

Abstract
A 3 day old infant with persistent severe hypoglycemia was found to have a cystic pancreatic tumor. Cessation of glucose infusion led to severe hypoglycemia. Pancreaticoduodenectomy was performed and revealed an intraductal papillary mucinous neoplasm (IPMN) with high-grade dysplasia. Sequencing of the IPMN revealed a KRAS gene mutation not present in surrounding normal tissues. Deep sequencing of the patient's blood for KRAS mutations showed no evidence of mosaicism. Whole exome sequencing of the blood of the patient and both parents revealed a de novo germline SKIL mutation in the child that was not present in either parent. This suggests a possible role for SKIL in the pathogenesis of pancreatic tumors.
Keywords: intraductal papillary mucinous neoplasm, neonatal hypoglycemia, KRAS, SKIL, pancreas
INTRODUCTION
Young age of onset is extremely unusual for intraductal papillary mucinous neoplasms (IPMNs). Neoplasms that occur at an extremely young age suggest a genetic predisposition. Genome-wide sequencing can be used to discover the genetic alteration driving this predisposition.[1] [2] We present the case of a newborn child with an intraductal papillary mucinous neoplasm of the pancreas with high-grade dysplasia. Whole exome sequencing of the blood of the patient, the blood of the patient's parents and the patient's tumor revealed a de novo germline mutation in the SKIL gene, suggesting a possible role for this gene in the genetic predisposition to pancreatic cancer.
METHODS
The patient was a term female infant who presented to the emergency department one day following discharge from the newborn nursery with lethargy, decreased oral intake, and a blood glucose level of 15 mg/dL. She subsequently developed seizures. The patient was reported to have fed vigorously from birth until the morning of presentation. She was immediately started on dextrose-containing intravenous fluids and was admitted to the pediatric intensive care unit. Evaluation for sepsis was unrevealing. Constant administration of 10% dextrose solution administered at a glucose infusion rate (GIR) of 10 mg/kg/min was required to maintain normoglycemia. Reduction in GIR led to severe hypoglycemia. Metabolic evaluation demonstrated elevated insulin levels with corresponding C peptide. Abdominal ultrasound showed a cystic mass adjacent to the right kidney. Abdominal magnetic resonance imaging (MRI) further characterized the mass as a multi-loculated 2.2 × 3.7 × 3.9cm cystic mass arising from the head of the pancreas. Given the coexistence of hyperinsulinism and a focal pancreatic mass, operative excision was indicated. During operative exploration the tumor was found to fully involve the head of the pancreas, precluding simple enucleation. A pancreaticoduodenectomy was required for complete resection. Blood glucose levels normalized immediately after the lesion was removed, while the patient was still in the operating room. MRI at one year post resection demonstrated no evidence of recurrence. The patient has continued to be normoglycemic with appropriate growth and development at 18 months of age.
Pathologic examination of the resected specimen was performed with hematoxylin and eosin staining as well as immunostains for chromogranin, insulin, somatostatin and glucagon. For genetic analyses the resected IPMN was microdissected to enrich for neoplastic cellularity. Whole exome sequencing of white blood cells from the patient and both parents was carried out using Illumina HiSeq2000 and Agilent SureSelect whole exome capture reagents as previously described. [3]
RESULTS
Examination of the resected pancreas revealed a 4.0 cm intraductal papillary mucinous neoplasm (IPMN) with high-grade dysplasia. The IPMN involved the main and branch ducts, and had a pancreatobiliary phenotype. The islets of Langerhans were enlarged within the tumor as compared to adjacent normal pancreas, confirmed by chromogranin immunostaining. Immunostaining for insulin and somatostatin was positive in the majority of the cells in the clustered islets, as well as in rare cells at the base of the neoplastic epithelial cells in the IPMN. The immunostain for glucagon was positive only in rare cells in the clustered islets of Langerhans, and not in the epithelium of the IPMN itself. (Figure 1)
Figure 1.



Immunostain demonstrating islet cells (below) and IPMN epithelium (top). Insulin and somatostatin are expressed in the islet cells and rare IPMN cells. Glucagon is expressed only in rare islet cells. 1a: insulin, 1b: somatostatin, 1c: glucagon.
