

Abstract
Healthy aging is associated with cognitive declines typically accompanied by increased task-related brain activity in comparison to younger counterparts. The Scaffolding Theory of Aging and Cognition (STAC) (Park and Reuter-Lorenz, 2009; Reuter-Lorenz and Park, 2014) posits that compensatory brain processes are responsible for maintaining normal cognitive performance in older adults, despite accumulation of aging-related neural damage. Cross-sectional studies indicate that cognitively intact elders at genetic risk for Alzheimer’s disease (AD) demonstrate patterns of increased brain activity compared to low risk elders, suggesting that compensation represents an early response to AD-associated pathology. Whether this compensatory response persists or declines with the onset of cognitive impairment can only be addressed using a longitudinal design. The current prospective, 5-year longitudinal study examined brain activation in APOE ε4 carriers (N=24) and non-carriers (N=21). All participants, ages 65–85 and cognitively intact at study entry, underwent task-activated fMRI, structural MRI, and neuropsychological assessments at baseline, 18, and 57 months. fMRI activation was measured in response to a semantic memory task requiring participants to discriminate famous from non-famous names. Results indicated that the trajectory of change in brain activation while performing this semantic memory task differed between APOE ε4 carriers and non-carriers. The APOE ε4 group exhibited greater activation than the Low Risk group at baseline, but they subsequently showed a progressive decline in activation during the follow-up periods with corresponding emergence of episodic memory loss and hippocampal atrophy. In contrast, the non-carriers demonstrated a gradual increase in activation over the 5-year period. Our results are consistent with the STAC model by demonstrating that compensation varies with the severity of underlying neural damage and can be exhausted with the onset of cognitive symptoms and increased structural brain pathology. Our fMRI results could not be attributed to changes in task performance, group differences in cerebral perfusion, or regional cortical atrophy.
Keywords: Aging, Alzheimer’s disease, APOE ε4, fMRI, Semantic memory
Introduction
Healthy aging is associated with mild and gradual declines in cognition functions, with the greatest aging-related changes involving memory, processing speed, and visuospatial skills (Salthouse, 2010). Such changes often occur in parallel with age-related alterations in brain structure, characterized by cortical atrophy and white matter abnormalities (Drachman, 2006; Kramer et al., 2007). Paradoxically, fMRI studies have consistently found increased regional brain activity in healthy elders relative to their younger counterparts during the performance of a cognitive task. This increased task-related brain activity in healthy elders typically occurs in brain regions also activated by younger participants, but can also be observed in homologous regions in the opposite hemisphere (Cabeza et al., 2002; Nielson et al., 2002, 2006). Some investigators have noted that age-related increases in brain activity occur most often in the frontal cortex; for reviews and discussion, see (Buckner, 2004; Eyler et al., 2011; Nielson et al., 2002). This increased neural activity is thought to serve as a compensatory function to support a high level of performance in older adults (Bangen et al., 2012; Carp et al., 2010; Grady, 2008; Han et al., 2009; Nielson et al., 2002, 2006; Prvulovic et al., 2005; Reuter-Lorenz and Cappell, 2008; Wierenga et al., 2008).
One prominent theory, the Scaffolding Theory of Aging and Cognition (STAC) (Park and Reuter-Lorenz, 2009), posits that compensatory brain processes are responsible for preserving cognitive performance in older adults, despite accumulation of neural changes in the context of healthy aging (e.g., mild white matter disease, age-related atrophy). This theory identifies neural factors that contribute to maintenance of a specific level of cognitive function and does not address dynamic longitudinal changes occurring during the aging process. More recently, these authors (Reuter-Lorenz and Park, 2014) revised the STAC theory (STAC-r) to account for both positive (e.g., physical activity) and negative (e.g., presence of brain amyloid) factors that contribute to the rate of change in cognitive function during aging. This revision provides a framework for tracking the trajectory of neural compensation (scaffolding) in response to rate of change in cognitive processes, but empirical validation of the theory is dependent on imaging data derived from extended longitudinal imaging studies.
In the current prospective, 5-year longitudinal fMRI study, we examined compensatory neural scaffolding processes in cognitively intact elders at varying genetic risk for developing Alzheimer’s disease (AD). The most important genetic risk factor for the sporadic form of AD (onset occurring after age 65) is the apolipoprotein E epsilon 4 (APOE ε4) allele (Farrer et al., 1997). Cross-sectional fMRI studies from our group (Seidenberg et al., 2009; Smith et al., 2011; Woodard et al., 2009, 2010) and others (Bookheimer et al., 2000; Borghesani et al., 2008; Filippini et al., 2011; Han et al., 2007; Trachtenberg et al., 2012; Wierenga and Bondi, 2007; Wierenga et al., 2010) have consistently demonstrated greater brain activation (neural scaffolding) in cognitively intact elders at higher genetic risk for AD (based on the presence of one or both APOE ε4 alleles and/or a family history of AD) than elders at lower genetic risk. Presumably, this increased activation occurs because the neuropathological changes associated with AD begin years or decades prior to symptom manifestation in persons at genetic risk for AD (Bateman et al., 2012; Jack et al., 2010). Indeed, alterations in task-related brain activity and cognitive performance have been reported in cross-sectional studies of APOE-ε4 positive individuals beginning in middle age and earlier (Evans et al., 2014; Reiman et al., 2004).
For this study, we recruited cognitively intact elders, APOE ε4 carriers and non-carriers, who underwent repeat cognitive testing, structural MRI, and task-activated fMRI on three occasions: study entry, 18 and 57 months. The fMRI task consisted of the Famous Name Recognition Task (FNRT) (Douville et al., 2005), a low-effort semantic memory task. The FNRT is performed with high accuracy even in patients with amnestic mild cognitive impairment (Woodard et al., 2009), thus removing high effort/low accuracy from complicating the interpretation of the longitudinal brain maps (Kennedy et al., 2014). Previous cross-sectional studies (Nielson et al., 2006, 2010; Seidenberg et al., 2009; Woodard et al., 2010) using this task have demonstrated a highly reproducible pattern of brain activation in regions that overlap with regions that comprise the “default-mode network” (Nielson et al., 2010; Sugarman et al., 2012).
Based on the STAC-r model, we hypothesized that the cognitively intact APOE ε4 carriers would exhibit greater task-related brain activation than non-carriers at study entry, presumably reflecting a compensatory response that may signal subsequent cognitive decline (Miller et al., 2008). Over time, however, a breakdown of neural scaffolding in the APOE ε4 carrier group is predicted to occur, characterized by the presence of age-inappropriate cognitive impairment. Decreased brain activity occurs in association with increased AD-related neural pathology (O’Brien et al., 2010) and is predicted to coincide with decreased episodic memory performance. Conversely, non-carriers, who maintain intact and stable episodic memory over the course of the 5 year follow-up period, should show a steady increase in brain activation reflecting the increasing demands of their scaffolding system to maintain memory performance with advancing age.
Material and methods
Participants
This study was approved by the Institutional Review Board of the Medical College of Wisconsin, which oversees the ethical standards of human research. Written informed consent was obtained from all subjects included in this study. All participants received financial compensation.
The recruitment strategy for this study, described in detail in Seidenberg et al. (2009), involved over-sampling persons at genetic risk for AD based on the presence of an APOE ε4 allele. Briefly, healthy older adults between the ages of 65 and 85 were recruited from newspaper advertisements. A telephone screen, used to determine study eligibility, was administered to 459 individuals. Participants were excluded if they reported a history of neurological disease, medical illnesses, major psychiatric disturbance meeting DSM-IV Axis I criteria, a Geriatric Depression Scale [GDS; (Yesavage et al., 1982)] score >20, Lawton Instrumental Activities of Daily Living [IADL; (Lawton and Brody, 1969)] scale <5, substance abuse meeting DSM-IV Axis I criteria, or were currently taking psychoactive medications. Additional exclusion criteria related to fMRI scanning included pregnancy, weight inappropriate for height, ferrous objects within the body, low visual acuity, and a history of claustrophobia. For purposes of fMRI scanning, only right-handed participants were included based on the Edinburgh Handedness Inventory (Oldfield, 1971). Of the individuals meeting inclusion/exclusion eligibility criteria, 109 agreed to undergo APOE genotype testing from blood samples, neuropsychological evaluation, and an fMRI scanning session. APOE genotype was determined using a polymerase chain reaction method. DNA was isolated with Gentra Systems Autopure LS for Large Sample Nucleic Acid Purification (O’Brien et al., 2001). Of the 109 enrolled participants, we excluded 31 participants with a family history of AD but without an APOE ε4 allele.
