

Abstract
Background
Reprints and permission: sagepub.com/journalsPermissions.nav Asthma is a complex heterogeneous disease process with mild, moderate, and severe classifications. Although the science of genomics has opened our understanding of the molecular underpinnings of asthma, epigenetics is emerging as a mechanism whereby the expression of disease-risk genes may be influenced by environmental exposure.
Objectives
The purpose of this article is to discuss the methodology of data collection and evaluation involved in genome-wide methylation profiling (epigenomic) through presentation of data generated for a population presenting with severe asthma.
Method
Over 14,000 gene promoter sites were analyzed for methylation status among six subjects with severe asthma and four normal controls in this pilot study. Two duplicate samples were used as technical replicates. Non-smoking case/control subjects were chosen based on similar gender and age. Blood samples were used for DNA extraction, and methylation data were collected utilizing the Illumina Infinium HumanMethylation27BeadChip platform.
Results
Technical replicates were highly concordant, and statistically significant differences were found in methylation profiles between subjects with severe asthma and normal controls (p < 10−8), some previously reported with pulmonary function and others never before reported. After correction for multiple testing, three gene promoter regions remained statistically different: FAM181A, ZNF718, and MRI1.
Discussion
This research supports the internal validity of the Illumina platform in methylation analysis of DNA from stored blood samples. Although significant differences in methylation were noted between subjects with severe asthma and controls, the small sample size warrants further investigation into these results.
Keywords: epigenetic, gene, methylation, severe asthma
Asthma is a complex and multifactorial disease involving multiple pathways that lead to wheezing, shortness of breath, chest tightness, and nighttime or early morning coughing (Centers for Disease Control and Prevention, 2011). Guidelines of the American Thoracic Society (ATS), Global Initiative for Asthma (GINA), and the National Asthma Education and Prevention Program (Moore et al., 2007; National Heart Lung and Blood Institute, 2002; National Institutes of Health, 2007) classify asthma as mild, moderate, or severe. While it is an emerging area of research, little is currently known about the genomic and epigenetic factors that contribute to the expression of asthma and severity of this disease process. Understanding the pathogenesis of these phenotypes may lead to better assessment, classification, and treatment of patients with asthma. Epigenetic differences likely arise from environmental exposures and inflammatory elements acting in concert with specific underlying genetics, which can lead to cellular responses that result in an augmentation or suppression of asthma (Kabesch & Adcock, 2012). The purpose of this article is to highlight the methodology of epigenetic research within the context of severe asthma and discuss some exploratory research findings.
Background
Research has revealed hundreds of genes associated with asthma or in pathways involved in asthma. Between 2007 and 2010, eight genome-wide association studies revealed 10 different genes with significant associations with asthma; however, their respective odds ratios ranged between .5 and 2.32, and the calculated attributable risk for these genes ranged between 3.9% and 24% (Lee, Park, & Park, 2011). Given this low attributable risk and evidence of environmental influences in the development and progression of asthma, scientists point to rare DNA variants, micro RNA, and the epigenome to explain other potential attributable risks in the development of this disease.
Epigenetics traditionally refers to heritable gene expression or cellular phenotype caused by mechanisms other than underlying DNA sequences (Waddington, 1942). Examples of epigenetic alterations include DNA methylation, modification of histone tails, and noncoding RNAs, each of which may be influenced by the environment, diet, lifestyle, diseases, and aging (Yang & Schwartz, 2011). DNA methylation, the focus of this article, is a chemical modification in and around repetitive cytosine nucleotide occurring next to a guanine nucleotide, also known as, cytosine - phosphate link - guanine (CpG) along a DNA sequence, and if methylation occurs in a promoter region it contributes to downregulation (decreased expression) of a gene (Kuriakose & Miller, 2010).
Without changing the DNA nucleotide sequence, epigenetic mechanisms may potentially integrate environmental factors (i.e., cadmium, arsenic, nickel, and benzene) with genetic polymorphisms in the complex model of severe asthma (Baccarelli & Bollati, 2009; Sofer et al., 2013). Most asthma methylation studies to date have been limited to few gene promoter regions and have not included genome-wide analysis.
