR
paired-pulse ratio
- RVM
rostral ventromedial medulla
- TTX
tetrodotoxin
- VLPAG
ventrolateral periaqueductal gray
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ligand-gated ion channels. Br J Pharmacol. 2013b;170:1582–1606. doi: 10.1111/bph.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ion channels. Br J Pharmacol. 2013c;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013d;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banning A, Sjogren P, Henriksen H. Pain causes in 200 patients referred to a multidisciplinary cancer pain clinic. Pain. 1991;45:45–48. doi: 10.1016/0304-3959(91)90163-R. [DOI] [PubMed] [Google Scholar]
- Basbaum AI, Fields HL. Endogenous pain control systems: brainstem spinal pathways and endorphin circuitry. Annu Rev Neurosci. 1984;7:309–338. doi: 10.1146/annurev.ne.07.030184.001521. [DOI] [PubMed] [Google Scholar]
- Becker R, Jakob D, Uhle EI, Riegel T, Bertalanffy H. The significance of intrathecal opioid therapy for the treatment of neuropathic cancer pain conditions. Stereotact Funct Neurosurg. 2000;75:16–26. doi: 10.1159/000048379. [DOI] [PubMed] [Google Scholar]
- Bercovitch M, Adunsky A. High dose controlled-release oxycodone in hospice care. J Pain Palliat Care Pharmacother. 2006;20:33–39. [PubMed] [Google Scholar]
- Bradaia A, Berton F, Ferrari S, Lüscher C. β-Arrestin2, interacting with phosphodiesterase 4, regulates synaptic release probability and presynaptic inhibition by opioids. Proc Natl Acad Sci U S A. 2005;102:3034–3039. doi: 10.1073/pnas.0406632102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budai D, Fields HL. Endogenous opioid peptides acting at μ-opioid receptors in the dorsal horn contribute to midbrain modulation of spinal nociceptive neurons. J Neurophysiol. 1998;79:677–687. doi: 10.1152/jn.1998.79.2.677. [DOI] [PubMed] [Google Scholar]
- Buvanendran A, Ali A, Stoub TR, Kroin JS, Tuman KJ. Brain activity associated with chronic cancer pain. Pain Physician. 2010;13:E337–E342. [PubMed] [Google Scholar]
- Caillard O, Moreno H, Schwaller B, Llano I, Celio MR, Marty A. Role of the calcium-binding protein parvalbumin in short-term synaptic plasticity. Proc Natl Acad Sci U S A. 2000;97:13372–13377. doi: 10.1073/pnas.230362997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Yu L. Differential regulation by cAMP-dependent protein kinase and protein kinase C of the μ opioid receptor coupling to a G protein-activated K+ channel. J Biol Chem. 1994;269:7839–7842. [PubMed] [Google Scholar]
- Coleman RE. How can we improve the treatment of bone metastases further? Curr Opin Oncol. 1998;10:S7–S13. [PubMed] [Google Scholar]
- Drew GM, Lau BK, Vaughan CW. Substance P drives endocannabinoid-mediated disinhibition in a midbrain descending analgesic pathway. J Neurosci. 2009;29:7220–7229. doi: 10.1523/JNEUROSCI.4362-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPen A, Shen D, Ersek M. Mechanisms of opioid-induced tolerance and hyperalgesia. Pain Manag Nurs. 2007;8:113–121. doi: 10.1016/j.pmn.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Felmy F, Neher E, Schneggenburger R. Probing the intracellular calcium sensitivity of transmitter release during synaptic facilitation. Neuron. 2003;37:801–811. doi: 10.1016/s0896-6273(03)00085-0. [DOI] [PubMed] [Google Scholar]
- Gimbel JS, Richards P, Portenoy RK. Controlled-release oxycodone for pain in diabetic neuropathy: a randomized controlled trial. Neurology. 2003;60:927–934. doi: 10.1212/01.wnl.0000057720.36503.2c. [DOI] [PubMed] [Google Scholar]
- Goitia B, Raineri M, González LE, Rozas JL, Garcia-Rill E, Bisagno V, et al. Differential effects of methylphenidate and cocaine on GABA transmission in sensory thalamic nuclei. J Neurochem. 2013;124:602–612. doi: 10.1111/jnc.12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hack SP, Vaughan CW, Christie MJ. Modulation of GABA release during morphine withdrawal in midbrain neurons in vitro. Neuropharmacology. 2003;45:575–584. doi: 10.1016/s0028-3908(03)00205-3. [DOI] [PubMed] [Google Scholar]
