

Abstract
OBJECTIVE
There is increasing pre-clinical evidence indicating that metformin, a medication commonly used for type 2 diabetes, may protect against cancer. Motivated by this emerging evidence we asked two questions: (a) can metformin prevent ovarian cancer growth by altering metabolism, and (b) will metformin increase sensitivity to chemotherapy.
STUDY DESIGN
The effect of metformin in ovarian cancer was tested in vitro and by using two different mouse models. In vitro, cell lines (n=6) were treated with metformin (10 to 40 mM) or PBS and cellular proliferation and metabolic alterations (AMP-activated protein kinase activity, glycolysis, lipid synthesis) were compared between the two groups. In mouse models, a prevention study was performed by treating mice with metformin (250 mg/kg/day intraperitoneal (i.p.)) or placebo for 2 weeks followed by i.p. injection of the SKOV3ip1 human ovarian cancer cell line and the mean number of tumor implants in each treatment group was compared. In a treatment study, the LSL-K-rasG12D/+/PTENfloxP/floxP genetic mouse model of ovarian cancer was used. Mice were treated with placebo, paclitaxel (3 mg/kg/week i.p. x 7 weeks), metformin (100 mg/kg/day in water x 7 weeks), or paclitaxel plus metformin and tumor volume was compared between treatment groups.
RESULTS
In vitro, metformin decreased proliferation of ovarian cancer cell lines and induced cell cycle arrest, but not apoptosis. Further analysis showed that metformin altered several aspects of metabolism including AMP-activated protein kinase activity, glycolysis, and lipid synthesis. In the prevention mouse model, mice pre-treated with metformin had 60 % fewer tumor implants compared to controls (p<0.005). In the treatment study, mice treated with paclitaxel plus metformin had a 60% reduction in tumor weight compared to controls (p=0.02); a level of tumor reduction greater than that resulting from either paclitaxel or metformin alone.
CONCLUSION
Based on these results, we conclude that metformin alters metabolism in ovarian cancer cells, prevents tumor growth, and increases sensitivity to chemotherapy in vitro and in mouse models. These pre-clinical findings suggest that metformin warrants further investigation for use as an ovarian cancer therapeutic.
Keywords: mouse models, ovarian cancer, metformin, cancer prevention, metabolism, chemosensitization
Introduction
Developing new cancer drugs is an endeavor with a high probability of failure and extraordinary costs. It is estimated that each new cancer therapeutic entering phase II clinical testing has only a 20% probability of gaining Federal Drug Administration (FDA) approval and this success comes after an average of 12 years of development and a capitalized cost of $1,042 million dollars.1 One tactic used to accelerate new cancer drug discovery is to focus on compounds that have anti-cancer effects and are already FDA approved for non-cancer indications. This approach, called drug repurposing, builds on the available clinical safety and pharmacologic data to reduce both the cost and the time expended introducing a new drug.2 For diseases, such as ovarian cancer (OvCa), in which there have been few new effective treatments, drug repurposing may increase the efficiency of moving therapy from bench to bedside.
A candidate for this drug development paradigm may be metformin. With 20 years of use for type 2 diabetes, metformin has a strong safety profile, is well tolerated, inexpensive, and there is evidence the drug has biological activity in cancer. A protective effect of metformin in cancer was first identified in epidemiologic studies that reported decreased cancer incidence and increased cancer survival among diabetics treated with metformin.3–6 The epidemiological data has now been supported by preclinical studies demonstrating anti-tumorigenic effects of metformin in breast, colon, lung and prostate cancer.7–10 Studies have linked metformin’s anti-cancer effects to activation of AMP-activated protein kinase (AMPK) through the LKB1 tumor suppressor.11 Upon activation, AMPK inhibits the mammalian target of rapamycin (mTOR) and decreases the proliferation of cancer cells. This promising epidemiologic and preclinical data have now culminated in the initiation of prospective randomized clinical trials of metformin in breast and colon cancer.12,13
We have reported an association between metformin use and improved survival in OvCa. In a retrospective cohort of OvCa patients, progression-free survival at 5 years was 51% for diabetic patients who used metformin compared with 23% for non-diabetic patients and 8% for diabetic patients who did not use metformin.14 In an independent study at Mayo Clinic, S. Kumar et al. also reported nearly identical findings in their OvCa cohort.15 The finding that diabetics on metformin had an improved outcome when compared to non-diabetics was unexpected in light of prior published evidence indicating that diabetics should have a worse outcome.16 Therefore, there was a strong indication that, in OvCa, metformin may have a beneficial anticancer effect.
Based on strong epidemiologic support, our own clinical findings, and evidence of biologic plausible mechanisms for a protective effect of metformin in OvCa, we evaluated the effect of metformin on ovarian carcinogenesis. Using two mouse models of OvCa and in vitro studies, we asked if metformin could prevent OvCa progression and/or increase sensitivity to chemotherapy. The answer to these questions will be important if we are to consider repurposing metformin as an agent for the prevention and/or adjuvant treatment of OvCa.
