

Abstract
Exposure to high levels of organophosphorus compounds (OP) can induce status epilepticus (SE) in humans and rodents via acute cholinergic toxicity, leading to neurodegeneration and brain inflammation. Currently there is no treatment to combat the neuropathologies associated with OP exposure. We recently demonstrated that inhibition of the EP2 receptor for PGE2 reduces neuronal injury in mice following pilocarpine-induced SE. Here, we investigated the therapeutic effects of an EP2 inhibitor (TG6-10-1) in a rat model of SE using diisopropyl fluorophosphate (DFP). We tested the hypothesis that EP2 receptor inhibition initiated well after the onset of DFP-induced SE reduces the associated neuropathologies. Adult male Sprague-Dawley rats were injected with pyridostigmine bromide (0.1 mg/kg, sc) and atropine methylbromide (20 mg/kg, sc) followed by DFP (9.5 mg/kg, ip) to induce SE. DFP administration resulted in prolonged upregulation of COX-2. The rats were administered TG6-10-1 or vehicle (ip) at various time points relative to DFP exposure. Treatment with TG6-10-1 or vehicle did not alter the observed behavioral seizures, however six doses of TG6-10-1 starting 80-150 min after the onset of DFP-induced SE significantly reduced neurodegeneration in the hippocampus, blunted the inflammatory cytokine burst, reduced microglial activation and decreased weight loss in the days after status epilepticus. By contrast, astrogliosis was unaffected by EP2 inhibition 4 d after DFP. Transient treatments with the EP2 antagonist 1 h before DFP, or beginning 4 h after DFP, were ineffective. Delayed mortality, which was low (10%) after DFP, was unaffected by TG6-10-1. Thus, selective inhibition of the EP2 receptor within a time window that coincides with the induction of cyclooxygenase-2 by DFP is neuroprotective and accelerates functional recovery of rats.
Keywords: DFP, PGE2, EP2, COX-2, organophosphorus, Hippocampus, Neurodegeneration, Inflammation, Status Epilepticus
Introduction
Organophosphorus pesticides and nerve agents potently inhibit acetylcholinesterase (AChE), leading to the accumulation of acetylcholine. Exposure to high levels of organophosphorus compounds induces status epilepticus (SE, an unremitting seizure lasting longer than 5 to 30 min, or a series of seizures without intervening regain of consciousness) in humans and rodents, leading to brain injury and long-term cognitive deficits. The current treatments following toxic OP exposure in man such as pralidoxime (2-PAM) and atropine, must be given soon after OP exposure for optimal effectiveness (Jett, 2007). Delay in atropine administration results in decreased heart rate, increased bronchial secretions, lacrimation, and other features of cholinergic stimulation. The administration of an oxime such as 2-PAM helps to remove OP agents from acetylcholinesterase and prevent aging in which the bond between the OP and AChE is strengthened thus destroying the enzyme (Jett, 2007). Diazepam is administered to terminate seizures that may develop following OP exposure (Jett, 2007). The current treatments are not logistically possible within a therapeutic timeframe for most civilian populations exposed to OPs and thus therapies that are effective hours to days after OP exposure are needed to mitigate the ensuing brain injury. One prominent feature of evolving brain injury is an inflammatory response mediated partly by cyclooxygenase-2 (COX-2), a rate limiting enzyme critical for the production of prostaglandins. COX-2 inhibitors can be helpful or harmful in various animal models of brain injury including epilepsy (Rojas et al., 2014). Prolonged treatment with COX-2 inhibitors can lead to deleterious side effects, recalling the problems with Vioxx (Rojas et al., 2014). Prostanoid receptors that bind prostaglandins are the main downstream effectors of COX-2 and offer alternative, more selective targets for drug development and disease modification. The prostaglandin EP2 receptor plays an important role in neuroinflammatory conditions (Andreasson, 2010; Bilak et al., 2004; Jiang et al., 2012; Jiang et al., 2013; Johanson et al., 2013; Liang et al., 2005; Liang et al., 2008; Liu et al., 2005; McCullough et al., 2004; Taniguchi et al., 2011). Recent studies demonstrated that inhibition of the EP2 receptor with a selective small molecule antagonist (TG6-10-1) affords neuroprotection after pilocarpine induced status epilepticus in mice, associated with reduced inflammation and accelerated functional recovery (Jiang et al., 2013). Here, we tested the hypothesis that EP2 receptor inhibition by TG6-10-1 reduces DFP induced neuropathologies. We ask the questions: What is the time course of up-regulation of COX-2, brain inflammation and neurodegeneration after DFP? Does EP2 inhibition by a selective small molecule antagonist oppose such neuropathologies, and what is the optimal timing of antagonist administration? To address these questions we optimized an adult rat model of status epilepticus following DFP exposure. Prior to or following DFP exposure rats were injected with either the EP2 receptor antagonist TG6-10-1 or its vehicle. The rats were allowed to recover and were examined for seizure associated neuropathologies within one week of the initial insult. The results support further investigation into therapeutic modalities for the inhibition of EP2 following exposure to organophosphorus nerve agents or pesticides.
Materials and Methods
Organophosphorus-induced status epilepticus (SE)
All procedures and experiments conformed to the guidelines of the Animal Care and Use Committee of Emory University. Adult male Sprague–Dawley rats (200-220 g body weight) were purchased from Charles River Labs (Wilmington, MA, USA) and housed in standard plastic cages (2 rats/cage) in a temperature controlled room (22 ± 2 °C) on a 12 h reverse light–dark cycle. Food and water were provided ad libitum. On the day of organophosphorus exposure the rats were weighed, placed individually into a plastic cage and moved into a ventilation hood. The rat DFP exposure model of status epilepticus is very similar to that described previously (Li et al., 2011). Awake rats were injected subcutaneously (sc) with the reversible acetylcholinesterase inhibitor pyridostigmine bromide (P1339, TCI America, Portland, OR) at 0.1 mg/kg in 0.9% saline. Twenty minutes later the rats were injected sc with the muscarinic receptor antagonist atropine methylbromide (A6883, Sigma, St. Louis, MO) at 20 mg/kg in 0.9% saline. Pyridostigmine bromide and atropine methylbromide were administered to rats prior to DFP to reduce peripheral OP toxicity and increase survival of the rats following DFP exposure without altering the development of seizures. Pyridostigmine bromide and atropine methylbromide are unable to cross the blood-brain barrier. Ten minutes after the atropine injection, rats were injected ip with diisopropyl fluorophosphate (DFP) (D0879, Sigma) at 9.5 mg/kg diluted in sterile distilled water. DFP was prepared fresh within 5 minutes of administration with thorough mixing. Control (non-seizure) rats were treated similarly except they were given sterile water instead of DFP. Each rat received a volume of compound based on weight (1 ml/kg).
Behavioral scoring of seizure activity
In rats, organophosphorus-induced seizures began within 10 minutes of DFP exposure and consisted of distinct motor behaviors that included forelimb clonus, tail extension, and whole body clonic seizures. Rats presenting these behaviors with increasing seizure intensity, duration, and frequency after exposure to DFP were declared to be in status epilepticus, which is characterized by non-intermittent whole body clonic seizures that persist. Approximately 21% of the rats injected with 9.5 mg/kg DFP (ip) experienced occasional seizures but did not enter status epilepticus. The seizure activity was scored and recorded every 5 minutes for 80-90 min using a modified Racine scale (Racine, 1972) (below). All rats monitored for at least 6 hours exhibited persistent non-intermittent seizure activity. The seizure intensity eventually waned after several hours. The rats were placed in clean plastic cages with fresh bedding, soft food and water and allowed to recover. To maintain hydration lactated Ringer's solution (2 ml, sc) was administered when the rats were placed into cages and then daily until rats were able to eat and drink on their own.
Modified Racine scale for DFP exposure
| Behavioral Score | Observed Motor Behavior | |
| 0 | Normal Behavior: | walking, exploring, sniffing, grooming |
| 1 | Freeze Behavior: | immobile, staring, heightened startle, curled-up posture |
| 2 | Repetitive Behavior: | blinking, chewing, head bobbing, scratching, face washing, whisker twitching |
| 3 | Early Seizure Behavior: | myoclonic jerks, partial body clonus |
| 4 | Advance Seizure Behavior: | whole body clonus |
| 5 | Status Epilepticus (SE): | repeated seizure activity (≥2 events in stages 3, 4 or 6 within a 5 minute window) |
| 6 | Intense Seizure Behavior: | repetitive jumping or bouncing, wild running, tonic seizures |
| 7 | Death | |
Modified Irwin test
A modified Irwin test (Irwin, 1968) was performed to access the health of rats prior to and after DFP-induced status epilepticus. The test comprised 12 parameters (ptosis, exophthalmus, lacrimation, body posture, bushy tail, tremors, running vs. walking, dragging body, hyper-/hypoactive, aggression when handled, muscle tone when handled and vocalization when handled) that can be measured simply by experimenter observation. Some parameters require agitating the rats by forcing them to move about their home cage. The test was given twice (once prior to DFP and again 24 hours after DFP exposure). Each parameter was scored on a three point scale (i.e., 0=normal, 1=mild to moderate impairment and 2=severe impairment) with a total score ranging from 0-24. A total score of 0 as the sum of all 12 parameters indicates a normal healthy rat. A total score ranging from 1-11 indicates a healthy animal that appears slightly impaired. A compromised animal would fail the modified Irwin test with a total score of ≥12.
Pharmacokinetics and administration of TG6-10-1
The novel EP2 receptor antagonist TG6-10-1 was synthesized in our laboratory (Jiang et al., 2012). Pharmacokinetic analysis and brain distribution of TG6-10-1 were performed in untreated healthy adult rats at Sai Life Sciences Limited (India). Male Sprague Dawley rats (8-12 weeks olds) weighing between 200 to 250 g were divided into groups of three. Rats were administered either a single intraperitoneal (10 mg/kg) or oral (20 mg/kg) dose of TG6-10-1 dissolved in a formulation solution consisting of 5% N-methyl pyrrolidone (NMP), 5% solutol HS in saline. Blood samples were collected from cannulated jugular veins in freely moving rats at 0.08, 0.025, 0.5, 1, 2, 4, 8, and 24 hours (ip), or 0.25, 0.5, 1, 2, 4, 6, 8 and 24 hours (po) for bioanalysis. Plasma was harvested by centrifugation at 4000 rpm for 10 min at 4 °C and stored frozen until analysis. A separate group of rats was used for brain collection at 1, 2, and 4 hours after TG6-10-1 dosing (ip and po). Brain samples were homogenized using ice-cold phosphate buffer saline (pH 7.4) and homogenates were kept frozen until analysis.
For DFP exposure experiments rats were assigned to a random number stream and received an intraperitoneal injection of: i) one dose (60 minutes prior to DFP exposure), ii) two doses (4 hours and 21 hours after DFP-induced status epilepticus onset) or iii) six doses (80-150 minutes, 5-6 hours, 9-21 hours, 24 hours, 31-42 hours, and 48 hours after DFP-induced status epilepticus onset) of vehicle (10% DMSO, 40% water, 50% polyethylene glycol) or the EP2 receptor antagonist, TG6-10-1 (5 mg/kg), dissolved in the same vehicle. The working concentration of TG6-10-1 was 2.5 mg/ml. Each rat received a volume of either vehicle or TG6-10-1 based on weight (2 ml/kg).