Of the remaining 78 participants, 33 were excluded because they were unable to complete all three testing sessions (baseline, 18, and 57 months) due to the following reasons: death (6), lost to follow-up (4), moved away (3), refusal (6), medical contraindications for MR scanning (6), MR scanner upgrade at 57months preventing comparison with baseline and 18-month scans (7), and motion corruption of MR scan (1). The final sample consisted of 45 participants divided into two groups: 1) APOE ε4 group (n = 24; mean age = 72.5 years. [SD = 4.1]; mean education = 15.7 years. [SD = 3.2]; 19 females [79%]) who were carriers of one or both ε4 alleles (22 ε3/ε4; 2 ε4/ε4) and 2) Low Risk group (n = 21; mean age = 73.2 years. [SD = 5.3]; mean education = 14.1 year. [SD = 1.8]; 17 females [81%]) who did not possess an APOE ε4 allele (1 ε2/ε3; 20 ε3/ε3). In the APOE ε4 group, 17 participants (70.8%) had a family history of dementia, while no participant in the Low Risk group had a family history of dementia. No significant group differences were observed for age or gender; a significant group difference in education, reflecting more years of education in the APOE ε4 group, was observed (p= 0.04).
For each session, neuropsychological testing and MR scanning were conducted on the same day. Participants were asked to refrain from alcohol use 24 h and caffeine use 12 h prior to testing. The neuropsychological test battery consisted of the Mini-Mental State Examination (Folstein et al., 1975), Mattis Dementia Rating Scale 2 [DRS-2; (Jurica et al., 2001)], and Rey Auditory Verbal Learning Test [RAVLT; (Rey, 1958)]. Alternate, equivalent test forms were used at each session to minimize practice effects.
Participants were evaluated for the presence of Mild Cognitive Impairment (MCI) or AD at the two follow-up sessions. The diagnosis of MCI and AD was based on a multidisciplinary consensus conference that reviewed each participant’s medical history and social history since the previous examination, cognitive test results, and activities of daily living competency. Each participant was queried regarding cognitive complaints (e.g., memory, planning ability, attention, language). If a complaint was expressed, performance on the DRS-2 and RAVLT was compared to an age-adjusted normative database; scores 1.5 SD below the mean on one or more subscales indicated MCI, as long as no functional impairment was reported on the IADL scale. If IADL scores were in the abnormal range (<5), a diagnosis of AD was made. No participant met the criteria for AD during the study period.
Imaging
MRI acquisition
Whole-brain, event-related fMRI was conducted on a General Electric (Waukesha, WI) Signa Excite 3.0 Tesla short bore scanner equipped with a quad split quadrature transmit/receive head coil. Echoplanar images were collected using an echoplanar pulse sequence (TE = 25 ms; flip angle = 77°; field of view (FOV) = 24 mm; matrix size = 64 × 64). Thirty-six contiguous axial 4-mm-thick slices were selected to provide coverage of the entire brain (voxel size = 3.75 × 3.75 × 4 mm). The interscan interval (TR) was 2 s.
High-resolution, three-dimensional spoiled gradient-recalled at steady-state (SPGR) anatomic images were acquired (TE = 3.9 ms; TR = 9.5 ms; inversion recovery (IR) preparation time = 450 ms; flip angle = 12°; number of excitations (NEX) = 2; slice thickness = 1.0 mm; FOV = 24 cm; resolution = 256 × 224).
Perfusion images were collected at the 57 month follow-up using pseudocontinuous ASL (pCASL) (Dai et al., 2008), using an echoplanar pulse sequence (flip angle=90°; field of view (FOV)=240mm; matrix size = 64 × 64). Scans were collected as two volumes (inferior and superior), each consisting of 12 axial 5-mm-thick slices (+1 mm skip), selected to provide whole brain coverage (voxel size = 3.75 × 3.75 × 4mm, TR=4 s, reps=90). In order to register the two volumes, a whole brain image of 24 slices was acquired (slices = 24, TR = 4 s, reps = 6). Foam padding was used to reduce head movement within the coil.
fMRI task
The task stimuli consisted of 30 names of famous persons and 30 names of unfamiliar individuals selected from an original pool of 784 names because of a high rate of identification (>90% correct; (Douville et al., 2005)). A trial consisted of the visual presentation of a single name for 4 s. Participants were instructed to make a right index finger key press if the name was famous and a right middle finger key press if the name was not famous. The 60 name trials were randomly interspersed with 30 4-sec. trials in which the participant was instructed to fixate on a single centrally placed crosshair. This condition was performed in order to introduce “jitter” into the fMRI time course. The imaging run began and ended with 12 s of fixation. Total time for the single imaging run was 5 min and 24 s.
Accuracy (% correct) and reaction time (RT) were recorded for identification of Famous Names and rejection of Non-Famous Names. In addition, a signal detection index of discriminability (d′) (Aaronson and Watts, 1987; Grier, 1971) was calculated along with a measure of intraindividual variability (IIV) calculated from the standard deviation of the Famous and Non-Famous RTs for each participant and time interval.
fMRI image analysis
Functional images were generated with the Analysis of Functional NeuroImages (AFNI) software package (Cox, 1996). fMRI time series data were time-shifted to coincide with the beginning of the TR, motion corrected, transformed to Talairach space, normalized to a whole brain average signal of 10,000, and Gaussian spatial filtered to 6 mm FWHM.
The hemodynamic response (HDR) was deconvolved using ordinary least squares regression with an impulse response model from stimulus onset to 16 s post-stimulus-onset, using regressors for three types of trials: famous names correct, non-famous names correct, and incorrect. Hemodynamic responses were shifted so that stimulus onset was 0. The unfamiliar names’ HDR was subtracted from the famous names’ HDR. The Famous-Non-Famous contrast was summarized by summing the points at 4, 6 and 8 s post-stimulus-onset (peak of the HDR curve).
The first phase of the fMRI analysis examined the spatial extent of neural activation with voxelwise t-tests performed on the Famous-Non-Famous Names subtraction for each group at each session. A voxel was deemed “active” with the following threshold: individual voxel p < 0.005 and minimum cluster volume >0.731 ml. These values were determined from a Monte Carlo simulation producing a familywise error of identifying a significant cluster at p < 0.05 (Cox, 1996).
The second analytic phase involved the identification of functional regions of interest (fROI). A disjunction mask was derived from the voxelwise maps generated in the previous phase. Any voxel deemed “significant” in any of the groups and at any of the scan sessions was included in the fROI map. For each participant and time period, the fMRI signal change was averaged across voxels within each fROI.
The resulting longitudinal fROI data were fit to a linear mixed effects (LME) model using R version number 3.0.2 (Team, 2008). LME modeling has a number of advantages over a traditional repeated measures analysis of variance (ANOVA) (Singer and Willett, 2003). First, LME permits the modeling of the actual time intervals between repeated assessments, whereas repeated measures ANOVA assumes equal intervals between evaluations. In this study, our intervals between evaluations were 18 and 57 months, necessitating the use of an analytic technique that can model these unequally spaced time intervals. Furthermore, LME permits the use of the actual number of days between scans as the time variable for individual subjects. Second, LME permits an unequal number of within-subject observations, making this technique quite flexible in cases where missing data may occur. In contrast, repeated measures ANOVA requires all participants to have observations at each measurement point. If a participant has a single missing data point, that participant could not be included in the repeated measures analysis.
The level one random effects model for this analysis consisted of a linear within-subject model of fMRI activation as a function of weeks post baseline. The level two fixed effects model estimated the slope and intercept of fMRI activation across groups and weeks post baseline, where the Low Risk group provided the base model and the APOE ε4 group differences were modeled with respect to the Low Risk group. Residuals were visually inspected using quantile–quantile plots to confirm the assumption of normality. A quadratic model was also considered, but comparison of the Akaike Information Criterion (AIC) for each model showed that the linear model was preferred.
