

Abstract
In addition to its thoroughly investigated role in bone formation, the osteoblast master transcription factor RUNX2 also promotes osteoclastogenesis and bone resorption. Here we demonstrate that 17β-estradiol (E2), which is known to attenuate bone turnover in vivo and RUNX2 activity in vitro, strongly inhibits RUNX2-mediated osteoblast-driven osteoclastogenesis in co-cultures. Towards deciphering the underlying mechanism, we induced premature expression of RUNX2 in primary murine pre-osteoblasts, which resulted in robust differentiation of co-cultured splenocytes into mature osteoclasts. This was attributable to RUNX2-mediated increase in RANKL secretion, determined by ELISA, as well as to RUNX2-mediated increase in RANKL association with the osteoblast membrane, demonstrated using confocal fluorescence microscopy. The increased association with the osteoblast membrane was recapitulated by transiently expressed GFP-RANKL. E2 abolished the RUNX2-mediated increase in membrane-associated RANKL and GFP-RANKL, as well as the concomitant osteoclastogenesis. RUNX2-mediated RANKL cellular redistribution was attributable in part to a decrease in Opg expression, but E2 did not influence Opg expression either in the presence or absence of RUNX2. Diminution of RUNX2-mediated osteoclastogenesis by E2 occurred regardless of whether the pre-osteoclasts were derived from wild type or estrogen receptor alpha (ERα)-knockout mice, suggesting that activated ERα inhibited osteoblast-driven osteoclastogenesis by acting in osteoblasts, possibly targeting RUNX2. Furthermore, the selective ER modulators (SERMs) tamoxifen and raloxifene mimicked E2 in abrogating the stimulatory effect of osteoblastic RUNX2 on osteoclast differentiation in the co-culture assay. Thus, E2 antagonizes RUNX2-mediated RANKL trafficking and subsequent osteoclastogenesis. Targeting RUNX2 and/or downstream mechanisms that regulate RANKL trafficking may lead to the development of improved SERMs and possibly non-hormonal therapeutic approaches to high turnover bone disease.
Keywords: Postmenopausal Osteoporosis, TRAP, Protein Trafficking, Secretion
1.0 - INTRODUCTION
Runx2 is as an osteoblast master regulator that is required for bone formation. Initially identified based on its interaction with the bone-specific Osteocalcin promoter in vitro [1–3], the pivotal role of Runx2 in osteogenesis in vivo was demonstrated by the absence of differentiated osteoblasts and failure of skeletal mineralization in Runx2-deficient mice [4, 5]. Furthermore, inhibition of Runx2 in vitro abrogates expression of osteoblast markers, and its forced expression in non-osteoblasts induces bone-like cellular phenotypes [2, 3]. Contrasting the role of Runx2 as a master regulator of osteoblast differentiation and embryonic bone development, its function in bone resorption is less appreciated. Over-expression of Runx2 in osteoblasts resulted in increased osteoclast differentiation from co-cultured pre-osteoclasts in vitro [6–8] and exaggerated bone resorption in vivo [6, 9]. Conversely, expression of a dominant negative RUNX2 decreased osteoclastogenesis in co-culture assays and decreased bone resorption in vivo [7]. Accordingly, osteoclast number decreased in mice with either global or osteoblast-specific Runx2 ablation [4, 10]. Thus, RUNX2 promotes both osteoblastogenesis and osteoblast-driven osteoclastogenesis.
Regulation of osteoclastogenesis by osteoblasts constitutes a fundamental principle in the coupling of bone resorption to bone formation [11]. Among osteoblast-borne signals mediating this coupling is the quintessential osteoclastogenic factor RANKL [12–14]. Although RUNX2 can increase RANKL mRNA levels in smooth muscle cells [15], stimulation of osteoclastogenesis by RUNX2 does not appear to involve the regulation of Rankl mRNA levels in osteoblasts [8, 16, 17]. Indeed, using primary osteoblast cultures, the present work demonstrates that RUNX2 influences RANKL through regulating its trafficking to the cell membrane without significantly affecting its mRNA expression.
Postmenopausal osteoporosis inflicts a pathological fracture on two in every five women over the age of fifty [18]. It is mostly attributable to reduced stimulation of estrogen receptor α (ERα) in osteoblasts and osteoclasts [19]. Accordingly, estrogens and selective estrogen receptor modulators (SERMs) constitute therapeutic options for the preservation of bone mass in postmenopausal women, and some SERMs have beneficial effects on the skeleton when used for the management of breast cancer [20]. Based on previous reports on inhibition of RUNX2 activity by ERα [21], as well as resistance to ovariectomy-induced bone loss in mice expressing of a dominant negative RUNX2 isoform [7], we hypothesized that activation of ERα in osteoblasts attenuates RUNX2-driven osteoclastogenic signal(s). Indeed, we show that estrogen signaling in osteoblasts abrogates RUNX2-mediated RANKL membrane association and differentiation of co-cultured splenocytes into mature osteoclasts.
2.0 - MATERIALS AND METHOD
2.1 - Animals
C57BL/6 JAX® mice from Jackson Laboratory (Sacramento, CA) were used for the extraction of both osteoblasts and splenocytes without regard to mouse gender. Splenocytes were isolated from either wild type or ERα knockout (ERKO) animals. Mice were housed in microisolator-type cages at the vivaria of University of Southern California (USC) or University of California Los Angeles (UCLA). The respective Institutional Animal Care and Use Committees approved all experimental procedures with animals.
2.2 - Reagents
Doxycycline (dox) was purchased from Calbiochem (La Jolla, CA) and used at a final concentration of 0.5 μg/ml. Estradiol (E2) and 1α,25-dihydroxy-vitamin D3 [1,25(OH)2D3], both from Sigma-Aldrich (St Louis, MO), were used at a final concentration of 10 nM. Tamoxifen (Tam) and raloxifene (Ral) were purchased from R&D Systems (Minneapolis, MN) and used at a final concentration of 100 nM. Collagenase P (1 mg/ml) and protease inhibitor cocktail tablets were purchased from Roche Diagnostics (Indianapolis, IN) and dissolved in phosphate-buffered saline (PBS). M-70 anti-RUNX2 antibody, FL-317 anti-RANKL antibody, I-19-R anti-ACTIN antibody, and goat anti-rabbit IgG-HRP secondary antibody were purchased from Santa Cruz Biotechnology (Dallas, Texas). The goat anti-rabbit IgG-DyLight 488 secondary antibody was purchased from Jackson ImmunoResearch (West Grove, Pennsylvania). Tissue culture media, penicillin/streptomycin (1% final concentration) and trypsin were purchased from Gibco (Carlsbad, CA). Both Fetal bovine serum (FBS) and Charcoal-Stripped FBS (CSS) were purchased from Gemini Bioproducts (West Sacramento, CA).
2.3 - Cell culture
Newborn Mouse Calvarial Osteoblasts (NeMCO) were extracted from 1- to 2-day old newborn wild-type mice by digestion of parietal bones, free of sutures, as previously described [22]. Cells were maintained in alpha minimal essential medium (αMEM) supplemented with 20% FBS. For treatment with estrogens, cells were cultured in phenol red-free αMEM containing 10% CSS. Primary splenocytes were prepared from 4 to 6-week old mice by digestion with 1 mM Tris-HCl lysis buffer containing 0.74% NH4Cl as previously described [23].
