

Abstract
Shp2 (Src homology 2 domain-containing protein tyrosine phosphatase 2) regulates neural cell differentiation. It is also expressed in postmitotic neurons, however, and mutations of Shp2 are associated with clinical syndromes characterized by mental retardation. Here we show that conditional-knockout (cKO) mice lacking Shp2 specifically in postmitotic forebrain neurons manifest abnormal behavior, including hyperactivity. Novelty-induced expression of immediate-early genes and activation of extracellular-signal-regulated kinase (Erk) were attenuated in the cerebral cortex and hippocampus of Shp2 cKO mice, suggestive of reduced neuronal activity. In contrast, ablation of Shp2 enhanced high-K+-induced Erk activation in both cultured cortical neurons and synaptosomes, whereas it inhibited that induced by brain-derived growth factor in cultured neurons. Posttetanic potentiation and paired-pulse facilitation were attenuated and enhanced, respectively, in hippocampal slices from Shp2 cKO mice. The mutant mice also manifested transient impairment of memory formation in the Morris water maze. Our data suggest that Shp2 contributes to regulation of Erk activation and synaptic plasticity in postmitotic forebrain neurons and thereby controls locomotor activity and memory formation.
INTRODUCTION
A nonreceptor-type protein tyrosine phosphatase (PTPase) Shp2 (Src homology 2 [SH2] domain-containing protein tyrosine phosphatase 2) is expressed in most cell types, including neurons, and plays an important role in the transduction of signaling initiated by growth factors (1, 2). In response to stimulation of growth factor receptors, Shp2 binds via its SH2 domains either to the autophosphorylated receptor or to docking proteins, such as insulin receptor substrates, Grb2-associated binder proteins, or fibroblast growth factor (FGF) receptor substrates, and it thereby positively regulates activation of the Ras–mitogen-activated protein kinase (MAPK) pathway. The Drosophila ortholog of Shp2, corkscrew, is required in multiple developmental pathways (3). Shp2 is also required for basic FGF-induced MAPK activation, mesodermal marker induction, and completion of gastrulation in Xenopus (4). In mice, homozygous deletion of the Shp2 gene leads to embryonic death as a result of abnormal mesoderm patterning (5). Mutations of the human Shp2 gene (PTPN11) that result in constitutive activation or inactivation of the encoded protein have been detected in ∼50% of individuals with Noonan syndrome and ∼90% of those with LEOPARD syndrome (LS), both of which are congenital malformation syndromes with similar pathologies and are characterized by facial dysmorphia and other developmental abnormalities, including learning difficulties and mental retardation (6, 7).
Knockdown of Shp2 or forced expression of a constitutively active (Noonan syndrome-type) mutant of Shp2 in mouse cerebrocortical precursor cells inhibited neurogenesis and gliogenesis, respectively (8). Mutations of the human Shp2 gene may thus result in perturbation of neural cell fate and thereby give rise to the cognitive impairments of Noonan syndrome (and LS) patients. On the other hand, Shp2 is highly expressed in mature neurons, and its dysfunction in the adult brain may thus also contribute to the pathogenesis of mental retardation in Noonan syndrome and LS patients. The functional roles of Shp2 in mature neurons have remained unclear, however.
Neuron-specific ablation of Shp2 by the crossing of mice homozygous for a floxed allele of the Shp2 gene (Ptpn11flox/flox mice; here referred to as Shp2flox/flox mice) with the mice that express a Cre recombinase in a neuron-specific manner resulted in the development of early-onset obesity (9, 10), suggesting that neuronal Shp2 is important for the control of energy balance, possibly as a result of enhancement of MAPK signaling downstream of the leptin receptor in the hypothalamus. However, the contribution of Shp2 to higher brain functions has not been investigated in detail.
We have here ablated Shp2 selectively in postmitotic forebrain neurons by crossing Shp2flox/flox mice with transgenic mice that express a Cre recombinase transgene under the control of the calmodulin-dependent kinase IIα gene promoter (CaMKIIα-Cre mice). Characterization of the resulting offspring revealed that Shp2 regulates MAPK activity in neurons and contributes to synaptic transmission, short-term synaptic plasticity, and memory formation in the mature brain.
MATERIALS AND METHODS
Animals.
Shp2flox/flox and CaMKIIα-Cre transgenic (CRE-159) mice were generated as described previously (11, 12). Homozygous floxed mice harboring the CaMKIIα-Cre transgene were crossed with homozygous floxed mice, and the resulting Shp2 cKO (Shp2flox/flox; CaMKIIα-Cre) mice and their littermates that did not harbor the Cre transgene (Shp2flox/flox mice) were studied. Age-matched (12- to 40-week-old) male mice were used in all experiments. Mice were bred and maintained at the Bioresource Center of the Gunma University Graduate School of Medicine under specific-pathogen-free conditions. They were housed in an air-conditioned room with a 12-h light, 12-h dark cycle. All animal experiments were approved by the Animal Care and Experimentation Committee of Gunma University (approval no. 09-064).
Primary antibodies and reagents.
Rabbit polyclonal antibodies to Shp2 and to doublecortin (DCX) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA), and those to Erk1/2 and to Thr183- and Tyr185-phosphorylated Erk1/2 were from Promega (Madison, WI). Mouse monoclonal antibodies to postsynaptic density protein 95 (PSD-95), to Tau-1, to NeuN, and to Tyr279-phosphorylated glycogen synthase kinase 3β (GSK3β) as well as rabbit polyclonal antibodies to microtubule-associated protein 2 (MAP2), to calbindin, and to GluA1 were obtained from Millipore (San Diego, CA). Rabbit polyclonal antibodies to Akt and to Ser473-phosphorylated Akt, as well as a rabbit monoclonal antibody to GSK3β, were obtained from Cell Signaling Technology (Beverly, MA). 4′,6-Diamidino-2-phenylindole (DAPI) was obtained from Nacalai Tesque (Kyoto, Japan). Rabbit polyclonal antibodies to GluN2B (13) were kindly provided by T. Yamamoto and T. Nakazawa (University of Tokyo, Tokyo, Japan). Rabbit polyclonal antibodies to parvalbumin, as well as methylphenidate hydrochloride, recombinant human brain-derived neurotrophic factor (BDNF), d,l-2-amino-5-phosphonovaleric acid (APV), and 3,5-dihydroxyphenylglycine (DHPG) were obtained from Sigma (St. Louis, MO). Rabbit polyclonal antibodies to glial fibrillary acidic protein (GFAP) were obtained from Dako (Glostrup, Denmark). A mouse monoclonal antibody to synaptophysin (171B5) (14) and rabbit polyclonal antibodies to vesicle-associated membrane protein 2 (VAMP2) were kindly provided by S. C. Fujita and M. Takahashi (Kitasato University, Kanagawa, Japan), respectively.
Measurement of blood glucose.
Blood samples were collected from the tail veins of mice, and blood glucose levels were determined with a Glutest Neo Super (Sanwa Kagaku, Nagoya, Japan).
Plasmids.
A lentiviral vector and packaging plasmids pCS-CDF-EG-PRERfA, pCAG-HIVgp, and pCMV-VSV-G-RSV-Rev were kindly provided by H. Miyoshi (BioResource Center, RIKEN Tsukuba Institute, Ibaraki, Japan).
Preparation of synaptosomes.
A synaptosome fraction was prepared as described previously (15). In brief, mice were killed by cervical dislocation and the cerebral cortex and hippocampus were immediately dissected into ice-cold phosphate-buffered saline (PBS) and homogenized together with a Teflon pestle and glass homogenizer (12 up-and-down strokes at 900 rpm) in 10 volumes (wt/vol) of an ice-cold solution containing 0.32 M sucrose and 5 mM HEPES-NaOH (pH 7.4). All subsequent isolation procedures were performed at 0 to 4°C. The homogenate was centrifuged at 3,000 × g for 3 min to remove nuclei, and the resulting supernatant was centrifuged at 9,400 × g for 12 min. The resulting pellet (a crude synaptosomal fraction) was resuspended in a solution containing 20 mM HEPES-NaOH (pH 7.4), 140 mM NaCl, 5 mM KCl, 10 mM glucose, 5 mM NaHCO3, 1.2 mM Na2HPO4, and 1 mM MgCl2 (saturated with 95% O2 and 5% CO2 for 2 h before use) to yield a protein concentration of 1 mg/ml as determined with a Pierce bicinchoninic acid protein assay kit (Thermo Fisher Scientific, Waltham, MA). Bovine serum albumin and CaCl2 were then added to final concentrations 0.7 mg/ml and 1.33 mM, respectively, and the suspension was incubated for 10 min at 37°C before the induction of depolarization of the synaptosomal membrane by the addition of KCl to a final concentration of 25 mM. After incubation for various times, the synaptosomes were collected by centrifugation at 3,000 × g for 5 min at 4°C. The synaptosomes were subsequently homogenized in a solution containing 20 mM Tris-HCl (pH 7.5), 140 mM NaCl, 1 mM EDTA, 1 mM EGTA, 50 mM NaF, 1% Nonidet P-40, 0.2% sodium deoxycholate, 1 mM phenylmethylsulfonyl fluoride, aprotinin (10 μg/ml), and 1 mM sodium vanadate and subjected to immunoblot analysis.
Immunoblot analysis.
Immunoblot analysis of brain homogenates or of cell or synaptosome lysates was performed as described previously (16).
Histological analysis.
For immunohistofluorescence analysis, mice were anesthetized with ether and then perfused transcardially with fixation buffer (4% paraformaldehyde in 0.1 M phosphate buffer [pH 7.4]). Brain tissue was dissected and fixed again in fixation buffer overnight at 4°C with gentle shaking. The tissue was then transferred sequentially to a series of sucrose solutions (7, 20, and 30% [wt/vol] in 0.1 M phosphate buffer [pH 7.4]) for cryoprotection, embedded in optimal cutting temperature compound (Sakura Fine Technical, Tokyo, Japan), and rapidly frozen in liquid nitrogen. Frozen sections with a thickness of 10 μm were prepared with a cryostat, mounted on glass slides, and air dried. All sections were then incubated for 1 h at room temperature in buffer G (PBS supplemented with 5% goat serum and 0.1% Triton X-100) and stained overnight at room temperature with primary antibodies diluted in the same buffer. They were then washed with PBS and exposed to corresponding secondary antibodies conjugated with the fluorescent dye Cy3 (Jackson Immuno Research Laboratories, West Grove, PA) or Alexa Fluor 488 (Invitrogen, Carlsbad, CA). Nuclei were also stained with DAPI. Fluorescence images were acquired with a fluorescence microscope (DM RXA; Leica, Wetzlar, Germany) equipped with a cooled charge-coupled device (CCD) camera (Cool SNAP HQ; Roper Scientific, Trenton, NJ) and IPLab Image analysis software (Scanalytics, Inc., Billerica, MA). For Nissl staining, anesthetized mice were perfused transcardially with fixation buffer, after which the brain was removed, reexposed to the same fixative, embedded in paraffin, and sectioned at a thickness of 10 μm. Sections were stained with 0.1% cresyl violet acetate to reveal neuronal structures.
Mouse behavioral tests.