Perfusion IMAGING
AFNI was used to process the perfusion data. Each subject’s whole brain perfusion image was aligned to the fMRI data. After discarding the first four images, each of the two perfusion volumes was corrected for head motion. For each tagged image in the time series, the preceding and subsequent untagged images were averaged, and this control image was subtracted from its respective tagged image. All volumes in the tagged-control time series were averaged (t-c), as were all volumes in the control time series (c). Voxelwise blood flow was calculated using Eq. (1) in Wang et al. (2005), with duration of labeling pulse = 1.5 s, post-labeling delay time=1 s and tagging efficiency=0.95. To obtain whole brain voxelwise blood flow data, each of the two volumes was aligned to the realigned whole brain volume and combined, using average values for any overlapping voxels, and transformed to Talairach space. Average blood flow for each of the fROIs was extracted, and each region for the two groups was compared using a two-sample Welch’s t-test.
Hippocampal volume
The left and right hippocampal volumes were measured from T1-weighted SPGR images using longitudinal Free surfer v.5.1 (Reuter et al., 2012) applied to all three scan sessions. No significant differences were observed between the left and right hippocampal volumes, so they were summed to create a single bilateral volume, which was normalized by dividing by the total intracranial volume.
Cortical thickness
Volume-defined cortical fROIs (see fMRI Image Analysis section) were projected onto the longitudinal Free surfer surfaces for each subject and scan session Mean cortical thickness was extracted for each of the fROIs on the cortical surface (https://surfer.nmr.mgh.harvard.edu/fswiki/VolumeRoiCorticalThickness). ROI cortical thickness measurements were subjected to the identical LME analysis as applied to the fMRI data.
Results
No group differences were observed in the time interval (in months) between the baseline and second session (Low Risk = 18.4, SD = 1.7; APOE ε4=18.2, SD=0.6) and between the baseline and the third session (Low Risk=58.0, SD=2.1; APOE ε4=56.4, SD=4.3). No significant group differences were observed for baseline neuropsychological testing, fMRI task performance, or hippocampal volume (Table 1).
Table 1.
Baseline characteristics of Low Risk and APOE ε4 groups.
| Variable | Low Risk (n = 21) | APOE e4 (n = 24) | p* | Cohen’s d |
|---|---|---|---|---|
| MMSE | 29.29 (0.85)** | 29.21 (0.98) | 0.78 | 0.08 |
| DRS-2 memory | 24.29 (0.96) | 24.04 (1.60) | 0.53 | 0.18 |
| DRS-2 total | 141.05 (1.99) | 140.33 (3.60) | 0.41 | 0.24 |
| RAVLT delayed recall | 9.90 (2.14) | 9.75 (2.92) | 0.84 | 0.06 |
| RAVLT trials 1–5 | 49.33 (8.39) | 48.50 (8.17) | 0.74 | 0.10 |
| fMRI task | ||||
| Famous (% correct) | 93.97 (5.23) | 91.39 (7.54) | 0.19 | 0.39 |
| Non-famous (% correct) | 95.87 (5.76) | 97.78 (3.63) | 0.20 | −0.40 |
| d′ | 3.29 (0.64) | 3.32 (0.62) | 0.85 | −0.06 |
| Famous (RT, msec) | 1236 (180) | 1249 (151) | 0.81 | −0.07 |
| Non-famous (RT, msec) | 1622 (354) | 1578 (358) | 0.68 | 0.13 |
| IIV famous (RT) | 351 (90) | 371 (131) | 0.57 | −0.17 |
| IIV non-famous (RT) | 347 (101) | 330 (94) | 0.56 | 0.18 |
| Hippocampal volume (% ICV) | 0.47 (0.06) | 0.46 (0.07) | 0.63 | 0.14 |
MMSE = Mini Mental State Examination; DRS-2 = Dementia Rating Scale-2; RAVLT = Rey Auditory Verbal Learning Test; RT = reaction time; IIV= intraindividual variability based on the average standard deviation of RTs for correct responses; % ICV = percent intracranial volume.
p-Values derived from Student t-test, except for gender (Fischer’s exact test).
Mean (standard deviation).
Table 2 summarizes LME analysis applied to neuropsychological testing, fMRI task performance, and hippocampal volume. No significant differences in the intercept (baseline) were observed between the Low Risk and APOE ε4 groups on these variables. Likewise, no changes over time (slope) were observed in neuropsychological testing, fMRI task performance, or hippocampal volume in the Low Risk group with the exception of a reduction in RT for the Non-Famous Names condition. Significant differences in slope were detected in the APOE ε4 group relative to the Low Risk group for a measure of episodic memory (RAVLT Delayed Recall) and hippocampal volume, with a pronounced decline observed in the APOE ε4 group over time (Fig. 1). No other differences in slope were detected between the Low Risk and APOE ε4 groups. Performance on the fMRI task remained above 88% correct for all groups and sessions (chance = 50%).
Table 2.
Coefficients from linear mixed effects analysis of neuropsychological test scores, fMRI task performance, and hippocampal volume.
| Variable | Intercept (baseline)
|
Slope (time)
|
||
|---|---|---|---|---|
| Low Riska | APOE ε4 vs. Low Riskb | Low Riskc | APOE ε4 vs. Low Riskd | |
| MMSE | 29.35 (0.18) | 0.014 (0.246) | 0.004 (.007) | −0.014 (.009) |
| DRS-2 memory | 24.30 (0.27) | −0.597 (0.367) | 0.008 (.008) | −0.019 (.011) |
| DRS-2 total | 140.19 (0.58) | −0.717 (0.790) | 0.007 (.016) | −0.019 (.022) |
| RAVLT delayed recall | 9.63 (0.56) | −0.397 (0.770) | 0.006 (.011) | −0.036 (.015) |
| RAVLT trials 1–5 | 48.84 (1.71) | −1.515 (2.339) | −0.005 (.032) | −0.028 (.044) |
| fMRI task | ||||
| Famous (% correct) | 94.46 (1.27) | −2.479 (1.738) | −0.099 (0.054) | 0.026 (0.074) |
| Non-famous (% correct) | 96.91 (1.09) | 1.456 (1.498) | −0.113 (0.071) | −0.030 (0.097) |
| d′ | 3.38 (0.12) | −0.011 (0.162) | −0.007 (0.005) | −0.003 (0.006) |
| Famous (RT, msec) | 1248.0 (36.0) | −0.893 (49.339) | −1.034 (0.757) | 1.688 (1.046) |
| Non-famous (RT, msec) | 1628.1 (74.1) | −46.960 (101.430) | −3.425 (1.675) | 3.997 (2.304) |
| IIV famous (RT, msec) | 353.4 (23.0) | 10.622 (31.475) | −0.237 (0.408) | 0.532 (0.564) |
| IIV non-famous (RT, msec) | 338.3 (21.4) | −5.952 (29.308) | −0.438 (0.593) | 1.394 (0.817) |
| Hippocampal volume (% ICV) | 0.47 (0.01) | −0.0100 (0.0203) | −0.0001 (0.0001) | −0.0003 (0.0001) |
Bolded values are statistically significant at p < 0.05.
MMSE=MiniMental State Examination; DRS-2=Dementia Rating Scale-2; RAVLT=Rey Auditory Verbal Learning Test; RT=reaction time; IIV=intraindivual variability based on the average standard deviation of RTs for correct responses; % ICV = percent intracranial volume.
Predicted mean intercept (baseline) value of each dependent variable for the Low Risk group. All values are statistically significant from 0.
Predicted difference between mean intercept (baseline) values for the Low Risk group and the APOE ε4 group.
Predicted average monthly rate of change (slope) for the Low Risk group.
Predicted difference in the average monthly rate of change (slope) between the Low Risk group and the APOE ε4 group. Standard errors of coefficients are in parentheses.
Fig. 1.
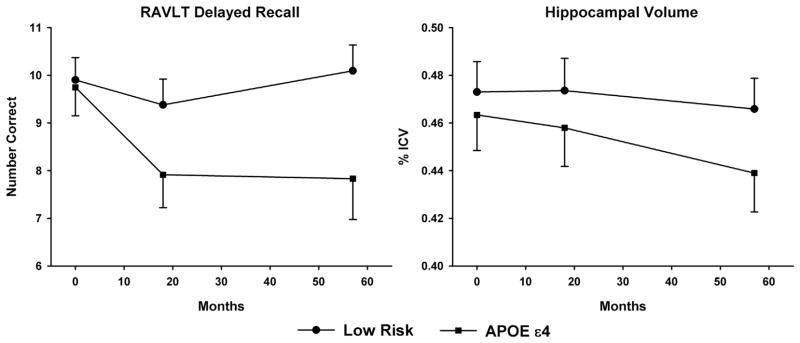
Longitudinal changes in RAVLT Delayed Recall (left) and ICV-corrected hippocampal volume (right) for the Low Risk and APOE ε4 groups at baseline (0 months), 18 months, and 57 months.