In an effort to strengthen our understanding of severe asthma, we performed an exploratory pilot study focusing on global epigenetic methylation methodology (i.e., across the whole genome) and differences between subjects with predefined severe asthma and healthy controls within a convenience sample from subjects recruited to the Severe Asthma Research Program (SARP) at the University of Pittsburgh Asthma Institute at the University of Pittsburgh Medical Center. We utilized previously collected and banked peripheral blood mononuclear cell specimens. Research included analysis of concordance between duplicate samples to determine internal validity of utilizing the Illumina Infinium platform to assess global methylation of these stored samples. Given the huge expense involved in epigenetic analysis, working through the methodology of this platform and internal validity was prudent.
Materials and Method
Setting and Participants
The parent SARP study, funded by the National Heart, Lung, and Blood Institute, is a comprehensive research program for the study of severe asthma in children and adults using a descriptive cross-sectional cohort study design. Its study population has been described previously, including medical history and physiological testing to determine phenotype (Moore et al., 2007).
We obtained approval from the Institutional Review Board at the University of Pittsburgh as well as informed consent for the use of banked blood specimens for genetic analysis. All subject data and specimens were de-identified with subject codes. The 10 European White participants in this pilot study research included nonsmokers (<5 pack years) who were more than 18 years of age. Although all participants were adults, there was an up to 15-year difference between the two male cases with severe asthma and two normal (i.e., no diagnosis of asthma) male controls and up to a 33-year difference between the four female cases with severe asthma and two normal female controls. It was not possible to age match cases and controls in these banked specimens, as participants with severe asthma were older than those without asthma for both males and females. We made no adjustment for covariates and chose two male subject samples for technical duplicate analysis.
Phenotypic Classification
As previously reported, each of the SARP centers defined severe asthma using the ATS definition (Moore et al., 2007; Wenzel & Busse, 2007). The SARP steering committee adopted the definition of severe asthma from proceedings from an ATS workshop of refractory asthma published in 2000 (Wenzel & Busse, 2007; Wenzel et al., 2000).
Epigenomic Collection
Whole blood had been collected previously, the cells had been lysed, and the samples had been labeled with a numerical code and stored in the lab for up to a year without refrigeration as outlined in the Qiagen-QIAmp DNA extraction reagent kit handbook (Qiagen, 2010). There was no control for time sequence of specimen collection in a consistent manner (i.e., same point in time after disease onset, initiation of specific treatment, etc.); however, blood samples were collected within 1 month of a participant’s entry into SARP. Quiagen indicates that DNA in lysed cells is stable for up to 2 years using reagents in this Quiagen kit. DNA extraction followed the Qiagen-QIAmp DNA extraction reagent kit protocol. We used 2 of the 10 samples as duplicate, technical replicate quality controls. Extracted DNA was provided to the University of Pittsburgh Genomics and Proteomics Core Laboratory for methylation data collection. Bisulfite conversion of the DNA was conducted using a kit and directions from Zymo Research (Catalog kit #D5001, Irvine, CA). Genome-wide methylation data were collected using the Illumina Infinium HumanMethylation27 BeadChip platform (San Diego, CA), which allowed interrogation of 27,578 highly informative CpG sites per sample at single-nucleotide resolution (Illumina, n.d.). This panel targets CpG sites located within the proximal promoter regions of transcription start sites of 14,475 consensus coding sequencing (CCDS) in the National Center for Biotechnology Information (NCBI) database (Genome Build 36). The CCDS is a collaborative effort to identify a core set of human and mouse protein-coding regions that are consistently annotated and represent real proteins (NCBI, n.d.; Pruitt et al., 2009). All samples were processed on the same chip.