Behavioral tests were performed with male littermates. The light-dark transition test, open-field test, thermal preference test, rotarod test, Crawley-type social interaction test, acoustic startle response and prepulse inhibition test, Porsolt forced-swim test, and tail suspension test were performed essentially as described previously (16–22). All tests were performed with equipment from O'Hara & Co. Ltd. (Tokyo, Japan). In all those requiring digital imaging of mice, data were recorded with a CCD camera and analyzed with NIH Image software (developed by W. Rasband at the National Institute of Mental Health, Bethesda, MD) modified appropriately for each test (17) (available through O'Hara & Co. Ltd.). A sequence of the behavioral tests described above was started with Shp2flox/flox and Shp2 cKO mice at 13 to 17 weeks of age and took 5 weeks to complete. For the open-field test, mice were placed at the corner of the open-field apparatus (50 by 50 by 40 cm; O'Hara & Co. Ltd., Tokyo, Japan), which was illuminated at 100 lx, and their behavior was recorded over a 60-min test period in digital images obtained with equipment from O'Hara & Co. Ltd. Traveled distance, vertical activity (rearing, measured by counting photobeam interruptions), and time spent in the central area were calculated by analysis of the images with NIH Image-based software. After the behavioral tests described above, monitoring of home cage activity was started with the same mice. For this test, mice were housed individually in a test cage under a 12-h light, 12-h dark cycle with free access to both food and water. The spontaneous activity of each mouse was monitored for 7 consecutive days with an apparatus equipped with a paired infrared pyroelectric sensor, which measures the radiant body heat of the animal (O'Hara & Co. Ltd.). At the end of the monitoring test, mice were 19 to 26 weeks of age. Morris water maze tests were performed with another set of male mice at 26 to 30 weeks of age as described previously (12), with some modifications. The tests were carried out in a pool (100 cm in diameter) filled with opaque water at 23°C that contained a hidden platform (10 cm in diameter) in a laboratory with prominent external cues. The top surface of the platform was 1 cm below the water surface. On days 1 to 3, mice were trained to find the hidden platform maintained in the same position, with six trials per day and an intertrial interval of ∼30 min. In each trial, the mouse was allowed to swim until it found the platform or until 120 s had elapsed. If a mouse failed to escape, it was guided to the platform and then allowed to sit on it for 30 s. On day 4, the platform was removed and the first probe trial was performed for 120 s. On days 4 to 6, beginning 1 h after the first probe trial, the platform was placed in the quadrant opposite to its original location and reversal learning was monitored with six trials per day. On day 40, the platform was removed and the second probe trial was performed for 120 s. On day 41, mice were subjected to six trials with a visible platform that was marked with a small yellow flag 15 cm above the surface of the water. The behavior of mice was recorded with a CCD camera, and the total distance traveled, the path traveled to reach the platform, swimming speed, escape latency to discovery of the platform, and time spent in the target and other quadrants were measured automatically (O'Hara & Co. Ltd.).
Exposure of mice to a novel environment.
Each mouse was housed in an isolated cage for 1 week and then transferred from the home cage to a new cage in another room and exposed to the novel environment for various times. The mice were then killed by cervical dislocation, and the cerebral cortex, hippocampus, and striatum were immediately dissected into ice-cold PBS and frozen with liquid nitrogen before analysis. As a control, mice were killed within 5 min after transfer from the home cage to the new cage.
Culture and treatment of primary cortical neurons.
The cerebral cortex was isolated from mouse embryos at gestational day 18, and cortical neurons were isolated and cultured as described previously (23). Cultured neurons were infected with a lentiviral vector encoding a short hairpin RNA (shRNA) specific for mouse Shp2 mRNA (shShp2) or a control scrambled shRNA (shCtrl) the day after plating (2 days in vitro [div]) and then cultured for 10 or 14 days before experiments. They were treated for various times either with BDNF (50 ng/ml) beginning at 14 div or with culture medium containing 25 mM KCl beginning at 10 div.
Depletion of Shp2 by RNAi in cultured neurons.
Two 19-nucleotide sequences, 5′-ACATCCTGCCCTTTGATCA-3′ and 5′-GGTGGTTCCATGGTCACTT-3′, corresponding to nucleotides 514 to 532 and 1222 to 1240 of the mouse Shp2 mRNA (NM011202.3), respectively, were selected as target sequences for RNA interference (RNAi). A random 19-nucleotide sequence (5′-GGTAACTTCCGGTTTGGTC-3′) was selected for a control scrambled shRNA. Two pairs of 64-bp oligonucleotides containing a target sequence and the reverse complementary sequence on the same strand (pair 1, 5′-GATCCCCGGTGGTTCCATGGTCACTTTTCAAGAGAAAGTGACCATGGAACCACCTTTTTGGAAA-3′ and 5′-AGCTTTTCCAAAAAGGTGGTTCCATGGTCACTTTCTCTTGAAAAGTGACCATGGAACCACCGGG-3′; pair 2, 5′-GATCCCCACATCCTGCCCTTTGATCATTCAAGAGATGATCAAAGGGCAGGATGTTTTTTGGAAA-3′ and 5′-AGCTTTTCCAAAAAACATCCTGCCCTTTGATCATCTCTTGAATGATCAAAGGGCAGGATGTGGG-3′) and one pair of oligonucleotides containing the random sequence and its reverse complementary sequence (5′-GATCCCCGGTAACTTCCGGTTTGGTCTTCAAGAGAGACCAAACCGGAAGTTACCTTTTTGGAAA-3′ and 5′-AGCTTTTCCAAAAAGGTAACTTCCGGTTTGGTCTCTTGAAGACCAAACCGGAAGTTACCGGG-3′) were synthesized. Double-stranded DNAs prepared by annealing each pair of oligonucleotides were used to construct lentiviral vectors that encode both the shRNA and enhanced green fluorescent protein (EGFP) as described previously (24). Lentivirus particles were also produced as described previously (25). Data are presented for one of the Shp2 shRNAs, but similar results were obtained with both.
Quantitative PCR analysis.
Total RNA was extracted from dissected brain areas or cultured neurons with Sepasol RNA I (Nacalai Tesque) and an RNeasy minikit (Qiagen, Hilden, Germany). The cerebral cortex (including the anterior portion of the primary and secondary motor cortex, as well as the cingulate cortex [areas 1 and 2]), the hippocampus, and the striatum (caudate putamen) were dissected according to the atlas of Paxinos and Franklin (26). First-strand cDNA was synthesized from 1 μg of total RNA with a QuantiTect reverse transcription kit (Qiagen), and cDNA fragments of interest were amplified by real-time PCR in 96-well plates (Roche Diagnostics, Mannheim, Germany) with a QuantiTect SYBR green PCR kit (Qiagen) and a LightCycler 480 instrument (Roche Applied Science). The amplification results were analyzed with LightCycler 480 software and then normalized on the basis of the Gapdh mRNA level in each sample. The sequences of the primers used (forward and reverse, respectively) were as follows: c-Fos, 5′-AGGGCAGCAGCAGCAACGAG-3′ and 5′-CTCGGGCAGTGGCACGTCTG-3′; Arc, 5′-GGCTCCCCGACCAGACCCTT-3′ and 5′-GCTTGACCTGGCAGGCAGGG-3′; Npas4, 5′-GGGGCGCGAAGAGACGCTAC-3′ and 5′-GTCCCGCACCAGGTACACGC-3′; Homer1a, 5′-AGCAGGCTGTGGAACAAGGAAGT-3′ and 5′-TGAGCATTAGCAGCAGAGTGTCCT-3′; Bdnf, 5′-TTGCCAGAGCCCCAGGTGTGAC-3′ and 5′-CTCAACGCCTGTCACTGAGCCCTA-3′; Zif268, 5′-GCAAGTACCCCAACCGGCCC-3′ and 5′-CGGCGATCGCAGGACTCGAC-3′; Drd1 (D1 dopamine receptor), 5′-ACCCCTTCCCGGTGTGTGTTGA-3′ and 5′-TACCCCCACCCGAGGGGACT-3′; Drd2 (D2 dopamine receptor), 5′-TGGCAAAACCCGGACCTCCCT-3′ and 5′-TGTGCGTGATGAAGAAGGGCAGC-3′; and Gapdh, 5′-TCCCACTCTTCCACCTTCGA-3′ and 5′-GTCCACCACCCTGTTGCTGTA-3′.
EEG and EMG recording.
The electroencephalogram (EEG) was recorded via four electrodes implanted in the skull of a mouse anesthetized by intraperitoneal injection of sodium pentobarbital (50 mg/kg). The electrodes were connected to a 5-mm2 computer circuit board and located at ±2.5 mm lateral and ±2.5 mm anterior or posterior relative to the bregma (26). The electromyogram (EMG) was recorded with electrodes implanted into the trapezius muscle of a mouse anesthetized as described above. The mouse was allowed to recover for at least 7 days after electrode implantation. EEG and EMG signals were amplified, filtered (EEG, 0.5 to 30 Hz; EMG, 15 to 300 Hz), and digitized at a sampling rate of 128 Hz with EEG4214 (Nihon Koden, Tokyo, Japan) and PowerLab8/30 (AD Instruments, Dunedin, New Zealand). The processed data were then analyzed with Labchart 5.0 software (AD Instruments).
Electrophysiology.
All electrophysiological experiments were performed in compliance with a protocol approved by the Ethics Review of Nippon Medical School. Animals were deeply anesthetized by halothane inhalation (∼2%, vol/vol, in air), and the brain was rapidly removed. Transverse slices (350 μm thick) were prepared from the mid-hippocampus with a tissue slicer (VT1200; Leica) at 4°C in a solution containing 299.2 mM sucrose, 3.4 mM KCl, 0.3 mM CaCl2, 3.0 mM MgCl2, 10 mM HEPES-NaOH (pH 7.4), 0.6 mM NaH2PO4, and 10 mM glucose. The slices were incubated at 30°C for 30 min and then maintained for >90 min in a submersion chamber containing artificial cerebrospinal fluid (ACSF; 138.6 mM NaCl, 3.4 mM KCl, 2.0 mM CaCl2, 1.3 mM MgCl2, 21.0 mM NaHCO3, 0.6 mM NaH2PO4, 10 mM glucose). Each slice was placed in a submersion-type recording chamber with ACSF at 28°C flowing at a rate of 1.5 ml/min. Extracellular field excitatory postsynaptic potentials (fEPSPs) evoked with a bipolar tungsten stimulating electrode (Unique Medical, Tokyo, Japan) were recorded in the stratum radiatum of area CA1 with a glass microelectrode (World Precision Instruments, Sarasota, FL) filled with 2 M NaCl (electrical resistance, 2 to 4 MΩ). The stimulus was adjusted so that the induced fEPSP did not reach saturation and was delivered at a frequency of 0.05 Hz. Evoked fEPSPs were acquired and controlled with an Axon 700B Multiclamp amplifier and pClamp acquisition software (Molecular Devices, Sunnyvale, CA). A baseline recording of >20 min was obtained before induction of long-term potentiation (LTP) or posttetanic potentiation (PTP) with a standard single-tetanus protocol for the CA1 region (100 Hz for 1 s in the absence or presence of 50 μM APV, respectively). The fEPSP slope was measured over ∼1-ms time windows positioned on the early rising phase. Paired-pulse facilitation (PPF) was induced by delivery of two stimuli with an interstimulus interval of 50 to 500 ms. Chemically induced, metabotropic glutamate receptor (mGluR)-dependent long-term synaptic depression (LTD) was elicited by bath application of 100 μM DHPG for 10 min. Data were stored in a personal computer for subsequent analysis.