FluoroJade B histochemistry
Four days following DFP-induced status epilepticus rats were euthanized, decapitated and the brains were removed rapidly and longitudinally bisected. One-half of each brain was rapidly frozen on dry ice and kept for RNA isolation (below). The other half was fixed overnight in a 4% paraformaldehyde solution at 4 °C and proces sed for immunohistochemistry and FluoroJade B histochemistry. The half brains post-fixed in 4% paraformaldehyde were transferred to 30% (w/v) sucrose in phosphate buffered saline (PBS) at 4 °C the next day until they sank. The brains were dehydrated, embedded in paraffin, sectioned (8 μm) coronally through the hippocampus and mounted onto slides. Every 20th hippocampal section was labeled with cresyl violet. Every 6th section from each rat brain was used for FluoroJade B staining to label degenerating cells according to the manufacturer protocol (Histo-chem Inc., Jefferson, AR) as described by Schmued et al. (1997). Briefly, hippocampal sections were immersed in 100% ethyl alcohol for 3 min followed by a 1 min change in 70% alcohol and a 1 min change in distilled water. The sections were then transferred to a solution of 0.06% potassium permanganate for 15 min and were gently shaken on a rotating platform at 25 °C. The sections were rinsed for 1 min in distilled water and then transferred to the 0.001% FluoroJade B staining solution where they were gently agitated for 30 min. Following staining, the sections were rinsed with three 1 min changes of Tris-buffered saline (TBS). The sections were made transparent with xylenes and then mounted under D.P.X. (Aldrich Chem. Co., Milwaukee, WI) mounting media. FluoroJade B labeling was visualized using an Axio Observer A1 epifluorescence microscope equipped with an AxioCam MRc 5 camera and a filter suitable for visualizing fluorescein or FITC (Zeiss, Oberkochen, Germany). Pictures were taken using the software AxioVision AC 4.7 (Zeiss).
Quantification of FluoroJade labeled cells
Following FluoroJade B labeling, images of three hippocampal areas (i.e., hilus, CA1, CA3) were taken at 50× and 100× total magnification using the AxioVision AC 4.1 software (Zeiss) from each of 8 sections in each rat. Sections used for FJB quantification were obtained from the dorsal hippocampus between bregma -2.56 and -4.16 mm (Paxinos and Watson, 1986) across all rats. The number of bright FluoroJade B positive cells in the cell layers was counted by another researcher unaware of the experimental conditions. The cell numbers were recorded and expressed as the mean number of positive FluoroJade B (+FJB) cells/section.
Immunohistochemistry
Rats were euthanized under deep isoflurane anesthesia at various time points after DFP exposure and their brains were removed rapidly and longitudinally bisected. One-half of each brain was rapidly frozen on dry ice to obtain protein lysates (below). The other half was fixed overnight in a 4% paraformaldehyde solution at 4 °C and processed for COX-2 immunohistochemistry. The half brains were mounted onto a tissue cutting block and frozen in optimum cutting temperature (OCT) compound (VWR, West Chester, PA). Coronal sections (40 μm) through the hippocampus were cut using a cryostat CM 1850 (Leica, Wetzlar, Germany) and placed in 0.05 M TBS. The free-floating sections were washed five times with PBS for 5 min each and then blocked with PBS containing: 1% bovine serum albumin (BSA), 10% normal goat serum (NGS), and 0.3% Triton X-100 at 25 °C for 2 hours to reduce nonspecific immunostaining. The sections were then incubated at 4 °C for 24 hrs in the primary antibody (rabbit anti-COX-2, 1:1000, Abcam, Cambridge, MA) diluted in antibody dilution solution (ADS), which contains 0.1% gelatin and 0.3% Triton X-100 in PBS. After washing three times with ADS, the sections were incubated with an Alexa Fluor Goat anti-rabbit 488-conjugated secondary antibody (Molecular Probes, Grand Island, NY) diluted 1:200 in ADS for 4 hrs at 25 °C. The sections were again washed with PBS, attached onto slides, allowed to dry and mounted under FluoroGel mount. Images were taken using an Axio Observer A1 fluorescence microscope and the AxioVision AC 4.1 software. Hippocampal sections in which the primary antibody was omitted showed no staining (data not shown).
For GFAP and Iba1 immunohistochemistry, rats that experienced status epilepticus four days earlier and had been administered the EP2 antagonist TG6-10-1 or vehicle were deeply anesthetized by inhalation with isoflurane and decapitated. Paraffin embedded hippocampal sections (8 μm) were prepared. The sections were dewaxed, washed three times with 1× PBS, blocked for 45 minutes in PBS containing 1% BSA, and incubated in the primary antibody [rabbit anti-GFAP, (1:3000), Dako, Carpinteria, CA; rabbit anti-Iba1 (1:3000), Wako, Richmond, VA] diluted in ADS containing 0.1% gelatin and 0.3% Triton X-100 in PBS at 4 °C for 24 hours. After washing three times with ADS, the sections were incubated with an Alexa Fluor fluorophore-conjugated secondary antibody [AlexaFluor goat anti-rabbit 594 (1:2000) for GFAP; AlexaFluor goat anti-rabbit 488 (1:2000) for Iba1; Molecular Probes, Eugene, OR) diluted in ADS for 4 hours at 25°C. The sections were again washed with PBS, allowed to dry, and finalized by the addition of FluoroGel mounting media and a cover slip. Images were taken at 200× (total magnification) using an Axio Observer A1 fluorescence microscope and the AxioVision AC 4.1 software. Sections treated in a similar manner except that the primary antibodies were omitted showed no staining. Quantification of GFAP and Iba1 immunohistochemistry was carried out by manually counting the number of positive labeled cells in a field. A single field image was taken from three dorsal hippocampal subregions (hilus, CA1 and CA3) in a total of four different sections (from every 20th coronal hippocampal section) per animal (4 fields/rat). All of the sections used for GFAP and Iba1 quantification were obtained from the dorsal hippocampus between bregma -2.56 and -4.16 mm (Paxinos and Watson, 1986) across all rats.
RNA isolation and quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was isolated using Trizol with the PureLink RNA Mini Kit (Invitrogen, Carlsbad, CA) from the frozen half brains minus the cerebellum of rats that experienced status epilepticus. RNA concentration and purity were measured by a SmartSpec 3000 spectrophotometer (Biorad, Hercules, CA) using the A260 value and the A260/A280 ratio, respectively. First-strand cDNA synthesis was performed with 1 μg of total RNA, 200 units of SuperScript II Reverse Transcriptase (Invitrogen), and 0.5 μg random primers in a reaction volume of 20 μl at 42 °C for 50 minutes. The reaction was terminated by heating at 70 °C for 15 minutes. qRT-PCR was performed by using 8 μl of 10× diluted cDNA, 0.1–0.5 μM primers, and B-R iQ SYBR Green Supermix (Quanta, Gaithersburg, MD) with a final volume of 20 μl in the iQ5 Multicolor Real-Time PCR Detection System (Bio-Rad). Cycling conditions were as follows: 95 °C for 2 minutes followed by 40 cycles of 95 °C for 15 seconds and 60 °C for 1 minute. Melting curve analysis was used to verify specificity of the primers by single-species PCR product. Fluorescent data were acquired at the 60 °C step. The geometric mean of cycle thresholds for β-actin, glyceraldehyde-3-phosphate (GAPDH), and hypoxanthine phosphoribosyltransferase 1 (HPRT1) (Table 1) was used as an internal control for relative quantification. Samples without cDNA template served as the negative controls. Real time PCR primer sequences are listed in Table 2. Of the 10 inflammatory mediators investigated (Table 2), four (IL-1β, IL-6, IL-15 and TNFα) are produced and secreted by cells involved in both innate and acquired immunity to stimulate inflammation; four (CCL2, CCL3, CCL4 and CXCL10) are chemokines that recruit peripheral leukocytes to sites of inflammation or injury, although CXCL10 can also exert direct excitatory effects on neurons (Nelson and Gruol, 2004). The delayed induction of IL-10 protein acts to resolve inflammation in a murine model of wound healing (Sato et al., 1999), and COX-2 is an intracellular enzyme that forms prostanoids that modify the inflammatory response. Analysis of quantitative real time PCR data was performed by subtracting the geometric mean of the three internal control genes from the measured cycle threshold value obtained from the log phase of each amplification curve of each gene of interest. The fold increase of each gene of interest was estimated for each animal 4 days after DFP-induced SE relative to the amount of RNA found in the control animals using the 2-ΔΔCT method (Livak and Schmittgen, 2001). All conditions for qRT-PCR were the same.
Table 1.
CT values and geometric means of housekeeping genes in four groups of rats (n≥8) for 4 day inflammatory mediator measurement.
| Genes: | Control Vehicle | Control TG6-10-1 | DFP Vehicle | DFP TG6-10-1 |
|---|---|---|---|---|
| HPRT1 | 20.8 ± 0.4 | 19.2 ± 0.1 | 20.7 ± 0.2 | 21.3 ± 0.1 |
| β-ACTIN | 18.3 ± 0.4 | 16.7 ± 0.2 | 17.7 ± 0.2 | 18.1 ± 0.1 |
| GAPDH | 16.5 ± 0.4 | 15.1 ± 0.2 | 16.4 ± 0.1 | 16.6 ± 0.1 |
| Geomean | 18.4 ± 0.4 | 16.9 ± 0.1 | 18.2 ± 0.1 | 18.6 ± 0.1 |
Table 2.