Methylation measurements collected represent absolute measurements for a given sample. The beta value (b) is calculated as a ratio (intensity of methylated probe)/(intensity of methylated probe + intensity of unmethylated probe); thus, the measurement scale is 0–1, or 0–100%, with 0 indicating that no methylated molecules were identified, and 1, or 100%, indicating that all identified molecules were methylated (Laird, 2010; Naumova et al., 2012). There are potential sources of bias in a large interrogation of thousands of genetic loci, including incomplete bisulphite conversion; however, as mentioned previously, technical replicates were included in this study to assess reliability (i.e., internal validity) of the data as well as illuminate the potential problems that might occur in an analysis of this magnitude.
Analysis
Statistical analysis was conducted utilizing SAS JMP software version 8.0.1, SPSS version 19, and GenomeStudio version 1.8. GenomeStudio is statistical analysis software specifically designed to process the Illumina methylation data. This package includes data input, quality control, variance stabilization, normalization, and gene annotation portions (Du, Kibbe, & Lin, 2008).
Before analysis, data quality diagnostics were examined to determine outliers and unrealistic data values and to understand the distributions, patterns of missing data, and relationships between the variables. Since most statistical tests perform better if certain assumptions about the variables are met (e.g., normality), descriptive and graphic analyses were performed. Due to the small sample size, missing data were not imputed for this study. Differences in proportions between severely asthmatic and nonasthmatic participants were determined using the Fisher’s exact test. Lumi was utilized for quality control and data manipulation (Du, Bourgon, Geng, & Lin, n.d.). A priori data exclusion criteria included samples with low bisulphate conversion efficiency or with <95% CpG coverage per sample; in addition, methylation total intensity or value distribution outliers were not to be included in further statistical analyses. No subject samples needed to be excluded, based on these criteria; thus, we analyzed data from all 10 subjects.
GenomeStudio was utilized to analyze the technical replicates for two samples. This program provides concordance (i.e., r2 value) and graphic display (i.e., scatter plots, histogram plots, heat maps, and dendrograms). GenomeStudio software was also utilized to calculate average differential methylation b values between the severe asthma and no asthma (i.e., reference) groups. Comparison was then made between the severe asthma group and normal controls to provide differential scores for over 14,000 gene promoter regions. A GenomeStudio differential score is calculated from difference in β scores between case and control (i.e., severe asthma and normal control: 10 sign [βsevere asthma –βnormal] log10p, where the p represents the p value from a t-test; Kibriya et al., 2011). This equation included b values and p value, as confirmed with Illumina techinical support (Stisser, K., personal communication, March 2013), which appeared redundant and motivated us to test this equation with a separate confirmatiory t-test analysis run in SPSS to assure statistical significance.
Analysis in GenomeStudio was followed by correction for false discovery rate (FDR) utilizing Benjamini and Hochberg FDR. Since this research was exploratory versus a pathway analysis or candidate approach and since beta scores are commonly used measures of methylation for CpG locus, average β scores for genes with a low p value derived from differential score analysis in GenomeStudio were exported and run as separate independent sample t-test analyses in SPSS.
Results
All subjects were self-reported European and White: four males and six females (Table 1). Average age was 37.3 years; however, as described previously, there was a statistically significant difference between ages of subjects with severe asthma and those of normal controls (severe asthma mean age = 44.4 years and normal control mean age = 26.6 years). Those with severe asthma had a mean age at asthma diagnosis of 5.5 years and had a mean asthma duration of 38.9 years. In this cohort, three subjects with severe asthma reported a history of smoking, with an average of 1.3 years; no subjects were smoking at the time of enrollment into SARP. Given the associations reported between body mass index (BMI) and asthma severity (Gibeon et al., 2013; Moore et al., 2010), for informational purposes on the participant population, we compared subjects’ BMI and found no statistically significant difference between groups. Our duplicate sample analysis with two male subjects met concordance with r2 values of .9988 and .9983, indicating good reproducibility of site-specific methylation level measurements (Figure 1).
Table 1.