- Hahm ET, Kim Y, Lee JJ, Cho YW. GABAergic synaptic response and its opioidergic modulation in periaqueductal gray neurons of rats with neuropathic pain. BMC Neurosci. 2011;12:41. doi: 10.1186/1471-2202-12-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen MS, Mathiesen O, Trautner S, Dahl JB. Intranasal fentanyl in the treatment of acute pain – a systematic review. Acta Anaesthesiol Scand. 2012;56:407–419. doi: 10.1111/j.1399-6576.2011.02613.x. [DOI] [PubMed] [Google Scholar]
- Heiskanen T, Kalso E. Controlled-release oxycodone and morphine in cancer related pain. Pain. 1997;73:37–45. doi: 10.1016/s0304-3959(97)00072-9. [DOI] [PubMed] [Google Scholar]
- Herman MA, Kallupi M, Luu G, Oleata CS, Heilig M, Koob GF, et al. Enhanced GABAergic transmission in the central nucleus of the amygdala of genetically selected Marchigian Sardinian rats: alcohol and CRF effects. Neuropharmacology. 2013;67:337–348. doi: 10.1016/j.neuropharm.2012.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram SL, Vaughan CW, Bagley EE, Connor M, Christie MJ. Enhanced opioid efficacy in opioid dependence is caused by an altered signal transduction pathway. J Neurosci. 1998;18:10269–10276. doi: 10.1523/JNEUROSCI.18-24-10269.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang IS. GABAA receptors facilitate spontaneous glutamate release in rat periaqueductal gray neurons. Neuroreport. 2011;22:834–838. doi: 10.1097/WNR.0b013e32834bc733. [DOI] [PubMed] [Google Scholar]
- Johnson EA, Oldfield S, Braksator E, Gonzalez-Cuello A, Couch D, Hall KJ, et al. Agonist-selective mechanisms of μ-opioid receptor desensitization in human embryonic kidney 293 cells. Mol Pharmacol. 2006;70:676–685. doi: 10.1124/mol.106.022376. [DOI] [PubMed] [Google Scholar]
- Kalso E. Oxycodone. J Pain Symptom Manage. 2005;29:S47–S56. doi: 10.1016/j.jpainsymman.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs reporting guidelines working group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Takei H, Yamamoto K, Hatanaka H, Koshikawa N. Kinetics of GABAB autoreceptor-mediated suppression of GABA release in rat insular cortex. J Neurophysiol. 2012;107:1431–1442. doi: 10.1152/jn.00813.2011. [DOI] [PubMed] [Google Scholar]
- Kohno T, Ji RR, Ito N, Allchorne AJ, Befort K, Karchewski LA, et al. Peripheral axonal injury results in reduced μ opioid receptor pre- and post-synaptic action in the spinal cord. Pain. 2005;117:77–87. doi: 10.1016/j.pain.2005.05.035. [DOI] [PubMed] [Google Scholar]
- Koyyalagunta D, Bruera E, Solanki DR, Nouri KH, Burton AW, Toro MP, et al. A systematic review of randomized trials on the effectiveness of opioids for cancer pain. Pain Physician. 2012;15:ES39–ES58. [PubMed] [Google Scholar]
- Lemberg KK, Kontinen VK, Siiskonen AO, Viljakka KM, Yli-Kauhaluoma JT, Korpi ER, et al. Antinociception by spinal and systemic oxycodone: why does the route make a difference? In vitro and in vivo studies in rats. Anesthesiology. 2006;105:801–812. doi: 10.1097/00000542-200610000-00027. [DOI] [PubMed] [Google Scholar]
- Liu ZL, Ma H, Xu RX, Dai YW, Zhang HT, Yao XQ, et al. Potassium channels underlie postsynaptic but not presynaptic GABAB receptor-mediated inhibition on ventrolateral periaqueductal gray neurons. Brain Res Bull. 2012;88:529–533. doi: 10.1016/j.brainresbull.2012.05.010. [DOI] [PubMed] [Google Scholar]
- Manabe T, Wyllie DJ, Perkel DJ, Nicoll RA. Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J Neurophysiol. 1993;70:1451–1459. doi: 10.1152/jn.1993.70.4.1451. [DOI] [PubMed] [Google Scholar]
- Marker CL, Stoffel M, Wickman K. Spinal G-protein-gated K+ channels formed by GIRK1 and GIRK2 subunits modulate thermal nociception and contribute to morphine analgesia. J Neurosci. 2004;24:2806–2812. doi: 10.1523/JNEUROSCI.5251-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercadante S, Arcuri E. Breakthrough pain in cancer patients: pathophysiology and treatment. Cancer Treat Rev. 1998;24:425–432. doi: 10.1016/s0305-7372(98)90005-6. [DOI] [PubMed] [Google Scholar]