In vivo microdialysis.
In vivo microdialysis was performed essentially as described previously (27). In brief, mice were anesthetized by intraperitoneal injection of sodium pentobarbital (40 mg/kg), and a guide cannula (one site per animal) for a dialysis probe (Eicom, Kyoto, Japan) was implanted stereotaxically in the striatum (A, +0.5 mm; L, −1.8 mm; and V, −2.7 mm relative to the bregma) (26). The cannula was cemented in place with dental acrylic, and the animals were maintained warm and allowed to recover from anesthesia. The active probe membrane was 1 mm in length. On the day after surgery, the probe was perfused with Ringer's solution (147.2 mM NaCl, 4.0 mM KCl, 2.2 mM CaCl2) (Fuso Pharmaceutical Industries, Osaka, Japan) at a constant flow rate of 2 μl/min. Experiments were initiated after a stabilization period of 3 h. Microdialysis samples (20 μl) were collected every 10 min and assayed for dopamine, norepinephrine, and serotonin by high-performance liquid chromatography with electrochemical detection.
Measurement of tissue monoamine content.
The monoamine content of the tissue in the striatum was measured as described previously (16).
Pharmacological experiment.
For examination of the effect of methylphenidate on locomotor activity, mice were placed in an open-field apparatus (45 by 45 by 30 cm, Acti-Track; Panlab, Barcelona, Spain) (28) and their behavior was recorded for 90 min (habituation period). The mice were then injected intraperitoneally with methylphenidate (3 mg/kg) or the same volume of saline, and their behavior was recorded continuously for 30 min.
Statistical analysis.
Data are presented as mean values ± the standard error of the mean (SEM) and were analyzed with Student's t test or repeated-measure analysis of variance (ANOVA). A P value of <0.05 was considered statistically significant.
RESULTS
Forebrain neuron-specific knockout of the Shp2 gene in mice.
To investigate the functions of Shp2 in the adult brain, we generated Shp2 conditional-knockout (Shp2flox/flox; CaMKIIα-Cre or cKO) mice by crossing Shp2flox/flox mice with CaMKIIα-Cre (CRE-159) transgenic mice, in the latter of which Cre recombinase is expressed specifically in forebrain neurons after postnatal day 20 (P20) under the control of the CaMKIIα gene promoter (12). In the Shp2flox/flox mice, exon 11 of the Shp2 gene, which encodes a region of the protein containing the cysteine residue in the catalytic center, is flanked by loxP sites (11).
The cKO mice were viable and fertile. Consistent with the results of a previous study in which the TrkB gene was deleted from the same CRE-159 mouse line (12), immunoblot analysis revealed that the amount of Shp2 protein in the hippocampus, striatum, and cerebral cortex of Shp2 cKO mice at P90 was reduced by 37 to 49% compared with that in control mice, whereas the abundance of Shp2 in the cerebellum did not differ between the two genotypes (Fig. 1A). Deletion of the Shp2 gene did not result in any apparent defects in brain structure, as revealed by Nissl staining (Fig. 1B). Immunohistofluorescence analysis revealed that Shp2 was expressed in NeuN-positive neurons in the hippocampus (CA1 region), striatum, and cerebral cortex and that the amount of Shp2 immunoreactivity was markedly reduced in the brains of Shp2 cKO mice (Fig. 1C; data not shown). In contrast, expression of Shp2 was not affected in parvalbumin-positive interneurons in the CA1 region of the hippocampus in Shp2 cKO mice (Fig. 1D), consistent with the previous study of TrkB (12). Immunostaining of Tau-1 (an axonal marker), MAP2 (a dendritic marker), calbindin (a mossy fiber marker), and GFAP (an astrocytic marker) revealed no apparent abnormalities in the abundance or distribution of these proteins in the hippocampi of Shp2 cKO mice (data not shown). The expression of DCX, a maker of neurogenesis, also appeared normal in the dentate gyri of Shp2 cKO mice (data not shown). Immunoblot analysis also revealed that the expression levels of neuronal proteins (Tau-1 and MAP2), glial proteins (GFAP), and synaptic proteins (synaptophysin, VAMP2, the GluA1 subunit of the α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid [AMPA]-sensitive glutamate receptor, the GluN2B subunit of the N-methyl-d-aspartate [NMDA]-sensitive glutamate receptor, and PSD-95) in the hippocampus and cerebral cortex did not differ between Shp2 cKO and control mice (data not shown).
FIG 1.

Generation of postmitotic forebrain neuron-specific Shp2 knockout mice. (A) Immunoblot analysis of Shp2 in homogenates of the hippocampus (Hip), cerebral cortex (Cbr), striatum (Str), or cerebellum (Cer) of Shp2flox/flox (control [Ctrl]) or Shp2 cKO mice at P90. The intensities of the Shp2 bands relative to the corresponding values for control mice were determined as mean ± SEM (n = 7 mice). **, P < 0.01 (Student's t test). NS, not significant. (B) Nissl staining of coronal sections prepared from the hippocampus or cerebral cortex of control or Shp2 cKO mice at 14 weeks of age. Scale bars, 200 μm. (C and D) Immunofluorescence staining of coronal sections of the hippocampi (CA1 region) of control or Shp2 cKO mice at 20 weeks of age with antibodies to Shp2 (red) and either antibodies to the neuronal nuclear antigen NeuN (green) (C) or antibodies to parvalbumin (green) (D). Merged images are also shown. The boxed areas in panel C are shown at a higher magnification in the insets. Scale bars, 50 μm. (E) The body weights of male and female Shp2flox/flox (control [Ctrl]) or Shp2 cKO mice maintained on normal chow were monitored weekly from 5 to 20 weeks of age. Data are mean values ± SEM (n = 20 male mice; n = 11 [control] or 18 [cKO] female mice). *, P < 0.05 versus the corresponding value for control mice (Student's t test). Data in panels B to D are representative of at least three experiments.
Neuron-specific ablation of Shp2 has been reported to induce severe obesity in mice (9, 10). The body weights of males and female Shp2 cKO mice were 13 and 22% greater than those of control (Shp2flox/flox) littermates, respectively, at 18 weeks of age (Fig. 1E), suggesting that the obesity phenotype of Shp2 cKO mice was mild. A similar but not significant trend was also observed in another set of experiment with a small number of male mice (30.3 ± 0.95 g [control] versus 33.6 ± 1.13 g [Shp2 cKO] [mean ± SEM; n = 3 for control mice and 4 for Shp2 cKO mice] at 18 weeks of age [P = 0.089 by Student's t test]). The greater subcutaneous white adipose tissue weight in these cKO mice than in control mice was not significant (1.09 ± 0.23 g [control] versus 1.33 ± 0.26 g [Shp2 cKO] [mean ± SEM; P = 0.535 by Student's t test]), suggesting that the obesity phenotype was mild in our cKO mice. Blood glucose levels in both the randomly fed (182 ± 21.4 mg/dl [control] versus 213.8 ± 20.1 mg/dl [Shp2 cKO] [mean ± SEM; P = 0.335 by Student's t test]) and 22-h-fasted (109.7 ± 12.9 mg/dl [control] versus 98.5 ± 10.2 mg/dl [Shp2 cKO] [mean ± SEM; P = 0.520 by Student's t test]) states were also similar between the two genotypes.
Abnormal behaviors of Shp2 cKO mice.
We next performed a battery of behavioral tests with Shp2 cKO mice. The mutant mice showed normal behavior in the tail suspension test, in which an increase in immobility time represents a state of despair (29); in the rotarod test, which evaluates both motor function and learning (30); or in the thermal preference test, which evaluates thermosensation (21; data not shown). These results suggested that Shp2 cKO mice had a normal stress response, normal motor coordination, and normal thermosensation and nociception. In contrast, the mutant animals exhibited a reduced stay time in a dark place, an index of anxiety-like behavior, as well as a greater number of transitions and a greater distance traveled, in the light-dark transition test (31), whereas their latency to light was normal (Fig. 2A). The mutant mice also exhibited reduced depression-like behavior (immobility) in the forced-swim test (32) (Fig. 2B) and an increased acoustic startle response (33), whereas prepulse inhibition, a measure of sensorimotor gating (34), was normal (Fig. 2C). In addition, Shp2 cKO mice exhibited a slight reduction in the interaction with a novel stranger mouse in the Crawley-type social interaction test (35) (Fig. 2D). Some of these abnormal behaviors are likely due to the hyperactivity of Shp2 cKO mice, because the mutant mice exhibited markedly increased locomotor activity in the open-field test (36) (Fig. 2E). The distance traveled by the mutant mice during each 5-min block of the 60-min test period was thus consistently greater than that traveled by Shp2flox/flox mice (Fig. 2E). The number of rearings was also greater for Shp2 cKO mice than for control mice (Fig. 2E), whereas the time spent in the center of the field, a common measure of anxiety-like behavior, did not differ significantly between the two genotypes (Fig. 2E). We also measured the home cage activity of Shp2 cKO and control mice at 19 to 26 weeks of age for 7 consecutive days (Fig. 2F). The mutant mice manifested hyperactivity during the dark period, but not in the light period, compared with control mice. We also investigated an age-dependent change in hyperactivity with the same data. The mean activity of control mice during the dark phase on the 7th day of home cage monitoring was reduced at 26 weeks of age compared with that at 19 weeks of age (13,854 ± 1,998 arbitrary units [A.U.] [n = 6 mice at 19 weeks of age] versus 8,316 ± 1,140 A.U. [n = 5 mice at 26 weeks of age] [mean ± SEM; P = 0.049 by Student's t test]). In contrast, age-dependent reduction of locomotor activity was not observed in Shp2 cKO mice (24,506 ± 5,573 A.U. [n = 6 mice at 19 weeks of age] versus 29,130 ± 5,544 A.U. [n = 5 mice at 26 weeks of age] [mean ± SEM; P = 0.575 by Student's t test]). Thus, the hyperactivity phenotype of Shp2 cKO mice became more prominent in an age-dependent manner than in control mice.
FIG 2.