For conditional expression of RUNX2, NeMCO were transduced with lentiviruses encoding doxycycline (dox)-inducible FLAG-RUNX2, which were produced as previously described [24] at the Vector Core of the UCLA Geffen School of Medicine. The GFP-RANKL plasmid [25], a gift from Dr. Masashi Honma and Dr. Hiroshi Suzuki, University of Tokyo, was introduced into the so-called NeMCO/Rx2dox cells using the Lipofectamine LTX with PLUS reagent and buffer (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. For functional analysis of osteoblast-driven activation of NFκB in osteoclasts, a RAW264.7/NFκB-Luc reporter cell line was constructed essentially as previously described [26]. Briefly, RAW 264.7 cells were stably transfected with an NFκB-luciferase plasmid, a gift from Dr. Ebrahim Zandi (USC) using 10 μg/mL puromycin for selection and the RAW264.7/NFκB-Luc reporter cells were added (30,000 cells per well in 24-well plates) to NeMCO cultures and subjected to luciferase assay after 24 hours. Long-term osteoblast/osteoclast co-cultures were prepared and analyzed using standard protocols [27]. Briefly, NeMCO were seeded in 96-well plates (5,000 cells/well) for at least 3 hours before splenocytes were added (150,000 cells/well). On Day 1, medium was supplemented with 1,25(OH)2D3 along with estrogens and/or dox as indicated, and the cell culture medium was replaced every 3 days. At the end of the culture period, osteoblasts were removed by 0.1% collagenase P digestion (1 mg/mL in PBS) for 15–20 min and osteoclasts were enumerated based on the activity of tartrate-resistant acid phosphatase (TRAP; detected with the TRAP assay kit from Sigma-Aldrich) and the presence of at least three nuclei.
2.4 - RNA Extraction and Analysis
Total RNA was extracted using Aurum total RNA mini kit (Bio-Rad Laboratories, Hercules, CA) according to the manufacturer’s protocol and 1 μg RNA was reverse-transcribed using iScript cDNA synthesis kit (Bio-Rad). The cDNA was subjected to quantitative PCR (qPCR) analysis using the CFX96 real time PCR system (Bio-Rad) and the iQTM SYBR Green Supermix (Bio-Rad) according to the manufacturer’s protocol. The primers used for qPCR are listed in Table 1. Data was normalized for the 18S rRNA levels, which themselves were not significantly affected by treatment.
Table 1.
Primers used for RT-qPCR
| Primer | Sequences (5′ → 3′) | |
|---|---|---|
| Runx2 | F | TCT TCC CAA AGC CAG AGT GG |
| R | ATC AGT TCC ATA GGT TGG ATT C | |
| Rankl | F | GGG GGC CGT GCA GAA GGA AC |
| R | CTC AGG CTT GCC TCG CTG GG | |
| Osteocalcin | F | ACA AGT CCC ACA CAG CAG CTT |
| R | GCC GGA GTC TGT TCA CTA CCT | |
| Osterix | F | GTACGGCAAGGCTTCGCATCTG |
| R | CTGA TGTTTGCTCAAGTGGTCGC | |
| Fasl | F | CTGGGTTGTACTTCGTGTATTCC |
| R | TGTCCAGTAGTGCAGTAGTTCAA | |
| TFF1 | F | TTGTGGTTTTCCTGGTGTCA |
| R | CCGAGCTCTGGGACTAATCA | |
| 18S | F | GTA ACC CGT TGA ACC CCA TT |
| R | CCA TCC AAT CGG TAG TAG CG | |
| Vwa7 | F | GCTGGTCTGGTGACTCTTCC |
| R | AGGACCTATGCCCTCCTCTG | |
| Nppb | F | CTGAAGGTGCTGTCCCAGATG |
| R | GACGGATCCGATCCGGTC | |
| Stx2 | F | CGG GGC AAG CTG |
| R | ACG TCC ACA AAG | |
| Prdm4 | F | AAAGCCAGGAACCGTGAA |
| R | ATGACCCATAAAGTGAACGTG | |
| Pstpip2 | F | ACTTCTGGAGCACGGACATT |
| R | AGGTTCAGCAGGTCTTTGCC | |
2.5 – Microarray Analysis
NeMCO/Rx2dox cells were treated in triplicates with dox and/or with E2. After 24 hours of treatment, RNA was extracted and submitted to the Southern California Genotyping Consortium (SCGC) for microarray analysis using MouseRef-8 v2.0 Expression BeadChips (Illumina, Inc). Raw data processing was performed by GenomeStudio (Illumina Inc) with background subtraction and quantile normalization. Differential expression analysis was performed by one-way ANOVA using Partek Genomics Suite™ (Partek). The microarray data has been deposited to the GEO database (accession code pending).
2.6 - RANKL Fluorescence Microscopy
Cells were fixed with formaldehyde, incubated with the FL-317 primary antibody (1:50) followed by the DyLight 488 secondary antibody (1:200) and mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA) containing DAPI, and images were captured using a ZEISS LSM 510 confocal system. A GFP-RANKL fusion protein was transiently expressed and visualized using a Nikon Eclipse Ti microscope. The proportion of the cell perimeter with RANKL or GFP-RANKL was quantified double-blindedly using the NIS-Elements AR 3.2 software.
2.7 - Western and ELISA
For Western blot analysis, cells were washed 3 times with PBS and lysed in a 50 mM Tris-HCl buffer (pH=7.0) containing 150 mM NaCl, 1 mM EDTA, 1% Triton X-100 and a protease inhibitor cocktail. Cell lysates were subjected to SDS-PAGE and proteins were transferred to Amersham Hybond-P PVDF membranes (Piscataway, NJ). After blocking with 5% milk, RUNX2 or RANKL were detected with the M-70 antibody (1:500 dilution) or the FL-317 antibody (1:200), respectively, and visualized using the Thermo Scientific ECL detection system (Waltham, MA). ACTIN was detected as a loading control using the I-19-R antibody (1:200). For RANKL ELISA, NeMCO/Rx2dox were cultured in 10 cm plates (500,000 cells/plate), initially in 10 ml of 10% CSS for 48 hours and then in 5 ml of 1% CSS for 12 hours. ELISA was performed using a mouse RANKL single plex Milliplex kit (MBN-41K-1RANKL; Millipore; Billerica, MA, USA).
3.0 - RESULTS
3.1 - Dox-inducible Runx2 expression in Newborn Mouse Calvarial Osteoblasts (NeMCO)
RUNX2 promotes not only osteoblast differentiation and bone formation, but also osteoblast-driven osteoclastogenesis [6–10, 28]. Because estrogens inhibit RUNX2 activity [21], we asked whether they would inhibit osteoclast differentiation driven by expression of RUNX2 in co-cultured osteoblasts. First, we transduced Newborn Mouse Calvarial Osteoblasts (NeMCO) with lentiviruses encoding doxycycline (dox)-inducible FLAG-RUNX2 [24] and treated the so-called NeMCO/Rx2dox cells in isolation with dox and/or estradiol (E2) for 48 hours. As demonstrated by Western blot analysis, dox induced RUNX2 expression in day-2 NeMCO/Rx2dox cultures from a hardly detectable level to a level several fold higher than that of endogenous RUNX2 on day 6 (Figure 1A). As expected, the induction of RUNX2 was accompanied with stimulation of the osteoblast marker genes Osteocalcin (Oc; Figure 1C) and Osterix (Osx; Figure 1D). RUNX2-mediated stimulation of these target genes was significantly attenuated by E2 (Figure 1C, D), consistent with previous reports on inhibition of RUNX2 activity and impediment of osteoblast differentiation [21, 29]. The inhibition by E2 occurred without any significant change to RUNX2 mRNA or protein levels (Figure 1B). Thus, we established a system for conditional, robust RUNX2 induction in primary osteoblast cultures, and documented antagonism of RUNX2-mediated stimulation of its target genes by E2.