Fig. 2 presents the results of the voxelwise analysis demonstrating changes in spatial extent of activation for the two groups over the three sessions. This figure demonstrates a pattern of increasing spatial extent of activation in the Low Risk group over time and decreasing spatial extent in the APOE ε4 group.
Fig. 2.

Voxelwise subtraction of the Famous and Non-Famous Name hemodynamic response functions for the Low Risk and APOE ε4 groups at baseline (0 months), 18 months, and 57 months.
To test this observation, a fROI disjunction mask was created from the voxelwise analysis (see Materials and methods section) and resulted in 16 regions shown in Fig. 3. Table 3 summarizes the LME analysis applied to the 16 fROIs. Intercept analysis indicated that the APOE ε4 group demonstrated greater activation at baseline than the Low Risk group in 10 of 16 fROIs. The Low Risk group demonstrated a significant increase in activation over time (slope) in 4 fROIs. The APOE ε4 group demonstrated a significant decline over time (slope) relative to the Low Risk group in 10 of 16 fROIs. Fig. 4 plots the longitudinal activation changes in these 10 regions. A crossover in activation was observed, characterized by greater activation of the APOE ε4 group relative to the Low Risk group at baseline that subsequently declined over time. In contrast, activation in the Low Risk group was lower than the APOE ε4 group at baseline but gradually increased over time.
Fig. 3.

Functional regions of interest (fROIs) generated from a disjunction mask derived from the Low Risk and APOE ε4 groups at baseline (0 months), 18 months, and 57 months (see Methods). fROI region numbers correspond to numbers in Tables 3–5. BA= Brodmann’s areas; R= right, L= left, B = bilateral; SMA = supplementary motor area.
Table 3.
Coefficients from linear mixed effects analysis of functional ROIs from famous name recognition task.
| No. | Side | Region | Intercept (baseline)
|
Slope (time)
|
||
|---|---|---|---|---|---|---|
| Low Risk a | APOE ε4 vs Low Risk b | Low Risk c | APOE ε4 vs Low Risk d | |||
| 1 | B | Precuneus, posterior cingulate cortex | 0.2610 (0.0828) | 0.2537 (0.1135) | 0.0029 (0.0024) | −0.0078 (0.0033) |
| 2 | L | Middle temporal, angular gyri | 0.2381 (0.0552) | 0.1839 (0.0756) | 0.0012 (0.0016) | −0.0061 (0.0022) |
| 3 | R | Middle temporal, angular gyri | 0.0875 (0.0520) | 0.2360 (0.0712) | 0.0030 (0.0014) | −0.0066 (0.0020) |
| 4 | L | Middle, superior frontal gyri | 0.2299 (0.0575) | 0.1063 (0.0788) | −0.0002 (0.0017) | −0.0041 (0.0024) |
| 5 | L | Inferior & middle occipital, fusiform gyri | −0.2994 (0.0586) | 0.2578 (0.0802) | 0.0071 (0.0017) | −0.0090 (0.0024) |
| 6 | L | Caudate nucleus | 0.0448 (0.0661) | 0.3274 (0.0905) | 0.0027 (0.0021) | −0.0080 (0.0029) |
| 7 | R | Superior medial gyrus, SMA | −0.3366 (0.0638) | 0.1339 (0.0874) | 0.0033 (0.0020) | −0.0027 (0.0027) |
| 8 | R | Superior occipital gyrus | −0.2401 (0.0638) | 0.2303 (0.0875) | 0.0037 (0.0018) | −0.0037 (0.0025) |
| 9 | R | Inferior occipital gyrus | −0.2962 (0.0565) | 0.3126 (0.0773) | 0.0055 (0.0017) | −0.0086 (0.0023) |
| 10 | R | Fusiform, lingual gyri | −0.1653 (0.0811) | 0.5732 (0.1111) | 0.0081 (0.0026) | −0.0160 (0.0035) |
| 11 | L | Fusiform, lingual gyri | −0.3607 (0.0836) | 0.5036 (0.1145) | 0.0107 (0.0025) | −0.0145 (0.0035) |
| 12 | L | Cerebellum (VII–VIII) | −0.00844 (0.0843) | 0.4364 (0.1154) | 0.0041 (0.0024) | −0.0128 (0.0033) |
| 13 | L | Superior medial gyrus, anterior cingulate | 0.3442 (0.1038) | 0.0979 (0.1422) | −0.0009 (0.0028) | −0.0044 (0.0038) |
| 14 | R | Hippocampus | 0.0990 (0.0708) | 0.2039 (0.0970) | 0.0030 (0.0020) | −0.0082 (0.0027) |
| 15 | L | Hippocampus | 0.3171 (0.1064) | −0.0314 (0.1458) | 0.0003 (0.0032) | −0.0017 (0.0044) |
| 16 | L | Parahippocampal, fusiform gyri | 0.1142 (0.1156) | 0.3340 (0.1584) | 0.0047 (0.0034) | −0.0089 (0.0047) |
Bolded values are statistically significant after control for multiple comparisons using false discovery rate. SMA = supplementary motor area.
Predicted mean intercept (baseline) value of each dependent variable for the Low Risk group.
Predicted difference between mean intercept (baseline) values for the Low Risk group and the APOE ε4 group.
Predicted average monthly rate of change (slope) for the Low Risk group.
Predicted difference in the average monthly rate of change (slope) between the Low Risk group and the APOE ε4 group. Values are percent signal change.
Fig. 4.

Percent MR signal intensity derived from subtraction of Famous and Non-Famous Names for 10 fROIs demonstrating significant differences between the Low Risk and APOE ε4 groups in slope (see Table 3). Numbers in brackets correspond to fROIs described in Fig. 3.
No participant was diagnosed with AD or any other form of dementia over the course of the study. At the 57-month follow-up, 8 of 24 APOE ε4 carriers (33.3%) converted to MCI, whereas only one noncarrier (4.8%)was diagnosed with MCI. This group difference was statistically significant (Fisher’s exact test, p = 0.027).
In light of the higher rate of conversion to MCI in the APOE ε4 group, it is conceivable that the decrease in brain activation pattern in this group was due to cortical atrophy. Table 4 summarizes the LME analysis applied to 13 cortical fROIs. The Low Risk group demonstrated an increase in cortical thickness in three fROIs over time. Importantly, the rate of change in cortical thickness was not different between the APOE ε4 carrier and non-carrier groups.
Table 4.
Coefficients from linear mixed effects analysis of cortical thickness in functional ROIs.
| No. | Side | Region | Intercept (baseline) | Slope (time) | ||
|---|---|---|---|---|---|---|
| Low Risk a | APOE ε4 vs. Low Risk b | Low Risk c | APOE ε4 vs. Low Risk d | |||
| 1 | B | Precuneus, posterior cingulate cortex | 2.532 (.024) | 0.0434 (.0331) | 0.0006 (.0003) | −0.0004 (.0004) |
| 2 | L | Middle temporal, angular gyri | 2.502 (.028) | 0.0223 (.0378) | −0.0003 (.0003) | −0.0008 (.0004) |
| 3 | R | Middle temporal, angular gyri | 2.490 (.030) | 0.0524 (.0408) | −0.0001 (.0003) | −0.0009 (.0004) |
| 4 | L | Middle, superior frontal gyri | 2.349 (.034) | 0.0297 (.0469) | 0.0012 (.0005) | −0.0009 (.0007) |
| 5 | L | Inferior & middle occipital, fusiform gyri | 1.976 (.033) | 0.0223 (.0460) | 0.0006 (.0004) | −0.0006 (.0006) |
| 7 | R | Superior medial gyrus, SMA | 2.649 (.040) | 0.0413 (.0475) | 0.0007 (.0004) | −0.0004 (.0006) |
| 8 | R | Superior occipital gyrus | 2.014 (.035) | 0.0413 (.0475) | 0.0003 (.0004) | −0.0006 (.0006) |
| 9 | R | Inferior occipital gyrus | 1.99 (.044) | 0.0346 (.0599) | 0.0005 (.0005) | −0.0002 (.0007) |
| 10 | R | Fusiform, lingual gyri | 2.064 (.041) | 0.0591 (.0555) | 0.0011 (.0004) | −0.0016 (.0006) |
| 11 | L | Fusiform, lingual gyri | 2.136 (.043) | −0.0112 (.0583) | 0.0015 (.0005) | −0.0013 (.0007) |
| 13 | L | Superior medial gyrus, anterior cingulate | 2.517 (.082) | 0.2154 (.1125) | 0.0028 (.0009) | −0.0032 (.0011) |
| 16 | L | Parahippocampal, fusiform gyri | 2.296 (.085) | 0.1826 (.1165) | 0.0007 (.0008) | −0.0014 (.0011) |
Standard errors of coefficients are in parentheses.