Behavioral analyses of Shp2 cKO mice. (A) Light-dark transition test. Shp2flox/flox (control [Ctrl]) or Shp2 cKO mice were allowed to move freely between two equal-size chambers (21 by 21 by 25 cm) connected by a door. One chamber was illuminated at 390 lx, whereas the other was dark (2 lx). At the start of the test, a mouse was placed in the dark chamber and its behavior was monitored for 10 min. The latency to the first entry into the light chamber, the total number of transitions between the two chambers, the time spent in each chamber, and the total distance traveled (left to right panels, respectively) were recorded automatically. (B) Porsolt forced-swim test. Mice were subjected to the forced-swim test in a Plexiglas cylinder filled with water to a height of 7.5 cm. The animals were subjected to the same test twice with a 24-h intertest interval (left and right panels, respectively), and their behavior was recorded with a CCD camera. Immobility was determined automatically from the images, and immobility time is expressed as a percentage of each 1-min interval. (C) Acoustic startle response (ASR) and prepulse inhibition (PPI). The ASR and PPI were measured with an automatic startle reflex recording system. Mice were placed in a Plexiglas cylinder and left for 10 min without stimulation. A 110- or 120-dB startle stimulus was then delivered for 40 ms either alone (left panel) or 40 ms after a 40-ms prepulse stimulus of 74, 78, or 82 dB (right panel). The amplitude of the ASR is expressed in A.U. The magnitude of PPI was determined as the percent decrease in the startle response after exposure to a prepulse compared with the startle response to the pulse alone. (D) Crawley-type (three-chamber) social interaction test. Social interaction of mice was measured automatically with an apparatus consisting of three chambers (left panel). A mouse was placed in the central chamber and allowed to move freely among the three chambers for 10 min. A lattice cage was then placed in a corner of both the left and right chambers, and a stranger mouse (stranger 1) was placed in one of the cages whereas the other cage was left empty. The time spent by the test mouse in the quadrant-like space around each cage (gray zones in the left panel) during a 10-min period was measured (center panel). Immediately after this first test, the mouse was tested again under the same conditions, except that a novel stranger mouse (stranger 2) was placed in one of the two cages and the familiar mouse used in the previous test was placed in the other (right panel). (E) Behavior of Shp2flox/flox (Ctrl) or Shp2 cKO mice in the open-field test. The total distance traveled (left panel), number of rearings (center panel), and stay time in the central area of the open field (right panel) were measured automatically every 5 min during a 60-min test period. (F) Locomotor activity of control or Shp2 cKO mice in the home cage. Activity was determined as the number of spontaneous movements measured automatically for 2-h intervals over 7 consecutive days. Vertical white and gray stripes represent light and dark periods, respectively. ZT, zeitgeber time. All data are mean values ± SEM (n = 17 to 20 mice). P values for comparisons between control and Shp2 cKO mice or for the comparisons indicated were determined by Student's t test (A, C, D) or repeated-measure ANOVA (B, E, F). *, P < 0.05; **, P < 0.01; NS, not significant.
Attenuated novelty-induced expression of immediate-early genes (IEGs) in the brains of Shp2 cKO mice.
Dysregulation of brain monoamine systems, in particular, that of the dopamine axis, has been shown to give rise to hyperactive behavior in animal models (37). However, the content of monoamines in the striatum, behavioral responses to methylphenidate (an inhibitor of monoamine transporters), and the abundance of D1 and D2 dopamine receptor mRNAs in the striatum did not differ between Shp2 cKO and control mice (data not shown). In vivo microdialysis also revealed that intraperitoneal administration of methylphenidate increased the extracellular levels of dopamine and norepinephrine in the striatum to similar extents in both Shp2 cKO and control mice (data not shown). Dysregulation of monoamine systems was thus not evident in the brains of Shp2 cKO mice.
Hyperactivity is frequently associated with reduced neural activity or neuronal loss after brain injury (37–40). We therefore next measured the expression of IEGs, including those for c-Fos, Zif268, Npas4, Arc, Homer1a, and Bdnf, in the brains of Shp2 cKO mice as an index of neuronal activity (41, 42). Exposure of control mice to a novel environment increased the expression of all of these IEGs in the cerebral cortex, hippocampus, and striatum within 1 h, with the expression levels of all of the genes, with the exception of that for Bdnf, having declined at 4 h in all three brain regions (Fig. 3). Exposure to a novel environment also increased the expression of all of the IEGs examined in the cerebral cortices, hippocampi, and striata of Shp2 cKO mice, but to a lesser extent than in control mice (Fig. 3). The basal expression levels of IEGs in Shp2 cKO mice were similar to those in control mice. These results thus suggested that the novelty-induced increase in neuronal activity was broadly suppressed in the forebrain area of Shp2 cKO mice.
FIG 3.
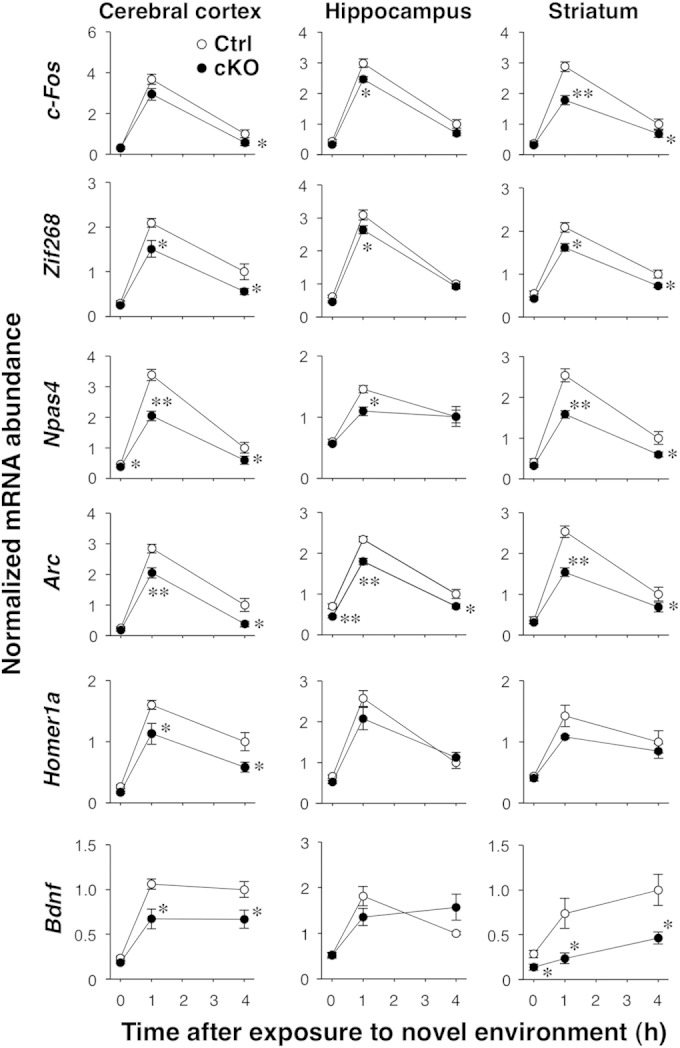
Novelty-induced IEG expression in the brains of Shp2 cKO mice. Shp2flox/flox (control [Ctrl]) or Shp2 cKO mice at 14 to 20 weeks of age were exposed to a novel environment for 0, 1, or 4 h, immediately after which the cerebral cortex, hippocampus, and striatum were dissected for isolation of total RNA. The expression of the IEGs indicated was then determined by quantitative PCR analysis. The amount of each mRNA was normalized to that of glyceraldehyde-3-phosphate dehydrogenase mRNA and is presented relative to the mean value of control mice at 4 h. Data are mean values ± SEM (n = 6 mice). *, P < 0.05; **, P < 0.01 (versus the corresponding value of control mice; Student's t test).
We also measured the neuronal activity of Shp2 cKO mice in vivo by recording the EEG from the cerebral cortex on 3 consecutive days. Simultaneous recording of an EMG from the trapezius muscle revealed that the muscle activity of Shp2 cKO mice was significantly higher than that of control mice in the awake state (mean spectral power of EMG of 450.9 ± 20.1 μV/S2 [control mice, n = 5] versus 1,261.2 ± 41.8 μV/S2 [Shp2 cKO mice] [n = 5, mean ± SEM; P = 1.2 × 10–7 by Student's t test]) but not during sleep (mean spectral power of EMG of 67.6 ± 3.4 μV/S2 [control mice, n = 5] versus 69.82 ± 3.7 μV/S2 [Shp2 cKO mice] [n = 5, mean ± SEM; P = 0.67 by Student's t test]), consistent with the hyperactivity of these animals in the awake state (Fig. 2F). Shp2 cKO mice also exhibited an increase in δ waves (0.5 to 4 Hz) on the EEG in the awake state (mean spectral power of EEG of 411.3 ± 26.3 μV/S2 [control mice, n = 5) versus 600.7 ± 25.8 μV/S2 [Shp2 cKO mice, n = 5] [mean ± SEM; P = 3.4 × 10–3 by Student's t test]) but not during sleep (mean spectral power of EEG of 747.1 ± 37.8 μV/S2 [control mice, n = 5] versus 758.0 ± 36.6 μV/S2 [Shp2 cKO mice, n = 5] [mean ± SEM; P = 0.42 by Student's t test]). These waves are characteristic of non-rapid-eye-movement sleep and are associated with a hyperpolarized “down” state of neurons (43). The EEG data thus also suggested that neuronal activity is reduced in the brains of Shp2 cKO mice.
Effects of Shp2 ablation on MAPK activation in vivo and in vitro.
We next examined the effect of genetic ablation of Shp2 on activation of the MAPK extracellular-signal-regulated kinase (Erk) in the brains of Shp2 cKO mice. Immunoblot analysis revealed that exposure of control mice to a novel environment induced the phosphorylation (activation) of Erk isoforms 1 and 2 (Erk1/2) in the hippocampus and cerebral cortex (Fig. 4A and B). In contrast, significant activation of Erk1/2 was not observed in the brains of Shp2 cKO mice after exposure to a novel environment (Fig. 4A and B). A previous study suggested that LS-type Shp2 mutants have a stimulatory effect on the phosphoinositol 3-kinase/Akt/GSK3β pathway (44); thus, we also examined the activation of Akt and GSK3β in the cerebral cortex of mice before and after exposure to a novel environment. However, phosphorylation of Akt (Ser473) and GSK3β (Tyr279) did not change significantly in response to stimulation, and no significant difference between the two genotypes was found (data not shown).
FIG 4.
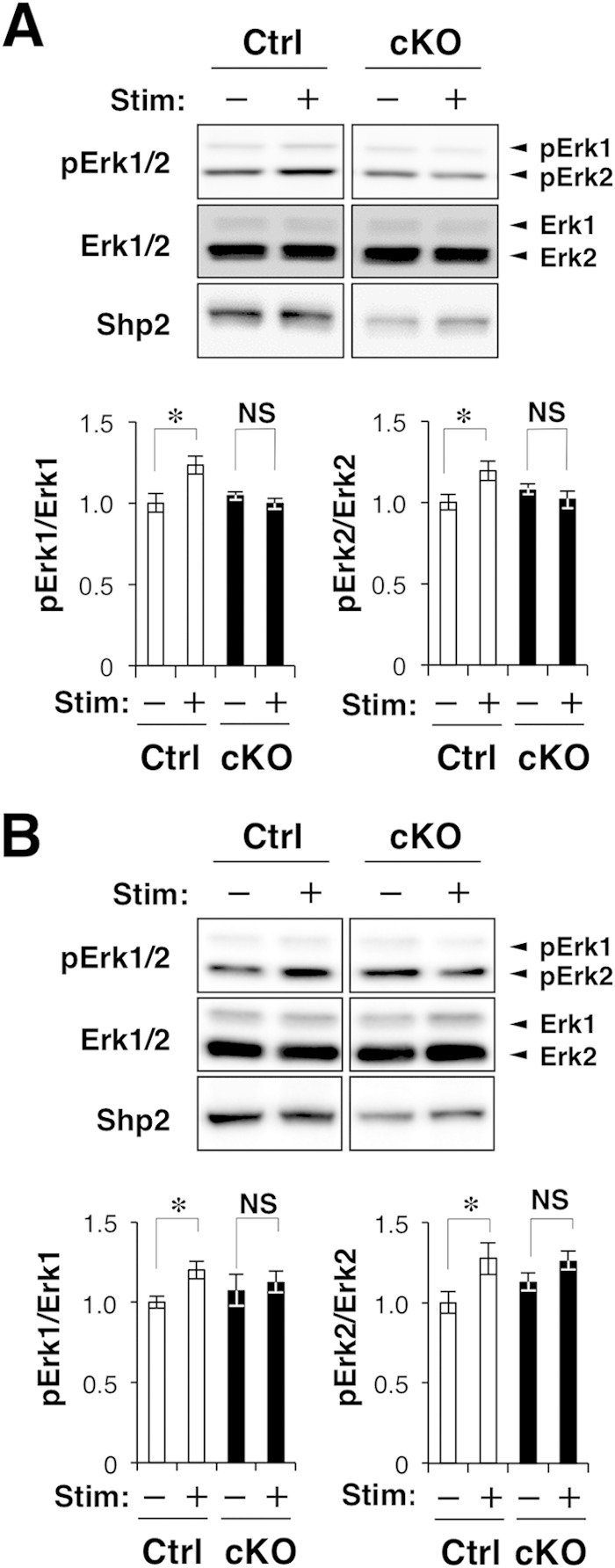
Novelty-induced activation of Erk in the brains of Shp2 cKO mice. (A, B) Shp2flox/flox (control [Ctrl]) or Shp2 cKO mice at 14 to 20 weeks of age were exposed to a novel environment for 0 (Stim –) or 0.5 h (Stim +), immediately after which homogenates of the hippocampus (A) and cerebral cortex (B) were prepared and subjected to immunoblot analysis with antibodies to the phosphorylated (p) or total form of Erk1/2 and to Shp2. The ratio of the intensity of the pErk1 or pErk2 bands to that of the Erk1 or Erk2 bands, respectively, was determined and expressed relative to the corresponding value for control mice without stimulation. Quantitative data are mean values ± SEM (n = 6 to 8 mice). *, P < 0.05 (Student's t test); NS, not significant.