Figure 1.

Newborn Mouse Calvarial Osteoblasts (NeMCO) were transduced with lentiviruses encoding dox-inducible RUNX2. NeMCO/Rx2dox cultures were treated for 48 hours with 0.5 μg/mL dox, 10 nM E2, and/or vehicle as indicated. (A) Western blot analyses with anti-RUNX2 antibody (top), demonstrating FLAG-RUNX2 (arrow) and endogenous RUNX2 (arrowhead). ACTIN was used as a loading control (bottom). (B–D) The mRNA levels of Runx2 (B), Osteocalcin (C) and Osterix (D) were determined by RT-qPCR (Mean±SD; n=3; *p < 0.05). Inset in B is Western blot performed with anti-RUNX2 antibodies as in A with Actin used as loading control (bottom panel). Abbreviations: C, Control; D, Dox; E, Estradiol; DE, Dox plus Estradiol.
3.2 - Estradiol Antagonizes RUNX2-Mediated Osteoblast-Driven Osteoclastogenesis
Inhibition of bone resorption by E2 in vivo could be mediated in part by antagonism of RUNX2-mediated osteoblast-driven osteoclastogenesis. We addressed this notion using the NeMCO/Rx2dox system by asking whether E2 would antagonize dox-driven osteoclastogenesis from co-cultured splenocytes. Consistent with previous observations [6, 7, 9], induction of RUNX2 in the pre-osteoblasts resulted in an increase in the number of differentiated osteoclasts, defined as TRAP-positive cells with ≥3 nuclei (Figure 2A). Remarkably, the RUNX2-mediated osteoblast-driven osteoclastogenesis was not observed in the presence of E2 (Figure 2A–B). Specifically, the number of osteoclasts that differentiated from their splenocytic precursors was increased by 2.7-fold in response to dox, and this stimulation was completely abolished in the presence of E2 (Figure 2C).
Figure 2.

NeMCO/Rx2dox were co-cultured with splenocytes from WT (A–C) or ERKO (D) mice in the presence of 10 nM 1,25 vitamin D3 and 0.5 μg/mL dox and/or 10 nM E2 as indicated. On day 19, osteoblasts were removed by 0.1% collagenase P digestion, and osteoclasts were identified by TRAP staining. TRAP-stained cultures are shown with a X-fold magnification in A (scale bar = 100 μm) and without magnification in B. Values in C and D (Osteoclast numbers per well) are from one of 3 experiments with similar results (Mean ± SD; n=3; *p < 0.05). Abbreviations: C, Control; D, Dox; E, Estradiol; DE, Dox plus Estradiol.
Inhibition of osteoclastogenesis by E2 may involve activation of ERα not only in osteoblasts but also in cells of the osteoclast lineage [30]. To test the potential contribution of ERα signaling in pre-osteoclasts, we re-examined the effect of E2 on RUNX2-mediated osteoblast-driven osteoclastogenesis in co-cultures of NeMCO/Rx2dox with splenocytes isolated from ERα knockout mice [31]. As shown in Figure 2D, treatment of these co-cultures with E2 again blocked RUNX2-mediated osteoblast-driven osteoclastogenesis even though ERα was absent in the splenocytes. These results suggest that E2 blocked RUNX2-mediated osteoclastogenesis specifically through crosstalk between osteoblastic RUNX2 and osteoblastic ERα.
3.3 - E2 Counteracts RUNX2-Driven RANKL Membrane Association
RANKL is a quintessential factor for osteoblast-driven osteoclastogenesis. However, RUNX2 did not stimulate Rankl mRNA or protein levels in our (Figure 3A,C) and other osteoblast culture systems [7, 8]. Opg mRNA, encoding the only identified endogenous RANKL antagonist, decreased by ~2-fold in response to RUNX2 in both the presence and absence of E2. However, unlike in other osteoblastic culture systems [32, 33], E2 did not significantly influence Opg expression in NeMCO (Figure 3B). In pursuit of an inclusive model that may explain both RUNX2-mediated osteoblast-driven osteoclastogenesis and its antagonism by E2, we considered the role recently ascribed to RUNX2 in protein trafficking and secretion [8, 34], and also the evidence that E2 inhibits RANKL presentation on human bone marrow stromal cells in vivo [35]. First, we performed ELISA of medium conditioned by cells treated with dox and/or E2. Although RUNX2 did not significantly affect Rankl mRNA or protein expression (Figure 3A, C), the ELISA disclosed a remarkable 8-fold increase in RANKL accumulation in medium conditioned by dox-treated as compared to control osteoblasts (Figure 3D). We then treated NeMCO/Rx2dox with dox and/or E2 for two days and added RAW264.7/NFκB-Luc reporter cells to the culture wells for the last 24 hours prior to harvest and luciferase assay. Dox-treated NeMCO/Rx2dox over-expressing RUNX2 stimulated luciferase activity in the RAW264.7/NFκB-Luc reporter cells to levels 8-fold greater than control NeMCO cultured without dox (Figure 3E). Both the ELISA and the luciferase results suggest that RUNX2 stimulates RANKL mobilization, contributing to the stimulation of osteoblast-mediated osteoclastogenesis (Figure 2). The anti-RUNX2 effect of E2, however, was only minimal in the ELISA (Figure 3D) and absent in the luciferase assay (Figure 3E). Thus, the robust RUNX-mediated osteoblast-driven osteoclastogenesis (Figure 2) is attributable to stimulation of RANKL secretion, but neither the ELISA nor the RAW 264.7/NFkB-Luc reporter assay provided an explanation for the 2.6-fold inhibition of osteoclastogenesis by E2 (Figure 2).
Figure 3.

NeMCO/Rx2dox were treated for 48 hours with 0.5 μg/ml dox and/or 10 nM E2 as indicated. (A,B) Rankl and Opg mRNA levels were measured using RT-qPCR. (C) RANKL expression was assessed by Western blot analysis using ACTIN as loading control. (D) RANKL concentration in conditioned medium was determined by ELISA. (E) RAW264.7/NFκB-Luc reporter cells were added to NeMCO for additional 24 hours and luciferase assay was performed as previously described (21). Abbreviations: C, Control; D, Dox; E, Estradiol; DE, Dox plus Estradiol.
It is believed that osteoblast-borne RANKL promotes osteoclastogenesis primarily through cell-cell interaction [36]. We therefore employed confocal immunofluorescence microscopy to address the possibility that E2 inhibited RUNX2-mediated RANKL association with the osteoblast membrane. As shown in Figure 4A, RUNX2 promoted localization of RANKL at the cell perimeter, with a 6-fold increase in membrane association in response to dox (Figure 4B). Furthermore, the RUNX2-mediated enrichment of the cell membrane for RANKL was diminished by E2 (Figure 4A,B). To confirm that the effects of RUNX2 and E2 on RANKL trafficking were post-translational, and not related to mechanisms of alternative RANKL transcription or mRNA splicing, we assessed by fluorescence microscopy the effects of RUNX2 and E2 on a transiently expressed GFP-RANKL fusion protein. Similar to the effects on endogenous RANKL, RUNX2 stimulated GFP-RANKL membrane association by 4-fold and again this was diminished by E2 (Figure 4C,D). Taken together, our results suggest that expression of RUNX2 in osteoblasts promotes osteoclastogenesis by increasing membrane association and/or secretion of RANKL, and that E2 antagonizes RUNX2-mediated osteoblast-driven osteoclastogenesis primarily by attenuating the presentation of RANKL on the osteoblast membrane.