Whole exome sequencing of the IPMN revealed a G12V KRAS gene mutation. Neither GNAS nor RNF43 gene mutations were observed. Sequencing of the nonneoplastic pancreas resected with the tumor and deep sequencing of the patient's blood for KRAS revealed only wild-type KRAS sequences suggesting that the KRAS mutation did not occur early in embryonic development. Blood samples were obtained from the infant and both parents, and the samples submitted for whole exome sequencing. The average read depth was 159.8 (range 137 to 187) with at least 89.7% of the targeted positions covered by at least 10 reads. This analysis revealed a de novo mutation in the SKIL gene (A512T) in the infant (Figure 2). This mutation changes the hydrophobic amino acid alanine to the polar amino acid threonine, and was one of only three point mutations or insertion/deletions (indels) in the exome in the baby's germline that was not present in either parent. The other two, one in MUC5B and the other in TUBB5, were felt to be unlikely disease-causing as MUC5B codes for a large mucin and TUBB5 codes for structural protein (a tubulin), although it should be noted that TUBB5 codes for a protein that has been linked to the Robo-Slit pathway.[4] Too many heterozygous variants in the parents that were homozygous in the child were identified for evaluation. We therefore cannot rule out a recessive trait as the cause of the child's IPMN.
Figure 2.
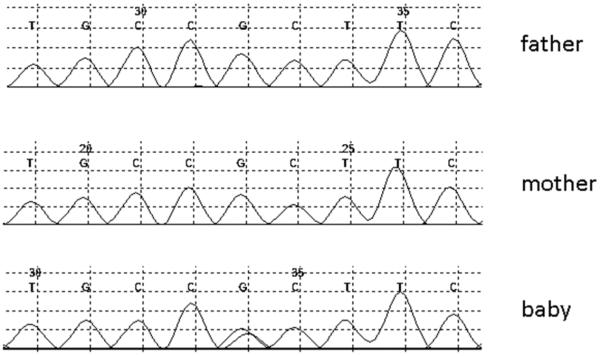
Traces from the index patient and both parents for the coding region of SKIL demonstrating de novo mutation in the baby that is not present in either parent.
DISCUSSION
Congenital hyperinsulinism (CHI) may occur in focal or diffuse forms, and the identification of focal hyperinsulinism generally relies on (18)F DOPA PET-CT scanning to identify a causative lesion as they are generally not evident on standard cross-sectional imaging.[5] Unusually, this patient had a large cystic neoplasm obviating the need for additional imaging studies. Although IPMN occurring concomitantly with neuroendocrine tumors have been reported,[6;7] IPMN in association with CHI is a novel finding.
This patient presented with an IPMN of the pancreas at an extremely young age of onset. It has been estimated that approximately 10% of pancreatic cancers have a familial basis.[8] Clues to a genetic predisposition include young age of onset, multiple cancers and a family history of cancer.[9] Sequencing of DNA isolated from the IPMN identified a KRAS mutation, while sequencing of the patient and the patient's parents germline DNA revealed a de novo germline SKIL mutation in the infant's germline. The SKIL gene codes for a protein that functions in the transforming growth factor β pathway (TGF β).[10] In the absence of TGF-β signaling, SKIL binds to the promoter regions of TGF β-responsive genes and recruits a nuclear repressor complex, turning off the expression of TGF-β responsive genes. When TGF-β signaling is activated, smad3 is translocated to the nucleus where it degrades SKIL, allowing TGF-β responsive genes to be expressed. Inactivating SKIL gene mutations would therefore be predicted to increase the expression of TGF-β responsive genes, and activating SKIL mutations would suppress TGF-β signaling.[11] Of interest, the SMAD4 gene, which also codes for a member of the TGF-β signaling pathway, is one of the most commonly somatically mutated genes in sporadic pancreatic cancer.[12] While we would favor the hypothesis that the germline SKIL gene mutation caused this patient's IPMN, we cannot exclude the possibility that the KRAS mutation occurred in a normal cell in the pancreas early during embryogenesis and initiated the IPMN, with the SKIL mutation playing no role.
The coexistence of an IPMN and congenital hyperinsulinism (CHI) is a unique feature of this case. Persistent CHI in the neonate is associated with mutations in a wide variety of genes leading to dysregulation of insulin production.[13] Sequencing of the baby's germline did not identify any mutations in KCNJ11, however, a missense V560M variant was identified in ABCC8. The minor allele frequency for this variant in the population is 0.005. The variant was inherited from the father who was phenotypically normal and did not have any reported blood glucose abnormalities. We were not able to identify a relationship between the mutation identified and beta cell function. The exact cause of the hyperinsulinism in this patient is therefore unclear, but the process resolved immediately after the IPMN was resected.