Given that Erk activation correlates with neuronal activity (45, 46), reduced activation of Erk in the brains of Shp2 cKO mice is thus consistent, in part, with the notion that novelty-induced neuronal activation is suppressed in the mutant mice. In contrast, the Ras-MAPK pathway is important for IEG expression in response to neuronal activity (41, 42), and Shp2 is indispensable for activation of the Ras-MAPK pathway (1, 2). So, it was also possible that the suppression of the IEG response in Shp2 cKO mice was due to dysregulation of the Ras-MAPK pathway rather than to a reduced level of neuronal activity. To address this possibility, we examined the effect of ablation of Shp2 on neuronal-activity-induced activation of Erk1/2 and IEG expression in vitro. Cultured mouse cortical neurons were infected with a lentivirus that encodes EGFP and either a shRNA specific for mouse Shp2 mRNA (shShp2) or a control scrambled shRNA (shCtrl), and the cells were then exposed to a high K+ concentration (25 mM) to induce membrane depolarization. Depletion of Shp2 with shShp2 did not affect the basal activity of Erk1/2 (Fig. 5A). Unexpectedly, however, the high-K+-induced activation of Erk1/2 was significantly enhanced by Shp2 depletion, with this effect being most pronounced at 5 to 10 min, suggesting that Shp2 negatively regulates high-K+-induced activation of Erk1/2. Neither basal nor high-K+-induced expression of the IEGs for c-Fos, Zif268, or Npas4 was affected by knockdown of Shp2, whereas the high-K+-induced expression of the Arc was attenuated in Shp2-depleted neurons (Fig. 5B). These data thus suggested that Shp2 does not contribute to the high-K+-induced expression of the mRNAs for c-Fos, Zif268, and Npas4.
FIG 5.

Effects of Shp2 ablation on membrane depolarization-induced Erk activation and IEG expression. (A) Immunoblot analysis of Erk1/2 phosphorylation in cultured mouse cortical neurons infected with a lentivirus encoding Shp2 or control shRNAs and incubated in culture medium containing 25 mM KCl for 0, 5, 10, or 30 min. Quantitative data are expressed relative to the value for shCtrl-treated cells at time zero. (B) Quantitative PCR analysis of IEG expression in cultured neurons infected as described for panel A and exposed to normal culture medium (5 mM KCl) or medium containing 25 mM KCl for 60 min. Data are expressed relative to the value for shCtrl-treated cells exposed to a high K+ concentration. (C) Immunoblot analysis of Erk1/2 phosphorylation in synaptosomes prepared from the brains of Shp2flox/flox (control [Ctrl]) or Shp2 cKO mice at 43 to 46 weeks of age and exposed to medium containing 25 mM KCl for the indicated times. Quantitative data are expressed relative to the control value at time zero. All quantitative data are the mean value ± SEM of three independent experiments (A, C) or six culture dishes in three independent experiments (B). *, P < 0.05; **, P < 0.01 (Student's t test) for comparisons between shCtrl and shShp2 neurons (A, B) or between Ctrl and Shp2 cKO synaptosomes (C); NS, not significant.
We also examined the effect of Shp2 ablation on Erk1/2 activation in synaptosomes. High-K+ treatment induced Erk1/2 phosphorylation in synaptosomes from control mice, consistent with previous observations (47), and this effect was significantly enhanced in synaptosomes from Shp2 cKO mice (Fig. 5C). The basal level of Erk2 phosphorylation, but not that of Erk1 phosphorylation, was also significantly increased in synaptosomes from Shp2 cKO mice (Fig. 5C). These data suggested that Shp2 negatively regulates membrane depolarization-induced activation of Erk1/2 in synaptic terminals.
Given that Shp2 acts as a positive regulator of the Ras-MAPK pathway downstream of various growth factor receptors (1, 2), we further examined the effect of Shp2 knockdown on BDNF-induced MAPK activation in cultured neurons. Phosphorylation of Erk1/2 was increased in control neurons by treatment with BDNF, and this effect was significantly attenuated by Shp2 depletion (Fig. 6A). BDNF also increased the abundance of the mRNAs for c-Fos, Zif268, and Arc but not that of the mRNA for Npas4 in control neurons, and these effects were attenuated by Shp2 depletion (Fig. 6B), suggesting that Shp2 positively regulates BDNF-induced Erk activation, as well as expression of the genes for c-Fos, Zif268, and Arc. We also found that BDNF significantly increased the level of the mRNA for Npas4 in Shp2 knockdown neurons, suggesting that Shp2 negatively regulates BDNF-induced expression of the gene for Npas4.
FIG 6.

Effects of Shp2 depletion on BDNF-induced Erk activation and IEG expression. (A) Immunoblot analysis of Erk1/2 phosphorylation in mouse cortical neurons infected with a lentivirus encoding Shp2 or control shRNA and stimulated with BDNF (50 ng/ml) for 0, 5, 10, or 30 min. Quantitative data are expressed relative to the value for shCtrl-treated cells at time zero. (B) Quantitative PCR analysis of IEG expression in cultured neurons infected as described for panel A and incubated in the absence or presence of BDNF for 60 min. Data are expressed relative to the value for shCtrl-treated cells stimulated with BDNF. Quantitative data are the mean value ± SEM of four independent experiments (A) or five or six culture dishes in three independent experiments (B). *, P < 0.05 (Student's t test) for comparisons between shCtrl- and shShp2-treated neurons; NS, not significant.
Reduced posttetanic potentiation in the hippocampal CA1 region of Shp2 cKO mice.
Although it remained possible that the attenuation of novelty-induced IEG expression in the brains of Shp2 cKO mice was due to dysregulation of the Ras-MAPK pathway, suppression of neuronal activity was the most likely explanation for this phenotype. We therefore next addressed the functional role for Shp2 in synaptic transmission, which directly regulates neuronal activity. We first examined the relationship between fiber volley amplitude and the extracellular fEPSP slope (the input-output [I/O] relationship) at Schaffer-CA1 synapses in the hippocampus, a brain structure in which Cre activity is predominant in CRE-159 mice (12). Fiber volley size reflects the strength of depolarization of presynaptic terminals, whereas the fEPSP slope represents the postsynaptic response. The efficiency of synaptic transmission (mean I/O slope) was significantly lower in Shp2 cKO mice than in Shp2flox/flox mice (0.838 ± 0.065 versus 1.722 ± 0.250; mean ± SEM [n = 10 slices; P = 0.003 by Student's t test]) (Fig. 7A). We also examined PPF in hippocampal CA1 neurons. PPF is thought to reflect presynaptic accumulation of Ca2+ after an initial stimulus that leads to an increase in transmitter release in response to a second stimulus applied after a short interval (48). The ratio of the second fEPSP slope to the first fEPSP slope, or the paired-pulse ratio (PPR), is inversely related to the initial release probability (49). Thus, in general, the smaller the probability of release in response to the first pulse, the more facilitated the response to the second pulse and the higher the PPR. Two consecutive stimuli with an appropriate interval (50 to 100 ms) induced PPF in the hippocampal CA1 region in both Shp2 cKO and Shp2flox/flox mice (Fig. 7B). The PPR was significantly greater in Shp2 cKO mice than in control mice, however, at these short interstimulus intervals, suggesting that the initial release probability was substantially smaller in Shp2 cKO mice.
FIG 7.

Abnormal synaptic transmission in the hippocampi of Shp2 cKO mice. Synaptic transmission was examined at Schaffer collateral-CA1 synapses in hippocampal slices prepared from Shp2flox/flox (control [Ctrl]) or Shp2 cKO mice at 14 to 20 weeks of age. (A) Relationship between fiber volley amplitude and fEPSP slope (I/O relationship) for basal synaptic transmission examined over a range of stimulus intensities (1 to 6 V). The slope of the I/O curve was calculated for each slice for statistical analysis. (B) Ratio of the second to the first fEPSP slope (PPR) for PPF evoked by delivery of two stimuli with a short interstimulus interval. Sample traces show averages of three fEPSPs at an interstimulus interval of 50 ms. Calibration: 20 ms, 0.2 mV. **, P < 0.01 versus the corresponding control value (Student's t test). (C) fEPSP slope before (baseline) and after induction of LTP by tetanic stimulation (100 Hz, 1 s). Data are expressed as percentages of the baseline value. Sample traces show averages of three fEPSPs recorded before (1, thin lines) and after (2, thick lines) LTP induction. Calibration: 5 ms, 0.5 mV. (D) fEPSP slope examined as in panel C but in the presence of 50 μM APV. Sample traces show fEPSPs recorded before (1, thin lines) and after (2, thick lines) PTP induction. Calibration: 5 ms, 0.5 mV. All data are mean values ± SEM (n = 6 to 18 slices from three or four mice).