- Minami K, Hasegawa M, Ito H, Nakamura A, Tomii T, Matsumoto M, et al. Morphine, oxycodone, and fentanyl exhibit different analgesic profiles in mouse pain models. J Pharmacol Sci. 2009;111:60–72. doi: 10.1254/jphs.09139fp. [DOI] [PubMed] [Google Scholar]
- Nakamura A, Hasegawa M, Minami K, Kanbara T, Tomii T, Nishiyori A, et al. Differential activation of the μ-opioid receptor by oxycodone and morphine in pain-related brain regions in a bone cancer pain model. Br J Pharmacol. 2013;168:375–388. doi: 10.1111/j.1476-5381.2012.02139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura A, Fujita M, Ono H, Hongo Y, Kanbara T, Ogawa K, et al. G protein-gated inwardly rectifying potassium (KIR3) channels play a primary role in the antinociceptive effect of oxycodone, but not morphine, at supraspinal sites. Br J Pharmacol. 2014;171:253–264. doi: 10.1111/bph.12441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassirpour R, Bahima L, Lalive AL, Lüscher C, Luján R, Slesinger PA. Morphine- and CaMKII dependent enhancement of GIRK channel signaling in hippocampal neurons. J Neurosci. 2010;30:13419–13430. doi: 10.1523/JNEUROSCI.2966-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen CK, Ross FB, Lotfipour S, Saini KS, Edwards SR, Smith MT. Oxycodone and morphine have distinctly different pharmacological profiles: radioligand binding and behavioural studies in two rat models of neuropathic pain. Pain. 2007;132:289–300. doi: 10.1016/j.pain.2007.03.022. [DOI] [PubMed] [Google Scholar]
- Olkkola KT, Kontinen VK, Saari TI, Kalso EA. Does the pharmacology of oxycodone justify its increasing use as an analgesic? Trends Pharmacol Sci. 2013;34:206–214. doi: 10.1016/j.tips.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Park C, Kim JH, Yoon BE, Choi EJ, Lee CJ, Shin HS. T-type channels control the opioidergic descending analgesia at the low threshold-spiking GABAergic neurons in the periaqueductal gray. Proc Natl Acad Sci U S A. 2010;107:14857–14862. doi: 10.1073/pnas.1009532107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel MK, Gonzalez MI, Bramwell S, Pinnock RD, Lee K. Gabapentin inhibits excitatory synaptic transmission in the hyperalgesic spinal cord. Br J Pharmacol. 2000;130:1731–1734. doi: 10.1038/sj.bjp.0703530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peckham EM, Traynor JR. Comparison of the antinociceptive response to morphine and morphine-like compounds in male and female Sprague-Dawley rats. J Pharmacol Exp Ther. 2006;316:1195–1201. doi: 10.1124/jpet.105.094276. [DOI] [PubMed] [Google Scholar]
- Quigley C. The role of opioids in cancer pain. Br Med J. 2005;331:825–829. doi: 10.1136/bmj.331.7520.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raehal KM, Bohn LM. The role of beta-arrestin2 in the severity of antinociceptive tolerance and physical dependence induced by different opioid pain therapeutics. Neuropharmacology. 2011;60:58–65. doi: 10.1016/j.neuropharm.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichling DB, Kwiat GC, Basbaum AI. Anatomy, physiology and pharmacology of the periaqueductal gray contribution to antinociceptive controls. Prog Brain Res. 1988;77:31–46. doi: 10.1016/s0079-6123(08)62777-6. [DOI] [PubMed] [Google Scholar]
- Ross FB, Smith MT. The intrinsic antinociceptive effects of oxycodone appear to be κ-opioid receptor mediated. Pain. 1997;73:151–157. doi: 10.1016/S0304-3959(97)00093-6. [DOI] [PubMed] [Google Scholar]
- Scanziani M, Capogna M, Gahwiler BH, Thompson SM. Presynaptic inhibition of miniature excitatory synaptic currents by baclofen and adenosine in the hippocampus. Neuron. 1992;9:919–927. doi: 10.1016/0896-6273(92)90244-8. [DOI] [PubMed] [Google Scholar]