Bolded values are statistically significant after control for multiple comparisons using false discovery rate.
Predicted mean intercept (baseline) value of each dependent variable for the Low Risk group. Though these values are statistically significant, they’re not indicated in bold text.
Predicted difference between mean intercept (baseline) values for the Low Risk group and the APOE ε4 group.
Predicted average monthly rate of change (slope) for the Low Risk group.
Predicted difference in the average monthly rate of change (slope) between the Low Risk group and the APOE ε4 group. Unless otherwise specified, values are cortical thickness in millimeters.
The fMRI results could be affected by changes in neurovascular coupling associated with AD pathology, presumed to be greater in APOE ε4 carriers. Resting cerebral blood flow was measured using ASL in the 16 fROIs during the 57-month follow-up session. Table 5 indicates that there were no significant differences in blood flow between the Low Risk and APOE ε4 groups for any of the fROIs. Furthermore, resting blood flow did not correlate with fMRI activation in any of the fROIs after correction for multiple comparisons.
Table 5.
Arterial spin labeling measurements of blood flow conducted at 57-month follow-up.
| No. | Side | Region | Blood flow | Correlation of fMRI activation with blood flow
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Low Risk | APOE ε4 | |||||||||
|
|
|
|
||||||||
| Low Risk | APOE ε4 | p* | Cohen’s d | r** | p | r** | p | |||
| 1 | B | Precuneus, posterior cingulate cortex | 31.16 (13.50)*** | 30.20 (9.58) | 0.69 | 0.10 | 0.00 | 0.98 | −0.13 | 0.26 |
| 2 | L | Middle temporal, angular gyri | 25.87 (8.68) | 25.43 (8.34) | 0.72 | 0.09 | −0.02 | 0.90 | −0.14 | 0.24 |
| 3 | R | Middle temporal, angular gyri | 30.89 (13.14) | 28.75 (8.18) | 0.37 | 0.22 | −0.09 | 0.48 | 0.00 | 1.00 |
| 4 | L | Middle, superior frontal gyri | 20.57 (10.08) | 20.25 (10.00) | 0.85 | 0.05 | −0.15 | 0.26 | 0.01 | 0.90 |
| 5 | L | Inferior & middle occipital, fusiform gyri | 14.66 (7.77) | 13.09 (7.59) | 0.44 | 0.20 | 0.08 | 0.55 | −0.08 | 0.51 |
| 6 | L | Caudate nucleus | 19.74 (8.15) | 17.56 (5.27) | 0.37 | 0.21 | −0.18 | 0.16 | 0.16 | 0.17 |
| 7 | R | Superior medial gyrus, SMA | 21.42 (8.87) | 23.02 (7.66) | 0.53 | 0.16 | −0.24 | 0.06 | 0.07 | 0.54 |
| 8 | R | Superior occipital gyrus | 20.12 (12.46) | 18.80 (9.36) | 0.62 | 0.13 | −0.03 | 0.80 | −0.05 | 0.65 |
| 9 | R | Inferior occipital gyrus | 20.18 (20.18) | 15.86 (10.99) | 0.34 | 0.24 | 0.01 | 0.92 | −0.24 | 0.04 |
| 10 | R | Fusiform, lingual gyri | 27.15 (11.84) | 25.34 (13.90) | 0.62 | 0.15 | 0.17 | 0.18 | 0.10 | 0.38 |
| 11 | L | Fusiform, lingual gyri | 25.22 (14.03) | 23.45 (11.67) | 0.48 | 0.19 | −0.02 | 0.86 | 0.03 | 0.79 |
| 12 | L | Cerebellum (VII–VIII) | 12.32 (17.51) | 9.10 (7.13) | 0.39 | 0.20 | −0.10 | 0.45 | −0.14 | 0.24 |
| 13 | L | Superior medial gyrus, anterior cingulate | 27.48 (8.45) | 28.34 (8.46) | 0.81 | 0.06 | −0.08 | 0.55 | −0.04 | 0.71 |
| 14 | R | Hippocampus | 21.49 (11.85) | 22.95 (9.31) | 0.76 | 0.08 | −0.02 | 0.89 | 0.22 | 0.07 |
| 15 | L | Hippocampus | 29.25 (15.00) | 32.05 (14.63) | 0.65 | 0.13 | −0.15 | 0.26 | 0.19 | 0.12 |
| 16 | L | Parahippocampal, fusiform gyri | 30.56 (18.12) | 33.52 (19.33) | 0.70 | 0.12 | −0.22 | 0.08 | −0.18 | 0.13 |
ASL blood flow values are ml/100 g/min.
SMA = supplementary motor area.
p-Values derived from Welch’s t-test.
Pearson correlations between fMRI activation and ASL blood flow (within group).
Mean (standard deviation).
Discussion
In this study, we investigated the longitudinal trajectory of semantic memory activation over a 5-year period among APOE ε4 carriers and non-carriers. At baseline, the non-carrier group showed uniformly lower task-related activation during the semantic memory paradigm compared to the APOE ε4 carriers. Over the 5-year interval, however, functional activation steadily decreased in multiple regions of interest in the APOE ε4 carriers, particularly in posterior cortical areas commonly associated with the default mode network (Raichle et al., 2001). In contrast, the non-carrier group demonstrated a consistent increase in activation in the same regions over the same time frame.
Overall, our results provided evidence in support of differential longitudinal trajectories for task-related brain activation as a function of genetic risk for development of AD. The findings provide support for the STAC-r theory that attempts to account for differences in the trajectory of neural compensatory scaffolding associated with changes in cognitive performance over time (Reuter-Lorenz and Park, 2014). Our study illustrated the differential trajectories associated with episodic memory performance, hippocampal volume, and fMRI activation. At baseline, APOE ε4 carriers demonstrate elevated functional activation at an earlier stage than non-carriers to compensate presumably for accelerated accumulation of neural pathology, likely related to AD pathology and possibly neurovascular risk factors (see below). The progressive decline in functional activation over time reflects reduced compensatory scaffolding associated with age-inappropriate episodic memory decline and accumulated AD-related pathology as reflected by atrophy of the hippocampus. In contrast, APOE ε4 non-carriers demonstrated a gradual increase in compensatory task-related brain activation over time in the context of age-appropriate changes in episodic memory and hippocampal volume, consistent with what would be expected based on prior cross-sectional studies (Bangen et al., 2012; Grady, 2008; Nielson et al., 2002, 2006).
The STAC-r model infers that compensatory activation responses are predominantly associated with dorsolateral frontal regions. This conclusion is derived primarily from fMRI studies that use effortful, mostly episodic memory, tasks that engage dorsolateral frontal regions. In contrast, we have demonstrated in a series of studies (Douville et al., 2005; Nielson et al., 2006, 2010; Seidenberg et al., 2009; Smith et al., 2011; Woodard et al., 2009, 2010) that compensatory brain activation can occur within non-frontal brain regions using the Famous Name Recognition Task, a low effort, high accuracy semantic memory task. As noted by Binder et al. (2009), there is considerable overlap between the semantic system and the default mode network (DMN), whose primary brain regions (hippocampus, posterior cingulate, temporoparietal junction, medial frontal) are particularly vulnerable to early AD-related neuropathological changes (Buckner, 2004). Our results suggest that the STAC-r model should be generalized to include compensatory processes occurring outside dorsolateral prefrontal regions.