Real Time PCR Primer Sequences. Receptors are denoted with “R” in gene name. The approved human gene nomenclature symbol is in parentheses if different from gene name.
| Genes: | Forward Primer (sequence 5′-3′): | Reverse Primer (sequence 5′-3′): |
|---|---|---|
| HPRT1 | GGTCCATTCCTATGACTGTAGATTTT | CAATCAAGACGTTCTTTCCAGTT |
| β-ACTIN (ACTB) | CCAACCGTGAAAAGATGACC | ACCAGAGGCATACAGGGACA |
| GAPDH | GGTGAAGGTCGGTGTGAAC | CCTTGACTGTGCCGTTGAA |
| CCL2 | CAGAAACCAGCCAACTCTCA | GTGGGGCATTAACTGCATCT |
| CCL3 | TCCACGAAAATTCATTGCTG | AGATCTGCCGGTTTCTCTTG |
| CCL4 | CATCGGAACTTTGTGATGGA | CACAGATTTGCCTGCCTTTT |
| CXCL10 | GTGCTGCTGAGTCTGAGTGG | TTGCAGGAATGATTTCAAGTTTT |
| IL-1β (IL1B) | CAGGAAGGCAGTGTCACTCA | TCCCACGAGTCACAGAGGA |
| IL-6 (IL6) | AACTCCATCTGCCCTTCAGGAACA | AAGGCAGTGGCTGTCAACAACATC |
| TNFα (TNF) | CGTAGCCCACGTCGTAGC | GGTTGTCTTTGAGATCCATGC |
| COX-2 (PTGS2) | ACCAACGCTGCCACAACT | GGTTGGAACAGCAAGGATTT |
| IL-10 (IL10) | ACAGATTCCTTACTGCAGGACTTT | CAAATGCTCCTTGATTTCTGG |
| IL-15 (IL15) | CGATCTGGAGAAAATTGAAAGTC | CTGTACTCGTGCAAAATAACCTGT |
| GFAP | CATCTCCACCGTCTTTACCAC | AACCGCATCACCATTCCTG |
| lba1 (AIF1) | TCGATATCTCCATTGCCATTCAG | GATGGGATCAAACAAGCACTTC |
|
| ||
| IL-1βR (IL1R1) | TCTGACAGGCAACCACTTAC | CATCCCATACACACGGACAA |
| TNFαR (TNFRSF1A) | GCTGTTGCCTCTGGTTATCT | AGGAGCTGAARCCCTACAAATG |
| CCR1 | CCCTAGCCATCTTAGCTTCTATTC | TGAAACCTCTTCCACGTCTTC |
| CCR2 | TGCCTCTCTACCAGGAATCA | GGGCAGGATCAAACTCAAGATA |
| CCR4 | AACCTAGCCATCTCGGATTTAC | GAAGAAGATCCCGCTGTAGAAG |
| CCR5 | CGCTGTAGGAATGAGAAGAAGAG | CAGGAGGAGGACAATGTTGTAG |
Quantification of proteins by ELISA
Protein lysates were generated from half brains (hemisphere without cerebellum) taken from DFP treated and control rats at 0, 0.5, 1, 2, 5, 24, 48 and 96 hours after DFP, by solubilizing the tissue in RIPA lysis and extraction buffer (Thermo Scientific, Rockford, IL) with proteinase and phosphatase inhibitors (Thermo Scientific) using a tissue homogenizer. The lysates were incubated on ice for two hours and then cleared by centrifugation at 14,000 × g for 15 min. The soluble fraction of the lysates was stored at −80 °C until further use. The total protein level was measured by spectrophotometry using a SmartSpec 3000. ELISA was performed according to the manufacturer protocol provided with the kit.
Acetylcholinesterase activity
Protein lysates were prepared from rat half brains (hemisphere without cerebellum) taken from DFP treated rats injected with pyridostigmine bromide (0.1 mg / kg in 0.9% saline, sc), atropine methylbromide (20 mg / kg in 0.9 % saline, sc), and DFP (9.5 mg / kg in water, ip) as well as control rats injected with water instead of DFP at various time points (0, 1, 5, 25 hrs) as described above for ELISA. AChE activity was measured using an acetylcholinesterase assay kit (colorimetric) (Abcam) according to the manufacturer's protocol provided with the kit. The assay uses 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB) to quantify thiocholine production by the hydrolysis of acetylcholine by AChE in brain lysates. On the day of the assay the brain lysates were diluted and plated onto a 96-well plate (50 μl / well). Immediately after plating the acetylcholinesterase standards and unknowns, an acetylthiocholine reaction mixture (containing DTNB and acetylthiocholine; 50 μl / well) was added to the samples. Within 4 minutes the AChE activity was determined by measuring the change in the absorbance at 405 nm using a microplate reader. The data from the brain samples were normalized using the acetylcholinesterase standard curve according to the manufacturer's protocol provided with the kit and compared to brain homogenates obtained from control rats.
Reagents and solutions
The rat COX-2 ELISA kit was purchased from IBL international (Hamburg, Germany). The rat COX-1 ELISA kit was obtained from MyBioSource (San Diego, CA). Rat ELISA kits for IL-10, IL-1β and TNFα were from R&D Systems (Minneapolis, MN). All plasticware and reagents were sterile and RNase and DNase-free.
Data analysis
Data are presented as means ± standard error. Statistical analysis was performed with Microsoft Excel or GraphPad Prism version 4 or 5 (GraphPad software, San Diego, CA). Student's t test or one-way ANOVA (with Bonferroni or Dunnett's posthoc tests) of selected means were performed as appropriate to examine differences of chemical effects. Fisher's exact test was used to compare mortality rates and the percent of rats that entered status epilepticus. The differences were considered to be statistically significant if p < .05.
Results
DFP-induced status epilepticus and pharmacokinetics of TG6-10-1 in rats
Experiments were performed to adapt a rat organophosphorus seizure model to investigate the importance of the EP2 receptor in seizure induced neuropathologies. Adult male Sprague-Dawley rats were injected subcutaneously with pyridostigmine bromide and atropine methylbromide followed 10 min later by a single dose of DFP (9.5 mg/kg, ip) according to the protocol in Figure 1A. This dose of DFP was optimized to effectively obtain status epilepticus in adult male rats. DFP administration has been shown to evoke electrographic seizure activity in male Sprague-Dawley rats (Deshpande et al., 2010; Todorovic et al., 2012). Rats were administered TG6-10-1 (EP2 receptor antagonist) prior, during and following status epilepticus according to the dosing paradigms in Figure 1A. The exposure schedules for TG6-10-1 were informed by pharmacokinetics analysis carried out in normal adult male Sprague-Dawley rats at Sai Life Sciences Ltd (India). Administration of TG6-10-1 (10 mg/kg ip or 20 mg/kg po) in adult male Sprague Dawley rats revealed a brain to plasma concentration ratio in the range of 0.34-1.12 from 1 to 4 hours after injection, and a terminal plasma half-life (T1/2) of 1.9-2.5 hours (Figure 1B, C and Table 3). The TG6-10-1 exposure profile is similar to that in C57Bl/6 mice (brain/plasma = 1.6; plasma half-life = 1.6 hr), including oral bioavailability in both species. The shorter plasma half-life after oral administration in rats (Table 3) is offset by a larger brain to plasma ratio (Figure 1C) following this route of administration. The pharmacokinetic studies were done on untreated rather than DFP-treated rats for logistical reasons. Based on the effects of other agents that induce SE (e.g, Serrano et al., 2011; Jiang et al., 2013; Van Vliet et al., 2014), it is likely that brain penetration of TG6-10-1 is even higher in the DFP-treated rat.
Figure 1.
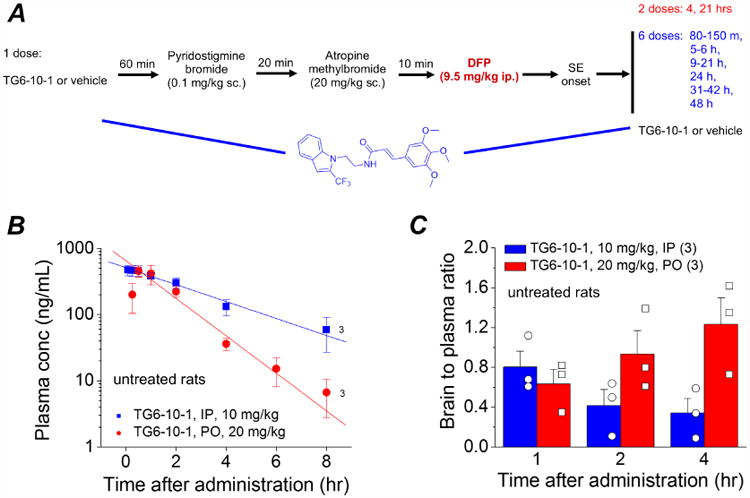
A, Experimental paradigm of chemical administration in a rat model of DFP-induced status epilepticus. For the pre-treatment experiment, rats were injected with 1 dose of the EP2 antagonist TG6-10-1 (5 mg/kg, ip) or vehicle one hour prior to DFP. For post-treatment following DFP, rats were injected with multiple doses (red = 2 doses and blue = 6 doses) of TG6-10-1 or vehicle beginning 80-150 minutes from the onset of SE. B, plasma concentration of untreated rats that received TG6-10-1 (n = 3 rats) ip (10 mg/kg) or po (20 mg/kg). The plasma levels of TG6-10-1 decreased over time. However, the brain to plasma ratio (C) increased in rats administered TG6-10-1 orally.
Table 3.
Individual plasma pharmacokinetic parameters of TG6-10-1 in male SD rats.
| Animal ID: | Tmax (hr) | Cmax (ng/mL) | AUClast (hr*ng/mL) | AUCINF (hr*ng/mL) | T1/2 (hr) |
|---|---|---|---|---|---|
| 1 | 0.25 | 595 | 1401 | 1463 | 1.89 |
| 2 | 0.08 | 567 | 1423 | 1502 | 1.92 |
| 3 | 2.00 | 404 | 1913 | 2528 | 3.69 |
| Mean (ip) | 0.78 | 522 | 1579 | 1831 | 2.50 |
|
| |||||
| 4 | 1.00 | 627 | 1252 | 1262 | 1.54 |
| 5 | 0.50 | 283 | 569 | 570 | 0.78 |
| 6 | 0.50 | 530 | 1102 | 1180 | 3.5 |
| Mean (po) | 0.67 | 480 | 974 | 1004 | 1.94 |
Rats 1-3 received a single intraperitoneal (10 mg/kg) administration of TG6-10-1. Rats 4-6 received a single oral (20 mg/kg) administration of TG6-10-1. Tmax = time to peak plasma concentration; Cmax = peak plasma concentration; AUClast = area under the plasma concentration versus time curve from zero to the last quantifiable concentration; AUCinf = estimated area under the plasma concentration versus time curve from zero to infinity; T1/2 = terminal half-life
DFP was administered to 126 adult male Sprague-Dawley rats, of which 100 entered status epilepticus resulting in a 79% success rate (Figure 2A). About 21% of rats displayed episodic seizure like activity but failed to develop non-intermittent seizure activity, and were given the label “No SE”. Unfortunately, a subset of rats succumbed within minutes to the effects of acute DFP exposure, mainly by respiratory cessation. In total, 43 of 126 rats died either within minutes of DFP administration, or during the ensuing SE, or within 7 days after SE had waned (34% total mortality) (Figure 2B). About 2/3 of the total mortality involved rats that succumbed before or within 8 hr of the onset of status epilepticus. The temporal evolution of seizure activity following DFP exposure revealed a rapid advancement towards status epilepticus with seizures progressively intensifying and status epilepticus being reached within 40-45 minutes after DFP exposure (Figure 2C, D). The proportion of rats that survived the initial DFP exposure, entered status epilepticus (Figure 2E) and recovered the days following was relatively high. We then investigated the effect of EP2 receptor inhibition by TG6-10-1 on seizures and seizure-induced neuropathologies.
Figure 2. DFP-induced status epilepticus.
