

Abstract
Objective
Mitochondrial (mt) DNA encodes the proteins of the electron transfer chain to produce ATP through oxidative phosphorylation, and is essential to sustain life. mtDNA is unique from the nuclear genome in so much as it is solely maternally inherited (non-Mendelian patterning), and shows a relatively high rate of mutation due to the absence of error checking capacity. While it is generally assumed that most new mutations accumulate through the process of heteroplasmy, it is unknown whether mutations initiate in the mother are inherited, occur in utero, or occur and accumulate early in life. The purpose of this study is to examine the maternally heritable and de novo mutation rate in the fetal mtDNA through high fidelity sequencing from amongst a large population-based cohort.
Study design
Samples were obtained from 90 matched maternal (blood) and fetal (placental) pairs. In addition, a smaller cohort (n=5) of maternal (blood)-fetal (placental)-and neonatal (cord blood) trios were subjected to DNA extraction and shotgun sequencing. The whole genome was sequenced on the Illumina HiSeq platform, andaplogroups and mtDNA variants were identified through mapping to reference mitochondrial genomes (NC_012920).
Results
We observed 665 single nucleotide polymorphisms (SNP) and 82 indel variants identified in the cohort at large. We achieved high sequencing depth of the mtDNA to an average depth of 65X (range 20–171X) coverage. The proportions of haplogroups identified in the cohort are consistent with the patient’s self-identified ethnicity (>90% Hispanic), and all maternal-fetal pairs mapped to the identical haplogroup. Only variants from samples with average depth >20X and allele frequency >1% were included for further analysis. While the majority of the maternal-fetal pairs (>90%) demonstrated identical variants at the single nucleotide level, we observed rare mtSNP discordance between maternal and fetal mitochondrial genomes.
Conclusion
In this first in depth sequencing analysis of mtDNA from maternal-fetal pairs at the time of birth, a low rate of de novo mutations appear in the fetal mitochondrial genome. This implies that thesemutations likely arise from the maternal heteroplasmic pool (such as in the oocyte), and accumulate later in the offsprings life. These findings have key implications for both the occurrence and screening for mitochondrial disorders.
Key words and phrases: maternal mitochondrial transmission, mitochondrial DNA heritability, mitochondrial DNA mutations, mitochondrial DNA variation, mitochondrial heteroplasmy
Introduction
The maternally transmitted mitochondria are the most abundant organelles in the human oocyte and early embryo, and serve as the primary generator of cellular energy and orchestrator of cellular metabolism via oxidative ATP production. Mitochondrial integrity is crucial to not only oxidative phosphorylation and generation of ATP, but plays a role in lipid and amino acid metabolism, cell proliferation, differentiation and apoptosis. Mitochondria harbor their own 16.5 kb circular, double-stranded DNA genomes (mtDNA) which contains a mere 37 genes encoding for 13 polypeptides for oxidative phosphorylation, as well as 22 tRNAs, and 2 rRNAs necessary for mitochondrial protein synthesis1–3. Despite its diminutive size, mtDNA is of major evolutionary and functional importance as it is a legacy of the endosymbiosis from prokaryotes that likely created the eukaryotic cell over 1.5 billion years ago4. Although the vast majority of proteins involved in mitochondrial function are encoded by the nuclear genome, the 13 mtDNA encoded proteins are crucial for human life. Underscoring its role in human health, over 200 mtDNA point mutations have been associated with a variety of human diseases (http://www.mitomap.org), including diabetes, cancer, and neurodegenerative disorders1. With respect to perinatal disorders, there are initial studies suggesting a role for nonsynonymous (protein coding) mtDNA mutations in preeclampsia5, oocyte wastage and early embryonic demise6,7, and preterm birth8. We have recently demonstrated that mtDNA variation of the human host significantly influences the structure and community members of the vaginal and gut human microbiome9. Despite its evident importance in both cellular function and human disease, genomic variation studies of mtDNA have largely been relatively underrepresented when compared to nuclear DNA analyses2,3.