Materials and Methods
Reagents and cell lines
The K-ras/PTEN cell line were established by us from ovarian tumors generated using a genetic mouse model.17 The SKOV3ip1 and HeyA8 cell lines were provided by Dr. Gordon Mills (MD Anderson Cancer Center, Houston, TX). The IOSE 397 cell line was kindly shared by Dr. Nelly Auersperg (University of British Columbia, Canada). The IGROV1 and Ovcar-5 cell lines were purchased from American Type Culture Collection (ATCC). The Kuramochi cell line was purchased from the Japanese Collection of Research Bioresources Cell Bank. Cell lines were validated by short tandem repeat (STR) DNA fingerprinting using the AMPF’STR Identifier kit (Applied Biosystems) and compared with ATCC and University of Texas MD Anderson Cancer Center fingerprints. Metformin obtained from Sigma-Aldrich (St Louis, MO). The Cdk4, cyclin D1, AMPK, EGFR, ErbB4, PDGFRα, FABP4 and FASN antibodies were from Cell Signaling Technologies (Beverly, MA) and the cyclin D1 antibody used for immunohistochemistry was from Novus Biologicals (Littleton, CO). The LKB1 and ACC antibodies were from Millipore (Billerica, MA), PARP 1/2 was from Santa Cruz Biotechnology (Santa Cruz, CA), FASN was from ATLAS (Stockholm, Sweden), phosphorylated RON was from R&D Systems (Minneapolis, MN), and RON was from Epitomics (Burlingame, CA).
MTT Assay
Cells were plated in quadruplicate into 96-well plates and treated with vehicle control, paclitaxel, and/or metformin for the designated amount of time and cellular proliferation was measured using MTT assays as previously described.17 The effect of treatment was calculated as a percentage of control cell growth obtained from vehicle-treated cells grown in the same plate. Each experiment was conducted in triplicate.
Apoptosis and cell cycle analysis
Cells were serum-starved for 24h, treated for 24h, fixed, and re-suspended in Propidium Iodide (PI)/RNase staining buffer or Annexin V and PI staining buffer and analyzed with a FACS Calibur (Becton Dickson, San Jose, CA). The percentage of cells in the G2/M, S-phase, and sub-G0-G1 population (apoptotic cells) was determined using FlowJo software. Staurosporine treatment served as a positive control. Each experiment was conducted in triplicate.
Western Blot Analysis
Cells were serum-starved for 24h and treated with metformin or vehicle control for the indicated amount of time. Western blots were performed as previously described.18,19 Briefly, the cells were lysed in radioimmunoprecipitation assay buffer, cell extract (30 μg) was separated by SDS-PAGE, and transferred to a nitrocellulose membrane. The membrane was incubated with respective primary antibodies and then with secondary horseradish peroxidase-conjugated IgG and visualized with enhanced chemoluminescence reagents.
Quantitative Real-Time RT-PCR
RNA was extracted from OvCa cells using TRIzol (Invitrogen, Carlsbad, CA) and was transcribed into cDNA using high capacity cDNA kit (Applied Biosystems). Quantitative real-time RT-PCR was performed as described20 using the Applied Biosystems 7500 Real Time PCR system (Applied Biosystems, Forest City, CA) and the following probes (Applied Biosystems, Forest City, CA): carnitine palmitoyltransferase 1a (CPT1a, Hs00912681_m1), fatty acid binding protein 4 (FABP4, Hs00609791_m1), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH, Hs00266705_gl). Relative levels of mRNA expression were calculated using the 2−ΔΔCT method.21
Receptor Tyrosine Kinase Array
The expression level of activated (phosphorylated) receptor tyrosine kinases were analyzed using the Proteome Profiler Human Phospho-RTK Array Kit (R&D Systems, Minneapolis, MN) according to the manufacturer’s protocol. Briefly, OvCa cells were serum-starved for 24 h and treated with metformin or vehicle control for 48 hours. The cells were then lysed in NP-40 lysis buffer, RTK array membranes were incubated cell lysates (500 μg), and then incubated with horseradish peroxidase conjugated anti-phosphotyrosine and visualized with enhanced chemoluminescence reagents. The level of phosphorylated RTK was densitometrically quantified with Image J software (NIH) and normalized to the internal phosphotyrosine-positive control spots.
Immunostaining
Mouse tumors were formalin-fixed, paraffin-embedded, sectioned, and mounted on slides. Immunohistochemistry staining of slides for TUNEL and Cyclin D1 was performed as previously described.17 TUNEL staining was performed using a TUNEL apoptosis detection kit (Millipore, Billerica, MA) according to the manufacturer’s protocol. Quantification of immunostaining was performed using the Automated Cellular Imaging System (ACIS, Clarient, San Juan Capistrano, CA) by setting color-specific thresholds to determine brown (positive) and blue (negative) nuclei within 12 representative regions per slide and by calculating the ratio of positively stained nuclei to all nuclei, expressed as a region score.19,22
Glycolysis
Glycolysis and glycolytic capacity were measured using the Seahorse Extracellular Flux (XF-96) analyzer (Seahorse Bioscience, North Billerica, MA). Cells were cultured in Seahorse XF-96 plates at a density of 30,000 cells per well and grown in complete growth media for three days. On the day of the experiment, cells were changed to unbuffered DMEM in the absence of glucose. Sequential injections were performed using D-glucose (2 g/L), oligomycin (1 μM), and 2-Deoxyglucose (100 mM). The ECAR after the injection of D-glucose is a measure of glycolysis and the ECAR following the injection of oligomycin represented maximal glycolytic capacity. Non-glycolytic activity was quantified by measuring ECAR after the injection of 2-deoxyglucose.