- Sillar KT, Simmers AJ. Presynaptic inhibition of primary afferent transmitter release by 5-hydroxytryptamine at a mechanosensory synapse in the vertebrate spinal cord. J Neurosci. 1994;14:2636–2647. doi: 10.1523/JNEUROSCI.14-05-02636.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestri B, Bandieri E, Del Prete S, Ianniello GP, Micheletto G, Dambrosio M, et al. Oxycodone controlled-release as first-choice therapy for moderate-to-severe cancer pain in Italian patients: results of an open-label, multicentre, observational study. Clin Drug Investig. 2008;28:399–407. doi: 10.2165/00044011-200828070-00001. [DOI] [PubMed] [Google Scholar]
- Spivak CE, Beglan CL. Kinetics of recovery from opioids at wild-type andmutant μ opioid receptors expressed in xenopus oocytes. Synapse. 2000;38:254–260. doi: 10.1002/1098-2396(20001201)38:3<254::AID-SYN4>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Stell BM. Biphasic action of axonal GABA-A receptors on presynaptic calcium influx. J Neurophysiol. 2011;105:2931–2936. doi: 10.1152/jn.01125.2010. [DOI] [PubMed] [Google Scholar]
- Trudeau LE, Emery DG, Haydon PG. Direct modulation of the secretory machinery underlies PKA-dependent synaptic facilitation in hippocampal neurons. Neuron. 1996;17:789–797. doi: 10.1016/s0896-6273(00)80210-x. [DOI] [PubMed] [Google Scholar]
- Vaughan CW, Christie MJ. Presynaptic inhibitory action of opioids on synaptic transmission in the rat periaqueductal grey in vitro. J Physiol. 1997;498:463–472. doi: 10.1113/jphysiol.1997.sp021872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virk MS, Williams JT. Agonist-specific regulation of μ-opioid receptor desensitization and recovery from desensitization. Mol Pharmacol. 2008;73:1301–1308. doi: 10.1124/mol.107.042952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson CP, Babul N. Efficacy of oxycodone in neuropathic pain: a randomized trial in post herpetic neuralgia. Neurology. 1998;50:1837–1841. doi: 10.1212/wnl.50.6.1837. [DOI] [PubMed] [Google Scholar]
- Watson CP, Moulin D, Watt-Watson J, Gordon A, Eisenhoffer J. Controlled-release oxycodone relieves neuropathic pain: a randomized controlled trial in painful diabetic neuropathy. Pain. 2003;105:71–78. doi: 10.1016/s0304-3959(03)00160-x. [DOI] [PubMed] [Google Scholar]
- Wu LG, Saggau P. Presynaptic calcium is increased during normal synaptic transmission and paired-pulse facilitation, but not in long-term potentiation in area CA1 of hippocampus. J Neurosci. 1994;14:645–654. doi: 10.1523/JNEUROSCI.14-02-00645.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Wang HY, Lin CC, Lu HC, Cheng SJ, Chen CC, et al. GABAB receptor-mediated tonic inhibition of noradrenergic A7 neurons in the rat. J Neurophysiol. 2011;105:2715–2728. doi: 10.1152/jn.00459.2010. [DOI] [PubMed] [Google Scholar]
- Xu C, Zhao MX, Poo MM, Zhang XH. GABAB receptor activation mediates frequency-dependent plasticity of developing GABAergic synapses. Nat Neurosci. 2008;11:1410–1418. doi: 10.1038/nn.2215. [DOI] [PubMed] [Google Scholar]
- Yaksh TL, Yeung JC, Rudy TA. Systematic examination in the rat of brain sites sensitive to the direct application of morphine: observation of differential effects within the periaqueductal gray. Brain Res. 1976;114:83–103. doi: 10.1016/0006-8993(76)91009-x. [DOI] [PubMed] [Google Scholar]
- Yanagisawa Y, Furue H, Kawamata T, Uta D, Yamamoto J, Furuse S, et al. Bone cancer induces a unique central sensitization through synaptic changes in a wide area of the spinal cord. Mol Pain. 2010;6:38. doi: 10.1186/1744-8069-6-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yum DS, Cho JH, Choi IS, Nakamura M, Lee JJ, Lee MG, et al. Adenosine A1 receptors inhibit GABAergic transmission in rat tuberomammillary nucleus neurons. J Neurochem. 2008;106:361–371. doi: 10.1111/j.1471-4159.2008.05400.x. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]