The mitochondrial genome is inherited in a non-Mendelian manner, vertically transmitted solely from the mitochondria of the oocyte. This so called “maternal inheritance” pattern, whereby a mother carrying a mtDNA mutation will pass it on to all of her children regardless of sex, but only her daughters will transmit it to their progeny, has been the clinical hallmark of mitochondrial disorders1. Since each cell contains varying number of mtDNA copies, some mtDNA genomes in any given cell or tissue are normal, while others contain mutations (defined as heteroplasmy, or intra-individual variation in mtDNA sequences)1–4. Homoplasmic mtDNA pathogenic mutations, when nearly 100% of mtDNA genomes in a given cell, tissue or organism are severely variant, happens rarely, and are thought to result in fetal or neonatal lethality or organ-specific mitochondrial disease (such as Leber hereditary optic neuropathy)10. Conversely, low level (1–10%) heteroplasmy is felt to be of little clinical significance and may actually represent a reservoir of genetic mtDNA variants that can increase the functional capacity in response to aging and environmental stressors11,12. The degree of mtDNA mutation has been reported to vary between generations, although it remains unclear the degree to which bottlenecking during oogenesis and accumulated lifetime mutation rates contribute to generational differences in the mitochondrial genomic sequence. While a recent study by Bebolledo-Jaramillo et al estimated the rate of heteroplasmy in 39 maternal-child pairs with known mutations in 1 in 8 subjects2, an accurate estimate of the mtDNA mutation rate at the time of birth from a healthy and non-diseased population is still lacking13,14.
In this study, we investigated the maternally inheritable and de novo mutation frequency in fetal mtDNA from a relatively large population-based cohort comprised of 90 maternal (blood)-fetal (placental) pairs as detected by NextGen sequencing technology (Illumina HiSeq platform). Because it was a formal possibility that neonatal cord blood and fetal (placental) detection of mutations may differ, we additionally sequenced 5 trios comprised of maternal (blood)-fetal (placental)-and neonatal (cord blood) mt genomes. With derived high-fidelity and high-depth sequence data we determined (i) 665 single nucleotide polymorphisms (SNP) and 82 indel variants in the cohort at large, (ii) the rate of haplogroup, SNP, and indel variation between maternal-fetal pairs, (iii) the rate of mtSNP discordance between maternal and fetal mitochondrial genomes, and (iv) an estimate of the mtDNA de novo mutation rate at the time of birth. These findings have the potential to not only inform human developmental and perinatal genomics, but also underscores the rarity with which de novo mitochondrial disease would be anticipated in the newborn and thus provides subsequent insights into mtDNA disease pathogenesis14–17.
Materials and Methods
Study Population
The index study was a prospective, observational single-center longitudinal cohort study of term and preterm births conducted in the Harris County Hospital system (August 2011–July 2014). The Institutional Review Board for Baylor College of Medicine and Affiliated Hospitals reviewed the study design and protocol, and approved the study along with the consent form (H-27393 and H-26589). After selection, informed consent was obtained. A nine page consent document including background information, purpose and procedures in detail was reviewed with the subject by a trained clinical registered nurse assigned to the study trial. Inclusion criteria included ability to sign informed consent, willing to provide blood samples, confirmation of singleton gestation, estimated due date established by ultrasound at or before 12 and 0/7 weeks or by LMP consistent with ultrasound at or before 14 and 0/7 weeks. Exclusion criteria included the following: use of vaginal or vulvar medications in the past, twin gestation, presence of acute medical illness (including pre-eclampsia, fever, gestational diabetes, type II diabetes), chronic disease (including pulmonary, cardiovascular, gastrointestinal, hepatic or renal disease), maternal history of cancer, positive maternal HCV, HIV or HBV (confirmed by immunoblot or molecular testing), history of major GI surgery except appendectomy or cholecystecomy in the last five years, confirmed or suspected condition of immunosuppression, history of uncontrolled GI disorders (including inflammatory bowel disease, ulcerative colitis, Crohn’s disease, irritable bowel syndrome, persistent, infectious gastroenteritis, colitis, gastritis, persistent or chronic diarrhea, C. difficile infection, untreated H. Pylori infection, or chronic constipation), urinary incontinence with use of incontinence protection garments, condloma or Human Papilloma Virus diagnosed within the previous two years, treatment for or suspicion of having Toxic Shock Syndrome, history of candidiasis, urinary tract infection, active STD within the previous 2 months, history of dysplasia (vulvar, vaginal or cervical) within the last 5 years, or history of recurrent rash in the past 6 months (including psoriasis, or recurrent eczema).