We next measured LTP of synaptic transmission, a model of activity-dependent synaptic plasticity, in CA1 neurons (Fig. 7C). High-frequency tetanic stimulation (100 Hz, 1 s) induced a rapid increase in the fEPSP slope that persisted for at least for 1 h in both Shp2 cKO and control mice. The average fEPSP slope at 40 to 60 min after tetanic stimulation was increased by 25 to 30% from the baseline and did not differ significantly between Shp2 cKO and control mice (130.7% ± 7.9% and 125.7% ± 7.2%, respectively [mean ± SEM, n = 10 slices; P = 0.6509 by Student's t test]). In contrast, in the early phase of LTP (0 to 5 min after tetanic stimulation), the potentiation of synaptic transmission was smaller in Shp2 cKO mice than in control mice (mean fEPSP slope of 186.6% ± 11.6% versus 218.7% ± 12.4% [mean ± SEM, n = 10 slices; P = 0.0875 by Student's t test]), although this difference did not achieve statistical significance. This early phase of synaptic potentiation includes PTP, a form of short-term synaptic plasticity, as a component. Presynaptic accumulation of Ca2+ during tetanic stimulation is thought to be important for PTP (48), whereas the induction of LTP at Schaffer-CA1 synapses depends on the function of postsynaptic NMDA receptors (50). Indeed, we found that PTP was apparent in the presence of the NMDA receptor antagonist APV, whereas LTP was blocked in the presence of this agent (Fig. 7D). Under this condition, the extent of PTP was significantly less in Shp2 cKO mice than in control mice (mean fEPSP slope at 0 to 5 min of 115.3% ± 2.1% [n = 6 slices] versus 132.8% ± 2.4% [n = 7 slices] [mean ± SEM; P = 2.9 × 10–4 by Student's t test]). These results suggested that short-term, but not long-term, synaptic plasticity is attenuated in the brains of Shp2 cKO mice. We also examined mGluR-dependent LTD, which was induced in CA1 neurons by exposure of hippocampal slices to DHPG, a group I mGluR agonist. However, the lack of Shp2 did not affect DHPG-induced LTD (data not shown).
Recall of premature spatial memory is specifically impaired in Shp2 cKO mice.
To investigate further the impact of the defect in hippocampal synaptic function in Shp2 cKO mice, we examined hippocampus-dependent learning behavior. The Morris water maze measures hippocampus-dependent spatial learning and memory (51). The maze consists of a water-filled pool with a hidden escape platform and multiple external visual cues, and mice have to swim in opaque water and learn to find the hidden platform in order to escape from the water. Mice were trained for 18 trials (6 trials per day for 3 days), and their learning ability was measured as the time required to reach the hidden platform. In addition, on the 4th day, each mouse was examined with a probe trial for which the platform was removed. In this trial, the capacity of the animal to retrieve and retain the memory of the platform's location was measured as the time spent in the region that previously contained the platform. On the first day of training (trials 1 to 6), the escape latency (time required to reach the platform) declined over time, and the performance of Shp2 cKO mice was indistinguishable from that of Shp2flox/flox (control) mice (Fig. 8A). On the 2nd day (trials 7 to 12), the escape latency of Shp2 cKO mice was markedly greater than that of control mice in the first trial of the day (trial 7), but it did not differ significantly between the two groups in the later trials (trials 8 to 12) (Fig. 8A). The performance of the two types of mice was also similar on the 3rd day (trials 13 to 18) and on the 4th day (probe trial) (Fig. 8A and B), which is suggestive of normal memory formation in Shp2 cKO mice after full training.
FIG 8.

Impairment of memory formation in Shp2 cKO mice. (A) Acquisition of spatial memory in the Morris water maze. Latency to escape during training of Shp2flox/flox (control [Ctrl]) or Shp2 cKO mice at 26 to 30 weeks of age to find a hidden platform in the water maze on 3 consecutive days (six trials per day). (B) Time spent in each of the four quadrants of the pool during a 2-min probe trial on day 4 for which the platform had been removed. The configuration of the four quadrants in the probe test is indicated (T, target quadrant; O, opposite quadrant; R, adjacent right quadrant; L, adjacent left quadrant). (C) Reversal of spatial learning after the first probe trial. Mice were trained for 3 consecutive days as for panel A, except that the hidden platform was placed in the opposite quadrant of the maze. (D) Five weeks after the reversal trials, the mice were subjected to a second probe trial as for panel B. All data are mean values ± SEM (n = 12 mice), and P values for comparisons of control and Shp2 cKO mice or for the comparisons indicated were determined by repeated-measure ANOVA (A, C) or Student's t test (B, D), respectively. *, P < 0.05; **, P < 0.01.
The probe trial on the 4th day was followed by reversal trials (six trials per day for 3 days; trials R1 to R18) that were identical to the training trials on days 1 to 3 (trials 1 to 18), except that the platform had been moved to the opposite quadrant of the pool in order to examine cognitive flexibility (Fig. 8C). A marked increase in the escape latency of Shp2 cKO mice was again apparent only in the first trial (trial R7) of the 2nd day of the reversal trials. Five weeks after the 3rd day of the reversal trials, the mice were subjected to a second probe trial in order to examine long-term memory retention (Fig. 8D). The time spent by mice of both genotypes in the region that previously contained the platform was significantly longer than that spent in the other regions, with essentially no apparent difference between the two groups, suggesting that retrieval and retention of established memory were normal in Shp2 cKO mice even after 5 weeks. No difference between the two groups was detected when the animals were tested with a visible platform after the second probe trial, and the swimming speeds of both types of mice were similar during trials 1 to 18 (data not shown). The hyperactivity phenotype of Shp2 cKO mice thus did not affect their overall performance in this test.
DISCUSSION
Forebrain neuron-specific ablation of Shp2 resulted in behavioral abnormalities, including hyperactivity. The hyperactive behavior of Shp2 cKO mice may contribute to some other behavioral abnormalities, such as an increased number of transitions and increased distance traveled in the light-dark transition test, reduced immobility in the forced-swim test, and an increased acoustic startle response. In contrast, a reduced stay time in the dark in the light-dark transition test or abnormal social interaction may be independent of the hyperactivity of the mutant mice. The obesity of Shp2 cKO mice seemed not to affect their behavior significantly, because it was very mild. In contrast to our present data, previous studies suggested that neuron-specific Shp2 cKO mice exhibited severe obesity (9, 10). In these studies, Shp2 cKO mice were generated by crossing other lines of Cre transgenic mice (CaMKIIα-Cre [R1ag5] or myosin light chain II promoter-Cre [CRE-3]) and Shp2flox/flox mice, in which exon 4 of the Shp2 gene was flanked by loxP sites. In contrast, we used another line of CaMKIIα-Cre mice (CRE-159) and Shp2flox/flox mice in which exon 11 of the Shp2 gene was flanked by loxP sites. CaMKIIα-Cre transgenic lines exhibit diversity of Cre expression levels or patterns, which directly affects the gene targeting pattern (52). Thus, the mild obesity of our Shp2 cKO mice is likely due to the difference between the mouse lines studied.
Depletion of Shp2 inhibited BDNF-dependent activation of Erk1/2 in cultured neurons, suggesting a positive role for Shp2 in BDNF-dependent activation of Ras-MAPK signaling. Forebrain neuron-specific ablation of BDNF or of its receptor TrkB results in hyperactive behavior of mice (53–55). Genetic ablation of Erk1 or administration of SL327, an ERK kinase (MEK) inhibitor, also induces hyperactivity in mice and rats, respectively (56–58). In contrast, knockin mice expressing an active mutant of H-Ras (H-RasG12V) exhibit reduced activity in a novel environment compared with control mice (59). Thus, downregulation of BDNF- and TrkB-dependent Ras-MAPK signaling in forebrain neurons may, at least in part, underlie the hyperactive behavior of Shp2 cKO mice; other mechanisms that do not involve Ras-MAPK pathway are not excluded, however.
Hyperactivity is often associated with a reduction in neuronal activity in both humans and animals (38–40). BDNF induces activation of Ras-MAPK signaling and a marked increase in synaptic transmission in mouse hippocampal slices (60), suggesting the importance of BDNF signaling in the regulation of neuronal activity. The downregulation of BDNF- and TrkB-dependent Ras-MAPK signaling in forebrain neurons of Shp2 cKO mice may therefore result in a reduction in neuronal activity in vivo. We found that the novelty-induced expression of IEGs, an index of neuronal activity, was attenuated in the brains of Shp2 cKO mice. These data are consistent with the notion that neuronal activity is reduced in these animals. The defects in the I/O relationship for the hippocampal CA1 region, the increased PPR in PPF, and the increased power spectrum for δ waves in the cortical EEG are also consistent with attenuation of synaptic efficiency and a reduction in neuronal activity in the brains of Shp2 cKO mice.
The lack of Shp2 resulted in enhancement of high-K+-induced Erk1/2 activation in cultured neurons and synaptosomes, which is suggestive of a negative role for Shp2 in membrane depolarization-induced Erk activation. Such a function of Shp2 was not reported previously and appears inconsistent with the reduced activation of Erk1/2 in the brains of Shp2 cKO mice after exposure to a novel environment. The activation of MAPK in the brain, however, might be expected to reflect the summation of responses to multiple stimuli, such as neuronal membrane depolarization, neurotransmitter release, and stress hormones, with the relative contribution of membrane depolarization to the total level of Erk activation in vivo thus remaining unclear.
Suppression of membrane depolarization-induced MAPK activation by Shp2 does not appear to affect the neuronal-activity-dependent expression of IEGs, such as those for c-Fos, Zif268, and Npas4, in cultured neurons. In contrast, it may influence the regulation of synaptic function, given that enhancement of high-K+-induced MAPK activation was also observed in synaptosomes prepared from Shp2 cKO mice. MAPK activation is thought to be important for the induction of LTP (57, 61). However, the induction of hippocampal LTP was normal in Shp2 cKO mice. The Shp2-dependent mechanism that controls membrane depolarization-induced MAPK activation thus does not appear to contribute to the induction of LTP. In contrast, PTP at CA1 synapses was attenuated in Shp2 cKO mice. Presynaptic Ca2+ remaining after a preceding stimulus and consequent Ca2+-dependent signaling events have been implicated in the expression of PTP (48). Ca2+ influx is also important for membrane depolarization-induced activation of the Ras-MAPK pathway (61). Shp2 was previously described as a Ca2+-responsive molecule that mediates activity-dependent neuronal excitotoxicity (62). Such Ca2+ responsiveness of Shp2 may contribute to the expression of PTP.
Although a role for Shp2-MAPK signaling in the regulation of PTP has not been previously described, high-frequency stimulation induced activation of Erk1/2 in presynaptic structures of mossy-fiber–CA3 synapses in the mouse hippocampus, and such activation of Erk1/2 inhibited high-frequency stimulation-induced posttetanic enhancement, a form of short-term synaptic plasticity at these synapses (63). Similarly, enhancement of membrane depolarization-induced MAPK activation by Shp2 ablation may contribute to the attenuation of PTP in the hippocampal CA1 region of Shp2 cKO mice. In contrast, dysregulation of BDNF- and TrkB-dependent Ras-MAPK signaling does not likely contribute to the abnormal short-term synaptic plasticity in Shp2 cKO mice, because a reduction in PTP was not reported in mice with forebrain-specific deficiency of TrkB (12, 64).
Shp2 has been shown to contribute to regulation of the spacing effect for long-term memory induction in the adult Drosophila brain (65). In contrast, the physiological roles of Shp2 in the mature mammalian brain have not been fully explored. Shp2 cKO mice exhibited mildly impaired performance in the Morris water maze. Whereas short-term memory (immediate memory) is lost within a minute without repetition, long-term memory is further divided into recent memory sustained by the minute, hour, and day and remote memory sustained for a longer period (66). The first recall of the “premature” recent spatial memory that was acquired in the trials of the Morris water maze test on the previous day was significantly impaired in Shp2 cKO mice. A Shp2-dependent mechanism may thus contribute to the recall of “premature” memory, and this process may differ from the recall of the “established” (or “consolidated”) memory after full training, given that the latter process was not affected by the lack of Shp2. The recall of acquired memories initially depends on hippocampal function (67, 68), and we found that short-term synaptic plasticity (PTP) was abnormal in the hippocampi of Shp2 cKO mice. Shp2-dependent regulation of PTP may be a key mechanism for the recall of premature memory.