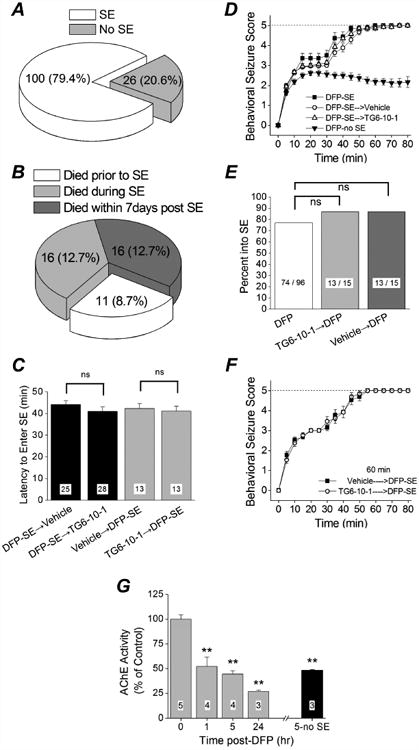
A, of 126 rats administered DFP, 100 entered status epilepticus. B, percent mortality before, during and after status epilepticus. Mortality prior to the onset of status epilepticus is attributed to acute respiratory arrest caused by DFP. C, latency to the onset of status epilepticus following a single intraperitoneal injection of DFP in different groups of rats. (ns = p > .05, one-way ANOVA with posthoc Bonferroni). The number inside the bar represent the number of rats from each group. D, the mean behavioral seizure scores of rats that received a single injection of DFP only (n = 14 rats), DFP followed by 6 doses of vehicle (n = 25 rats) and DFP followed by 6 doses of TG6-10-1 (n = 28 rats) are plotted as a function of time. Also shown is the behavioral seizure activity of rats that did not enter status epilepticus (n = 16 rats). The dashed line indicates the behavioral seizure activity score at the onset of status epilepticus. E, percent of rats entering status epilepticus following a single intraperitoneal injection of DFP was not reduced by a prior injection of TG6-10-1 (p = 0.52, Fisher's exact test). F, the mean behavioral score of rats that received a single injection of TG6-10-1 (n = 13 rats) or vehicle (n = 13 rats) followed by a single injection of DFP is plotted as a function of time. The dashed line indicates the behavioral seizure activity score at the onset of status epilepticus. G, inhibition of acetylcholinesterase in rat forebrain at the given times after DFP exposure. ** = p < .01, one-way ANOVA with posthoc Dunnett's. The number inside the bar represent the number of rats in each group.
TG6-10-1 does not modify status epilepticus per se
EP2 is a Gαs-coupled receptor that is activated by prostaglandin E2 and signals through generation of cyclic AMP. In mice inhibition of EP2 by a single dose of TG6-10-1 administered prior to pilocarpine did not alter the development of electrographic seizures, the temporal evolution of status epilepticus or the ability to enter status epilepticus, suggesting that TG6-10-1 is not an acute anticonvulsant in the mouse pilocarpine model (Jiang et al., 2013). To determine whether EP2 receptor inhibition attenuates DFP-induced seizures we injected rats with the selective EP2 antagonist TG6-10-1 or vehicle, then 60 minutes later (approaching the Tmax for plasma concentration, Table 3) we exposed the rats to DFP. The dose of TG6-10-1 chosen (5 mg/kg ip) is efficacious and has similar pharmacokinetics in mice (Jiang et al., 2013). The percent of rats that entered status epilepticus was identical (13 out of 15, 87%) regardless of whether they received TG6-10-1 or vehicle prior to DFP (Figure 2E), and similar to that of rats not injected prior to DFP (74 out of 96, 77%). The latency to enter status epilepticus following DFP exposure was similar for rats injected with either TG6-10-1 (41.2 ± 2.2 minutes, n = 13) or vehicle (42.3 ± 2.2 minutes, n = 13) 60 minutes prior to DFP (Figure 2C). The mean behavioral seizure score plotted as a function of time revealed an almost identical temporal evolution for rats injected with TG6-10-1 or vehicle 60 minutes prior to DFP (Figure 2F). Similarly, treatment with TG6-10-1 after SE began did not influence the temporal evolution of behavioral seizures or the latency to enter status epilepticus (Figure 2C, D). Although the behavioral seizure activity was scored and recorded for 80-90 minutes all rats administered DFP were observed for at least 6 hours and rats administered TG6-10-1 displayed no apparent difference in the duration of behavioral seizure activity compared to rats administered vehicle. Acetylcholinesterase activity was reduced in the brain of rats exposed to DFP relative to water controls (Figure 2G). Significant reduction was detected 1 hr after DFP exposure. Further inhibition was detected 24 hours after DFP exposure. Acetylcholinesterase activity was also inhibited in rats that were administered the same dose of DFP but did not experience status epilepticus as measured 5 hours after DFP administration, which demonstrates that failure to enter SE is not due to low brain exposure to DFP in these experiments (Figure 2G).
TG6-10-1 facilitates weight regain in rats following DFP exposure
Like mice (Jiang et al., 2013; Serrano et al., 2011), rats that experience more than 60 minutes of status epilepticus usually lose body weight for the first two days and then may start to regain weight the days following. Weight change following status epilepticus has been shown to correlate with functional recovery in mice (Jiang et al., 2013), so we weighed the rats daily for 4 days after DFP-induced status epilepticus. All rats exposed to DFP lost a similar amount of weight on day 1 (∼10% average) (Figure 3A-C). However, by day 2, DFP-injected rats that did not experience status epilepticus (No SE) stabilized their weight and by day 4 they returned close to their original weight prior to DFP (Figure 3B, C solid squares). By contrast, rats experiencing status epilepticus continued to lose weight if they were administered TG6-10-1 or the vehicle 60 minutes prior to DFP (Figure 3A), or 4 hours and then again 21 hours after DFP-induced status epilepticus (Figure 3B). However, in rats injected with 6 doses of TG6-10-1 beginning 80-150 min after onset of status epilepticus, there was a very noticeable improvement in weight by day 3 compared to rats administered 6 doses of the vehicle. The TG6-10-1 treated rats started to regain weight whereas the vehicle injected rats continued to lose weight (Figure 3C), and by day 4 this difference was significant (72.5 ± 3.1% of control for vehicle, n = 8; 91.2 ± 1.9% for TG6-10-1, n = 8; p < .0001, one-way ANOVA with posthoc Bonferroni) (Figure 3C), suggesting that multiple doses are necessary within a specified temporal therapeutic window (see discussion). Taken together, these data suggest a beneficial role of inhibiting the EP2 receptor early and repeatedly that allows rats to regain weight faster following DFP-induced status epilepticus, consistent with the recent finding in mice following pilocarpine-induced status epilepticus (Jiang et al., 2013).
Figure 3. Therapeutic window of TG6-10-1 after DFP-induced status epilepticus.
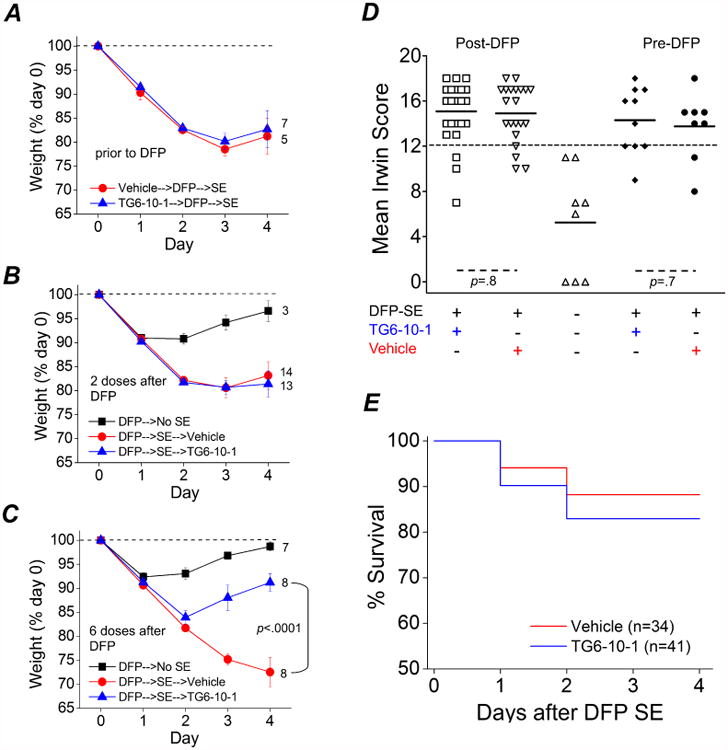
A, a single dose of TG6-10-1 (5 mg/kg, ip) (n = 7 rats) or vehicle (n = 5 rats) administered prior to DFP-induced status epilepticus failed to influence weight regain over the subsequent 4 d. B, two doses of TG6-10-1 (n = 13 rats) or vehicle (n = 14 rats) administered 4 h and 21 h following onset of DFP-induced status epilepticus also failed to elicit weight regain by day four. Rats that were administered DFP, but did not enter status epilepticus (n = 3 rats) returned to their initial weight just prior to DFP exposure by day four. C, six injections of TG6-10-1 (n = 8 rats) beginning 80-150 min after SE onset significantly accelerated weight regain compared to vehicle (n = 8 rats) administration (p < .0001 by one-way ANOVA with posthoc Bonferroni). Rats administered DFP that did not enter status epilepticus (n = 7) returned to the initial weight just prior to DFP exposure by day four. D, rats injected with 2 or 6 doses of TG6-10-1 or vehicle following DFP were combined and labeled “Post-DFP”. Rats that were injected with a single dose of TG6-10-1 or vehicle 1 hour prior to DFP are labeled “Pre-DFP”. There was no difference in the mean Irwin score of rats administered TG6-10-1 (n = 25) following DFP (open squares) compared to rats administered vehicle (down facing open triangle) (n = 21) (p = .8, t test). Similarly, no difference was detected in the mean Irwin score of rats injected with a single dose of TG6-10-1 (closed diamonds) (n = 10) compared to rats injected with vehicle prior to DFP (closed circles) (n = 8) (p = .7, t test). The up facing open triangles are rats that did not enter status epilepticus (n = 8 rats) following DFP administration. The short horizontal bold lines represent the average of the individual animals within the group. The long horizontal dashed line represents the cutoff for determining whether an animal was healthy or impaired. The modified Irwin test score for all rats was 0 prior to drug administration (not shown). E, survival rates of rats that received TG6-10-1 (n = 41 rats) or vehicle (n = 34 rats) up to day 4 after DFP-induced status epilepticus. No difference was detected in the survival rate for rats administered TG6-10-1 (34 of 41 rats survived) compared to vehicle (30 of 34 rats survived) on days 1-4 after DFP-induced status epilepticus (p = .7, Fisher's exact test).