Sample size estimation
A cohort size of 90 maternal fetal pairs (180 samples) and 5 trios (15 samples) was based on published observations of others employing 39 maternal offspring pairs, of which 1 in 8 were carriers of disease-associated mtDNA mutations in at least one of the two tissues analyzed were found2. In these authors study, multiplexing of samples to 12/run on the MiSeq resulted in 106 read pairs per sample.
Biologic sample attainment
Samples were collected under IRB approved consent from subjects in the form of maternal blood, placental tissue, and fetal cord blood if available. Samples were placed in EDTA, heparanized, or PureGene (Qiagen, Mansfield, MA) tubes (as appropriate) or flash frozen, and labeled by study ID number and by maternal, cord or placental (M, C, P) source respectively.
Whole genome sequencing
DNA from each sample was extracted using QIAGEN Gentra® Purgene® (Qiagen, Mansfield, MA) following manufacturer’s protocols. Ilumina NextGen sequencing platform was utilized to perform paired-end whole genome sequencing on all samples as previously described and detailed9,18–21.
Variant calling on assembled mitochondrial genomes
Sequence alignment, quality control, and variant calling were performed with BWA22 SAMTools23 and Altlas224 as previously described9. Prior to variant calling, the mtDNA sequence from each sample was aligned by BWA with Cambridge Reference Sequence (NC_012920), an established Genebank sequence. SAMtools was used to remove duplicate reads, convert, sort, and index the aligned data files. Atlas2 suite was adopted for variant calling to generate vcf files. Atlas2 suite employs logistic regression models in conjunction with adjustable cutoffs for accurately separation of true SNPs and INDELS with sequencing errors. This combined with the probability score and a minimal heuristic filters enabled us to filter out sequencing errors, and meanwhile generate highly accurate variants calls. As such, default parameters were used for variant analysis. Sequence variations found in both maternal and fetal samples were scored as germ-line variations. Any DNA sequence differences beyond anticipated by sequence and platform read2 between maternal and placental samples were scored as de novo mtDNA mutation. Each was then checked against the MITOMAP (www.mitomap.org) database and mtDB (www.genpat.uu.se/mtDB)25 for frequencies in general population. SNPs with higher than 1% allele frequency were considered common2,9.
Haplogroup Analysis
HaploGrep26 was used to define the haplogroup of each individual. HaploGrep is a web tool using the latest version of Phylotree as reference to determine haplogroup. It is reported to provide greater accuracy for assignment of haplogroup using NextGen Sequencing reads. 2,26,27.
Results
Patient population
There are ninety maternal-fetal pairs in the cohort providing adequate power for analysis of de novo mutation estimates (Table 1). The maternal serum sample and newborn’s placenta tissue sample were collected from each pair. In addition, the fetal cord blood sample was additionally collected for five individuals, to yield in total 195 samples for analysis from the time of birth. This constitutes the largest maternal-offspring cohort analyzed to date, and the only with subject samples acquired immediately at the time of birth. Consistent with the overall study design aimed at a term and preterm comparison, the mean gestational age was 34.05 weeks (SD of 4.06 weeks; Table I). Consistent with the largely Latino patient demographic of Harris County, TX the majority of subjects self-identified as Hispanic ethnicity, and these findings were entirely supported by the haplogroup assignment as described further below.
Table 1.
Baseline characteristics of the entire case-cohort of study subjects (n 90 maternal-fetal (placental) pairs)
| Maternal characteristics | Mean (SD) | Range |
|---|---|---|
| BMI (kg/m2) | 29.56 (6.14) | 17~45 |
| Height (inches) | 62.1 (2.9) | 56~70 |
| Weight (lb) | 163.18 (39.4) | 97~308 |
| Age (years) | 29.32 (7) | 17~45 |
| Infant characteristics | ||
| Gestational age (weeks) | 34.05 (4.06) | 24~41 |
| Weight (gram) | 2499.25 (873.2) | 635–4310 |
Variant calling and mt haplogroup assignment
The mitochondrial SNPs and INDELs were identified as described in Methods. We achieved high sequencing depth of the mitochondrial genome to on average depth 65X (compared with 12X depth of sequencing for most of human genome sequencing), which enabled accurate variant calling2,9. The increasing sequencing depth is independent from the number of variants found in each sample, further supporting our confidence in accuracy of variant calls (Figure 1). To reduce risk of error and maximize detection of potential significant heteroplasmy, only samples with greater than 20X sequencing depth were selected for further analysis employing the Atlas2 suite was used for variant calling (on default parameters). Haplogroup was assigned based on a combination of specific variants (HaploGrep). As shown in Figure 2, the majority of subjects were sequenced assigned to haplogroups A, B, C and D, which are unique to indigenous peoples of the Americas2,26. These results are consistent with the patient’s self-identified ethnicity (Hispanic), global distribution (southern Texas, with recent migration patterns from Mexico, Central, and South America) as well as migration trends of haplogroups. Of note, we observed 100% identical haplogroup assignments between each subjects maternal blood and the identified fetal placental sample, further supporting the methodology of our approach and validity of sequencing.