Animals
The xenograft model was described previously.19,23 Mice were treated for two weeks with metformin, 250 mg/kg/day, or control injected i.p. and then SKOV3ip1 cells (1×106) were injected i.p.. Treatment was continued until mice were sacrificed four weeks later. The LSL-K-rasG12D/+PTENloxP/loxP genetic mouse model was obtained from the Massachusetts Institute of Technology (Boston, MA)24 and used as previously described.17,22 OvCa was initiated by intrabursal injection of AdCre virus (2.5×107 plaque-forming units) in the right ovary; the left ovary was not injected and served as an internal control. Two weeks after initiation of OvCa, treatment began with control, paclitaxel 3 mg/kg/week injected i.p., or metformin added to water for an estimated final dose of 100 mg/kg/day per mouse. Mice were sacrificed 9 weeks after the injection of the virus and tumors were excised, weighed, measured, and the number of metastatic nodules and volume of ascites were recorded. All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Chicago. All animal studies were performed adhering to the ARRIVE guidelines for reporting animal research.25 This study did not involve human subjects and did not require Institutional Review Board approval. According to published pharmacokinetic analysis of metformin treated mice26 the doses of oral and i.p. metformin administered here would result in serum levels of 0.16 μg/mL and 4 μg/mL, respectively. In humans receiving 1 gm of metformin twice daily the metformin plasma concentrations range between 0.05 μg/mL to 4 μg/mL.27
Statistical analysis
Data was analyzed by an unpaired, two-tailed Student’s t-test of significance, assuming equal variance of the test and the control populations. For non-parametric data, a Mann-Whitney U test was completed to compare the medians for all groups. Data are presented as mean ± standard deviations. A p < 0.05 level was considered significant. All data analysis was performed with STATA 13 (StataCorp, College Station, TX).
Results
Metformin decreases proliferation of OvCa cells through induction of cell cycle arrest
To evaluate the effect of metformin in OvCa, five different OvCa cell lines that are commonly used in pre-clinical studies and immortalized human ovarian surface epithelial (IOSE) cells were treated with metformin and the proliferation rate was measured. Metformin treatment resulted in a significant dose-dependent decrease in proliferation in 4 out of 6 cell lines; the exceptions were the Kuramochi and IOSE lines both of which were much less sensitive to metformin treatment (Figure 1A). Metformin may inhibit carcinogenesis, in part, through activation of AMPK.11 To evaluate the effect of metformin on AMPK signaling in our model systems, we first demonstrated that LKB1, a tumor suppressor that regulates AMPK function, was expressed in our cell lines (Figure 1B). Metformin treatment produced a dose-dependent increase in the levels of phosphorylated AMPK at Thr172 in all cell lines, except the Kuramochi and IOSE lines (Figure 1C).
Figure 1.

Metformin inhibits growth and activates AMPK in ovarian cancer cells. (A) MTT proliferation assays showing that Ovcar-5, Kras/PTEN, SKOV3ip1, Kuramochi, HeyA8 and IOSE cell lines treated with 10, 20 or 40 mM metformin exhibit a dose-dependent reduction in proliferation. Bars represent fold change in proliferation. In 4 out of 6 cell lines the reduction in proliferation is statistically significant (p <0.05) with all metformin concentrations when compared to control, the exceptions are the Kuramochi and IOSE cell lines. (B) Western blots. Protein expression of LKB1 in cell lines. (C) Western blots. Protein expression of phosphorylated (activated) AMPK (Thr 172). B-actin done for each blot, but only shown for the IOSE blot. The indicated cell lines were serum-starved for 24 hours and then treated with 10, 20 or 40 mM metformin or vehicle for 24 hours.
To understand the mechanism by which metformin inhibited proliferation of OvCa cells, we tested whether metformin induced apoptosis. First, cleaved and total PARP 1/2 was evaluated using western blots and no change was noted with metformin treatment (Figure 2A). Consistent with this finding, flow cytometry for Annexin V/PI revealed no evidence of increased apoptosis in metformin-treated cells (Figure 2B). Therefore, to find an alternative explanation for metformin’s inhibition of proliferation, we evaluated the drug’s effect on cell cycle. Analysis with flow cytometry demonstrated arrest of the cell cycle in the G0/G1 phase with metformin treatment in two cell lines (Figure 3A). In the HeyA8 cell line, the cell cycle arrest induced by metformin was dose and time dependent (data not shown). To understand how metformin induced cell cycle arrest in OvCa cells Cdk4 and Cyclin D1 were evaluated. It was noted that metformin treatment resulted in a time-dependent decrease in the expression of Cdk4 and Cyclin D1 (Figure 3B).
Figure 2.

Metformin’s anti-proliferative effect is not a result of apoptosis. (A) Western blot. Protein expression of cleaved and total PARP 1/2. The Ovcar-5 OvCa cell line was treated with 10, 20 or 40 mM metformin or vehicle for 48 hours. (B) Apoptosis analysis. SKOV3ip1 cells were serum-starved for 24 hours before treating with 40 mM metformin for 24 hours. Cells were then stained with propidium iodine (PI) and Annexin V and analyzed using a flow cytometer. The percentage of apoptotic cells is shown.
Figure 3.