A recent study demonstrated that expression of Noonan syndrome-associated gain-of-function (GOF) Shp2 mutants resulted in impaired hippocampal LTP and learning deficits associated with increased Erk signaling and excitatory synaptic function in the mouse brain (69). This study demonstrated that expression of Noonan syndrome-type GOF mutant Shp2 led to postsynaptic changes in AMPA receptor trafficking, which contributed to the increase in excitatory synaptic function (69). The authors suggested that the expression of a GOF mutant form of Shp2 increased the number of synapses with postsynaptic AMPA receptors, thus occluding LTP and therefore impairing learning (69). However, Shp2-dependent control of AMPA receptor trafficking and hippocampal LTP may be specific for an Noonan syndrome model or human Noonan syndrome and not function in a normal brain, because lack of endogenous neuronal Shp2 in cKO mice did not affect the induction of hippocampal LTP. Cognitive deficits in Noonan syndrome model mice and Shp2 cKO mice could be thus caused by different mechanisms, and our present study provides clues to how a novel Shp2-dependent mechanism that contributes to the regulation of synaptic functions in the adult brain works.
ACKNOWLEDGMENTS
We thank B. G. Neel and W. Yang for Shp2flox/flox mice; E. Kramer and R. Klein for CRE-159 mice; T. Yamamoto and T. Nakazawa for antibodies to GluN2B; M. Takahashi for antibodies to VAMP2; S. C. Fujita for a monoclonal antibody to synaptophysin; H. Miyoshi for the lentiviral vector and packaging plasmids; M. Kobayashi, D. Kohno, T. Sasaki, and T. Kitamura for technical advice; and H. Kobayashi, E. Urano, Y. Kusakari, and Y. Hayashi for technical assistance.
This work was supported by a Grant-in-Aid for Scientific Research (C), a Grant-in-Aid for Young Scientists (B), and a grant-in-aid for Scientific Research on Innovative Areas (Brain Environment) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, as well as by grants from the Takeda Science Foundation, the Naito Foundation, the Japan Foundation for Neuroscience and Mental Health, and the Life Science Foundation of Japan. This work was carried out by the joint research program of the Institute for Molecular and Cellular Regulation, Gunma University (project 13017).
REFERENCES
- 1.Neel BG, Gu H, Pao L. 2003. The ‘Shp'ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci 28:284–293. doi: 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- 2.Matozaki T, Murata Y, Saito Y, Okazawa H, Ohnishi H. 2009. Protein tyrosine phosphatase SHP-2: a proto-oncogene product that promotes Ras activation. Cancer Sci 100:1786–1793. doi: 10.1111/j.1349-7006.2009.01257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perkins LA, Johnson MR, Melnick MB, Perrimon N. 1996. The nonreceptor protein tyrosine phosphatase corkscrew functions in multiple receptor tyrosine kinase pathways in Drosophila. Dev Biol 180:63–81. doi: 10.1006/dbio.1996.0285. [DOI] [PubMed] [Google Scholar]
- 4.Tang TL, Freeman RM Jr, O'Reilly AM, Neel BG, Sokol SY. 1995. The SH2-containing protein-tyrosine phosphatase SH-PTP2 is required upstream of MAP kinase for early Xenopus development. Cell 80:473–483. doi: 10.1016/0092-8674(95)90498-0. [DOI] [PubMed] [Google Scholar]
- 5.Saxton TM, Henkemeyer M, Gasca S, Shen R, Rossi DJ, Shalaby F, Feng GS, Pawson T. 1997. Abnormal mesoderm patterning in mouse embryos mutant for the SH2 tyrosine phosphatase Shp-2. EMBO J 16:2352–2364. doi: 10.1093/emboj/16.9.2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tartaglia M, Gelb BD, Zenker M. 2011. Noonan syndrome and clinically related disorders. Best Pract Res Clin Endocrinol Metab 25:161–179. doi: 10.1016/j.beem.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sarkozy A, Digilio MC, Dallapiccola B. 2008. Leopard syndrome. Orphanet J Rare Dis 3:13. doi: 10.1186/1750-1172-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gauthier AS, Furstoss O, Araki T, Chan R, Neel BG, Kaplan DR, Miller FD. 2007. Control of CNS cell-fate decisions by SHP-2 and its dysregulation in Noonan syndrome. Neuron 54:245–262. doi: 10.1016/j.neuron.2007.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang EE, Chapeau E, Hagihara K, Feng GS. 2004. Neuronal Shp2 tyrosine phosphatase controls energy balance and metabolism. Proc Natl Acad Sci U S A 101:16064–16069. doi: 10.1073/pnas.0405041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krajewska M, Banares S, Zhang EE, Huang X, Scadeng M, Jhala US, Feng GS, Krajewski S. 2008. Development of diabesity in mice with neuronal deletion of Shp2 tyrosine phosphatase. Am J Pathol 172:1312–1324. doi: 10.2353/ajpath.2008.070594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fornaro M, Burch PM, Yang W, Zhang L, Hamilton CE, Kim JH, Neel BG, Bennett AM. 2006. SHP-2 activates signaling of the nuclear factor of activated T cells to promote skeletal muscle growth. J Cell Biol 175:87–97. doi: 10.1083/jcb.200602029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Minichiello L, Korte M, Wolfer D, Kuhn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp HP, Bonhoeffer T, Klein R. 1999. Essential role for TrkB receptors in hippocampus-mediated learning. Neuron 24:401–414. doi: 10.1016/S0896-6273(00)80853-3. [DOI] [PubMed] [Google Scholar]
- 13.Nakazawa T, Komai S, Watabe AM, Kiyama Y, Fukaya M, Arima-Yoshida F, Horai R, Sudo K, Ebine K, Delawary M, Goto J, Umemori H, Tezuka T, Iwakura Y, Watanabe M, Yamamoto T, Manabe T. 2006. NR2B tyrosine phosphorylation modulates fear learning as well as amygdaloid synaptic plasticity. EMBO J 25:2867–2877. doi: 10.1038/sj.emboj.7601156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Obata K, Kojima N, Nishiye H, Inoue H, Shirao T, Fujita SC, Uchizono K. 1987. Four synaptic vesicle-specific proteins: identification by monoclonal antibodies and distribution in the nervous tissue and the adrenal medulla. Brain Res 404:169–179. doi: 10.1016/0006-8993(87)91368-0. [DOI] [PubMed] [Google Scholar]
- 15.Huttner WB, Schiebler W, Greengard P, De Camilli P. 1983. Synapsin I (protein I), a nerve terminal-specific phosphoprotein. III. Its association with synaptic vesicles studied in a highly purified synaptic vesicle preparation. J Cell Biol 96:1374–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohnishi H, Murata T, Kusakari S, Hayashi Y, Takao K, Maruyama T, Ago Y, Koda K, Jin FJ, Okawa K, Oldenborg PA, Okazawa H, Murata Y, Furuya N, Matsuda T, Miyakawa T, Matozaki T. 2010. Stress-evoked tyrosine phosphorylation of signal regulatory protein alpha regulates behavioral immobility in the forced swim test. J Neurosci 30:10472–10483. doi: 10.1523/JNEUROSCI.0257-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miyakawa T, Yamada M, Duttaroy A, Wess J. 2001. Hyperactivity and intact hippocampus-dependent learning in mice lacking the M1 muscarinic acetylcholine receptor. J Neurosci 21:5239–5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miyakawa T, Leiter LM, Gerber DJ, Gainetdinov RR, Sotnikova TD, Zeng H, Caron MG, Tonegawa S. 2003. Conditional calcineurin knockout mice exhibit multiple abnormal behaviors related to schizophrenia. Proc Natl Acad Sci U S A 100:8987–8992. doi: 10.1073/pnas.1432926100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morishima Y, Miyakawa T, Furuyashiki T, Tanaka Y, Mizuma H, Nakanishi S. 2005. Enhanced cocaine responsiveness and impaired motor coordination in metabotropic glutamate receptor subtype 2 knockout mice. Proc Natl Acad Sci U S A 102:4170–4175. doi: 10.1073/pnas.0500914102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shibata M, Yamasaki N, Miyakawa T, Kalaria RN, Fujita Y, Ohtani R, Ihara M, Takahashi R, Tomimoto H. 2007. Selective impairment of working memory in a mouse model of chronic cerebral hypoperfusion. Stroke 38:2826–2832. doi: 10.1161/STROKEAHA.107.490151. [DOI] [PubMed] [Google Scholar]
- 21.Moqrich A, Hwang SW, Earley TJ, Petrus MJ, Murray AN, Spencer KS, Andahazy M, Story GM, Patapoutian A. 2005. Impaired thermosensation in mice lacking TRPV3, a heat and camphor sensor in the skin. Science 307:1468–1472. doi: 10.1126/science.1108609. [DOI] [PubMed] [Google Scholar]
- 22.Nadler JJ, Moy SS, Dold G, Trang D, Simmons N, Perez A, Young NB, Barbaro RP, Piven J, Magnuson TR, Crawley JN. 2004. Automated apparatus for quantitation of social approach behaviors in mice. Genes Brain Behav 3:303–314. doi: 10.1111/j.1601-183X.2004.00071.x. [DOI] [PubMed] [Google Scholar]
- 23.Murata T, Ohnishi H, Okazawa H, Murata Y, Kusakari S, Hayashi Y, Miyashita M, Itoh H, Oldenborg PA, Furuya N, Matozaki T. 2006. CD47 promotes neuronal development through Src- and FRG/Vav2-mediated activation of Rac and Cdc42. J Neurosci 26:12397–12407. doi: 10.1523/JNEUROSCI.3981-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maruyama T, Kusakari S, Sato-Hashimoto M, Hayashi Y, Kotani T, Murata Y, Okazawa H, Oldenborg PA, Kishi S, Matozaki T, Ohnishi H. 2012. Hypothermia-induced tyrosine phosphorylation of SIRPα in the brain. J Neurochem 121:891–902. doi: 10.1111/j.1471-4159.2012.07748.x. [DOI] [PubMed] [Google Scholar]
- 25.Yamamoto T, Miyoshi H, Yamamoto N, Yamamoto N, Inoue J, Tsunetsugu-Yokota Y. 2006. Lentivirus vectors expressing short hairpin RNAs against the U3-overlapping region of HIV nef inhibit HIV replication and infectivity in primary macrophages. Blood 108:3305–3312. doi: 10.1182/blood-2006-04-014829. [DOI] [PubMed] [Google Scholar]
- 26.Paxinos G, Franklin KBJ. 1997. The mouse brain in stereotaxic coordinates. Academic Press, San Diego, CA. [Google Scholar]