Figure 1. Number of SNPs discovered in samples is independent of sequencing depth.
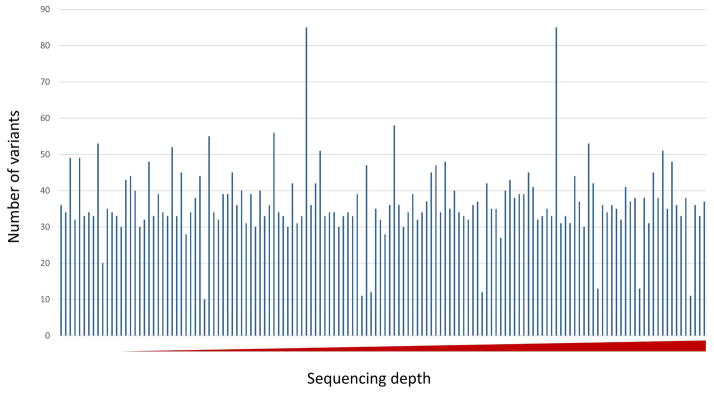
The X axis shows the sequencing depth from low to high (min 20, max 171). The Y axis depicts the number of SNPs discovered per subject sample.
Figure 2. Cladogram projection depicting the relationship and proportion of subjects for each haplogroup.

No variation in the mtDNA haplogroup was observed to be discordant or variant between any maternal-fetal pair tested. As anticipated by our subjects self-identified race and ethnicity, the majority of subjects are in haplogroup A, B, C, D which are the dominate haplogroups in the Hispanic population.
SNP analysis within maternal (blood)-fetal (placental)-and fetal (cord blood)
The initial five sets of subject trios were analyzed for concordance of identified SNPs among maternal blood, cord blood and placental tissues (Figure 3)28. The mtDNA variants discovered among each member of the trio were nearly identical, except for several de novo variants in the D-Loop promoter region and COXII. The D-loop is in a non-coding and hypervariable region, and thus changes that occur here do not cause amino acid substitutions or codon disruptions2,9,13. The mtSNPs identified between placental and cord blood specimens were nearly identical for most of sets as well; therefore, we limited all subsequent sequencing to maternal blood and fetal (placental) tissue.
Figure 3. Comparisons among five sets of trio subject samples.

The outer most circle represents the 16.569 Kb circular mitochondrial reference genome. Trio subject samples include specimens from paired cord blood (red), maternal blood (yellow) and fetal placental tissue (green). mtSNPs were identified for each sample and are depicted as non-variant or variant from the reference mitochondrial genome. Each set of trio subject samples clustered together, and are thus shown in parallel.
De novo SNP analysis in maternal-fetal pairs at the time of birth
We identified 665 SNP and 82 indel variants from the cohort at large. To ensure high quality of SNPs and filter out potential sequencing errors, only variants having allele frequencies greater than 1% based on mtDB, were included in all further analysis. There are 204 variants meeting this criteria. As shown in Figure 4, maternal-fetal sample pairs within the same haplogroup were clustered together (shown as the same color). In this projection of data, black dots indicated mtSNPs identified in both maternal and fetal samples such that haplogroup-associated mtSNPs which appear in every sample with the same haplogroup assignment can be readily identified (Figure 4). Conversely, blue dots represent de novo mtSNPs which were only detected among fetal samples (but not the corresponding maternal sample), and red dots represent mtSNPs only detected among maternal samples (but not in the corresponding fetal samples). As anticipated, the hypervariable D-loop region retains the largest number of variants compared with other regions on the mitochondrial genome. Table 2 summarizes this mtSNP variant data, and details the resultant mtSNP. Although most of the de novo variants are synonymous (i.e., not resulting in an amino acid change so not of relevance), several variants in maternal cohort were both non-synonymous (i.e., affecting protein function) and may potentially be related to disease-associated mtDNA mutations and notably only those with later in life manifestation. Specifically, C7256T located in COX1 (cytochrome c oxidase 1) has a higher frequency of appearance in patients at endometrioid cancer stage III29. However, de novo mutations found in the fetal cohort are all synonymous.