Using a modified Irwin test we determined the effect of EP2 inhibition on a selective subset of normal physiological characteristics of rats prior to any injections on day zero and again 24 hours after DFP-induced status epilepticus. In one group of rats TG6-10-1 or vehicle was administered (5 mg/kg, ip) one hour prior to DFP exposure. Other subsets of rats given DFP that endured status epilepticus were randomized and administered TG6-10-1 or vehicle (5 mg/kg, ip) at various time points after DFP exposure. All rats subjected to the modified Irwin test prior to any injections on day zero scored 0 (not shown) indicative of normal rat health and behavior. However, a large majority of rats failed (score ≥12) the modified Irwin test measured 24 hours after DFP induced status epilepticus (Figure 3D), consistent with compromised health. None of the rats that failed the Irwin test showed signs of infection from the multiple injections on day zero, but instead they displayed lethargy. The average modified Irwin test score for rats administered a single dose of TG6-10-1 prior to DFP was very similar to that of rats that received a single injection of the vehicle (13.8 ± 3 for vehicle, n = 8; 14.3 ± 2.9 for TG6-10-1, n = 10; p = .7, t test) (Figure 3D). Furthermore, no difference was detected in the 24 h modified Irwin test score of rats that were administered TG6-10-1 or the vehicle after DFP (combined 2 and 6 doses post-DFP dosing protocols) (14.9 ± 2.6 for vehicle, n = 21; 15.1 ± 2.7 for TG6-10-1, n = 25; p = .8, t test) (Figure 3D), suggesting that inhibition of the EP2 receptor does not alter the 12 characteristics measured at 24 hours after DFP-induced status epilepticus. This is consistent with the lack of effect of TG6-10-1 on weight loss measured at 1 day after status epilepticus (Figure 3B, C). Rats administered DFP that did not enter status epilepticus (DFP-no SE) had an average 24 hour modified Irwin test score of 5.3 ± 1.7 (n = 8) despite losing the same amount of weight as rats that endured status epilepticus (Figure 3A-C). Unlike rats that endured status epilepticus the “DFP-no SE” rats recovered quickly from the acute effects of DFP exposure without medical intervention, suggesting that compromised health is a result of status epilepticus following DFP administration.
In contrast to the high delayed mortality observed with pilocarpine (Jiang et al. 2013), rats that survived the acute episode of status epilepticus elicited by DFP usually survived the next four days (Figure 3E). All dosing protocols were combined for percent survival analysis as individually there was no difference in delayed mortality between TG6-10-1 and vehicle administered rats with the three different dosing paradigms. For example, in the 6-dose paradigm 9 of 10 rats administered TG6-10-1 survived over the 4 day period after status epilepticus whereas all 9 rats administered vehicle survived (p=1, Fisher's exact test). For the 2-dose paradigm 15 of 18 rats administered TG6-10-1 survived over the 4 day period after status epilepticus whereas 14 of 16 rats administered vehicle survived.
Expression profile of inflammatory mediators and receptors following DFP-induced status epilepticus
Induction of cyclooxygenase-2 (COX-2) occurs rapidly in the brains of rodents that experience at least one hour of status epilepticus (Jarvela el al., 2008; Lee et al., 2007; Pernot et al., 2011; Serrano et al., 2011; Voutsinos-Porche et al., 2004). COX-2 induction is an important component of an inflammatory cascade involving multiple genes that are altered following status epilepticus (Jiang et al., 2013; Serrano et al., 2011). Rats administered DFP were euthanized from 30 m to 96 h after the onset of status epilepticus; one hemisphere was processed for immunohistochemistry and the other hemisphere (without the cerebellum) was processed to obtain protein lysates that were subjected to ELISA for quantification of COX-1 (constitutive cyclooxygenase) and COX-2 (inducible cyclooxygenase). Fluorescence immunohistochemistry performed on coronal hippocampal sections (40 μm) revealed COX-2 upregulation in the forebrain during and following DFP-induced status epilepticus. Clear COX-2 induction in hippocampal pyramidal neurons could be detected as early as 2 hr after onset of status epilepticus of rats (Figure 4A-E). The COX-2 induction profile was recapitulated by ELISA. COX-2 in the brain was significantly increased within 2 hours following DFP exposure (201 ± 25% of control, n = 3) and peaked around 24-48 hours (∼300% of control, n = 3) (Figure 4F). The COX-2 level waned by 96 hours, however it remained significantly elevated (195 ± 28% of control, n = 4) (Figure 4F). The COX-1 level remained near the baseline at all times following DFP-induced status epilepticus (Figure 4F). These results show that exposure of rats to DFP and subsequent status epilepticus rapidly induces COX-2.
Figure 4. COX-2 is induced in principal neurons following DFP exposure.
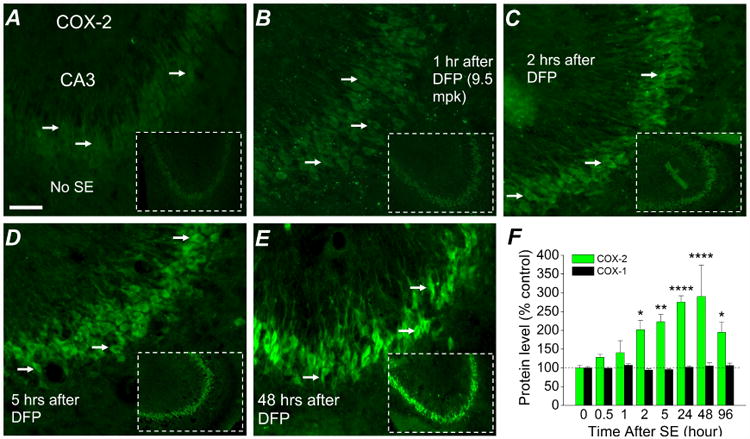
Fluorescent images taken from the CA3 region in the hippocampus (200× magnification) reveals basal expression of neuronal COX-2 in rats that did not experience status epilepticus (A); COX-2 elevation begins 1 h (B) and is prominent by 2 h after DFP-exposure (C). Neuronal COX-2 in the CA3 region is greatly induced 5 hours after DFP exposure (D) and appears to reach a peak 48 hours after DFP exposure (E), but declines back towards the basal level 96 hours after DFP induced status epilepticus (not shown). The images shown were representative of 5 sections each from 3 rats. F, changes in COX-1 and COX-2 protein in the brains of rats following DFP exposure at various times measured by ELISA. (n = 3-5 rats for each time point; * p < .05, ** p < .01, **** p < .0001, two-way ANOVA with Dunnett's post hoc test compared to time 0). Scale bar, 30 μm.
In addition to COX-2 the levels of other inflammatory mediators are also altered by increased seizure activity. For example, IL-1β and TNFα levels are rapidly augmented in the cortex and hippocampus of rats following exposure to sarin (Chapman et al., 2006). However, unlike IL-1β and TNFα, the mRNA expression of IL-10 remains relatively unchanged following pilocarpine induced status epilepticus (Gouveia et al., 2014). ELISA was performed to measure the level of IL-1β, TNFα and IL-10 in protein homogenates prepared from the forebrains of rats administered DFP and euthanized at various time points after the onset of status epilepticus. Like COX-2, IL-1β and TNFα displayed a transient time-dependent induction profile. The level of IL-1β and TNFα was significantly increased within 1 hour following DFP-induced status epilepticus (213 ± 14% of control for IL-1β; 212 ± 25% of control for TNFα) and appeared to reach a peak around 5 hours (358 ± 46% of control for IL-1β; 429 ± 90% of control for TNFα), whereas the level of IL-10 remained near the basal level at all time points (Figure 5A) similar to results in sarin-treated rats (Chapman et al., 2006). By 96 hours the levels of the induced inflammatory mediators had returned to the baseline (Figure 5A).
Figure 5. Induction of inflammatory cytokines and receptors 4 days after DFP induced status epilepticus in rats is attenuated by TG6-10-1.
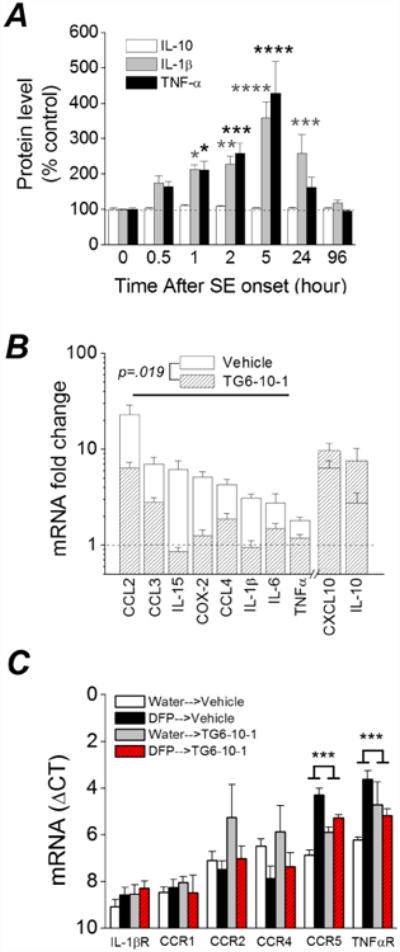
A, changes in IL-1β, TNFα and IL-10 protein in the forebrain at various times after DFP exposure measured by ELISA. (* p < .05, ** p < .01, *** p < .001, **** p < .0001, two-way ANOVA with Dunnett's post hoc test compared to time 0; n = 3-5 rats at each time point). B, change in abundance of 10 inflammatory mediator mRNAs from the forebrain of rats 4 d after injection with water or DFP to induce status epilepticus. Post-DFP treatment was 6 doses of TG6-10-1 (n = 8 rats) or vehicle (n = 9 rats). Following DFP induced status epilepticus, the mRNA fold change for 8 mediators as a group was significantly reduced by TG6-10-1 compared to vehicle (p = .019, paired t test). C, qRT-PCR was also performed to assess the abundance of 6 inflammatory cytokine and interleukin receptors (Table 2) (n = 8 rats per treatment group). The mRNA fold change for 2 of the receptors (TNFαR and CCR5) was significantly reduced by TG6-10-1 compared to vehicle (p < .001, one-way ANOVA with posthoc Bonferroni).
qRT-PCR was then carried out to measure expression of a selected panel of ten inflammatory mediators (Table 2), a majority of which were previously shown to be up-regulated in the mouse brain following pilocarpine induced status epilepticus (Jiang et al., 2013; Serrano et al., 2011). For this experiment only DFP-administered rats that experienced greater than 3 hours of behavioral status epilepticus were used. Four days after DFP exposure, rats administered six doses of vehicle following DFP status epilepticus showed increased brain mRNA levels in all ten of the inflammatory mediators, but not the three housekeeping genes (β-actin, GAPDH and HPRT1; Table 1), including 23-fold for CCL2, 7-fold for CCL3, 6-fold for CXCL10, 6-fold for IL-15, 5-fold for COX-2 and 4-fold for CCL4 (Figure 5B). However, rats administered 6 doses of the EP2 receptor antagonist TG6-10-1 following DFP displayed a broadly blunted inflammatory response (Figure 5B). The average change of eight inflammatory mediators combined was lower in the TG6-10-1 treated rats, with the seizure-induced up-regulation being reduced by almost 70% 4 days after DFP-induced status epilepticus (6.7 ± 2.2 fold induction for vehicle vs. 2.1± 0.6 fold induction for TG6-10-1, p = .019, paired t test). CXCL10 and IL-10 were not included in this analysis as these inflammatory mediators have pleiotropic (CXCL10) or opposing (IL-10) effects to the other eight inflammatory mediators (see discussion).