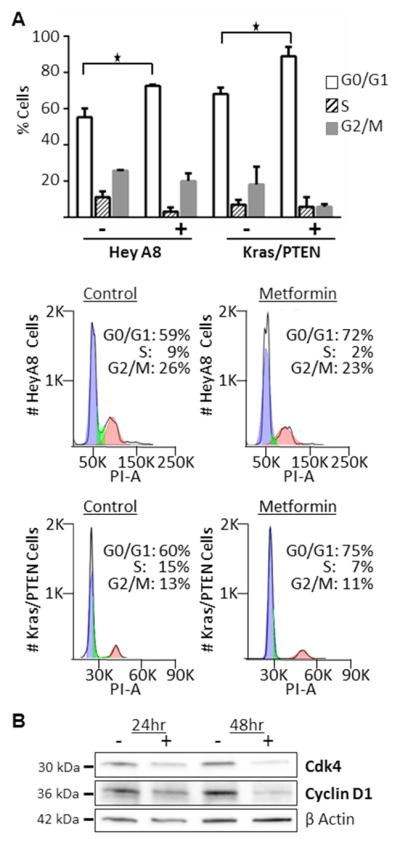
Cell cycle arrest with metformin treatment. (A) Cell cycle analysis. K-ras/PTEN and HeyA8 cells were serum-starved for 24 hours before treating with 20 mM metformin for 24 hours. Cells were then stained with propidium iodine and analyzed using flow cytometry. Columns, mean percent of cells in each phase of cell cycle; *, P<0.5. Histograms from each cell line are shown. Blue: G0/G1 phase, green: S phase, red: G2/M phase. (B) Western blots. Protein expression of Cdk4 and cyclin D1. The SKOV3ip1 ovarian cancer cell line was treated with 40 mM metformin or vehicle in serum free media for the indicated times.
Pleotropic effects of metformin in OvCa
When used as a treatment for diabetes metformin acts on the canonical AMPK pathway, altering cellular glucose and lipid metabolism.28 When activated, AMPK inactivates (phosphorylates) a key regulatory enzyme in lipid metabolism, acetyl CoA carboxylase (ACC) resulting in decreased malonyl-CoA production. When malonyl-CoA levels are depleted, fatty acid synthesis is decreased (regulated by fatty acid synthase, FASN) and fatty acid oxidation is increased (regulated by carnitine palmitoyltransferase, CPT-1).29 We tested whether metformin would induce similar metabolic changes in OvCa.
When we evaluated glucose and lipid metabolism, it was noted that metformin increased glycolytic capacity in the OvCa cells (Figure 4A), increased levels of phosphorylated (inactivated) acetyl CoA carboxylase, and decreased fatty acid synthase (Figure 4B). In addition, metformin increased mRNA levels of carnitine palmitoyltransferase (CPT-1), suggesting increased fatty acid oxidation (Figure 4C). Previously, our lab has shown that adipocytes contribute to ovarian tumorigenesis by transferring free fatty acids to adjacent cancer cells and that this tumor-promoting effect of adipocytes can be interrupted by pharmacologically inhibiting a protein involved in fatty acid transport, FABP4.30 Therefore, we asked if FABP4 could be a unique target of metformin in OvCa. Interestingly, we found that metformin decreased both the mRNA and protein levels of FABP4 in OvCa cells (Figure 4B, C).
Figure 4.

In ovarian cancer cells, metformin alters glucose and lipid metabolism. (A) Glycolysis levels in cancer cells treated with metformin are increased compared to control. IGROV cells were plated in complete growth medium and treated for 4 hours with 20 mM metformin or vehicle. Glycolytic response of the cell line to pharmacological inhibitors of metabolism was determined by a Seahorse SF96 Extracellular Flux Analyzer. Extracellular acidification rates (ECAR) for the two groups are depicted. (B) Western blots. Protein expression of phosphorylated ACC, fatty acid synthase, and fatty acid binding protein 4. Serum starved Ovcar-5 ovarian cancer cells were treated with 20 mM metformin or vehicle for 48 hours. (C) Quantitative real-time RT-PCR. mRNA levels of the key regulatory enzyme involved in fatty acid oxidation, carnitine palmitoyltransferase, was significantly increased and fatty acid binding protein 4 was significantly decreased with metformin treatment. Ovcar-5 ovarian cancer cells were treated with 20 mM metformin or vehicle for 48 hours, RNA was extracted, and quantitative real-time PCR was performed to determine the level of mRNA. ECAR: extracellular acidification rates, p-ACC: phosphorylated acetyl-coA carboxylase, FASN: fatty acid synthase, FABP4: fatty acid binding protein 4, CPT-1: carnitine palmitoyltransferase. *P<0.05
While metformin induced alterations in glucose and lipid metabolism are recognized mechanisms of action of the drug in diabetes, glucose and lipid metabolism are not well-established targets for OvCa therapeutics. Therefore, we also asked whether metformin, independent of its traditional metabolic effects, could also alter validated cancer signaling pathways, specifically receptor tyrosine kinases (RTK). To test this, phosphorylated RTK arrays were completed in two different OvCa cell lines. The arrays indicated that metformin treatment decreases activity of RON, ErbB4, and PDGFRα in SKOV3ip1 cells and reduces the activity of EGFR in HeyA8 cells (Figure 5A). The RTK array findings for RON, ErbB4, and EGFR were confirmed using western blots, the effect of metformin on PDGFR activity was difficult to interpret because metformin treatment reduced total and phosphorylated PDGFR (Figure 5B).
Figure 5.