Figure 4. Detailed analysis of maternal-fetal mitochondrial genomes.

The outer most circle represents 16.569 Kb circular mitochondrial reference genome, with inner circles representing each maternal-fetal pair in parallel sequence. The black dots depicts identical mtSNPs as detected in both subjects comprising the maternal-fetal pair. The red dots show mtSNPs which were observed in the mom but not the fetus, and the blue dots represent mtSNPs that only appear in the fetus (and conversely not the mom). The color of the inner circle is based on the haplogroup assignment as previously depicted in Figure 2, and enables visualization of non-haplogroup defining mtSNPs.
Table 2.
List of de novo mtSNPs.
| SNP | Allele Frequency | Encoded Region | Type of Mutation | Amino Acid changes | |
|---|---|---|---|---|---|
| de novo mtSNPs in fetal mtDNA | |||||
| T2352C | 1.923% | 16S rRNA | - | - | |
| G4655A | 1.775% | ND2 | Synonymous | Thr->Thr | |
| G6023A | 1.257% | COI | Synonymous | Glu->Glu | |
| T6776C | 2.551% | COI | Synonymous | His->His | |
| C7256T | 5.51% | COI | Synonymous | Asn->Asn | |
| C8655T | 3.18% | ATPase6 | Synonymous | Ile->Ile | |
| A10819G | 1.109% | ND4 | Synonymous | Lys->Lys | |
| T14212C | 1.109% | ND6 | Synonymous | Val->Val | |
| mtSNPs found in maternal but not in fetal mtDNA | |||||
| A813G | 1.62% | 12S rRNA | - | - | |
| T825A | 3.18% | 12S rRNA | - | - | |
| G930A | 2.256% | 12S rRNA | - | - | |
| T14470C | 2.514% | ND6 | Synonymous | Gly->Gly | |
Comment
Mitochondrial disorders are projected to affect >1 in 7500 live births30. The efficient functioning of mitochondria depends on the integrity of both the nuclear genome (with its Mendelian inheritance patterning) and mitochondrial genome (with its clonal maternal inheritance patterning). Because of its unique inheritance pattern and the occurrence of heteroplasmy, the precise mechanisms and timing with which new cases of mitochondrial encoded disorders arise have historically been poorly understood1–4. The recent work of Bebolledo-Jaramillo et al estimated the rate of heteroplasmy in 39 maternal-child pairs with known mutations in 1 in 8 subjects2, and found that on average an individual at a later stage of life carries one heteroplasmic variant with allele frequency ≥1%, suggesting a rather ubiquitous occurrence. Of interest, this study also found that the number of heteroplasmies in the maternal buccal and blood samples tripled among women >30 years of age. By employing transition to transversion ratios, these authors were able to estimate that transversion-causing oxidative damage was unlikely to be a cause of these mutations. Despite this studies significant work and findings, they did not provide an accurate estimate of the mtDNA mutation occurrence estimates at the time of birth from a healthy and non-diseased population at-large13,14. Closing this gap in our knowledge is the focus of the current analysis reported herein.
Our study design and execution has several significant strengths. First, our study cohort is a relatively homogenous Hispanic population which increases the likelihood of detection and estimation of de novo mutations. Although it may be argued that this also limits generalizability of our findings, the rate of de novo mutation is not reflective of the haplogroup and has been shown to be independent30. Second, since the mutation rate of mtDNA is known to exceed that of nuclear DNA, we adopted stringent criteria to ensure only high quality mtDNA variants were included in the study. Third, we achieved a high depth of sequencing (65X) on the mitochondrial genome and only included variants from samples with greater than 20X depth and having greater than 1% allele frequency. Fourth, we employed both maternal blood and two sources of fetal nucleic acid (cord blood and placenta). Since we obtained identical mtDNA variants, we surmise that it is not necessary to sequence both placenta and cord blood as fetal sources in ours and future analysis. Given the cost of sequencing and often limitation of available cord blood, this observation has potential practical implications.