Chemokine receptors in addition to chemokine ligands may play a role in neuropathological processes and neuroinflammation following seizures. Therefore, qRT-PCR was also carried out to measure expression levels of a selected panel of six inflammatory mediator and cytokine receptors (IL-1βR, TNFαR1, CCR1, CCR2, CCR4 and CCR5; Table 2). These receptors bind the inflammatory mediators described above that have increased expression following DFP induced status epilepticus. Rats administered six doses of vehicle following DFP-induced status epilepticus showed increased mRNA levels in only 2 of the 6 inflammatory mediator receptors investigated four days after DFP-induced status epilepticus, including 7.2-fold for CCR5 and 6.6-fold for TNFαR1 (Figure 5C). The induction of these two receptors was blunted in rats administered 6 doses of the EP2 receptor antagonist TG6-10-1 following DFP induced status epilepticus (Figure 5C). These data suggest that the EP2 receptor contributes significantly to the inflammatory reaction observed 4 days following DFP induced status epilepticus and that inhibition of this prostanoid receptor blunts the inflammatory response.
TG6-10-1 blunts gliosis 4 days after DFP
Experiments were then performed to determine the features of glial activation as it is known that astrogliosis and microgliosis are prominent in the hippocampus following status epilepticus (Borges et al., 2003; Serrano et al., 2011). mRNA of astrocytic GFAP and microglial Iba1 measured in rat brains after status epilepticus can be indicative of the level of gliosis. Compared to saline controls, rats that experienced status epilepticus four days earlier and were administered 6 doses of the vehicle showed increases in both GFAP and Iba1 mRNA (Figure 6A). In rats administered 6 doses of TG6-10-1 after DFP induced status epilepticus, the induction of GFAP showed a trend towards reduction [3.5 ± 0.5 (n = 7) for vehicle vs. 2.4 ± 0.2 (n = 7) for TG6-10-1; ns, one-way ANOVA with posthoc Bonferroni] (Figure 6A). However, the induction of Iba1 was greatly attenuated in rats administered TG6-10-1 compared to vehicle treated rats [4.9 ± 0.8 fold (n = 7) for vehicle vs. 1.6 ± 0.2 fold (n = 7) for TG6-10-1; p < .001, one-way ANOVA with posthoc Bonferroni] (Figure 6A), suggesting that TG6-10-1 reduces microgliosis much more than astrogliosis 4 days after DFP.
Figure 6. DFP induced microgliosis is reduced by TG6-10-1.
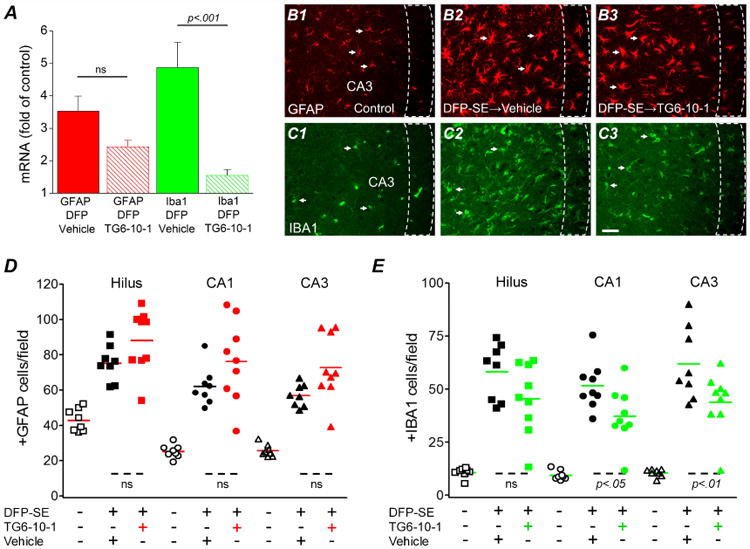
A, induction of GFAP and Iba1 mRNA in the forebrain four days following DFP status epilepticus in vehicle treated (n = 7 rats) and TG6-10-1 treated rats (n = 7 rats) (one-way ANOVA with posthoc Bonferroni). Representative fluorescence images (200× total magnification) showing positive GFAP immunostaining (red) (B1) as an astrocyte marker and Iba1 immunostaining (green) (C1) as a microglial marker in the hippocampal CA3 region. Four days after DFP-induced status epilepticus, astrogliosis and microgliosis were obvious in the sections obtained from rats as defined by the increased number of positively labeled cells in rats treated with 6 injections of TG6-10-1 (B3, C3) compared to sections taken from rats injected with 6 doses of vehicle (B2, C2). The arrows indicate typical astrocytes and microglia in each group. Scale bar, 20 μm. The dash boxes outline the CA3 pyramidal cell layer. Quantification of astrogliosis (D) and microgliosis (E) defined by the number of positive GFAP and Iba1 cells counted in three hippocampal areas (i.e. hilus, CA1 and CA3). The average number of astrocytes and microglia from rats that endured DFP-induced status epilepticus followed by vehicle treatment (6 injections) (n = 8 rats) or TG6-10-1 (n = 9 rats) treatment (6 injections) was compared by one-way ANOVA with posthoc Bonferroni. Each symbol represents data from an individual rat. The short horizontal bold lines (colored) represent the average of the individual animals within the group. The short horizontal dashed lines at the bottom show the groups that are compared.
It mice it has been shown that astrogliosis and microgliosis, which are prominent in the hippocampus following status epilepticus, can be attenuated by TG6-10-1 administration (Jiang et al., 2013). Fluorescent GFAP and Iba1 immunohistochemistry performed on coronal hippocampal sections taken from rat half brains four days after DFP induced status epilepticus confirmed the qRT-PCR results. No difference in the number of activated astrocytes was found in three hippocampal regions (hilus, CA1 and CA3) of rats administered 6 doses of vehicle compared to rats administered 6 doses of TG6-10-1 (Figure 6B, D), but significantly fewer activated microglia were present in the CA1 and CA3 regions of rats treated with the EP2 antagonist (Figure 6C, E).
Hippocampal neurodegeneration is attenuated by TG6-10-1
Status epilepticus induced by DFP produces a characteristic pattern of neuron loss in the hippocampus of adult rats (Kadriu et al., 2011b; Li et al., 2011; Li et al., 2012). To determine the extent of neuronal injury, FluoroJade B (FJB) staining was performed on coronal hippocampal sections (8 μm) taken from the brains of rats four days following DFP-induced status epilepticus. DFP produced a temporal pattern of neurodegeneration in the hippocampus that could be detected as early as 24 hours, but not 5 hours after DFP exposure (not shown). Robust neurodegeneration in the CA1 region of hippocampus was observed four days after status epilepticus (Figure 7A). Jiang et al. (2013) demonstrated that TG6-10-1 reduces hippocampal neurodegeneration in mice following pilocarpine-induced status epilepticus. To determine whether TG6-10-1 administration alters hippocampal neurodegeneration in DFP-treated rats, FJB positive cells were counted in the hilus, CA1 and CA3 and compared to vehicle treated rats. In the CA1 region the total number of FJB positive cells per section was significantly lower in rats administered 6 doses of TG6-10-1 starting 80-150 min after DFP-induced status epilepticus compared to rats administered the vehicle (151 ± 43 FJB positive cells per hippocampal section for vehicle, n = 6 vs. 68 ± 14 for TG6-10-1, n = 7; p < .05, oneway ANOVA with Bonferroni post hoc test) (Figure 7A-C). A strong trend for reduced neurodegeneration was also observed in the CA3 region of TG6-10-1 treated rats (76 ± 16 FJB positive cells per hippocampal section for vehicle, n = 6 vs. 35 ± 12 for TG6-10-1, n = 7), although statistical significance was not attained (Figure 7C). A comparison of the total number of FJB positive pyramidal cells in the combined CA1 and CA3 regions revealed significant neuroprotection in rats injected with 6 doses of TG6-10-1 following DFP-induced status epilepticus (228 ± 52 FJB positive cells per hippocampal section for vehicle, n = 6 vs. 103 ± 25 for TG6-10-1, n = 7) (p < .05, t test) (Figure 7D). Although neurodegeneration was detected in the hilus using FluoroJade B, there was no difference in the number of FJB positive cells per section taken from vehicle treated rats compared to TG6-10-1 treated rats (40 ± 2 cells per section for vehicle treated rats, n = 6 vs. 35 ± 6 for TG6-10-1 treated rats, n =7) (Figure 7C). No neurodegeneration was observed in rats that were injected with DFP but did not enter into status epilepticus or non-seizure control rats that were administered sterile water instead of DFP (not shown). Taken together, these data indicate that delayed inhibition of the EP2 receptor following DFP status epilepticus significantly reduces hippocampal neurodegeneration in the pyramidal cell layers.
Figure 7. Neuroprotection by TG6-10-1 following DFP-induced status epilepticus.
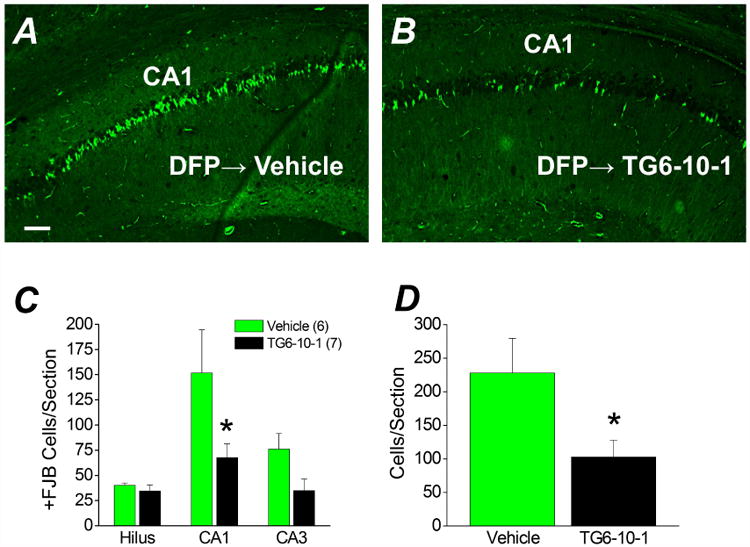
Representative images of FluoroJade B staining in hippocampal sections (8 μm) in the CA1 region for rats treated with 6 doses of vehicle (n = 6 rats) (A) four days after DFP-induced status epilepticus and rats injected with 6 doses of TG6-10-1 (n = 7 rats) (B). Images were taken at a total magnification of 100×. The images are representative of 8 sections per rat. Scale bar, 100 μm. C, the average number of injured neurons per section in three hippocampal regions of rats treated with 6 doses of vehicle (n = 6 rats) and rats injected with 6 doses of TG6-10-1 (n = 7 rats) four days after DFP-induced status epilepticus. (* p < .05 in CA1, one-way ANOVA with posthoc Bonferroni). D, the average number of injured pyramidal neurons from CA1 and CA3 combined per section. (* p < .05, t test). +FJB, positive FluoroJade B.