Figure 4.
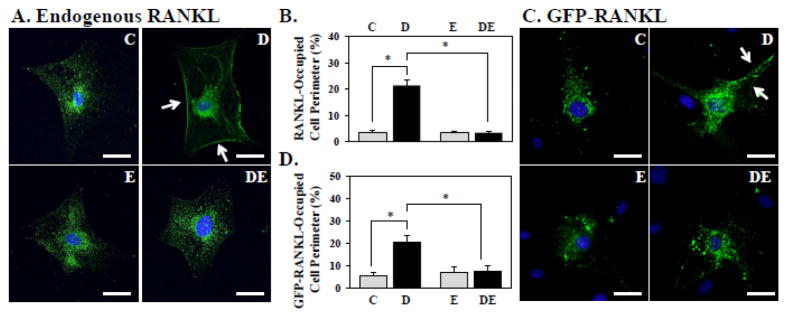
NeMCO/Rx2dox were treated for 48 hours with 0.5 μg/ml dox and/or 10 nM E2 as indicated, and endogenous RANKL (A,B) or transiently expressed GFP-RANKL (C,D) were imaged by indirect or direct immunofluorescence, respectively. (A,C) Representative microcgraphs (Scale bar = 50 μm), with arrows marking membrane-associated RANKL. (B,D) Percentage of cell perimeter containing RANKL or GFP-RANKL was determined in a double-blinded fashion for ≥10 randomly selected cells per condition. Results are from one of 3 experiments with similar results (Mean±SD; *p < 0.05). Abbreviations: C, Control; D, Dox; E, Estradiol; DE, Dox plus Estradiol.
3.4 - E2 blocks RUNX2-mediated stimulation of Pstpip2
To identify potential genes that mediate RUNX2-driven RANKL membrane trafficking and its antagonism by estrogens, we profiled mRNA expression in NeMCO cultures treated by dox (to induce RUNX2) and/or E2. Consistent with previous reports [21, 29], E2 globally attenuated the response to RUNX2 (Figure 5, scatter plot). We selected candidate genes that could potentially explain the reciprocal effects of RUNX2 and E2 on RANKL trafficking by first ranking all genes based on the sum absolute fold-stimulation by dox (RUNX2; Supplemental Table 1, column B) and fold-repression by E2 in the presence of dox (Supplemental Table 1; column D). We next validated the microarray results by RT-qPCR analysis of the five genes with the highest sum absolute response and with at least 1.3-fold response to RUNX2 alone (Supplemental Table 1, column F). The RT-qPCR results, obtained in an independent experiment (Figure 5) generally recapitulated the microarray results (Supplemental Table 1). Among the five genes with a >1.2 response to RUNX2 and the highest sum absolute response to RUNX2 and E2 was Proline-Serine-Threonine Phosphatase Interacting Protein 2 (Pstpip2) (Figure 5), whose DNA sequence is 48% identical to Fission Yeast Imp2 and is therefore predicted to play a critical role in membrane trafficking [37].
Figure 5.

Triplicate NeMCO/Rx2dox cultures were treated with dox and/or E2 for 24 hours and global mRNA expression was profiled using Beadchip arrays (Ilumina). Scatter plot shows that the response to dox plus E2 is generally weaker than the response to dox alone. RT-qPCR analysis of the indicated genes was performed to confirm the results for the 5 genes with the highest sum absolute response to dox and E2 (Supplemental Table 1). *p < 0.05 Abbreviations: C, Control; D, Dox; E, Estradiol; DE, Dox plus Estradiol.
3.5 - Selective Estrogen Receptor Modulators (SERMs) Mimic Antagonistic Action of E2 on RUNX2-Induced Osteoclastogenesis
The efficacy of SERMs such as tamoxifen (Tam) and raloxifene (Ral) for the treatment of breast cancer is predicated on their ability to antagonize estrogen signaling in mammary epithelial cells [20]. SERMs also have the unique feature of acting in osteoblasts as partial ER agonists, accounting for their bone-sparing properties [38]. We therefore set out to test the effects of SERMs on RUNX2-mediated osteoblast-driven osteoclastogenesis. First, we confirmed the transcriptional regulatory properties of SERMs in NeMCO and in MCF7 breast cancer cells. Tam and Ral antagonized E2-mediated stimulation of the classical ER target gene TFF1 (pS2) in MCF7 cells without significantly regulating gene expression on their own (Figure 6A, B). In contrast, Tam and Ral did not have a lasting anti-estrogenic effect on the ERα target gene Fasl in NeMCO cultures (Figure 6D). In fact, they mimicked E2 in NeMCO (Figure 6C), similar to observations previously made in U2OS-ERα cells [39]. Next, to test whether the established bone-sparing properties of SERMs could be mediated in part by mimicking E2 in attenuating RUNX2-mediated osteoblast-driven osteoclastogenesis, we treated co-cultures of splenocytes and NeMCO/Rx2dox cells with dox and/or SERMs. Similar to E2, both Tam and Ral abolished osteoclast differentiation when driven by RUNX2 expression in the co-cultured NeMCO (Figure 6E), and this was not associated with alterations to expression of Runx2 itself (Figure 6F). These results suggest that the bone sparing properties of SERMs are attributable in part to antagonism of RUNX2-mediated osteoblast-driven osteoclastogenesis.
Figure 6.

(A–D) MCF7 (A,B) and NeMCO/Rx2dox cultures (C,D) were treated for 6, 14, 24 and 48 hours with E2, Raloxifene, or Tamoxifen, alone (A, C) or with the indicated combinations (B, D), and expression of the indicated ER-target genes was measured by RT-qPCR (Mean ± SD; n=3). (E) Co-cultures of NeMCO/Rx2dox with splenocytes were treated as indicated, and differentiated osteoclasts were enumerated on day 14. Results are from one of 3 experiments with similar results (Mean ± SD; n=3). (F) NeMCO/Rx2dox were treated as indicated and Runx2 mRNA levels were measured by RT-qPCR (Mean ± SD, n=3; *p < 0.05). Abbreviations: C, Control; D, Dox; E, Estradiol; R, Raloxifene; T, Tamoxifen.
4.0 - DISCUSSION
It is well established that accelerated bone turnover increases fracture risk, with postmenopausal osteoporosis serving a prime example. Bone loss that occurs at physiological turnover rates is slow and usually transpires without pathological consequences because coupling mechanisms secure the replacement of most of the resorbed bone with newly deposited material. Adding to classical coupling mechanisms of signaling from osteoblasts to osteoclasts and back, the regulation of both osteoblast differentiation and osteoblast-driven osteoclastogenesis by the same transcription factor, RUNX2, likely contributes to balanced bone remodeling. Our results suggest that estrogens may regulate bone turnover rate by antagonizing RUNX2 in osteoblasts. If this is correct, then attenuation of bone turnover and bone loss in postmenopausal women may be achieved through novel therapeutic approaches that restore the anti-RUNX2 function of estrogens.