Metformin inhibits activity of several receptor tyrosine kinases. (A) Receptor tyrosine kinases array. SKOV3ip1 or HeyA8 cells were serum starved for 24 hours and then treated with control or 40 mM metformin for 48 hours prior to performing the arrays. Columns, protein expression level quantified by image J and expressed as percent of control. Image of control and metformin treated arrays. (B) Western blots. Protein expression of phosphorylated and total: RON (Y1238/Y1239), ErbB4 (Tyr1284), PDGFR (Tyr849), and EGFR (Tyr1045). B-actin done for each blot, but only shown for the ErbB4 and EGFR blots. Serum starved SKOV3ip1 and HeyA8 cells were treated with control or 40 mM metformin for 48 hours.
Metformin inhibits OvCa cell growth and metastasis
To investigate whether these promising in vitro findings would translate into a protective effect conferred by metformin in animal models of OvCa, we used two approaches: a preventive and treatment study design.31 In the preventive study, SKOV3ip1 xenograft mice were pre-treated with metformin or placebo for two weeks prior to initiation of OvCa, with continuation of metformin treatment until completion of the study (Figure 6A). In this experiment, at the time of sacrifice mice treated with metformin had significantly fewer tumor implants (mean number of tumors; placebo: 116 and metformin: 47, p<0.005) (Figure 6B).
Figure 6.

In a preventive study design, metformin inhibits ovarian cancer cell growth. (A) Study timeline. Female athymic nude mice were pretreated with metformin (250 mg/kg/day, n=9) or placebo (100 μl sterile PBS, n=9) by i.p. injection for 2 weeks followed by i.p. injection of SKOV3ip1 ovarian cancer cells. Treatment with metformin or placebo continued until completion of the study. (B) At the end of the experiment tumors are counted to determine tumor number. Each diamond indicates tumor number for an individual mouse and the line indicates mean tumor number for the group. (C) TUNEL staining of tumors from placebo- and metformin- treated mice. (D) Immunohistochemistry for cycling D1 in tumors from control and metformin treated mice. Representative staining from each group. Original magnification, x200. Scale bar, 50μm. Columns, mean percent positive for group; *, P<0.5.
To determine if the in vitro findings, indicating that metformin caused cell cycle arrest but did not induce apoptosis, were recapitulated in the animal model, the tumor samples were stained for TUNEL and cyclin D1. Consistent with the in vitro findings, TUNEL staining showed no increase in apoptosis in tumors from metformin-treated mice (Figure 6C). Also consistent with the in vitro findings, tumors from metformin-treated mice had significantly lower expression of cyclin D1 compared to controls (Figure 6D).
Metformin increases sensitivity to standard chemotherapy
It is possible that the increase in progression-free survival among metformin users in our OvCa patient cohort analysis14 may have been mediated, in part, by improved response to chemotherapy. In pre-clinical studies, it has been reported that metformin increases the cytotoxic effect of cisplatin32, here we asked if metformin also improves response to paclitaxel. In vitro testing demonstrated a reduction in the IC50 for paclitaxel with the addition of metformin in the HeyA8 and SKOV3ip1 human OvCa cell lines (Figure 7A). To test the combination of metformin and paclitaxel further, first we confirmed that metformin had paclitaxel-sensitizing effects in a cell line (K-ras/PTEN) established from the LSL-K-rasG12D/+PtenloxP/loxP genetic mouse model of OvCa (Figure 7B). Next, a treatment study was conducted that mimics treatment of advanced OvCa in humans. Here, ovarian cancer was initiated in LSL-K-rasG12D/+PtenloxP/loxP mice and two weeks later mice were treated with paclitaxel plus metformin or the individual drugs alone. At the time of sacrifice, mean tumor weight was reduced by 60% in mice treated with paclitaxel plus metformin compared to placebo treated mice (p=0.02) (Figure 7B). The reduction in tumor burden was 40% and 42% in mice treated with paclitaxel or metformin alone, respectively. The changes in tumor volume in these groups were not significantly different when compared to placebo or when compared to other treatments.
Figure 7.

Metformin increases sensitivity to paclitaxel. (A) IC50 curves for human ovarian cancer cell lines. HeyA8 and SKOV3ip1 human ovarian cancer cells were plated in complete medium and treated with paclitaxel concentrations ranging from 1 to 30 nM alone or with 5 mM metformin. MTT assay was performed to determine the level proliferation. (B) Genetic mouse model of ovarian cancer. A cancer cell line was established from the LSL-K-rasG12D/+PtenloxP/loxP genetic mouse model and proliferation was measured after treatment with paclitaxel alone or plus metformin. In a in vivo treatment study design, cancer was initiated in virginal LSL-K-rasG12D/+PtenloxP/loxP mice and two weeks later mice were treated with placebo (100 μl sterile PBS, n=21), paclitaxel alone (3 mg/kg/week injected i.p., n=25), metformin alone administered in drinking water (100 mg/kg/day, n=18) or paclitaxel plus metformin (n=29). Columns, fold change in tumor volume, normalized to control group; *, P=0.02. Dotted line, indicates 50% reduction in percent of proliferating cells.
Comment
The findings of this study provide pre-clinical data indicating that metformin inhibits OvCa growth in animal models using both a preventive and treatment study design. In the preventive study, metformin-treated mice developed significantly fewer tumors. In the treatment study, metformin had a chemosensitizing effect and, independent of chemotherapy, inhibited progression of OvCa. The findings also demonstrate that in addition to inducing protective metabolic changes in OvCa cells, metformin also inhibits the activity of several receptor tyrosine kinases.