In summary, this infant presented with an unusual congenital pancreatic mass causing hyperinsulinism. Pancreaticoduodenectomy proved curative, and revealed an IPMN with high-grade dysplasia. Sequencing revealed a somatic KRAS gene mutation in the IPMN and a de novo germline SKIL gene mutation in the infant, suggesting but not establishing a possible role for SKIL mutations in the genesis of IPMN.
ACKNOWLEDGEMENTS
Supported by the Lustgarten Foundation for Pancreatic Cancer Research, The Virginia and D. K. Ludwig Fund for Cancer Research; Susan Wojcicki and Dennis Troper; Sol Goldman Pancreatic Cancer Research Center, NIH grants CA62924, CA057345, CA123483, CA121113 and RO1CA97075, the Michael Rolfe Pancreatic Cancer Foundation, The Stringer Foundation, and the family and friends of Dick Knox and Cliff Minor.
K.W. Kinzler, N. Papadopoulos, and B.Vogelstein are founders of Personal Genome Diagnostics, Inc., a company focused on the identification of genetic alterations in human cancer for diagnostic and therapeutic purposes. K.W. Kinzler and B. Vogelstein are also members of the Consultants of Sysmex-Inostics, a company that is developing technologies for the molecular diagnosis of cancer. These companies and others have licensed patent applications from Johns Hopkins relevant to the current study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The terms of these arrangements are being managed by the university in accordance with its conflict of interest policies. No potential conflicts of interest were disclosed by the other authors.
References
- 1.Roberts NJ, Jiao Y, Yu J, Kopelovich L, Petersen GM, Bondy ML, et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2012;2:41–46. doi: 10.1158/2159-8290.CD-11-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324:217. doi: 10.1126/science.1171202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoang ML, Chen CH, Sidorenko VS, He J, Dickman KG, Yun BH, et al. Mutational Signature of Aristolochic Acid Exposure as Revealed by Whole-Exome Sequencing. Science Translational Medicine. 2013;5:102ra102. doi: 10.1126/scitranslmed.3006200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banerjee I, Avatapalle B, Padidela R, Stevens A, Cosgrove KE, Clayton PE, et al. Integrating genetic and imaging investigations into the clinical management of congenital hyperinsulinism. Clin Endocrinol (Oxf) 2013;78:803–813. doi: 10.1111/cen.12153. [DOI] [PubMed] [Google Scholar]
- 6.Kadota Y, Shinoda M, Tanabe M, Tsujikawa H, Ueno A, Masugi Y, et al. Concomitant pancreatic endocrine neoplasm and intraductal papillary mucinous neoplasm: a case report and literature review. World J Surg Oncol. 2013;11:75. doi: 10.1186/1477-7819-11-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Larghi A, Stobinski M, Galasso D, Lecca PG, Costamagna G. Concomitant intraductal papillary mucinous neoplasm and pancreatic endocrine tumour: Report of two cases and review of the literature. Dig Liver Dis. 2009;41:759–761. doi: 10.1016/j.dld.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 8.Hruban RH, Canto MI, Goggins M, Schulick R, Klein AP. Update on familial pancreatic cancer. Adv Surg. 2010;44:293–311. doi: 10.1016/j.yasu.2010.05.011. 293–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brune KA, Lau B, Palmisano E, Canto M, Goggins MG, Hruban RH, et al. Importance of age of onset in pancreatic cancer kindreds. J Natl Cancer Inst. 2010;102:119–126. doi: 10.1093/jnci/djp466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deheuninck J, Luo K. Ski and SnoN, potent negative regulators of TGF-beta signaling. Cell Res. 2009;19:47–57. doi: 10.1038/cr.2008.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hagerstrand D, Tong A, Schumacher SE, Ilic N, Shen RR, Cheung HW. Systematic interrogation of 3q26 identifies TLOC1 and SKIL as cancer drivers. Cancer Discov. 2013;3:1044–1057. doi: 10.1158/2159-8290.CD-12-0592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271:350–353. doi: 10.1126/science.271.5247.350. [DOI] [PubMed] [Google Scholar]
- 13.Palladino AA, Stanley CA. Nesidioblastosis no longer! It's all about genetics. J Clin Endocrinol Metab. 2011;96:617–619. doi: 10.1210/jc.2011-0164. [DOI] [PubMed] [Google Scholar]