RUNX2-mediated osteoblast-driven osteoclastogenesis has been well documented [6–10, 28] but does not necessarily involve regulation of Rankl gene expression in osteoblasts [7, 17]. Similarly in the present study, induction of RUNX2 in primary pre-osteoblasts strongly stimulated differentiation of co-cultured splenocytes into mature osteoclasts (Figure 2) without significantly increasing Rankl mRNA (Figure 3A) or protein levels (Figure 3C). We demonstrate, however, a marked increase in RANKL secretion (Figure 3D) and membrane association in response to RUNX2 (Figure 4), reminiscent of the recently suggested role of RUNX2 in regulating membrane trafficking [8, 34]. Given that RANKL trafficking is regulated by OPG [36], the RUNX2-mediated increase in RANKL membrane association could be mediated by the demonstrated inhibition of Opg expression (Figure 3B). We cannot rule out additional RUNX2-driven osteoclastogenic mechanisms such as stimulation of Sema7a and Ltc4s expression [8]. It will also be interesting to investigate whether RUNX2 controls RANKL trafficking in non-osteoblasts, such as breast cancer and vascular smooth muscle cells, in which ectopic expression of these two regulators has been linked to human disease [15, 40, 41].
RUNX2-mediated osteoblast-driven osteoclastogenesis may have important implications for postmenopausal osteoporosis. Indeed, E2 diminished the RUNX2-mediated differentiation of co-cultured splenocytes into mature osteoclasts (Figure 2). This anti-osteoclastogenic effect of E2 must now be further investigated, as it may represent a fundamental mechanism underlying the bone-sparing property of estrogens in vivo. In the present study, E2 antagonized RUNX2-mediated membrane association of both endogenous RANKL and transiently expressed GFP-RANKL (Figure 4), but it did not antagonize RUNX2-mediated inhibition of Opg expression (Figure 3B). Thus, whereas RUNX2-mediated RANKL membrane localization and secretion may be mediated by inhibition of Opg expression, other mechanisms remain to be delineated that explain the anti-RUNX2 effect of E2 with respect to RANKL membrane association. That E2 decreased RANKL membrane association (Figure 4) and osteoclastogenesis (Figure 2) without significantly decreasing RANKL measured in conditioned media (Figure 3D) is consistent with the idea that RANKL is most effective in promoting osteoclastogenesis when anchored in the membrane of presenting cells [42]. Indeed, RANKL presentation was greater on the surface of pre-osteoblasts isolated from the bone marrow of hypogonadal postmenopausal women as compared to either age-matched eugonadal pre-menopausal or hormone-repleted postmenopausal controls [35].
The bone sparing property of E2 in vivo is mediated by activation of ERα in cells of both the osteoblast and the monocyte/osteoclast linages. The former is responsible for protection of cortical bone in females and cortical and trabecular bone in males, whereas the latter is responsible for protection of female trabecular bone [19, 43, 44]. In our in vitro co-culture assay, E2 antagonized RUNX2-mediated osteoblast-driven osteoclastogenesis by activating ERα in osteoblasts, not osteoclasts, because the antagonism was fully recapitulated with ERα-deficient osteoclast precursors (Figure 2C). Given that RUNX2-mediated osteoclastogenesis in vivo primarily occurred at the endosteal aspect of cortical bone [6, 9], the anti-RUNX2 activity of estrogens with respect to regulating RANKL membrane association and osteoclastogenesis is likely most relevant to protection of cortical bone in females and both bone compartments in males.
Although RANKL expression has been reported in cells of different stages of the osteoblast lineage, from early bone marrow mesenchymal progenitors to matrix-embedded osteocytes [45–47], it is predominant in stages that precede the onset of DMP-1 expression [48]. Accordingly, early ablation of RANKL in vivo (with Prx1-Cre, Osx1-Cre and Osteocalcin-Cre) completely abrogated osteoclastogenesis, whereas DMP-1-Cre-driven ablation of RANKL from mature osteoblasts and osteocytes [49] resulted in only partial loss of osteoclastogenesis [48]. Like RANKL, RUNX2 is also predominantly expressed in cells early in the osteoblast lineage [50–52, 54, 55], and although it does not necessarily control RANKL gene expression [8, 16, 17], the present study suggests that it likely controls RANKL trafficking in these cells. As a result, RUNX2-driven RANKL trafficking to the surface of bone marrow pre-osteoblasts [35] could engage its receptor, RANK, on neighboring monocytic cells to promote osteoclast differentiation. RUNX2 is also expressed in matrix-embedded osteocytes and hypertrophic chondrocytes, where it may drive RANKL trafficking to cellular projection to attract and activate osteoclasts on the mineralized surfaces [45, 47–51, 53]. [50–52] [53, 54] [55]
Mechanisms by which estrogens antagonize RUNX2-driven RANKL membrane trafficking remain to be investigated. Plausibly, they involve the physical interaction between ERα and RUNX2 [21], which typically results in the inhibition of RUNX2 target genes. Some of these genes likely promote osteoblast differentiation and bone formation, and indeed treatment of isolated osteoblasts with estrogens has been shown to inhibit the osteoblast phenotype [29]. For example, similar to Osteoclacin [21], we report here that E2 antagonizes RUNX2-mediated stimulation of, Prdm4 and Nppb (Figure 5), which may play a role in RUNX2-driven osteoblastogenesis [56–58] and possibly in postmenopausal osteoporosis [59]. We also report here that E2 ablates RUNX2-mediated stimulation of Pstpip2 (Figure 5), which is highly homologous to Imp2, the product of which plays a critical role in Fission Yeast membrane trafficking [60]. The role of PSTPIP2 in RUNX2-driven and E2-mediated antagonism of RANKL trafficking remains to be investigated. Interestingly, PSTPIP1 interacts with calcineurin, the target of FK506 that causes high bone resorption in vivo [61]. Possibly, then, FK506 and RUNX2 (via PSTPIP2) may share a common target, calcineurin, leading to bone resorption. Additionally, given the bone phenotype of the mouse model for Platelet-type von Willebrand Disease [57], Vwa7 may also be involved in the reciprocal regulation of bone resorption by RUNX2 and estrogens (Figure 5). Future studies are warranted to directly test the role of RUNX2 and its target genes (e.g., Pstpip2, Vwa7) as mediators of the anti-osteoclastogenic property of estrogens, and weigh them against alternative anti-resorptive mechanisms of action of estrogens, such as the stimulation of Fasl and Mmp3 [39, 56]. [56] [57] [58] [59] [60] [61]
The present and previous studies demonstrating RUNX2-mediated osteoblast-driven osteoclastogenesis [6–9, 28] advocate the development of therapeutic approaches for the treatment of high turnover bone disease by targeting either RUNX2 or the downstream mechanisms by which it regulates bone resorption. Although such anti-RUNX2 agents might be deleterious at high doses and during accelerated bone formation in young individuals, their pursuit for the treatment of high-turnover osteoporosis is justifiable by the low and high bone mass phenotypes observed in mice where RUNX2 activity is manipulated upwards and downwards, respectively [4, 6, 9, 62]. In fact, our work suggests that the bone-sparing effect of E2 is attributable in part to antagonism of RUNX2-mediated osteoblast-driven osteoclastogenesis. Future targeting of RUNX2, or the RUNX2-regulated mechanisms responsible for RANKL membrane association, may therefore provide tissue-specific solutions to functionally restore at least part of the protective role of E2 in the skeleton. In fact, we show herein that SERMs inhibit both RUNX2-driven transcription [21] and RUNX2-mediated osteoblast-driven osteoclastogenesis (Figure 6), suggesting that their anti-RUNX2 activity may facilitate their bone sparing properties.