Understanding the molecular mechanisms by which metformin inhibits ovarian carcinogenesis is of critical importance if the drug is to be used as a cancer therapeutic. Activation of AMPK with subsequent decrease in protein production and ultimate cell death is the molecular mechanism most widely postulated for metformin’s effects in cancer.11 Consistent with the findings of other investigators32–34, we noted a dose-dependent activation of AMPK (increased pAMPK Thr172) in metformin-treated OvCa cells and alteration of several downstream targets of AMPK. Specifically, evidence of decreased fatty acid synthesis, increased fatty acid oxidation, and increased glycolysis was noted. In OvCa, we show that metformin induces cell cycle arrest in the G0/G1 phase, a finding that has also been reported in prostate cancer.10
Two previously unreported molecular targets of metformin in cancer are reported here. First, metformin decreased levels of FABP4, which has been identified as a mediator of the transport of free fatty acids to OvCa cells from adjacent adipocytes.30 Prior reports have shown metformin reduces FABP4 expression in macrophages.35 Adipocytes promote tumor growth through the production of cytokines and by providing lipids to adjacent tumor.36 Developing agents that target the interactions between cancer cells and adipocytes represents the next frontier in cancer therapeutics and understanding whether metformin may represent such an agent is an area of active investigation by our group and others. A second novel effect of metformin identified here is the drug’s ability to reduce the activity of several receptor tyrosine kinases, each of which plays a role in ovarian carcinogenesis.37–39 This finding is provocative as several RTK inhibitors have already been tested as OvCa treatments40, however, additional investigation will be needed to understand the mechanism by which metformin inhibits RTK activity. Identifying the molecular mechanisms of action of metformin in cancer is hindered by the fact that the findings reported here, and most other published reports, utilize supra-physiologic doses of metformin in vitro (10–40 mmol/L); plasma concentrations of metformin in patients are estimated to be 50 μmol/L.41 However, the glucose and growth factor conditions in vitro are also supra- physiologic and likely contribute to the need for high metformin concentrations.
Should metformin prove effective in OvCa, one could envision the drug being used either as primary prevention in high-risk women, such as BRCA mutation carriers, or in addition to standard chemotherapy for newly diagnosed OvCa. Improving response to chemotherapy could significantly decrease OvCa mortality, since up to 76% of patients have recurrence of disease after first line therapy.42 Here we report in vitro and in a genetic mouse model that the combination of metformin and paclitaxel has a greater effect than paclitaxel alone. Our findings of a possible paclitaxel sensitization effect of metformin in OvCa augments pre-clinical findings by other investigators indicating that metformin increases carboplatin sensitivity32,34,43 and our finding of a trend toward increased platinum sensitivity among OvCa patients that use metformin.14 In a prospective study in breast cancer, investigators also found that metformin users had a significantly improved response to neo-adjuvant chemotherapy.44
In conclusion, the findings reported here provide strong pre-clinical data indicating that metformin inhibits the growth of ovarian tumors and improves response to paclitaxel. In addition, we present clear evidence that, in OvCa, metformin alters both traditional metabolism targets (glucose and lipids) and cancer signaling pathways (RTKs). These findings add to the growing body of evidence of novel anti-cancer effects of a drug widely used for diabetic treatment45–47 and provide further evidence that metformin may be a worthwhile candidate for repurposing as an OvCa chemopreventive agent or therapeutic. In fact, prospective clinical testing of metformin as adjuvant treatment is ongoing for ovarian and endometrial cancer.48
Acknowledgments
Financial support: This work was supported by grants from the National Institutes of Health, Eunice Kennedy Shriver National Institute of Child Health and Human Development (2K12HD000849-26), The American Board of Obstetrics and Gynecology, The Gynecologic Cancer Foundation- St. Louis Ovarian Cancer Awareness Research, and The Prevent Cancer Foundation (to I.L.R). Ernst Lengyel is supported by grants from the National Cancer Institute (5R01CA111882-07 and 1R01CA169604-01A1).
Footnotes
Disclosure statement: The authors report no conflict of interest.