Our study also has several limitations. First, we did not sequence beyond the point of measuring 1–10% heteroplasmy nor did we sequence multiple maternal tissues. However, others have recently demonstrated that this is unlikely to be necessary2. Second, it is possible that some of our variant calls may represent microsatellite sequencing errors. Although we attempted to limit by employing high stringent analytic pipelines, this remains a formal possibility which would have overestimated our mutation call frequency. However, given that the primary finding of our study is the absolute rarity of de novo mutations, it does not alter our conclusions.
In sum, we have employed in depth massively parallel sequencing to provide the first population-based large cohort analysis of maternal-fetal pairs and the occurrence of de novo mtDNA variation at birth. We have confirmed that mom and fetus bear identical haplogroup assignments, and shown the existence of very low frequency of de novo mutants in utero. The effect of these mutations would be anticipated to be null to minimal at the time of birth, since none of the de novo variants detected in this study were non-synonymous. The practical and clinical implications of this study are multi-fold. First, they suggest that in an era in which an increasing frequency of genetic screening tests are offered by commercial entities, testing for mtDNA variants in non-affected women or their fetuses is unlikely to be of clinical value among low-risk individuals. Second, if fetal or newborn testing is warranted, it can likely be performed on either placental or cord blood. Third, with suggestions of mtDNA variation playing a potential role in perinatal disorders, there is unlikely benefit to deep sequencing beyond the maternal blood or the placental tissue. Fourth, it significantly extends the very recent literature on the biology underlying the inheritance of human mitochondrial DNA. Future studies aiming to decipher the impact of mtDNA on other aspects of reproductive biology will benefit from both the methodology and findings reported herein, and inform our collective understanding of the diversity of genomic sources underlying heritability.
Acknowledgments
Financial support: The Burroughs Welcome Fund Preterm Birth Initiative (K.A., 1008819.01) and the National Institute of Nursing Research Grant (K.A., R01NR014792). All sequencing reactions were performed by the Baylor College of Medicine Human Genome Sequencing Center (BCM-HGSC), which is funded by direct support from the National Human Genome Research Institute (NHGRI) at NIH [U54HG004973 (BCM), Dr. Richard Gibbs, P.I.]
Footnotes
Disclosure statement: The authors report no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wallace DC, Chalkia D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb Perspect Biol. 2013;5(11):a021220. doi: 10.1101/cshperspect.a021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rebolledo-Jaramillo B, Su M, Stoler N, et al. Maternal age effect and severe germ line bottleneck in the inheritance of human mitochondrial DNA. Proc Nation Acad Sci. 2014;111(43):15474–15479. doi: 10.1073/pnas.1409328111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pesolo G, Allen JF, Lane N, Martin W, Rand DM, et al. The neglected genome. EMBO. 2012;13:473–474. doi: 10.1038/embor.2012.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lane N, Marin W. The energetics of genome complexity. Nature. 2010;467:929–934. doi: 10.1038/nature09486. [DOI] [PubMed] [Google Scholar]
- 5.Ding D, Scott NM, Thompson EE, et al. Increased protein coding mutations in the mitochondrial genome of African American women with preeclampsia. Repro Sci. 19(2):1343–1351. doi: 10.1177/1933719112450337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chao HT1, Lee SY, Lee HM, Liao TL, Wei YH, Kao SH. Repeated ovarian stimulations induce oxidative damage and mitochondrial DNA mutations in mouse ovaries. Ann N Y Acad Sci. 2005 May;1042:148–56. doi: 10.1196/annals.1338.016. [DOI] [PubMed] [Google Scholar]
- 7.Colagar AH, Mosaieby E, Seyedhassani SM, Mohajerani M, Arasteh A, Kamalidehghan B, Houshmand M. T4216C mutation in NADH dehydrogenase I gene is associated with recurrent pregnancy loss. Mitochondrial DNA. 2013 Oct;24(5):610–2. doi: 10.3109/19401736.2013.772150. [DOI] [PubMed] [Google Scholar]