Demographic Comparison Between Normal Control and Severe Asthma Groups.
| Characteristic | Normal control, n = 4 |
Severe asthma, n = 6 |
p
Valuea |
|---|---|---|---|
| Gender, male (n) | 2 | 2 | .548 |
| Age at enrollment, mean (SD) | 26.6 (9.1) | 44.4 (12.8) | .044 |
| Ever smoked, no | 4 | 3 | NA |
| Body mass index, kg/m2, mean (SD) | 24.9 (4.2) | 27.9 (4.9) | .338 |
p Value based on Fisher’s exact test.
Figure 1.
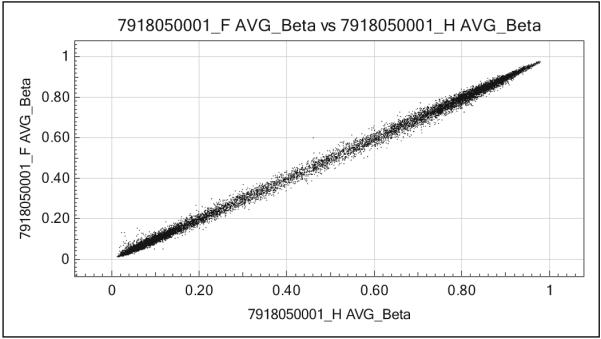
Scatter plot example of one of the duplicate methylation analyses. Analysis of 27,577 total CpG loci revealed good concordance in duplicate samples, r2 = .9988.
There was no mean difference in global methylation values between those with severe asthma versus normal controls. We found significantly different methylomic profiles (i.e., differential scores) between groups in the regions associated with 13 genes at a significance level of p < 10−8 utilizing the GenomeStudio Illumina program; however, after Benjamini and Hochberg FDR correction, only three gene methylation profiles remained significantly different between severe asthmatics and normal controls: FAM181A (p = .028), ZNF718 (p = .036), and MRI1 (p = .017; Table 2). Each of the three methylation probes was in the promoter region of their respective genes. The significant methylomic profile differences between the severe asthma group and the control group included both increased and decreased methylation. Since there were previous smokers in the severe asthma group, we performed an analysis to determine whether there was a significant difference between previous smokers and nonsmokers in these three gene methylomic profiles within that group and found no significant difference.
Table 2.
Genes Found to Have Significantly Different GenomeStudio Methylation Differential Scores Between Asthmatics and Normal Subjects (p < 10−8).
| Gene symbol (name) |
Methylation in severe asthma group as compared to controls |
Chromosome | Product/biological effect |
|---|---|---|---|
| FAM181A | Decreased | 14 | Family with sequence similarity 181, member A, is considered a hypothetical protein LOC90050. Gene associated with asthma (Yang et al., 2012; Yoon, Lee, & Bae, 2011) |
| ZNF718 | Increased | 4 | Zinc finger protein 718 involved in transcription regulation; considered a small interfering RNA (siRNA). Gene associated with asthma (Tsitsiou et al., 2012; Yoon et al., 2011) |
| MRI1 | Increased | 19 | Also known as methylthioribose-1-phosphate isomerase 1, it is an enzyme involved in conversion of MTR-1-P into MTRu-1-P and is also involved in promoting cell invasion (Kabuyama et al., 2009) |
Note. MTR-1-P = methylthioribose-1-phosphate; MTRu-1-P = methylthioribulose-1-phosphate. Gene chromosome location and protein product and/or biological effect descriptions are from GeneCards (Weizmann Institute of Science, n.d.).
Discussion
The focus of this project was to investigate the feasibility of collecting genome-wide DNA methylation data from stored blood samples and to perform pilot analyses to determine if any methylation data survived correction for multiple testing in our small sample of severe asthmatics. We were able to extract quality DNA from stored blood samples and analyze whole genome methylation profiles comparing subjects with well-characterized severe asthma to normal controls. Through this investigation, we found concordance between duplicate samples run using the Illumina Infinium platform, which supported internal validity. We found significant differences in methylation profiles for those with severe asthma compared to those with no asthma in three novel chromosome regions.