Supplementary Material
HIGHLIGHTS.
Estradiol Antagonizes RUNX2-Mediated Osteoblast-Driven Osteoclastogenesis
RUNX2 Regulates RANKL Membrane Association
E2 Counteracts RUNX2-Driven RANKL Membrane Association
SERMs Mimic Antagonistic Action of E2 on RUNX2-Induced Osteoclastogenesis
Acknowledgments
This work was supported by grants RO1 DK071122 and RO1 DK071122S1 from NIDDK to BF, holder of the J. Harold and Edna L. LaBriola Chair in Genetic Orthopaedic Research. AM was supported by training grant T32 DE021982 from NIDCR, and YG was supported by a Meyer Young Investigator Fellowship from The Arthritis Foundation. We thank Drs. Ebrahim Zandi (USC), Dr. Masashi Honma and Dr. Hiroshi Suzuki (University of Tokyo) for reagents, the UCLA Vector Core (supported by CURE/P30DK041301) for lentiviral preparation, and the Cell and Tissue Imaging Core of the USC Research Center for Liver Diseases (supported by NIH grants P30 DK048522 and S10 RR022508) for help with confocal microscopy. AM thanks USC’s Michael Paine, Gillian Little and Sanjeev Baniwal for mentorship and helpful discussions.
ABBREVIATIONS
- SERMs
selective estrogen receptor modulators
- ERα
estrogen receptor alpha
- NeMCO
newborn mouse calvarial osteoblasts
- Dox
Doxycycline
- E2
estradiol
- Tam
tamoxifen
- Ral
raloxifene
- DHT
dihydrotestosterone
Footnotes
CONFLICT OF INTEREST:
PK and CH are employed by and have stock in Amgen. The other authors have nothing to disclose
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Anthony Martin, Email: martinan@usc.edu.
Jian Xiong, Email: silviaxiong0923@gmail.com.
Theodora Koromila, Email: tkoromila@yahoo.gr.
Jie S. Ji, Email: jieji@usc.edu.
Stephanie Chang, Email: schan061@ucr.edu.
Yae S. Song, Email: yaesamso@usc.edu.
Jonathan L. Miller, Email: mill252@usc.edu.
Chun-Ya Han, Email: ehan@amgen.com.
Paul Kostenuik, Email: paulk@amgen.com.
Susan A. Krum, Email: skrum@mednet.ucla.edu.
Nyam-Osor Chimge, Email: chimgee@usc.edu.
Yankel Gabet, Email: yagabet@gmail.com.
Baruch Frenkel, Email: frenkel@usc.edu.
6.0 - REFERENCES
- 1.Ducy P, Karsenty G. Two distinct osteoblast-specific cis-acting elements control expression of a mouse osteocalcin gene. Mol Cell Biol. 1995;15:1858–69. doi: 10.1128/mcb.15.4.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–54. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 3.Banerjee C, McCabe LR, Choi JY, Hiebert SW, Stein JL, Stein GS, Lian JB. Runt homology domain proteins in osteoblast differentiation: AML3/CBFA1 is a major component of a bone-specific complex. J Cell Biochem. 1997;66:1–8. doi: 10.1002/(sici)1097-4644(19970701)66:1<1::aid-jcb1>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 4.Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, Kishimoto T. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–64. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- 5.Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Stamp GW, Beddington RS, Mundlos S, Olsen BR, Selby PB, Owen MJ. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–71. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 6.Geoffroy V, Kneissel M, Fournier B, Boyde A, Matthias P. High bone resorption in adult aging transgenic mice overexpressing cbfa1/runx2 in cells of the osteoblastic lineage. Mol Cell Biol. 2002;22:6222–33. doi: 10.1128/MCB.22.17.6222-6233.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maruyama Z, Yoshida CA, Furuichi T, Amizuka N, Ito M, Fukuyama R, Miyazaki T, Kitaura H, Nakamura K, Fujita T, Kanatani N, Moriishi T, Yamana K, Liu W, Kawaguchi H, Komori T. Runx2 determines bone maturity and turnover rate in postnatal bone development and is involved in bone loss in estrogen deficiency. Dev Dyn. 2007;236:1876–90. doi: 10.1002/dvdy.21187. [DOI] [PubMed] [Google Scholar]
- 8.Baniwal SK, Shah PK, Shi Y, Haduong JH, Declerck YA, Gabet Y, Frenkel B. Runx2 promotes both osteoblastogenesis and novel osteoclastogenic signals in ST2 mesenchymal progenitor cells. Osteoporos Int. 23:1399–413. doi: 10.1007/s00198-011-1728-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu W, Toyosawa S, Furuichi T, Kanatani N, Yoshida C, Liu Y, Himeno M, Narai S, Yamaguchi A, Komori T. Overexpression of Cbfa1 in osteoblasts inhibits osteoblast maturation and causes osteopenia with multiple fractures. J Cell Biol. 2001;155:157–66. doi: 10.1083/jcb.200105052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adhami MD, Rashid H, Chen H, Clarke JC, Yang Y, Javed A. Loss of Runx2 in Committed Osteoblasts Impairs Postnatal Skeletogenesis. J Bone Miner Res. 2014 doi: 10.1002/jbmr.2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suda T, Takahashi N, Martin TJ. Modulation of osteoclast differentiation. Endocr Rev. 1992;13:66–80. doi: 10.1210/edrv-13-1-66. [DOI] [PubMed] [Google Scholar]
- 12.Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G, Itie A, Khoo W, Wakeham A, Dunstan CR, Lacey DL, Mak TW, Boyle WJ, Penninger JM. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–23. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- 13.Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, Hsu H, Sullivan J, Hawkins N, Davy E, Capparelli C, Eli A, Qian YX, Kaufman S, Sarosi I, Shalhoub V, Senaldi G, Guo J, Delaney J, Boyle WJ. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–76. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- 14.Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A, Tsuda E, Morinaga T, Higashio K, Udagawa N, Takahashi N, Suda T. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A. 1998;95:3597–602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Byon CH, Sun Y, Chen J, Yuan K, Mao X, Heath JM, Anderson PG, Tintut Y, Demer LL, Wang D, Chen Y. Runx2-upregulated receptor activator of nuclear factor kappaB ligand in calcifying smooth muscle cells promotes migration and osteoclastic differentiation of macrophages. Arterioscler Thromb Vasc Biol. 31:1387–96. doi: 10.1161/ATVBAHA.110.222547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Brien CA, Kern B, Gubrij I, Karsenty G, Manolagas SC. Cbfa1 does not regulate RANKL gene activity in stromal/osteoblastic cells. Bone. 2002;30:453–62. doi: 10.1016/s8756-3282(01)00692-5. [DOI] [PubMed] [Google Scholar]
- 17.O’Brien CA. Control of RANKL gene expression. Bone. 46:911–9. doi: 10.1016/j.bone.2009.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Services USDoHaH. Bone Health and Osteoporosis: A Report Of The Surgeon General. Rockville, MD: U.S. Department of Health and Human Services, Office of the Surgeon General; 2004. 2010/10/15 ed. [PubMed] [Google Scholar]