To our knowledge, there have been no other longitudinal fMRI studies comparing cognitively intact older individuals with varying genetic risks for AD. However, one study (Smith et al., 2005) examined middle-aged women (mean ages = 53 and 54) divided into two groups: high genetic risk based on a positive family history of AD and possession of an APOE ε4 allele (n=14) and low risk (n=10). Changes in brain activation in response to a covert object-naming task were measured at baseline and at four-year follow-up. The high risk group did not demonstrate greater activation than the low risk group at baseline, but showed a greater reduction in brain activation over time. These results are difficult to interpret since the covert task used in the Smith et al. study yielded no behavioral data to verify that participants were in fact performing the task. A decline in activation may readily be explained by a lack of task compliance during the follow-up scan session. In contrast, the FNRT used in this study was performed at a high level of performance at all three scan sessions.
The APOE ε4 carriers demonstrated a longitudinal decline in performance on the RAVLT Delayed Recall measure (see Table 2 and Fig. 1), whereas the non-carriers’ performance remained intact and stable across the five year interval. We did not expect to see performance changes in the Famous Name Recognition Task, which was designed to be performed at greater than 85% accuracy, even in patients with amnestic MCI (Woodard et al., 2009). It is important to note that most fMRI studies of normal aging employ effortful episodic memory tasks whose performance declines with age. It is exceedingly difficult to interpret longitudinal fMRI activation maps when task performance is also declining. A brain map generated from an individual performing at near chance levels will invariably be different from a person performing well above chance. Removing incorrect trials does not entirely correct the problem because the resulting brain maps are generated from a sparse number of correct trials in individuals performing near chance. Moreover, the BOLD signals that result from greater task difficulty or effort in the poor performing group will be inseparable from activation related to memory retrieval-related processes, thus confounding any sound interpretation of the activation maps (Hantke et al., 2013).
The APOE ε4 allele is present in approximately 20% of the general population (Eichner et al., 2002) and 50% of patients diagnosed with AD (Ward et al., 2012). Implicit in our study is the assumption that a larger percentage of our APOE ε4 carriers will have developed AD-related pathology over the course of the 5-year follow-up period than the percentage of non-carriers. Although none of our participants met the criteria for AD at 5 years, 8 of 24 APOE ε4 carriers (33.3%) converted to MCI, whereas only one non-carrier (4.8%) converted (p=0.027). To appreciate these results, it is important to emphasize that all of our participants were cognitively intact at study entry. Long-term prospective studies suggest that the majority of the study participants meeting the criteria for MCI will eventually convert to AD (Petersen et al., 1999). It is conceivable that additional APOE ε4 carriers will convert to MCI/AD if examined over a longer follow-up interval. The statistically higher percentage of MCI converters among the carrier group supports our assumption that our APOE ε4 participants were more likely to be acquiring AD-related pathology than non-carriers.
One may speculate as to the neuropathological processes that result in increased compensatory scaffolding in APOE ε4 carriers at an earlier age than non-carriers, followed by diminished scaffolding as cognitive performance declines and hippocampal volumes diminish. Plausible theories suggest that ε4 disrupts lipid homeostasis, amyloid precursor protein function, and the handling of brain amyloid, cholinergic function, and neuroinflammation (Lane and Farlow, 2005; Poirier, 2000). Our studies (Smith et al., 2011, 2014) and those of others (Head et al., 2012) suggest that the negative effects of possessing an ε4 allele on functional and structural imaging may be offset by the positive effects of physical activity. Additional work is needed to identify the precise mechanisms that underlie the trajectory of neural compensation in APOE ε4 carriers.
It is also possible that the fMRI results could be affected by changes in neurovascular coupling associated with AD pathology, which is presumed to be greater in the APOE ε4 carriers. Our ASL measurements of resting cerebral blood flow at the 5-year follow-up session did not reveal group differences in any of the fROIs. Furthermore, blood flow did not correlate with fMRI activation in any of the fROIs after correction for multiple comparisons. Finally, it is possible that our results were related to brain atrophy. However, the rate of change in cortical thickness did not differ between the carrier and non-carrier groups in any of the fROIs.
Within the Low Risk group, unexpected positive slopes were observed for cortical thickness in the right and left fusiform/lingual gyri and the left superior medial gyrus/anterior cingulate. Similar findings, however, have been reported in prior neuroimaging studies of normal aging. Thambisetty et al. (2010) observed significant longitudinal increases in cortical thickness within the left fusiform and lingual gyri. Similarly, Salat et al. (2004) observed increases in cortical thickness within medial frontal regions, including the anterior cingulate. These increases have been attributed to decreases in gray/white matter contrast during aging (Thambisetty et al., 2010) and signal dropout and anatomical distortions at the base of the brain (Salat et al., 2004). Both explanations could potentially lead to distortions in regionally-specific estimates of cortical thickness.
In our task-activated fMRI analyses, we chose not to control for age since the two groups did not differ statistically in mean age. Nevertheless, we conducted a supplementary analysis in which age-corrected residual scores were generated for the mean fMRI signal change for each fROI, participant, and time period and then repeated the LME analyses. Notably, none of the intercept and slope results reported in Table 3 changed statistically.
The current study has its limitations. It is possible that the subset of participants who completed all three examination sessions are not representative of the entire sample of participants enrolled into this longitudinal study. We were unable to identify, however, any demographic or baseline neuropsychological or neuroimaging variables that differentiated participants who completed the study versus those who did not. This study did not use any other imaging biomarkers to measure AD-related pathology, such as CSF analyses of tau and amyloid proteins or amyloid deposition using PET imaging. The ASL measurement of perfusion was only conducted at the 5-year follow-up. Finally, the relatively small number of APOE ε4 carriers who converted to MCI limited our ability to conduct post-hoc imaging analyses comparing converters to non-converters.
Conclusions
Compensatory brain activation is commonly seen in the fMRI scans of cognitively intact elders. We have demonstrated that this compensatory response is accelerated in cognitively intact individuals at genetic risk for AD and declines precipitously once cognitive dysfunction and hippocampal atrophy become apparent. In contrast, elders with a lower risk of developing AD and age-appropriate cognition employ increased brain activation to maintain functionality; this compensatory response increases with age as long as the low risk individual continues to demonstrate age-appropriate cognition. Our fMRI results could not be attributed to changes in task performance, group differences in cerebral perfusion, or regional cortical atrophy. The results provide prospective, empirical evidence of differential longitudinal trajectories based on AD risk, supporting the theoretical propositions underlying the STAC-r neural compensation theory (Reuter-Lorenz and Park, 2014).