Specifically, we identified significant differential methylation profiles in two genes previously reported in association with asthma: FAM181A and ZNF718. FAM181A, family with sequence similarity 181 member A, is expressed in blood and liver; however, the role of this protein is unknown (Weizmann Institute of Science, n.d.). In previous research, the expression of FAM181A was increased in male subjects with severe asthma (3.28-fold, p = .0236) compared to normal male subjects; however, when comparing male patients with asthma to healthy subjects, findings were not significant (Yoon, Lee, & Bae, 2011). Bronchoscopy airway lavage (BAL)-derived epithelial cells had lower expression of FAM181A in atopic asthma subjects 4 hr after saline exposure versus normal subjects (1.35-fold decrease, p = .0221; Yang et al., 2012). This expression of FAM181A in BAL fluid of asthma patients trends in the same direction as in the present epigenetic study, in which the methylation profile of subjects with severe was increased.
ZNF718, zinc finger protein 718, is considered a small interfering RNA (siRNA) involved in transcription regulation (UniProt Consortium, n.d.; Weizmann Institute of Science, n.d.). Yoon, Lee, and Bae (2011) reported increased expression of this gene in male subjects with severe asthma (4.36-fold, p = .009) compared to normal male subjects; however, this trend was reversed when comparing male patients with asthma to healthy subjects (1.68-fold decrease in expression, p = .0317). Tsitsiou et al. (2012) reported increased expression of ZNF718 in isolated CD8+ T cells of patients with severe asthma compared with healthy subjects (1.75-fold increase, p = .0072); however, this trend was reversed when they compared subjects with nonsevere asthma to healthy subjects, with subjects with nonsevere asthma exhibiting a 1.38-fold decrease in expression (p = .0043). Methylation of ZNF718 in severe asthmatics was increased in this study, suggesting reduced expression in our population of subjects with severe asthma. We did not, however, measure actual expression of this gene. Divergence in findings here may be related to the small sample size or other mechanisms contributing to this phenotype.
By contrast, there are no previous reports of a genetic association between methylthioribose-1-phosphate isomerase (MRI1) and asthma, as we found in the present research. MRI1 catalyzes the conversion of methylthioribose-1-phosphate (MTR-1-P) into methylthioribulose-1-phosphate (MTRu-1-P), which promotes cell invasion in response to RhoA in cancer cells (Kabuyama et al., 2009). Although there has been no report of an association with asthma, Tolgyesi et al. (2009) did observe significant downregulation of MRI1 in a murine model of asthma (3.62-fold decrease, p = .0249).
The noticeably extreme methylation profile in normal controls in this study may suggest that key genes involved in normal airway function are upregulated in individuals without asthma and genes that may contribute to pathways leading to poor airway function are downregulated. This finding may further suggest that in asthma, and especially in severe asthma, there may be methylation changes in specific genes that dampen expression of those proteins or, alternatively, increase the chance those proteins will be expressed, either of which will lead to molecular pathophysiological changes.
Limitations
The most obvious limitation to this research is the small sample size and younger mean age in the normal controls versus older mean age in the group with severe asthma, and any findings should be interpreted with caution. Evidence is unclear as to whether smoking influences methylation profiles and which genes are most affected. However, none of the participants in this study had smoked more than five pack years. It is also unknown whether there are differences in methylation based on dose exposure and timing of exposure related to smoking (as in the case of the few subjects in this research who were not smoking when blood samples were obtained but reported a history of smoking). Thus, history of smoke exposure in an epigenetic research program could be a limitation and impetus for control and further analysis in determining if and how this exposure may influence disease processes such as severe asthma.