- 19.Manolagas SC, O’Brien CA, Almeida M. The role of estrogen and androgen receptors in bone health and disease. Nat Rev Endocrinol. 9:699–712. doi: 10.1038/nrendo.2013.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khosla S. Update on estrogens and the skeleton. J Clin Endocrinol Metab. 95:3569–77. doi: 10.1210/jc.2010-0856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khalid O, Baniwal SK, Purcell DJ, Leclerc N, Gabet Y, Stallcup MR, Coetzee GA, Frenkel B. Modulation of Runx2 activity by estrogen receptor-alpha: implications for osteoporosis and breast cancer. Endocrinology. 2008;149:5984–95. doi: 10.1210/en.2008-0680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gabet Y, Noh T, Lee C, Frenkel B. Developmentally regulated inhibition of cell cycle progression by glucocorticoids through repression of cyclin A transcription in primary osteoblast cultures. J Cell Physiol. 226:991–8. doi: 10.1002/jcp.22412. [DOI] [PubMed] [Google Scholar]
- 23.Udagawa N, Takahashi N, Akatsu T, Tanaka H, Sasaki T, Nishihara T, Koga T, Martin TJ, Suda T. Origin of osteoclasts: mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells. Proc Natl Acad Sci U S A. 1990;87:7260–4. doi: 10.1073/pnas.87.18.7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baniwal SK, Khalid O, Gabet Y, Shah RR, Purcell DJ, Mav D, Kohn-Gabet AE, Shi Y, Coetzee GA, Frenkel B. Runx2 transcriptome of prostate cancer cells: insights into invasiveness and bone metastasis. Mol Cancer. 9:258. doi: 10.1186/1476-4598-9-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kariya Y, Honma M, Aoki S, Chiba A, Suzuki H. Vps33a mediates RANKL storage in secretory lysosomes in osteoblastic cells. J Bone Miner Res. 2009;24:1741–52. doi: 10.1359/jbmr.090409. [DOI] [PubMed] [Google Scholar]
- 26.Singh PP, van der Kraan AG, Xu J, Gillespie MT, Quinn JM. Membrane-bound receptor activator of NFkappaB ligand (RANKL) activity displayed by osteoblasts is differentially regulated by osteolytic factors. Biochem Biophys Res Commun. 2012;422:48–53. doi: 10.1016/j.bbrc.2012.04.103. [DOI] [PubMed] [Google Scholar]
- 27.Cope AP. Arthritis research: methods and protocols. Springer Science & Business Media; 2007. [Google Scholar]
- 28.Enomoto H, Shiojiri S, Hoshi K, Furuichi T, Fukuyama R, Yoshida CA, Kanatani N, Nakamura R, Mizuno A, Zanma A, Yano K, Yasuda H, Higashio K, Takada K, Komori T. Induction of osteoclast differentiation by Runx2 through receptor activator of nuclear factor-kappa B ligand (RANKL) and osteoprotegerin regulation and partial rescue of osteoclastogenesis in Runx2−/− mice by RANKL transgene. J Biol Chem. 2003;278:23971–7. doi: 10.1074/jbc.M302457200. [DOI] [PubMed] [Google Scholar]
- 29.Almeida M, Martin-Millan M, Ambrogini E, Bradsher R, 3rd, Han L, Chen XD, Roberson PK, Weinstein RS, O’Brien CA, Jilka RL, Manolagas SC. Estrogens attenuate oxidative stress and the differentiation and apoptosis of osteoblasts by DNA-binding-independent actions of the ERalpha. J Bone Miner Res. 2010;25:769–81. doi: 10.1359/jbmr.091017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakamura T, Imai Y, Matsumoto T, Sato S, Takeuchi K, Igarashi K, Harada Y, Azuma Y, Krust A, Yamamoto Y, Nishina H, Takeda S, Takayanagi H, Metzger D, Kanno J, Takaoka K, Martin TJ, Chambon P, Kato S. Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts. Cell. 2007;130:811–23. doi: 10.1016/j.cell.2007.07.025. [DOI] [PubMed] [Google Scholar]
- 31.Dupont S, Krust A, Gansmuller A, Dierich A, Chambon P, Mark M. Effect of single and compound knockouts of estrogen receptors alpha (ERalpha) and beta (ERbeta) on mouse reproductive phenotypes. Development. 2000;127:4277–91. doi: 10.1242/dev.127.19.4277. [DOI] [PubMed] [Google Scholar]
- 32.Hofbauer LC, Khosla S, Dunstan CR, Lacey DL, Spelsberg TC, Riggs BL. Estrogen stimulates gene expression and protein production of osteoprotegerin in human osteoblastic cells. Endocrinology. 1999;140:4367–70. doi: 10.1210/endo.140.9.7131. [DOI] [PubMed] [Google Scholar]
- 33.Saika M, Inoue D, Kido S, Matsumoto T. 17beta-estradiol stimulates expression of osteoprotegerin by a mouse stromal cell line, ST-2, via estrogen receptor-alpha. Endocrinology. 2001;142:2205–12. doi: 10.1210/endo.142.6.8220. [DOI] [PubMed] [Google Scholar]
- 34.Little GH, Noushmehr H, Baniwal SK, Berman BP, Coetzee GA, Frenkel B. Genome-wide Runx2 occupancy in prostate cancer cells suggests a role in regulating secretion. Nucleic Acids Res. 40:3538–47. doi: 10.1093/nar/gkr1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eghbali-Fatourechi G, Khosla S, Sanyal A, Boyle WJ, Lacey DL, Riggs BL. Role of RANK ligand in mediating increased bone resorption in early postmenopausal women. J Clin Invest. 2003;111:1221–30. doi: 10.1172/JCI17215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jimi E, Nakamura I, Amano H, Taguchi Y, Tsurukai T, Tamura M, Takahashi N, Suda T. Osteoclast function is activated by osteoblastic cells through a mechanism involving cell-to-cell contact. Endocrinology. 1996;137:2187–90. doi: 10.1210/endo.137.5.8612568. [DOI] [PubMed] [Google Scholar]
- 37.Kita A, Higa M, Doi A, Satoh R, Sugiura R. Imp2, the PSTPIP homolog in fission yeast, affects sensitivity to the immunosuppressant FK506 and membrane trafficking in fission yeast. Biochem Biophys Res Commun. 2015 doi: 10.1016/j.bbrc.2014.12.100. [DOI] [PubMed] [Google Scholar]
- 38.Das S, Crockett JC. Osteoporosis - a current view of pharmacological prevention and treatment. Drug Des Devel Ther. 7:435–48. doi: 10.2147/DDDT.S31504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krum SA, Miranda-Carboni GA, Hauschka PV, Carroll JS, Lane TF, Freedman LP, Brown M. Estrogen protects bone by inducing Fas ligand in osteoblasts to regulate osteoclast survival. EMBO J. 2008;27:535–45. doi: 10.1038/sj.emboj.7601984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chimge NO, Frenkel B. The RUNX family in breast cancer: relationships with estrogen signaling. Oncogene. 32:2121–30. doi: 10.1038/onc.2012.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schramek D, Leibbrandt A, Sigl V, Kenner L, Pospisilik JA, Lee HJ, Hanada R, Joshi PA, Aliprantis A, Glimcher L, Pasparakis M, Khokha R, Ormandy CJ, Widschwendter M, Schett G, Penninger JM. Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature. 468:98–102. doi: 10.1038/nature09387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakashima T, Kobayashi Y, Yamasaki S, Kawakami A, Eguchi K, Sasaki H, Sakai H. Protein expression and functional difference of membrane-bound and soluble receptor activator of NF-kappaB ligand: modulation of the expression by osteotropic factors and cytokines. Biochem Biophys Res Commun. 2000;275:768–75. doi: 10.1006/bbrc.2000.3379. [DOI] [PubMed] [Google Scholar]