Acknowledgments
We thank Piero Antuono, Alissa M. Butts, Kelli L. Douville, Christina M. Figueroa, Malgorzata Franczak, Amelia Gander, Evan Gross, Leslie M. Guidotti-Breting, Nathan C. Hantke, Kathleen E. Hazlett, Emily Hoida, Cassandra Kandah, Christina D. Kay, Dana Kelly, Melissa A. Lancaster, Monica Matthews, Sarah K. Miller, Andria L. Norman, Katherine Reiter, Michael A. Sugarman, and Qi Zhang for their assistance. This work was supported by the National Institutes of Health Grants R01 AG022304 and M01 RR00058. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute on Aging or the National Institutes of Health.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- Aaronson D, Watts B. Extensions of Grier’s computational formulas for A′ and B″ to below-chance performance. Psychol Bull. 1987;102:439–442. [PubMed] [Google Scholar]
- Bangen KJ, Kaup AR, Mirzakhanian H, Wierenga CE, Jeste DV, Eyler LT. Compensatory brain activity during encoding among older adult with better recognition memory for face-name pairs: an integrative functional, structural, and perfusion imaging study. J Int Neuropsychol Soc. 2012;18:402–413. doi: 10.1017/S1355617712000197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman RJ, Xiong C, Benzinger TLS, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder JR, Desai RH, Graves WW, Conant LL. Where is the semantic system? A critical review and meta-analysis of 120 functional neuroimaging studies. Cereb Cortex. 2009;19:2767–2796. doi: 10.1093/cercor/bhp055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bookheimer SYS, MH, Cohen MS, Saunders AM, Pericak-Vance MA, Mazziotta JC, Small GW. Patterns of brain activation in people at risk for Alzheimer’s disease. N Engl J Med. 2000;343:450–456. doi: 10.1056/NEJM200008173430701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghesani PR, Johnson LC, Shelton AL, Peskind ER, Aylward EH, Schellenberg GD, Cherrier MM. Altered medial temporal lobe responses during visuospatial encoding in healthy APOE*4 carriers. Neurobiol Aging. 2008;29:981–991. doi: 10.1016/j.neurobiolaging.2007.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner RL. Memory and executive function in aging and AD:multiple factors that cause decline and reserve factors that compensate. Neuron. 2004;44:195–208. doi: 10.1016/j.neuron.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Cabeza R, Anderson ND, Locantore JK, McIntosh AR. Aging gracefully: compensatory brain activity in high-performing older adults. Neuroimage. 2002;17:1394–1402. doi: 10.1006/nimg.2002.1280. [DOI] [PubMed] [Google Scholar]
- Carp J, Gmeindl L, Reuter-Lorenz PA. Age differences in the neural representation of working memory revealed by multi-voxel pattern analysis. Front Hum Neurosci. 2010;4:1–10. doi: 10.3389/fnhum.2010.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox R. AFNI: software for analysis and visualization of functional magnetic resonance neuroimages. Comput Biomed Res. 1996;29:162–173. doi: 10.1006/cbmr.1996.0014. [DOI] [PubMed] [Google Scholar]
- Dai W, Garcia D, de Bazelaire C, Alsop DC. Continuous flow-driven inversion for arterial spin labeling using pulsed radio frequency and gradient fields. Magn Reson Med. 2008;60:1488–1497. doi: 10.1002/mrm.21790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douville KL, Woodard JL, Seidenberg M, Miller SK, Leveroni CL, Nielson KA, Franczak M, Antuono P, Rao SM. Medial temporal lobe activity for recognition of recent and remote famous names: an event-related fMRI study. Neuropsychologia. 2005;43:693–703. doi: 10.1016/j.neuropsychologia.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Drachman DA. Aging of the brain, entropy, and Alzheimer disease. Neurology. 2006;67:1340–1352. doi: 10.1212/01.wnl.0000240127.89601.83. [DOI] [PubMed] [Google Scholar]
- Eichner JE, Dunn ST, Perveen G, Thompson DM, Stewart KE, Stroehla BC. Apolipoprotein E polymorphism and cardiovascular disease: a HuGE review. Am J Epidemiol. 2002;155:487–495. doi: 10.1093/aje/155.6.487. [DOI] [PubMed] [Google Scholar]
- Evans S, Dowell NG, Tabet N, Tofts PS, King SL, Rusted JM. Cognitive and neural signatures of the APOE E4 allele in mid-aged adults. Neurobiol Aging. 2014;35:1615–1623. doi: 10.1016/j.neurobiolaging.2014.01.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyler LT, Sherzai A, Kaup AR, Jeste DV. A review of functional brain imaging correlates of successful cognitive aging. Biol Psychiatry. 2011;70:115–122. doi: 10.1016/j.biopsych.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. J Am Med Assoc. 1997;278:1349–1356. [PubMed] [Google Scholar]
- Filippini N, Ebmeier KP, MacIntosh BJ, Trachtenberg AJ, Frisoni GB, Wilcock GK, Beckmann CF, Smith SM, Matthews PM. Differential effects of the APOE genotype on brain function across the lifespan. Neuroimage. 2011;54:602–610. doi: 10.1016/j.neuroimage.2010.08.009. [DOI] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR. ‘Mini-mental state’: a practical method for grading the cognitive state of patients for the clinician. J Psychiatry Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- Grady CL. Cognitive neuroscience of aging. Ann N Y Acad Sci. 2008;1124:127–144. doi: 10.1196/annals.1440.009. [DOI] [PubMed] [Google Scholar]
- Grier JB. Nonparametric indexes for sensitivity and bias: computing formulas. Psychol Bull. 1971;75:424–429. doi: 10.1037/h0031246. [DOI] [PubMed] [Google Scholar]
- Han SD, Houston WS, Jak AJ, Eyler LT, Nagel BJ, Fleisher AS, Brown GG, Corey-Bloom J, Salmon DP, Thal LJ, Bondi MW. Verbal paired associate learning by APOE genotype in non-demented older adults: fMRI evidence of a right hemispheric compensatory response. Neurobiol Aging. 2007;28:238–247. doi: 10.1016/j.neurobiolaging.2005.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SD, Bangen KJ, Bondi MW. Functional magnetic resonance imaging of compensatory neural recruitment in aging and risk for Alzheimer’s disease: review and recommendations. Dement Geriatr Cogn Disord. 2009;27:1–10. doi: 10.1159/000182420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hantke N, Nielson KA, Woodard JL, Breting LM, Butts A, Seidenberg M, Carson Smith J, Durgerian S, Lancaster M, Matthews M, Sugarman MA, Rao SM. Comparison of semantic and episodic memory BOLD fMRI activation in predicting cognitive decline in older adults. J Int Neuropsychol Soc. 2013;19:11–21. doi: 10.1017/S1355617712000951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head D, Bugg JM, Goate AM, Fagan AM, Mintun MA, Benzinger T, Holtzman DM, Morris JC. Exercise engagement as a moderator of the effects of APOE genotype on amyloid deposition. Arch Neurol. 2012;69:636–643. doi: 10.1001/archneurol.2011.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Wiste HJ, Vemuri P, Weigand SD, Senjem ML, Zeng G, Bernstein MA, Gunter JL, Pankratz VS, Aisen PS, Weiner MW, Petersen RC, Shaw LM, Trojanowski JQ, Knopman DS, Initiative AsDN. Brain beta-amyloid measures and magnetic resonance imaging atrophy both predict time-to-progression from mild cognitive impairment to Alzheimer’s disease. Brain. 2010;133:3336–3348. doi: 10.1093/brain/awq277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurica PJ, Leitten CL, Mattis S. Dementia Rating Scale-2 Professional Manual. Psychological Assessment Resources; Lutz, FL: 2001. [Google Scholar]
- Kennedy KM, Rodrigue KM, Bischof GN, Hebrank AC, Reuter-Lorenz PA, Park DC. Age trajectories of functional activation under conditions of low and high processing demands: an adult lifespan fMRI study of the aging brain. Neuroimage. 2014 doi: 10.1016/j.neuroimage.2014.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer JH, Mungas D, Reed BR, Wetzel ME, Burnett MM, Miller BL, Weiner MW, Chui HC. Longitudinal MRI and cognitive change in healthy elderly. Neuropsychology. 2007;21:412–418. doi: 10.1037/0894-4105.21.4.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane RM, Farlow MR. Lipid homeostasis and apolipoprotein E in the development and progression of Alzheimer’s disease. J Lipid Res. 2005;46:949–968. doi: 10.1194/jlr.M400486-JLR200. [DOI] [PubMed] [Google Scholar]
- Lawton MP, Brody EM. Assessment of older people: self-maintaining and instrumental activities of daily living. Gerontologist. 1969;9:179–186. [PubMed] [Google Scholar]