- 8.Velez DR, Menon R, Simhan H, Fortunato S, Canter JA, Williams SM. Mitochondrial DNA variant A4917G, smoking and spontaneous preterm birth. Mitochondrion. 2008 Mar;8(2):130–5. doi: 10.1016/j.mito.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 9.Ma J, Coarfa C, Qin X, et al. mtDNA haplogroup and single nucleotide polymorphisms structure human microbiome communities. BMC Genomics. 2014;15(1):257. doi: 10.1186/1471-2164-15-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McFarland R, Clark KM, Morris AA, Taylor RW, Macphail S, Lightowlers RN, Turnbull DM. Multiple neonatal deaths due to a homoplasmic mitochondrial DNA mutation. Nat Genet. 2002;30:145–146. doi: 10.1038/ng819. [DOI] [PubMed] [Google Scholar]
- 11.Rose G, Romeo G, Dato S, et al. for the GEoHAP. Somatic point mutations in mtDNA control region are influenced by genetic background and associated with healthy aging: a GEHA study. PLoS One. 2010;5:e13395. doi: 10.1371/journal.pone.0013395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giuliani C, Barbieri C, Li M, et al. Transmission from centenarians to their offspring of mtDNA heteroplasmy revealed by ultra-deep sequencing. AGING. 2014;6 (6):454–467. doi: 10.18632/aging.100661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parsons TJ, Muniec DS, Sullivan K, Woodyatt N, Alliston-Greiner R, Wilson MR, Berry DL, Holland KA, Weedn VW, Gill P, Holland MM. A high observed substitution rate in the human mitochondrial DNA control region. Nat Genet. 1997 Apr;15(4):363–8. doi: 10.1038/ng0497-363. [DOI] [PubMed] [Google Scholar]
- 14.Henn BM, Gignoux CR, Feldman MW, Mountain JL. Characterizing the time dependency of human mitochondrial DNA mutation rate estimates. Mol Biol Evol. 2009;26:217–230. doi: 10.1093/molbev/msn244. [DOI] [PubMed] [Google Scholar]
- 15.Autere J, Moilanen JS, Finnilä S, et al. Mitochondrial DNA polymorphisms as risk factors for Parkinson’s disease and Parkinson’s disease dementia. Hum Genet. 2004;115(1):29–35. doi: 10.1007/s00439-004-1123-9. [DOI] [PubMed] [Google Scholar]
- 16.Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148(6):1145–59. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nesbitt V, Alston CL, Blakely EL, et al. A national perspective on prenatal testing for mitochondrial disease. Eur J Hum Genet. 2014;22(11):1255–1259. doi: 10.1038/ejhg.2014.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–14. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aagaard K, Petrosino J, Keitel W, Watson M, Katancik J, Garcia N, et al. The Human Microbiome Project strategy for comprehensive sampling of the human microbiome and why it matters. FASEB J. 2012;27(3):1012–22. doi: 10.1096/fj.12-220806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.The Human Microbiome Project Consortium. A framework for human microbiome research. Nature. 2012;486(7402):215–21. doi: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aagaard K, Riehle K, Ma J, Segata N, Mistretta T-AA, Coarfa C, et al. A metagenomic approach to characterization of the vaginal microbiome signature in pregnancy. PLoS One. 2012;7(6):e36466. doi: 10.1371/journal.pone.0036466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evani US, Challis D, Yu J, et al. Atlas2 Cloud: a framework for personal genome analysis in the cloud. BMC Genomics. 2012;13 (Suppl 6):S19. doi: 10.1186/1471-2164-13-S6-S19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ingman M, Gyllensten U. mtDB: Human Mitochondrial Genome Database, a resource for population genetics and medical sciences. Nucleic Acids Res. 2006;34(Database issue):D749–51. doi: 10.1093/nar/gkj010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kloss-Brandstätter A, Pacher D, Schönherr S, et al. HaploGrep: a fast and reliable algorithm for automatic classification of mitochondrial DNA haplogroups. Hum Mutat. 2011;32(1):25–32. doi: 10.1002/humu.21382. [DOI] [PubMed] [Google Scholar]
- 27.Segata N, Börnigen D, Morgan XC, Huttenhower C. PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat Commun. 2013;4:2304. doi: 10.1038/ncomms3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krzywinski M, Schein J, Birol I, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–45. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aikhionbare FO, Mehrabi S, Thompson W, Yao X, Grizzle W, Partridge E. mtDNA sequence variants in subtypes of epithelial ovarian cancer stages in relation to ethnic and age difference. Diagn Pathol. 2008;3(1):32. doi: 10.1186/1746-1596-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet. 2005;6(5):389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]