Presentations: These findings were presented at The Society of Gynecologic Oncology 43rd Annual Meeting, March 24–27, 2012, Austin, Texas.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Adams CP, Brantner VV. Estimating the cost of new drug development: Is it really 802 million dollars? Health Aff (Millwood) 2006;25(2):420–8. doi: 10.1377/hlthaff.25.2.420. [DOI] [PubMed] [Google Scholar]
- 2.Chong CR, Sullivan DJ. New uses for old drugs. Nature. 2007;448:645–6. doi: 10.1038/448645a. [DOI] [PubMed] [Google Scholar]
- 3.Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care. 2006;29(2):254–8. doi: 10.2337/diacare.29.02.06.dc05-1558. [DOI] [PubMed] [Google Scholar]
- 4.Evans JMM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. Br Med J. 2005;530:1304–5. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JMM. New users of metformin are at low risk of incident cancer: A cohort study among people with type 2 diabetes. Diabetes Care. 2009;32(9):1620–5. doi: 10.2337/dc08-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Landman GW, Kleefstra N, van Hateren KJJ, Groenier KH, Gans RO, Bilo HJ. Metformin associated with lower cancer mortality in type 2 diabetes: ZODIAC-16. Diabetes Care. 2010;33(2):322–6. doi: 10.2337/dc09-1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Memmott RM, Mercado JR, Maier CR, Kawabata S, Fox SD, Dennis PA. Metformin prevents tobacco carcinogen- induced lung tumorigenesis. Cancer Prevention Research. 2010;3(9):1066–76. doi: 10.1158/1940-6207.CAPR-10-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buzzai M, Jones RG, Amaravadi RK, et al. Systemic treatment with the anti-diabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67(14):6745–52. doi: 10.1158/0008-5472.CAN-06-4447. [DOI] [PubMed] [Google Scholar]
- 9.Cufi S, Corominas-Faja B, Vazquez-Martin A, et al. Metformin-induced preferential killing of breast cancer initiating CD44+CD24−/low cells is sufficient to overcome primary resistance to trastuzumab in HER2+ human breast cancer xenografts. Oncotarget. 2012;3(4):395–8. doi: 10.18632/oncotarget.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sahra IB, Laurent K, Loubat A, et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008;27:3576–86. doi: 10.1038/sj.onc.1211024. [DOI] [PubMed] [Google Scholar]
- 11.Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108(8):1167–74. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goodwin PJ, Stambolic V, Lemieux J, et al. Evaluation of metformin in early breast cancer: A modification of the traditional paradigm for clinical testing of anti-cancer agents. Breast Cancer Res Treat. 2011;126:215–20. doi: 10.1007/s10549-010-1224-1. [DOI] [PubMed] [Google Scholar]
- 13.Higurashi T, Takahasi H, Endo H, et al. Metformin efficacy and safety for colorectal polyps: A double-blind randomized controlled trial. BMC Cancer. 2012;12(118):1–7. doi: 10.1186/1471-2407-12-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Romero IL, McCormick A, McEwen KA, et al. Relationship of type II diabetes and metformin use to ovarian cancer progression, survival, and chemosensitivity. Obstet Gynecol. 2012;119(1):61–7. doi: 10.1097/AOG.0b013e3182393ab3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar S, Meuter A, Thapa P, et al. Metformin intake is associated with better survival in ovarian cancer: A case-control study. Cancer. 2013;119(3):555–62. doi: 10.1002/cncr.27706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bakhru A, Buckanovich RJ, Griggs JJ. The impact of diabetes on survival in women with ovarian cancer. Gynecol Oncol. 2011;121:106–11. doi: 10.1016/j.ygyno.2010.12.329. [DOI] [PubMed] [Google Scholar]
- 17.Romero IL, Gordon I, Jagadeeswaran S, et al. Effects of oral contraceptives or a gonadotropin-releasing hormone agonist on ovarian carcinogenesis in genetically engineered mice. Cancer Prevention Research. 2009;2(9):792–9. doi: 10.1158/1940-6207.CAPR-08-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kenny HA, Leonhardt P, Ladanyi A, et al. Targeting the urokinase plasminogen activator receptor inhibits ovarian cancer metastasis. Clin Cancer Res. 2011;17(3):459–71. doi: 10.1158/1078-0432.CCR-10-2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sawada K, Radjabi AR, Shinomiya N, et al. c-Met overexpression is a prognostic factor in ovarian cancer and an effective target for inhibition of peritoneal dissemination and invasion. Cancer Res. 2007;67(4):1670–80. doi: 10.1158/0008-5472.CAN-06-1147. [DOI] [PubMed] [Google Scholar]
- 20.Sawada K, Mitra AK, Radjabi AR, et al. Loss of E-cadherin promotes ovarian cancer metastasis via alpha 5-integrin, which is a therapeutic target. Cancer Res. 2008;68(7):2329–39. doi: 10.1158/0008-5472.CAN-07-5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shell S, Park SM, Radjabi AR, et al. Let-7 expression defines two differentiation stages of cancer. Proceedings of the National Academy of Sciences USA. 2007;104(27):11400–5. doi: 10.1073/pnas.0704372104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Romero IL, Lee W, Mitra AK, et al. The effects of 17β-estradiol and a selective estrogen receptor modulator, bazedoxifene, on ovarian carcinogenesis. Gynecol Oncol. 2012;124(1):134–41. doi: 10.1016/j.ygyno.2011.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zillhardt M, Christensen J, Lengyel E. An orally available small molecule inhibitor of c-Met, PF-2341066, reduces tumor burden in a pre-clinical model of ovarian cancer metastasis. Neoplasia. 2010;12(1):1–10. doi: 10.1593/neo.09948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dinulescu D, Ince TA, Quade B, Shafer S, Crowley D, Jacks T. Role of K-ras and PTEN in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat Med. 2005;11(1):63–70. doi: 10.1038/nm1173. [DOI] [PubMed] [Google Scholar]