Future Direction of Epigenetic Analysis in Severe Asthma
Due to the small sample size in this study, further research with a larger sample size is warranted before drawing conclusions about these associations with severe asthma. Longitudinal cohort studies are needed to examine the association of epigenetic regulation with time-dependent disease susceptibility and disease progression (i.e., asthma) associated with epigenetic regulation following environmental exposures (Kuriakose & Miller, 2010). There is evidence that level of maternal care, timing of delivery, parental abuse, and parental stress during the early stages of life may have an influence on epigenetic change in a child’s genome (Chen et al., 2013; McGowan et al., 2009; Schlinzig, Johansson, Gunnar, Ekstrom, & Norman, 2009). Thus, timing of other environmental exposures in early life may also have an influence on epigenetic change with consequences later in life. Knowing the dose and temporal relationship of these exposures may also help us to determine cause and effect for disease.
Exposure to specific chemicals and other compounds may increase or decrease methylation of key genes associated with severe asthma. Increasing our knowledge of these relationships may have utility for novel treatments. When exploring the epigenetic effects of environmental exposure to cigarette smoke for extended periods of time in the model of severe asthma, researchers should enter both timing of cigarette smoke exposure and length of this exposure into the analyses. The ability to integrate methylation analysis with RNA-expression profiling experiments and genome-wide association study/polymorphism data using GenomeStudio software provides an opportunity for analyzing potential cause and effect.
Since genes are expressed differentially in different cell types, future epigenetic analysis in severe asthma should consider different cell types to determine if there are epigenetic differences in these cells and which genes are affected. In this research, we analyzed blood cells. Our findings illustrate potential epigenetic changes that may contribute to severe asthma from the perspective of circulating blood; however, findings may be different at the cellular level in, for example, the upper and lower airways, and this possibility warrants future comparative analysis. Likewise, future epigenetic research should compare different asthma phenotypes (i.e., mild/moderate, exercise induced, etc.).
Conclusion
Although previous research involving genes on all chromosomes has identified genetic associations with asthma, these genetic variants contribute to a small-to-moderate effect. The fact that a large portion of the multifactorial process involved in asthma remains unexplained has led researchers to suggest that epigenetic research may help to elucidate the regulation of the inflammatory pathways involved in this disease. Given the wealth of data collected from previous candidate gene association studies, genome-wide association studies and longitudinal studies that included clinical data, epigenetic analysis, and genetic sequencing hold promise as the next important phase of genetics research.
In this study, some of our methylomic profile findings in subjects with severe asthma were congruent with published gene expression data. Further methylation analyses in populations with severe asthma in conjunction with expression analyses are warranted. Such research could lead to innovative clinical assessment tools and personalized preventive strategies and may have important pharmacogenetic implications for treatment of the disease.
Acknowledgments
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by NIH [Grant nos.: NIH-NINR # T32NR009759 (KW), NIH-NINR # T32NR009759 (YC), NIH # HL-69174 (SW).
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- Baccarelli A, Bollati V. Epigenetics and environmental chemicals. Current Opinion in Pediatrics. 2009;21:243–251. doi: 10.1097/mop.0b013e32832925cc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention Asthma. 2011 Retrieved from http://www.cdc.gov/asthma/
- Chen W, Boutaoui N, Brehm JM, Han YY, Schmitz C, Cressley A, Celedon JC. ADCYAP1R1 and asthma in Puerto Rican children. American Journal of Respiratory and Critical Care Medicine. 2013;187:584–588. doi: 10.1164/rccm.201210-1789OC. doi:10.1164/rccm. 201210-1789OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du P, Bourgon R, Geng G, Lin S. BeadArray specific methods for Illumina methylation and expression microarrays. Retrieved from http://www.bioconductor.org/packages/2.12/bioc/html/lumi.html.
- Du P, Kibbe WA, Lin SM. lumi: A pipeline for processing Illumina microarray. Bioinformatics. 2008;24:1547–1548. doi: 10.1093/bioinformatics/btn224. doi: 10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
- Gibeon D, Batuwita K, Osmond M, Heaney LG, Brightling CE, Niven R, Menzies-Gow A. Obesity-associated severe asthma represents a distinct clinical phenotype: Analysis of the British Thoracic Society Difficult Asthma Registry patient cohort according to BMI. Chest. 2013;143:406–414. doi: 10.1378/chest.12-0872. doi:10.1378/chest.12-0872. [DOI] [PubMed] [Google Scholar]
- Illumina Infinium HumanMethylation27 BeadChip. Retrieved from http://www.illumina.com.