Discussion
We optimized a model of organophosphorus exposure involving status epilepticus in adult male rats and investigated the role of the EP2 receptor in the associated neuropathologies. In the rat DFP model of status epilepticus, TG6-10-1 does not act as a conventional acute anticonvulsant like benzodiazepines, as judged by latency to enter status, temporal evolution of seizure behavior, and proportion of rats that enter status epilepticus. However, multiple doses of TG6-10-1 beginning 80-150 min after the onset of DFP status epilepticus accelerated recovery from weight loss. Similar to observations with other chemiconvulsants such as pilocarpine and kainate, the rat model of DFP exposure displayed a rapid and robust inflammatory reaction characterized by an early time-dependent increase in brain protein levels of COX-2, IL-1β and TNFα, but not COX-1 or IL-10. Quantification of brain mRNA from rats administered the vehicle following DFP-induced status epilepticus revealed upregulation of all inflammatory mediators investigated on day 4. However, rats injected with six doses of TG6-10-1 showed an attenuated inflammatory response characterized by reduced expression of 8 inflammatory mediators, less microgliosis and less hippocampal neurodegeneration 4 days after DFP exposure. Based on the PK profile (Figure 1B,C), the 6-dose paradigm would have maintained elevated brain levels of TG6-10-1 from about 2h to 15 h after SE onset and then periodically for 48 h, corresponding to the period of rapid COX-2 induction (Figure 4). Taken together, these results suggest that delayed activation of EP2 receptors exacerbates the neuropathological response to DFP induced status epilepticus without affecting the development of seizures and status epilepticus per se.
Approximately 21% of rats succumbed to organophosphorus exposure prior to or during status epilepticus. The rats that survived status epilepticus tend to survive the duration of the experiment as there appeared to be minimal delayed mortality within the first four days and no difference in delayed mortality for rats administered TG6-10-1 compared to rats injected with vehicle. This pattern of mortality is unlike that in the mouse pilocarpine model, which displays a low mortality prior to status epilepticus onset but high delayed mortality within the week following status epilepticus (Jiang et al., 2013). However, high acute mortality is common following organophosphorus exposure in rats (Misik et al., 2014), and is likely due to a multifaceted effect of organophosphorus agents on respiratory and other systems. A late occurring common neuropathology associated with DFP induced status epilepticus is neurodegeneration in multiple brain regions (Kadriu et al., 2011a; Kadriu et al., 2011b; Li et al., 2011; Li et al., 2012; Liu et al., 2012). We measured hippocampal neurodegeneration up to four days following DFP exposure. Neurodegeneration in hippocampal CA1 and CA3 regions was undetectable at 5 hr, modest at 24 hr and robust at 4 days after status epilepticus. We found significant neuroprotection of hippocampal neurons in the CA1 layer and a similar trend in CA3 after status epilepticus in rats administered 6 doses of TG6-10-1 compared to vehicle, even though both groups of rats displayed a similar behavioral seizure phenotype (Figure 2D). Therefore, EP2 activation during or after status epilepticus appears to contribute to delayed pyramidal cell neurodegeneration in hippocampal CA1 and CA3 in the rat DFP model.
Investigation of the time profile for induction of a small subset of inflammatory mediators following DFP exposure revealed a transient upregulation for IL-1β and TNFα whereas COX-2 shows a more sustained upregulation. By day four TNFα and IL-1β protein levels had returned to the basal level but COX-2 remained elevated. This persistent induction of COX-2 days following status epilepticus appears to be common after organophosphorus exposure as COX-2 levels remained elevated above the basal level even seven days following soman exposure in rats (Angoa-Perez et al., 2010). It will be important to determine whether such prolonged induction of this activity-dependent enzyme is caused by the appearance of spontaneous seizures after soman. Unlike COX-2, IL-1β and TNFα, the level of IL-10 protein remained unchanged during status epilepticus and up to 4 days following DFP exposure, although IL-10 mRNA level was elevated at 4 d. We also observed induction of a panel of inflammatory mediator mRNAs (CCL2, CCL3, CCL4, CXCL10, IL-1β, IL-6, COX-2, IL-15 and TNFα) in rat forebrain four days after DFP-induced status epilepticus. The induction of 8 inflammatory mediators was strongly attenuated in rats injected with 6 doses of TG6-10-1 following DFP, suggesting that delayed EP2 activation in the forebrain promotes a robust inflammatory response after organophosphorus exposure. Although CXCL10 mRNA was induced following DFP exposure it remained unchanged in rats injected with 6 doses of TG6-10-1, which indicates a selective rather than general modulation of inflammatory processes by EP2. qRT-PCR also revealed induction of the TNFα and CCR5 receptor mRNAs following status epilepticus, and that TG6-10-1 blunted these effects. Although mRNA levels of these two receptors was altered by inhibition of EP2, this was not the case for the other cytokine receptors. More experiments are necessary to further elucidate the role of activation of different cytokine receptors in neuroinflammation following organophosphorus exposure.
Gliosis is another prominent feature that can be detected late following brain injury induced by organophosphorus based nerve agents (Angoa-Perez, 2010; Liu 2012). Quantification of the degree of gliosis four days after DFP-induced status epilepticus revealed an increase in the number of astrocytes and microglia in the CA3 and CA1 hippocampal regions. Four days following DFP we detected a significant decrease in the number of Iba1 positive microglia in both CA1 and CA3 of rats administered 6 doses of TG6-10-1. This effect in the hippocampus coincides with a decrease in Iba1 mRNA level in the forebrain. Reduced microgliosis by TG6-10-1 is also found in the mouse pilocarpine model (Jiang et al., 2013) and recapitulates the effect of COX-2 ablation in principal forebrain neurons (Serrano et al., 2011). Although COX-2 mRNA induction by DFP is eliminated 4 days after administration of TG6-10-1 (Figure 5B), the enzyme is still present in principal forebrain neurons.
The degree of astrogliosis as measured by the number of GFAP positive cells in the hippocampus four days after DFP-induced status epilepticus was similar for rats injected with 6 doses of TG6-10-1 or vehicle. However, it should be noted that there was a trend for reduction in brain GFAP mRNA in TG6-10-1 treated rats, suggesting that there may be a delay in the effect of TG6-10-1 on astrogliosis and perhaps measuring astrogliosis 4 days post DFP-induced SE may be too early. Additional experiments utilizing other organophosphorus based nerve agents are necessary to determine whether astrogliosis and microgliosis are indeed regulated differently following EP2 activation. Nevertheless, these results reinforce the conclusion that the EP2 receptor contributes to robust brain inflammation following organophosphorus exposure.
A beneficial effect of reduced neurodegeneration, neuroinflammation and gliosis following status epilepticus is expected to be improved functional recovery. We used the rate at which body weight is regained as a surrogate indicator of health. Weight gain is a good indicator of improving health as rats that regained weight after status epilepticus went on to continue gaining weight whereas the health of rats that failed to regain weight continued to decline until they became morbid. TG6-10-1 dosing optimization revealed that one dose administered 1 hour prior to DFP, or 2 doses administered 4 and 21 hours after DFP, were ineffective in improving weight regain following status epilepticus. Only 6 doses administered beginning 80-150 minutes after DFP exposure was sufficient to accelerate weight regain of rats (Figure 3), suggesting that multiple exposures of TG6-10-1 administered within a delayed time window following insult is necessary to protect the brain from neuropathologies associated with organophosphorus exposure. The therapeutic window coincides with the time-dependent induction of COX-2 following brain injury. COX-2 is induced within two hours after status epilepticus onset (Figure 4) resulting in the production of prostaglandins including PGE2, the natural ligand for the EP2 receptor. Considering the pharmacokinetic profile of TG6-10-1 in rats, only the 6 dose paradigm would result in inhibition of EP2 in the brain throughout the period of COX-2 induction.
COX-2 inhibition appears to be beneficial or harmful in SE models depending on the time of inhibition and the model used (discussed in Rojas et al., 2013). Data presented here in the rat DFP model confirm those in the mouse pilocarpine model (Jiang et al., 2013) and suggest that targeting the pro-inflammatory prostanoid EP2 receptor downstream of COX-2 can potentially be more effective than COX-2 inhibition itself. An effective therapeutic window for TG6-10-1 following organophosphorus induced status epilepticus involves EP2 inhibition coinciding with the time-dependent induction of COX-2. Whether EP2 inhibition also ameliorates the long-term consequences of SE (cognitive deficits, appearance of spontaneous seizures) is worthy of study.
Conclusion
DFP causes a rapid induction of cyclooxygenase-2 in hippocampal neurons, which is expected to result in rapid synthesis of PGE2 and consequent activation of prostaglandin receptors on neurons and glia, including EP2. The EP2 antagonist, TG6-10-1, reduced morbidity in rats exposed to DFP, as manifested by reduced neuroinflammation, neurodegeneration and microgliosis, and accelerated weight regain within 4 days after DFP exposure. One somewhat trivial explanation for the beneficial effect of TG6-10-1 in the DFP and pilocarpine models, that the compound simply aborts or truncates status epilepticus, has been ruled out. We conclude that delayed inhibition of EP2 receptors by TG6-10-1 administered beginning 80-150 min after DFP exposure has a number of beneficial consequences. Together, these studies give insight into therapeutic modalities for the inhibition of EP2 in epilepsy. It will be important to determine whether this beneficial effect of TG6-10-1 occurs following exposure to other more potent organophosphorus neurotoxins, and to elucidate the EP2 signaling pathway(s) that mediate these effects.
Highlights.
DFP-induced SE in rats causes neuroinflammation, neurodegeneration and weight loss.
TG6-10-1 blunted neuroinflammation and microgliosis, but not astrogliosis.
Neurodegeneration and weight loss were attenuated by TG6-10-1.
EP2 antagonist TG6-10-1 did not alter DFP-induced behavioral seizures.
Acknowledgments
This work is supported by NIH UO1 NS058158-08 (RD), T32 DA15040 (AR) and P30 NS055077.
Abbreviations
- PGE2
prostaglandin E2
- EP2
prostaglandin E2 receptor 2
- EC50
half maximal effective concentration
- SE
status epilpeticus
- GFAP, CA1
Cornu Ammonis 1
- CA3
Cornu Ammonis 3
- CT
cycle threshold
- Con
control
- COX-2
cyclooxygenase 2
- COX-1
cyclooxygenase 1
- DFP
diisopropyl fluorophosphate
- OP
organophosphorus compound
- IP
intraperitoneal
- SC
subcutaneous
- PO
per oral
- FJB
FluoroJade B
- AchE
acetylcholinesterase
- qRT-PCR
quantitative real time polymerase chain reaction
- IL-1β
interleukin -1β
- TNFα
tumor necrosis factor alpha
- CXCL10
C-X-C motif chemokine 10
- CCL2
chemokine (C-C motif) ligand 2
- CCL3
chemokine (C-C motif) ligand 3
- CCL4
chemokine (C-C motif) ligand 4
- IL-6
interleukin 6
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- HPRT1
hypoxanthine phosphoribosyltransferase 1
- Veh
vehicle
- mpk
mg/kg
- ELISA
enzyme-linked immunosorbent assay
- TG6-10-1
potent and selective EP2 receptor antagonist
Footnotes
Participated in research design: Rojas and Dingledine.
Conducted experiments: Rojas, Gueorguieva and Lelutiu.
Performed data analysis: Rojas, Gueorguieva, Lelutiu and Dingledine.