- Miller SL, Fenstermacher E, Bates J, Blacker D, Sperling RA, Dickerson BC. Hippocampal activation in adults with mild cognitive impairment predicts subsequent cognitive decline. J Neurol Neurosurg Psychiatry. 2008;79:630–635. doi: 10.1136/jnnp.2007.124149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielson KA, Langenecker SA, Garavan H. Differences in the functional neuroanatomy of inhibitory control across the adult life span. Psychol Aging. 2002;17:56–71. doi: 10.1037//0882-7974.17.1.56. [DOI] [PubMed] [Google Scholar]
- Nielson KA, Douville KL, Seidenberg M, Woodard JL, Miller SK, Franczak M, Antuono P, Rao SM. Age-related functional recruitment for famous name recognition: an event-related fMRI study. Neurobiol Aging. 2006;27:1494–1504. doi: 10.1016/j.neurobiolaging.2005.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielson KA, Seidenberg M, Woodard JL, Durgerian S, Zhang Q, Gross WL, Gander A, Guidotti LM, Antuono P, Rao SM. Common neural systems associated with the recognition of famous faces and names: an event-related fMRI study. Brain Cogn. 2010;72:491–498. doi: 10.1016/j.bandc.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien DP, Campbell KA, Morken NW, Bair RJ, Heath EM. Automated nucleic acid purification for large samples. J Lab Autom. 2001;6:67–70. [Google Scholar]
- O’Brien JL, O’Keefe KM, LaViolette PS, DeLuca AN, Blacker D, Dickerson BC, Sperling RA. Longitudinal fMRI in elderly reveals loss of hippocampal activation with clinical decline. Neurology. 2010;74:1969–1976. doi: 10.1212/WNL.0b013e3181e3966e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldfield RC. The assessment of handedness: the Edinburgh inventory. Neuropsychologia. 1971;9:97–111. doi: 10.1016/0028-3932(71)90067-4. [DOI] [PubMed] [Google Scholar]
- Park DC, Reuter-Lorenz PA. The adaptive brain: aging and neurocognitive scaffolding. Annu Rev Psychol. 2009;60:173–196. doi: 10.1146/annurev.psych.59.103006.093656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol. 1999;56:303–308. doi: 10.1001/archneur.56.3.303. [DOI] [PubMed] [Google Scholar]
- Poirier J. Apolipoprotein E and Alzheimer’s disease. A role in amyloid catabolism. Ann N Y Acad Sci. 2000;924:81–90. doi: 10.1111/j.1749-6632.2000.tb05564.x. [DOI] [PubMed] [Google Scholar]
- Prvulovic D, Van de Ven V, Sack AT, Maurer K, Linden DE. Functional activation imaging in aging and dementia. Psychiatry Res Neuroimaging. 2005;140:97–113. doi: 10.1016/j.pscychresns.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Raichle ME, MacLeod AM, Snyder AZ, Powers WJ, Gusnard DA, Shulman GL. A default mode of brain function. Proc Natl Acad Sci U S A. 2001;98:676–682. doi: 10.1073/pnas.98.2.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, Saunders AM, Hardy J. Functional brain abnormalities in young adults at genetic risk for lateonset Alzheimer’s dementia. Proc Natl Acad Sci U S A. 2004;101:284–289. doi: 10.1073/pnas.2635903100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter M, Schmansky NJ, Rosas HD, Fischl B. Within-subject template estimation for unbiased longitudinal image analysis. Neuroimage. 2012;61:1402–1418. doi: 10.1016/j.neuroimage.2012.02.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter-Lorenz PA, Cappell KA. Neurocognitive aging and the compensation hypothesis. Curr Dir Psychol Sci. 2008;17:177–182. [Google Scholar]
- Reuter-Lorenz PA, Park DC. How does it STAC up? Revisiting the scaffolding theory of aging and cognition. Neuropsychol Rev. 2014;24:355–370. doi: 10.1007/s11065-014-9270-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey A. L’examen clinique en psychologie. Presses Universitaires de France; Paris: 1958. [Google Scholar]
- Salat DH, Buckner RL, Snyder AZ, Greve DN, Desikan RSR, Busa E, Morris JC, Dale AM, Fischl B. Thinning of the cerebral cortex in aging. Cereb Cortex. 2004;14:721–730. doi: 10.1093/cercor/bhh032. [DOI] [PubMed] [Google Scholar]
- Salthouse TA. Selective review of cognitive aging. J Int Neuropsychol Soc. 2010;16:754–760. doi: 10.1017/S1355617710000706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidenberg M, Guidotti L, Nielson KA, Woodard JL, Durgerian S, Antuono P, Zhang Q, Rao SM. Semantic memory activation in individuals at risk for developing Alzheimer disease. Neurology. 2009;73:612–620. doi: 10.1212/WNL.0b013e3181b389ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer JD, Willett JB. Applied Longitudinal Data Analysis. Oxford; New York: 2003. [Google Scholar]
- Smith CD, Kryscio RJ, Schmitt FA, Lovell MA, Blonder LX, Rayens WS, Andersen AH. Longitudinal functional alterations in asymptomatic women at risk for Alzheimer’s disease. J Neuroimaging. 2005;15:271–277. doi: 10.1177/1051228405277340. [DOI] [PubMed] [Google Scholar]
- Smith JC, Nielson KA, Woodard JL, Seidenberg M, Durgerian S, Antuono P, Butts AM, Hantke NC, Lancaster MA, Rao SM. Interactive effects of physical activity and APOE-e4 on BOLD semantic memory activation in healthy elders. Neuroimage. 2011;54:635–644. doi: 10.1016/j.neuroimage.2010.07.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JC, Nielson KA, Woodard JL, Seidenberg M, Durgerian S, Hazlett KE, Figueroa CM, Kandah CC, Kay CD, Matthews MA, Rao SM. Physical activity reduces hippocampal atrophy in elders at genetic risk for Alzheimer’s disease. Front Aging Neurosci. 2014;6:61. doi: 10.3389/fnagi.2014.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugarman MA, Woodard JL, Nielson KA, Seidenberg M, Smith JC, Durgerian S, Rao SM. Functional magnetic resonance imaging of semantic memory as a presymptomatic biomarker of Alzheimer’s disease risk. Biochim Biophys Acta Mol basis Dis. 2012;1822:442–456. doi: 10.1016/j.bbadis.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team, R.D.C. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; Vienna, Austria: 2008. [Google Scholar]
- Thambisetty M, Wan J, Carass A, An Y, Prince JL, Resnick SM. Longitudinal changes in cortical thickness associated with normal aging. Neuroimage. 2010;52:1215–1223. doi: 10.1016/j.neuroimage.2010.04.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachtenberg AJ, Filippini N, Mackay CE. The effects of APOE-e4 on the BOLD response. Neurobiol Aging. 2012;33:323–334. doi: 10.1016/j.neurobiolaging.2010.03.009. [DOI] [PubMed] [Google Scholar]
- Wang J, Zhang Y, Wolf RL, Roc AC, Alsop DC, Detre JA. Amplitude-modulated continuous arterial spin-labeling 3.0-T perfusion MR imaging with a single coil: feasibility study. Radiology. 2005;235:218–228. doi: 10.1148/radiol.2351031663. [DOI] [PubMed] [Google Scholar]
- Ward A, Crean S, Mercaldi CJ, Collins JM, Boyd D, Cook MN, Arrighi HM. Prevalence of apolipoprotein E4 genotype and homozygotes (APOE e4/4) among patients diagnosed with Alzheimer’s disease: a systematic review and meta-analysis. Neuroepidemiology. 2012;38:1–17. doi: 10.1159/000334607. [DOI] [PubMed] [Google Scholar]
- Wierenga CE, Bondi MW. Use of functional magnetic resonance imaging in the early identification of Alzheimer’s disease. Neuropsychol Rev. 2007;17:127–143. doi: 10.1007/s11065-007-9025-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga CE, Benjamin M, Gopinath K, Perlstein WM, Leonard CM, Gonzalez Rothi LJ, Conway T, Cato MA, Briggs R, Crosson B. Age-related changes in word retrieval: role of bilateral frontal and subcortical networks. Neurobiol Aging. 2008;29:436–451. doi: 10.1016/j.neurobiolaging.2006.10.024. [DOI] [PubMed] [Google Scholar]
- Wierenga CE, Stricker NH, McCauley A, Simmons A, Jak AJ, Chang YL, Delano-Wood L, Bangen KJ, Salmon DP, Bondi MW. Increased functional brain response during word retrieval in cognitively intact older adults at genetic risk for Alzheimer’s disease. Neuroimage. 2010;51:1222–1233. doi: 10.1016/j.neuroimage.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodard JL, Seidenberg M, Nielson KA, Antuono P, Guidotti L, Durgerian S, Zhang Q, Lancaster M, Hantke N, Butts A, Rao SM. Semantic memory activation in amnestic mild cognitive impairment. Brain. 2009;132:2068–2078. doi: 10.1093/brain/awp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodard JL, Seidenberg M, Nielson KA, Smith JC, Antuono P, Durgerian S, Guidotti L, Zhang Q, Butts A, Hantke N, Lancaster M, Rao SM. Prediction of cognitive decline in healthy older adults using fMRI. J Alzheimers Dis. 2010;21:871–885. doi: 10.3233/JAD-2010-091693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yesavage JA, Brink TL, Rose TL, Lum O, Huang V, Adey M, Leirer VO. Development and validation of a geriatric depression screening scale: a preliminary report. J Psychiatry Res. 1982;17:37–49. doi: 10.1016/0022-3956(82)90033-4. [DOI] [PubMed] [Google Scholar]