- Kabesch M, Adcock IM. Epigenetics in asthma and COPD. Biochimie. 2012;94:2231–2241. doi: 10.1016/j.biochi.2012.07.017. doi:10.1016/j.biochi.2012.07.017. [DOI] [PubMed] [Google Scholar]
- Kabuyama Y, Litman ES, Templeton PD, Metzner SI, Witze ES, Argast GM, Ahn NG. A mediator of Rho-dependent invasion moonlights as a methionine salvage enzyme. Molecular and Cellular Proteomics. 2009;8:2308–2320. doi: 10.1074/mcp.M900178-MCP200. doi:10.1074/mcp.M900178-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibriya MG, Raza M, Jasmine F, Roy S, Paul-Brutus R, Rahaman R, Ahsan H. A genome-wide DNA methylation study in colorectal carcinoma. BioMed Central Medical Genomics. 2011;4:50. doi: 10.1186/1755-8794-4-50. doi:10.1186/1755-8794-4-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriakose JS, Miller RL. Environmental epigenetics and allergic diseases: Recent advances. Clinical and Experimental Allergy. 2010;40:1602–1610. doi: 10.1111/j.1365-2222.2010.03599.x. doi:10.1111/j.1365-2222.2010.03599.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nature Reviews Genetics. 2010;11:191–203. doi: 10.1038/nrg2732. doi:10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- Lee SH, Park JS, Park CS. The search for genetic variants and epigenetics related to asthma. Allergy, Asthma and Immunology Research. 2011;3:236–244. doi: 10.4168/aair.2011.3.4.236. doi:10.4168/aair.2011.3.4.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan PO, Sasaki A, D’Alessio AC, Dymov S, Labonte B, Szyf M, Meaney MJ. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nature Neuroscience. 2009;12:342–348. doi: 10.1038/nn.2270. doi:10.1038/nn.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore WC, Bleecker ER, Curran-Everett D, Erzurum SC, Ameredes BT, Bacharier L, Wenzel SE. Characterization of the severe asthma phenotype by the National Heart, Lung, and Blood Institute’s Severe Asthma Research Program. Journal of Allergy and Clinical Immunology. 2007;119:405–413. doi: 10.1016/j.jaci.2006.11.639. doi: 10.1016/j.jaci.2006.11.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore WC, Meyers DA, Wenzel SE, Teague WG, Li H, Li X, Bleecker ER. Identification of asthma phenotypes using cluster analysis in the Severe Asthma Research Program. American Journal of Respiratory and Critical Care Medicine. 2010;181:315–323. doi: 10.1164/rccm.200906-0896OC. doi:10.1164/rccm.200906-0896OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Center for Biotechnology Information. National Heart Lung and Blood Institute . Global initiative for asthma: Global strategy for asthma management and prevention (Rev. 2002. ed.) U.S. Dept. of Health and Human Services, Public Health Service; Bethsda, MD: 2002. CCDS database. Retrieved from http://www.ncbi.nlm.nih.gov/CCDS/CcdsBrowse.cgi. [Google Scholar]
- National Institutes of Health Expert panel report 3: Guidelines for the diagnosis and management of asthma (NIH publication no. 08-4051) 2007 Retrieved from http://www.nhlbi.nih.gov/guidelines/asthma/asthgdln.pdf.
- Naumova OY, Lee M, Koposov R, Szyf M, Dozier M, Grigorenko EL. Differential patterns of wholegenome DNA methylation in institutionalized children and children raised by their biological parents. Development and Psychopathology. 2012;24:143–155. doi: 10.1017/S0954579411000605. doi:10.1017/S0954579411000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Harrow J, Harte RA, Wallin C, Diekhans M, Maglott DR, Lipman D. The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes. Genome Research. 2009;19:1316–1323. doi: 10.1101/gr.080531.108. doi:10.1101/gr.080531.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiagen QIAamp DNA blood midi/maxi handbook. (3rd) 2010 Retrieved from http://www.qiagen.com/resources/resourcedetail? id=bf32146a-77fd-40c2-8743-c28974f7935b.