Wrote or contributed to the writing of the manuscript: Rojas and Dingledine
Synthesized TG6-10-1: Ganesh
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andreasson K. Emerging roles of PGE2 receptors in models of neurological disease. Prostaglandins & other lipid mediators. 2010;91:104–12. doi: 10.1016/j.prostaglandins.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angoa-Perez M, Kreipke CW, Thomas DM, Van Shura KE, Lyman M, McDonough JH, Kuhn DM. Soman increases neuronal COX-2 levels: possible link between seizures and protracted neuronal damage. Neurotoxicology. 2010;31:738–46. doi: 10.1016/j.neuro.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilak M, Wu L, Wang Q, Haughey N, Conant K, St Hillaire C, Andreasson K. PGE2 receptors rescue motor neurons in a model of amyotrophic lateral sclerosis. Annals of neurology. 2004;56:240–8. doi: 10.1002/ana.20179. [DOI] [PubMed] [Google Scholar]
- Borges K, Gearing M, McDermott DL, Smith AB, Almonte AG, Wainer BH, Dingledine R. Neuronal and glial pathological changes during epileptogenesis in the mouse pilocarpine model. Experimental neurology. 2003;182:21–34. doi: 10.1016/s0014-4886(03)00086-4. [DOI] [PubMed] [Google Scholar]
- Chapman S, Kadar T, Gilat E. Seizure duration following sarin exposure affects neuro-inflammatory markers in the rat brain. Neurotoxicology. 2006;27:277–283. doi: 10.1016/j.neuro.2005.11.009. [DOI] [PubMed] [Google Scholar]
- Deshpande LS, Carter DS, Blair RE, DeLorenzo RJ. Development of a prolonged calcium plateau in hippocampal neurons in rats surviving status epilepticus induced by the organophosphate diisopropylfluorophosphate. Toxicol Sci. 2010;116:623–631. doi: 10.1093/toxsci/kfq157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouveia TL, Scorza FA, Iha HA, Frangiotti MI, Perosa SR, Cavalheiro EA, Silva JA, Jr, Feliciano RS, de Almeida AC, Naffah-Mazzacoratti MG. Lovastatin decreases the synthesis of inflammatory mediators during epileptogenesis in the hippocampus of rats submitted to pilocarpine-induced epilepsy. Epilepsy Behav. 2014;36:68–73. doi: 10.1016/j.yebeh.2014.04.009. [DOI] [PubMed] [Google Scholar]
- Irwin S. Comprehensive observational assessment: Ia. A systematic, quantitative procedure for assessing the behavioral and physiologic state of the mouse. Psychopharmacologia. 1968;13:222–57. doi: 10.1007/BF00401402. [DOI] [PubMed] [Google Scholar]
- Jarvela JT, Lopez-Picon FR, Holopainen IE. Age-dependent cyclooxygenase-2 induction and neuronal damage after status epilepticus in the postnatal rat hippocampus. Epilepsia. 2008;49:832–41. doi: 10.1111/j.1528-1167.2007.01454.x. [DOI] [PubMed] [Google Scholar]
- Jett DA. Neurological aspects of chemical terrorism. Annals of Neurology. 2007;61:9–13. doi: 10.1002/ana.21072. [DOI] [PubMed] [Google Scholar]
- Jiang J, Ganesh T, Du Y, Quan Y, Serrano G, Qui M, Speigel I, Rojas A, Lelutiu N, Dingledine R. Small molecule antagonist reveals seizure-induced mediation of neuronal injury by prostaglandin E2 receptor subtype EP2. Proceedings of the National Academy of Sciences. 2012;109:3149–54. doi: 10.1073/pnas.1120195109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Quan Y, Ganesh T, Pouliot WA, Dudek FE, Dingledine R. Inhibition of the prostaglandin receptor EP2 following status epilepticus reduces delayed mortality and brain inflammation. Proceedings of the National Academy of Sciences. 2013;110:3591–6. doi: 10.1073/pnas.1218498110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson JU, Pradhan S, Lokteva LA, Woodling NS, Ko N, Brown HD, Wang Q, Loh C, Cekanaviciute E, Buckwalter M, Manning-Bog AB, Andreasson KI. Suppression of inflammation with conditional deletion of the prostaglandin E2 EP2 receptor in macrophages and brain microglia. The Journal of neuroscience. 2013;33:16016–32. doi: 10.1523/JNEUROSCI.2203-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadriu B, Gocel J, Larson J, Guidotti A, Davis JM, Nambiar MP, Auta J. Absence of tolerance to the anticonvulsant and neuroprotective effects of imidazenil against DFP-induced seizure and neuronal damage. Neuropharmacology. 2011a;61:1463–9. doi: 10.1016/j.neuropharm.2011.08.043. [DOI] [PubMed] [Google Scholar]
- Kadriu B, Guidotti A, Costa E, Davis JM, Auta J. Acute imidazenil treatment after the onset of DFP-induced seizure is more effective and longer lasting than midazolam at preventing seizure activity and brain neuropathology. Toxicological sciences. 2011b;120:136–45. doi: 10.1093/toxsci/kfq356. [DOI] [PubMed] [Google Scholar]
- Lee B, Dziema H, Lee KH, Choi YS, Obrietan K. CRE-mediated transcription and COX-2 expression in the pilocarpine model of status epilepticus. Neurobiology of disease. 2007;25:80–91. doi: 10.1016/j.nbd.2006.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Lein PJ, Liu C, Bruun DA, Giulivi C, Ford GD, Tewolde T, Ross-Inta C, Ford BD. Neuregulin-1 is neuroprotective in a rat model of organophosphate-induced delayed neuronal injury. Toxicology and applied pharmacology. 2012;262:194–204. doi: 10.1016/j.taap.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Lein PJ, Liu C, Bruun DA, Tewolde T, Ford G, Ford BD. Spatiotemporal pattern of neuronal injury induced by DFP in rats: a model for delayed neuronal cell death following acute OP intoxication. Toxicology and applied pharmacology. 2011;253:261–9. doi: 10.1016/j.taap.2011.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Wang Q, Hand T, Wu L, Breyer RM, Montine TJ, Andreasson K. Deletion of the prostaglandin E2 EP2 receptor reduces oxidative damage and amyloid burden in a model of Alzheimer's disease. The Journal of neuroscience. 2005;25:10180–7. doi: 10.1523/JNEUROSCI.3591-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Wang Q, Shi J, Lokteva L, Breyer RM, Montine TJ, Andreasson K. The prostaglandin E2 EP2 receptor accelerates disease progression and inflammation in a model of amyotrophic lateral sclerosis. Annals of neurology. 2008;64:304–14. doi: 10.1002/ana.21437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Li Y, Lein PJ, Ford BD. Spatiotemporal patterns of GFAP upregulation in rat brain following acute intoxication with diisopropyl-fluorophosphate (DFP) Current neurobiology. 2012;3:90–97. [PMC free article] [PubMed] [Google Scholar]
- Liu D, Wu L, Breyer R, Mattson MP, Andreasson K. Neuroprotection by the PGE2 EP2 receptor in permanent focal cerebral ischemia. Annals of neurology. 2005;57:758–61. doi: 10.1002/ana.20461. [DOI] [PubMed] [Google Scholar]
- Livak K, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- McCullough L, Wu L, Haughey N, Liang X, Hand T, Wang Q, Breyer RM, Andreasson K. Neuroprotective function of the PGE2 EP2 receptor in cerebral ischemia. The Journal of neuroscience. 2004;24:257–68. doi: 10.1523/JNEUROSCI.4485-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misik J, Pavlikova R, Cabal J, Kuca K. Acute toxicity of some nerve agents and pesticides in rats. Drug and chemical toxicology. 2014 doi: 10.3109/01480545.2014.900070. In press. [DOI] [PubMed] [Google Scholar]
- Nelson TE, Gruol DL. The chemokine CXCL10 modulates excitatory activity and intracellular calcium signaling in cultured hippocampal neurons. J Neuroimmunol. 156:74–87. doi: 10.1016/j.jneuroim.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 2. New York: Academic; 1986. [DOI] [PubMed] [Google Scholar]
- Pernot F, Heinrich C, Barbier L, Peinnequin A, Carpentier P, Dhote F, Baille V, Beaup C, Depaulis A, Dorandeu F. Inflammatory changes during epileptogenesis and spontaneous seizures in a mouse model of mesiotemporal lobe epilepsy. Epilepsia. 2011;52:2315–25. doi: 10.1111/j.1528-1167.2011.03273.x. [DOI] [PubMed] [Google Scholar]
- Quan Y, Jiang J, Dingledine R. EP2 receptor signaling pathways regulate classical activation of microglia. J Biol Chem. 2013;288:9293–302. doi: 10.1074/jbc.M113.455816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalography and clinical neurophysiology. 1972;32:281–94. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Rojas A, Jiang J, Ganesh T, Yang MS, Lelutiu N, Gueorguieva P, Dingledine R. Cyclooxygenase-2 in epilepsy. Epilepsia. 2014;55:17–25. doi: 10.1111/epi.12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Ohshima T, Kondo T. Regulatory role of endogenous interleukin-10 in cutaneous inflammatory response of murine wound healing. Biochem Biophys Res Commun. 1999;265:194–199. doi: 10.1006/bbrc.1999.1455. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Albertson C, Slikker W., Jr Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain research. 1997;751:37–46. doi: 10.1016/s0006-8993(96)01387-x. [DOI] [PubMed] [Google Scholar]
- Serrano GE, Lelutiu N, Rojas A, Cochi S, Shaw R, Makinson CD, Wang D, FitzGerald GA, Dingledine R. Ablation of cyclooxygenase-2 in forebrain neurons is neuroprotective and dampens brain inflammation after status epilepticus. The Journal of neuroscience. 2011;31:14850–60. doi: 10.1523/JNEUROSCI.3922-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi H, Anacker C, Suarez-Mier GB, Wang Q, Andreasson K. Function of prostaglandin E2 EP receptors in the acute outcome of rodent hypoxic ischemic encephalopathy. Neuroscience letters. 2011;504:185–90. doi: 10.1016/j.neulet.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorovic MS, Cowan ML, Balint CA, Sun C, Kapur J. Characterization of status epilepticus induced by two organophosphates in rats. Epilepsy Res. 2012;101:268–276. doi: 10.1016/j.eplepsyres.2012.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Vliet EA, Otte WM, Gorter JA, Dijkhuizen RM, Wadman WJ. Longitudinal assessment of blood-brain barrier leakage during epileptogenesis in rats: A quantitative MRI study. Neurobiol Dis. 2014;63:74–84. doi: 10.1016/j.nbd.2013.11.019. [DOI] [PubMed] [Google Scholar]
- Voutsinos-Porche B, Koning E, Kaplan H, Ferrandon A, Guenounou M, Nehlig A, Motte J. Temporal patterns of the cerebral inflammatory response in the rat lithium-pilocarpine model of temporal lobe epilepsy. Neurobiology of disease. 2004;17:385–402. doi: 10.1016/j.nbd.2004.07.023. [DOI] [PubMed] [Google Scholar]