- 25.Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8(6):1–5. doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Memmott RM, Mercado JR, Maier CR, Kawabata S, Fox SD, Dennis PA. Metformin prevents tobacco carcinogen-induced lung tumorigenesis. Cancer Prev Res (Phila Pa) 2010;3(9):1066–76. doi: 10.1158/1940-6207.CAPR-10-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christensen MM, Brasch-Andersen C, Green H, et al. The pharmacogenetics of metformin and its impact on plasma metformin steady-state levels and glycosylated hemoglobin A1c. Pharmacogenet Genomics. 2011;21(12):837–50. doi: 10.1097/FPC.0b013e32834c0010. [DOI] [PubMed] [Google Scholar]
- 28.Hardie DG. AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nature Reviews. 2007;8:774–85. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 29.Swinnen JV, Brusselmans K, Verhoeven G. Increased lipogenesis in cancer cells: new players, novel targets. Curr Opin Clin Nutr Metab Care. 2006;9:358–65. doi: 10.1097/01.mco.0000232894.28674.30. [DOI] [PubMed] [Google Scholar]
- 30.Nieman KM, Kenny HA, Penicka CV, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011;17(11):1498–503. doi: 10.1038/nm.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clarke R. Issues in experimental design and endpoint analysis in the study of experimental cytotoxic agents in vivo in breast cancer and other models. Breast Cancer Res Treat. 1997;46(2–3):255–78. doi: 10.1023/a:1005938428456. [DOI] [PubMed] [Google Scholar]
- 32.Rattan R, Graham RP, Maguire JL, Giri S, Shridhar V. Metformin suppresses ovarian cancer growth and metastasis with enhancement of cisplatin cytotoxicity in vivo. Neoplasia. 2011;13(5):483–91. doi: 10.1593/neo.11148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rattan R, Giri S, Hartmann LC, Shridhar V. Metformin attenuates ovarian cancer cell growth in an AMP-kinase dispensable manner. J Cell Mol Med. 2011;15(1):166–78. doi: 10.1111/j.1582-4934.2009.00954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gotlieb WH, Saumet J, Beauchamp MC, et al. In vitro metformin anti-neoplastic activity in epithelial ovarian cancer. Gynecol Oncol. 2008;110:246–50. doi: 10.1016/j.ygyno.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 35.Song J, Ren P, Zhang L, Wang X, Li C, Shen YH. Metformin reduces lipid accumulation in macrophages by inhibiting FOXO1-mediated transcription of fatty acid-binding protein 4. Biochem Biophys Res Commun. 2010;393:89–94. doi: 10.1016/j.bbrc.2010.01.086. [DOI] [PubMed] [Google Scholar]
- 36.Nieman KM, Romero IL, Van Houten B, Lengyel E. Adipocyte tissue and adipocytes support tumorigenesis and metastasis. Biochim Biophys Acta. 2013;1831(10):1533–41. doi: 10.1016/j.bbalip.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matei D, Emerson RE, Lai YC, et al. Autocrine activation of PDGFRalpha promotes the progression of ovarian cancer. Oncogene. 2006;25(14):2060–9. doi: 10.1038/sj.onc.1209232. [DOI] [PubMed] [Google Scholar]
- 38.Ferrandina G, Martinelli E, Petrillo M, et al. Prognostic role of the recepteur d’origine nantais (RON) expression in ovarian cancer patients. Gynecol Oncol. 2008;111(2):237–43. doi: 10.1016/j.ygyno.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 39.Lafky JM, Wilken JA, Baron AT, Maihle NJ. Clinical implications of the ErbB/epidermal growth factor (EGF) receptor family and its ligands in ovarian cancer. Biochim Biophys Acta. 2008;1785(2):232–65. doi: 10.1016/j.bbcan.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 40.Klempner SJ, Myers AP, Mills GB, Westin SN. Clinical investigation of receptor and non-receptor tyrosine kinase inhibitors for the treatment of epithelial ovarian cancer. Expert Opin Pharmacother. 2013;14(16):2171–82. doi: 10.1517/14656566.2013.826650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin-Castillo B, Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. Metformin and cancer: doses, mechanisms and the dandelion and hormetic phenomena. Cell cycle. 2010;9(6):1057–64. doi: 10.4161/cc.9.6.10994. [DOI] [PubMed] [Google Scholar]
- 42.Ozols RF, Bundy B, Greer BE, et al. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: A Gynecologic Oncology Group study. J Clin Oncol. 2003;21(17):3194–200. doi: 10.1200/JCO.2003.02.153. [DOI] [PubMed] [Google Scholar]
- 43.Erices R, Bravo ML, Gonzalez P, et al. Metformin, at concentrations corresponding to the treatment of diabetes, potentiates the cytotoxic effects of carboplatin in cultures of ovarian cancer cells. Reprod Sci. 2013:1–14. doi: 10.1177/1933719113488441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiralerspong S, Palla SL, Giordano SH, et al. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J Clin Oncol. 2009;27(20):3297–302. doi: 10.1200/JCO.2009.19.6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kisfalvi K, Moro A, Sinnett-Smith J, Eibl G, Rozengurt E. Metformin inhibits the growth of human pancreatic cancer xenografts. Pancreas. 2013;42(5):781–5. doi: 10.1097/MPA.0b013e31827aec40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hirsch HA, Iliopoulos D, Struhl K. Metformin inhibits the inflammatory response associated with cellular transformation and cancer stem cell growth. Proc Natl Acad Sci U S A. 2013;110(3):972–7. doi: 10.1073/pnas.1221055110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bao B, Wang Z, Ali S, et al. Metformin inhibits cell proliferation, migration and invasion by attenuating CSC function mediated by deregulating miRNAs in pancreatic cancer cells. Cancer Prev Res (Phila) 2012;5(3):355–64. doi: 10.1158/1940-6207.CAPR-11-0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.NIH. http://clinicaltrials.gov/