- 27.Ago Y, Araki R, Tanaka T, Sasaga A, Nishiyama S, Takuma K, Matsuda T. 2013. Role of social encounter-induced activation of prefrontal serotonergic systems in the abnormal behaviors of isolation-reared mice. Neuropsychopharmacology 38:1535–1547. doi: 10.1038/npp.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishihama T, Ago Y, Shintani N, Hashimoto H, Baba A, Takuma K, Matsuda T. 2010. Environmental factors during early developmental period influence psychobehavioral abnormalities in adult PACAP-deficient mice. Behav Brain Res 209:274–280. doi: 10.1016/j.bbr.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 29.Cryan JF, Mombereau C, Vassout A. 2005. The tail suspension test as a model for assessing antidepressant activity: review of pharmacological and genetic studies in mice. Neurosci Biobehav Rev 29:571–625. doi: 10.1016/j.neubiorev.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 30.Brooks SP, Dunnett SB. 2009. Tests to assess motor phenotype in mice: a user's guide. Nat Rev Neurosci 10:519–529. doi: 10.1038/nrn2652. [DOI] [PubMed] [Google Scholar]
- 31.Bourin M, Hascoet M. 2003. The mouse light/dark box test. Eur J Pharmacol 463:55–65. doi: 10.1016/S0014-2999(03)01274-3. [DOI] [PubMed] [Google Scholar]
- 32.Porsolt RD, Le Pichon M, Jalfre M. 1977. Depression: a new animal model sensitive to antidepressant treatments. Nature 266:730–732. doi: 10.1038/266730a0. [DOI] [PubMed] [Google Scholar]
- 33.Koch M, Schnitzler HU. 1997. The acoustic startle response in rats—circuits mediating evocation, inhibition and potentiation. Behav Brain Res 89:35–49. doi: 10.1016/S0166-4328(97)02296-1. [DOI] [PubMed] [Google Scholar]
- 34.Braff DL, Geyer MA. 1990. Sensorimotor gating and schizophrenia. Human and animal model studies. Arch Gen Psychiatry 47:181–188. doi: 10.1001/archpsyc.1990.01810140081011. [DOI] [PubMed] [Google Scholar]
- 35.Moy SS, Nadler JJ, Perez A, Barbaro RP, Johns JM, Magnuson TR, Piven J, Crawley JN. 2004. Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice. Genes Brain Behav 3:287–302. doi: 10.1111/j.1601-1848.2004.00076.x. [DOI] [PubMed] [Google Scholar]
- 36.Walsh RN, Cummins RA. 1976. The open-field test: a critical review. Psychol Bull 83:482–504. doi: 10.1037/0033-2909.83.3.482. [DOI] [PubMed] [Google Scholar]
- 37.Viggiano D. 2008. The hyperactive syndrome: metanalysis of genetic alterations, pharmacological treatments and brain lesions which increase locomotor activity. Behav Brain Res 194:1–14. doi: 10.1016/j.bbr.2008.06.033. [DOI] [PubMed] [Google Scholar]
- 38.Kadam SD, Smith-Hicks CL, Smith DR, Worley PF, Comi AM. 2010. Functional integration of new neurons into hippocampal networks and poststroke comorbidities following neonatal stroke in mice. Epilepsy Behav 18:344–357. doi: 10.1016/j.yebeh.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Levine JB, Youngs RM, MacDonald ML, Chu M, Leeder AD, Berthiaume F, Konradi C. 2007. Isolation rearing and hyperlocomotion are associated with reduced immediate early gene expression levels in the medial prefrontal cortex. Neuroscience 145:42–55. doi: 10.1016/j.neuroscience.2006.11.063. [DOI] [PubMed] [Google Scholar]
- 40.Liston C, Malter Cohen M, Teslovich T, Levenson D, Casey BJ. 2011. Atypical prefrontal connectivity in attention-deficit/hyperactivity disorder: pathway to disease or pathological end point? Biol Psychiatry 69:1168–1177. doi: 10.1016/j.biopsych.2011.03.022. [DOI] [PubMed] [Google Scholar]
- 41.Lyons MR, West AE. 2011. Mechanisms of specificity in neuronal activity-regulated gene transcription. Prog Neurobiol 94:259–295. doi: 10.1016/j.pneurobio.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Greer PL, Greenberg ME. 2008. From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron 59:846–860. doi: 10.1016/j.neuron.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 43.Buzsáki G, Anastassiou CA, Koch C. 2012. The origin of extracellular fields and currents—EEG, ECoG, LFP and spikes. Nat Rev Neurosci 13:407–420. doi: 10.1038/nrn3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Edouard T, Combier JP, Nédélec A, Bel-Vialar S, Métrich M, Conte-Auriol F, Lyonnet S, Parfait B, Tauber M, Salles JP, Lezoualc'h F, Yart A, Raynal P. 2010. Functional effects of PTPN11 (SHP2) mutations causing LEOPARD syndrome on epidermal growth factor-induced phosphoinositide 3-kinase/AKT/glycogen synthase kinase 3β signaling. Mol Cell Biol 30:2498–2507. doi: 10.1128/MCB.00646-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hardingham GE, Arnold FJ, Bading H. 2001. A calcium microdomain near NMDA receptors: on switch for ERK-dependent synapse-to-nucleus communication. Nat Neurosci 4:565–566. doi: 10.1038/88380. [DOI] [PubMed] [Google Scholar]
- 46.Wiegert JS, Bading H. 2011. Activity-dependent calcium signaling and ERK-MAP kinases in neurons: a link to structural plasticity of the nucleus and gene transcription regulation. Cell Calcium 49:296–305. doi: 10.1016/j.ceca.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 47.Pereira DB, Carvalho AP, Duarte CB. 2002. Non-specific effects of the MEK inhibitors PD098,059 and U0126 on glutamate release from hippocampal synaptosomes. Neuropharmacology 42:9–19. doi: 10.1016/S0028-3908(01)00162-9. [DOI] [PubMed] [Google Scholar]
- 48.Fioravante D, Regehr WG. 2011. Short-term forms of presynaptic plasticity. Curr Opin Neurobiol 21:269–274. doi: 10.1016/j.conb.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dobrunz LE, Stevens CF. 1997. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron 18:995–1008. doi: 10.1016/S0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- 50.Malenka RC. 1994. Synaptic plasticity in the hippocampus: LTP and LTD. Cell 78:535–538. doi: 10.1016/0092-8674(94)90517-7. [DOI] [PubMed] [Google Scholar]
- 51.D'Hooge R, De Deyn PP. 2001. Applications of the Morris water maze in the study of learning and memory. Brain Res Rev 36:60–90. doi: 10.1016/S0165-0173(01)00067-4. [DOI] [PubMed] [Google Scholar]
- 52.Gavériaux-Ruff C, Kieffer BL. 2007. Conditional gene targeting in the mouse nervous system: Insights into brain function and diseases. Pharmacol Ther 113:619–634. doi: 10.1016/j.pharmthera.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 53.Monteggia LM, Luikart B, Barrot M, Theobold D, Malkovska I, Nef S, Parada LF, Nestler EJ. 2007. Brain-derived neurotrophic factor conditional knockouts show gender differences in depression-related behaviors. Biol Psychiatry 61:187–197. doi: 10.1016/j.biopsych.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 54.Kernie SG, Liebl DJ, Parada LF. 2000. BDNF regulates eating behavior and locomotor activity in mice. EMBO J 19:1290–1300. doi: 10.1093/emboj/19.6.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zörner B, Wolfer DP, Brandis D, Kretz O, Zacher C, Madani R, Grunwald I, Lipp HP, Klein R, Henn FA, Gass P. 2003. Forebrain-specific trkB-receptor knockout mice: behaviorally more hyperactive than “depressive.” Biol Psychiatry 54:972–982. doi: 10.1016/S0006-3223(03)00418-9. [DOI] [PubMed] [Google Scholar]
- 56.Engel SR, Creson TK, Hao Y, Shen Y, Maeng S, Nekrasova T, Landreth GE, Manji HK, Chen G. 2009. The extracellular signal-regulated kinase pathway contributes to the control of behavioral excitement. Mol Psychiatry 14:448–461. doi: 10.1038/sj.mp.4002135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mazzucchelli C, Vantaggiato C, Ciamei A, Fasano S, Pakhotin P, Krezel W, Welzl H, Wolfer DP, Pages G, Valverde O, Marowsky A, Porrazzo A, Orban PC, Maldonado R, Ehrengruber MU, Cestari V, Lipp HP, Chapman PF, Pouyssegur J, Brambilla R. 2002. Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron 34:807–820. doi: 10.1016/S0896-6273(02)00716-X. [DOI] [PubMed] [Google Scholar]
- 58.Einat H, Yuan P, Gould TD, Li J, Du J, Zhang L, Manji HK, Chen G. 2003. The role of the extracellular signal-regulated kinase signaling pathway in mood modulation. J Neurosci 23:7311–7316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Viosca J, Schuhmacher AJ, Guerra C, Barco A. 2009. Germline expression of H-Ras(G12V) causes neurological deficits associated to Costello syndrome. Genes Brain Behav 8:60–71. doi: 10.1111/j.1601-183X.2008.00443.x. [DOI] [PubMed] [Google Scholar]
- 60.Ji Y, Lu Y, Yang F, Shen W, Tang TT, Feng L, Duan S, Lu B. 2010. Acute and gradual increases in BDNF concentration elicit distinct signaling and functions in neurons. Nat Neurosci 13:302–309. doi: 10.1038/nn.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thomas GM, Huganir RL. 2004. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci 5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- 62.Rusanescu G, Yang W, Bai A, Neel BG, Feig LA. 2005. Tyrosine phosphatase SHP-2 is a mediator of activity-dependent neuronal excitotoxicity. EMBO J 24:305–314. doi: 10.1038/sj.emboj.7600522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vara H, Onofri F, Benfenati F, Sassoe-Pognetto M, Giustetto M. 2009. ERK activation in axonal varicosities modulates presynaptic plasticity in the CA3 region of the hippocampus through synapsin I. Proc Natl Acad Sci U S A 106:9872–9877. doi: 10.1073/pnas.0900077106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Minichiello L, Calella AM, Medina DL, Bonhoeffer T, Klein R, Korte M. 2002. Mechanism of TrkB-mediated hippocampal long-term potentiation. Neuron 36:121–137. doi: 10.1016/S0896-6273(02)00942-X. [DOI] [PubMed] [Google Scholar]
- 65.Pagani MR, Oishi K, Gelb BD, Zhong Y. 2009. The phosphatase SHP2 regulates the spacing effect for long-term memory induction. Cell 139:186–198. doi: 10.1016/j.cell.2009.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nadel L, Hardt O. 2011. Update on memory systems and processes. Neuropsychopharmacology 36:251–273. doi: 10.1038/npp.2010.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Holt W, Maren S. 1999. Muscimol inactivation of the dorsal hippocampus impairs contextual retrieval of fear memory. J Neurosci 19:9054–9062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Riedel G, Micheau J, Lam AG, Roloff EL, Martin SJ, Bridge H, de Hoz L, Poeschel B, McCulloch J, Morris RG. 1999. Reversible neural inactivation reveals hippocampal participation in several memory processes. Nat Neurosci 2:898–905. doi: 10.1038/13202. [DOI] [PubMed] [Google Scholar]
- 69.Lee YS, Ehninger D, Zhou M, Oh JY, Kang M, Kwak C, Ryu HH, Butz D, Araki T, Cai Y, Balaji J, Sano Y, Nam CI, Kim HK, Kaang BK, Burger C, Neel BG, Silva AJ. 2014. Mechanism and treatment for learning and memory deficits in mouse models of Noonan syndrome. Nat Neurosci 17:1736–1743. doi: 10.1038/nn.3863. [DOI] [PMC free article] [PubMed] [Google Scholar]