- 43.Manolagas SC, O’Brien CA, Almeida M. The role of estrogen and androgen receptors in bone health and disease. Nat Rev Endocrinol. 2013;9:699–712. doi: 10.1038/nrendo.2013.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Windahl SH, Borjesson AE, Farman HH, Engdahl C, Moverare-Skrtic S, Sjogren K, Lagerquist MK, Kindblom JM, Koskela A, Tuukkanen J, Divieti Pajevic P, Feng JQ, Dahlman-Wright K, Antonson P, Gustafsson JA, Ohlsson C. Estrogen receptor-alpha in osteocytes is important for trabecular bone formation in male mice. Proc Natl Acad Sci U S A. 2013;110:2294–9. doi: 10.1073/pnas.1220811110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kartsogiannis V, Zhou H, Horwood NJ, Thomas RJ, Hards DK, Quinn JM, Niforas P, Ng KW, Martin TJ, Gillespie MT. Localization of RANKL (receptor activator of NF kappa B ligand) mRNA and protein in skeletal and extraskeletal tissues. Bone. 1999;25:525–34. doi: 10.1016/s8756-3282(99)00214-8. [DOI] [PubMed] [Google Scholar]
- 46.Mueller RJ, Richards RG. Immunohistological identification of receptor activator of NF-kappaB ligand (RANKL) in human, ovine and bovine bone tissues. J Mater Sci Mater Med. 2004;15:367–72. doi: 10.1023/b:jmsm.0000021103.39114.cd. [DOI] [PubMed] [Google Scholar]
- 47.Silvestrini G, Ballanti P, Patacchioli F, Leopizzi M, Gualtieri N, Monnazzi P, Tremante E, Sardella D, Bonucci E. Detection of osteoprotegerin (OPG) and its ligand (RANKL) mRNA and protein in femur and tibia of the rat. J Mol Histol. 2005;36:59–67. doi: 10.1007/s10735-004-3839-1. [DOI] [PubMed] [Google Scholar]
- 48.Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med. 2011;17:1235–41. doi: 10.1038/nm.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kalajzic I, Matthews BG, Torreggiani E, Harris MA, Divieti Pajevic P, Harris SE. In vitro and in vivo approaches to study osteocyte biology. Bone. 2013;54:296–306. doi: 10.1016/j.bone.2012.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Inada M, Yasui T, Nomura S, Miyake S, Deguchi K, Himeno M, Sato M, Yamagiwa H, Kimura T, Yasui N, Ochi T, Endo N, Kitamura Y, Kishimoto T, Komori T. Maturational disturbance of chondrocytes in Cbfa1-deficient mice. Dev Dyn. 1999;214:279–90. doi: 10.1002/(SICI)1097-0177(199904)214:4<279::AID-AJA1>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 51.Bronckers AL, Sasaguri K, Engelse MA. Transcription and immunolocalization of Runx2/Cbfa1/Pebp2alphaA in developing rodent and human craniofacial tissues: further evidence suggesting osteoclasts phagocytose osteocytes. Microsc Res Tech. 2003;61:540–8. doi: 10.1002/jemt.10377. [DOI] [PubMed] [Google Scholar]
- 52.Amir LR, Jovanovic A, Perdijk FB, Toyosawa S, Everts V, Bronckers AL. Immunolocalization of sibling and RUNX2 proteins during vertical distraction osteogenesis in the human mandible. J Histochem Cytochem. 2007;55:1095–104. doi: 10.1369/jhc.6A7162.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Honma M, Ikebuchi Y, Kariya Y, Hayashi M, Hayashi N, Aoki S, Suzuki H. RANKL subcellular trafficking and regulatory mechanisms in osteocytes. J Bone Miner Res. 2013;28:1936–49. doi: 10.1002/jbmr.1941. [DOI] [PubMed] [Google Scholar]
- 54.Yu J, Adisetiyo H, Little GH, Vangsness CT, Jr, Jiang J, Sternberg H, West MD, Frenkel B. Initial Characterization of Osteoblast Differentiation and Loss of RUNX2 Stability in the Newly Established SK11 Human Embryonic Stem Cell-Derived Cell Line. J Cell Physiol. 2014 doi: 10.1002/jcp.24773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Komori T. Regulation of osteoblast differentiation by Runx2. Adv Exp Med Biol. 2010;658:43–9. doi: 10.1007/978-1-4419-1050-9_5. [DOI] [PubMed] [Google Scholar]
- 56.Huang S, Shao G, Liu L. The PR domain of the Rb-binding zinc finger protein RIZ1 is a protein binding interface and is related to the SET domain functioning in chromatin-mediated gene expression. J Biol Chem. 1998;273:15933–9. doi: 10.1074/jbc.273.26.15933. [DOI] [PubMed] [Google Scholar]
- 57.Galli GG, Honnens de Lichtenberg K, Carrara M, Hans W, Wuelling M, Mentz B, Multhaupt HA, Fog CK, Jensen KT, Rappsilber J, Vortkamp A, Coulton L, Fuchs H, Gailus-Durner V, Hrabe de Angelis M, Calogero RA, Couchman JR, Lund AH. Prdm5 regulates collagen gene transcription by association with RNA polymerase II in developing bone. PLoS Genet. 2012;8:e1002711. doi: 10.1371/journal.pgen.1002711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Suda M, Ogawa Y, Tanaka K, Tamura N, Yasoda A, Takigawa T, Uehira M, Nishimoto H, Itoh H, Saito Y, Shiota K, Nakao K. Skeletal overgrowth in transgenic mice that overexpress brain natriuretic peptide. Proc Natl Acad Sci U S A. 1998;95:2337–42. doi: 10.1073/pnas.95.5.2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kajita M, Ezura Y, Iwasaki H, Ishida R, Yoshida H, Kodaira M, Suzuki T, Hosoi T, Inoue S, Shiraki M, Orimo H, Emi M. Association of the −381T/C promoter variation of the brain natriuretic peptide gene with low bone-mineral density and rapid postmenopausal bone loss. J Hum Genet. 2003;48:77–81. doi: 10.1007/s100380300010. [DOI] [PubMed] [Google Scholar]
- 60.Mankin HJ, Dorfman H, Lippiello L, Zarins A. Biochemical and metabolic abnormalities in articular cartilage from osteo-arthritic human hips. II. Correlation of morphology with biochemical and metabolic data. J Bone Joint Surg Am. 1971;53:523–37. [PubMed] [Google Scholar]
- 61.Cvetkovic M, Mann GN, Romero DF, Liang XG, Ma Y, Jee WS, Epstein S. The deleterious effects of long-term cyclosporine A, cyclosporine G, and FK506 on bone mineral metabolism in vivo. Transplantation. 1994;57:1231–7. doi: 10.1097/00007890-199404270-00016. [DOI] [PubMed] [Google Scholar]
- 62.He N, Xiao Z, Yin T, Stubbs J, Li L, Quarles LD. Inducible expression of Runx2 results in multiorgan abnormalities in mice. J Cell Biochem. 112:653–65. doi: 10.1002/jcb.22968. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.