- Schlinzig T, Johansson S, Gunnar A, Ekstrom TJ, Norman M. Epigenetic modulation at birth-altered DNA-methylation in white blood cells after Caesarean section. Acta Paediatrica. 2009;98:1096–1099. doi: 10.1111/j.1651-2227.2009.01371.x. doi:10.1111/j.1651-2227.2009.01371.x. [DOI] [PubMed] [Google Scholar]
- Sofer T, Baccarelli A, Cantone L, Coull B, Maity A, Lin X, Schwartz J. Exposure to airborne particulate matter is associated with methylation pattern in the asthma pathway. Epigenomics. 2013;5:147–154. doi: 10.2217/epi.13.16. doi:10.2217/epi.13.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolgyesi G, Molnar V, Semsei AF, Kiszel P, Ungvari I, Pocza P, Szalai C. Gene expression profiling of experimental asthma reveals a possible role of paraoxonase-1 in the disease. International Immunology. 2009;21:967–975. doi: 10.1093/intimm/dxp063. doi:10.1093/intimm/dxp063. [DOI] [PubMed] [Google Scholar]
- Tsitsiou E, Williams AE, Moschos SA, Patel K, Rossios C, Jiang X, Lindsay MA. Transcriptome analysis shows activation of circulating CD8+ T cells in patients with severe asthma. Journal of Allergy and Clinical Immunology. 2012;129:95–103. doi: 10.1016/j.jaci.2011.08.011. doi:10.1016/j.jaci.2011.08.011. [DOI] [PubMed] [Google Scholar]
- UniProt Consortium UniProt knowledgebase: A hub of integrated protein data, 2013. doi: 10.1093/database/bar009. Retrieved from http://www.uniprot.org/ [DOI] [PMC free article] [PubMed]
- Waddington CH. Canalization of development and the inheritance of acquired characters. Nature. 1942;150:563–565. doi: 10.1038/1831654a0. doi:10.1038/150563a0. [DOI] [PubMed] [Google Scholar]
- Weizmann Institute of Science Gene cards: The human gene compendium. Retrieved from http://www.genecards.org.
- Wenzel SE, Busse WW. Severe asthma: Lessons from the Severe Asthma Research Program. Journal of Allergy and Clinical Immunology. 2007;119:14–21. doi: 10.1016/j.jaci.2006.10.025. doi:10.1016/j.jaci.2006.10.025. [DOI] [PubMed] [Google Scholar]
- Wenzel SE, Fahy JV, Irvin CG, Peters SP, Spector S, Szelfler SJ. Proceedings of the ATS workshop on refractory asthma: Current understanding, recommendations, and unanswered questions. American Thoracic Society [Consensus Development Conference Review]. American Journal of Respiratory and Critical Care Medicine. 2000;162:2341–2351. doi: 10.1164/ajrccm.162.6.ats9-00. [DOI] [PubMed] [Google Scholar]
- Yang IV, Schwartz DA. Epigenetic control of gene expression in the lung. American Journal of Respiratory and Critical Care Medicine. 2011;183:1295–1301. doi: 10.1164/rccm.201010-1579PP. doi:10.1164/rccm.201010-1579PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang IV, Tomfohr J, Singh J, Foss CM, Marshall HE, Que LG, Schwartz DA. The clinical and environmental determinants of airway transcriptional profiles in allergic asthma. American Journal of Respiratory and Critical Care Medicine. 2012;185:620–627. doi: 10.1164/rccm.201108-1503OC. doi:10.1164/rccm.201108-1503OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon Y, Lee H, Bae S. In vivo and in vitro study of asthma. Gene expression omnibus (GEO) 2011 Retrieved from http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE27876.