

Preface
For a long time lysosomes were considered merely to be cellular “incinerators” involved in the degradation and recycling of cellular waste. However, there is now compelling evidence indicating that lysosomes have a much broader function and that they are involved in fundamental processes such as secretion, plasma membrane repair, signaling and energy metabolism. Furthermore, the essential role of lysosomes in the autophagic pathway puts these organelles at the crossroads of several cellular processes, with significant implications for health and disease. The identification of a master gene, transcription factor EB (TFEB), that regulates lysosomal biogenesis and autophagy, has revealed how the lysosome adapts to environmental cues, such as starvation, and suggests novel therapeutic strategies for modulating lysosomal function in human disease.
Lysosomes are membrane-bound organelles that were first described in 1955 by Christian de Duve1. They have an acidic lumen, which is limited by a single-bilayer lipid membrane and contains several types of hydrolases that are devoted to the degradation of specific substrates. The lysosomal membrane contains proteins that are involved in transport of substances into and out of the lumen, acidification of the lysosomal lumen, and fusion of the lysosome with other cellular structures2. Extracellular material that is destined for degradation reaches the lysosome through the endocytic pathway3, while intracellular components are transported to the lysosome by autophagy4-6. Lysosomes can also secrete their contents by fusing with the plasma membrane7, 8. This process, known as lysosomal “exocytosis”, is very active in particular cell types, such as cells from the hematopoietic lineage9, osteoclasts10 and melanocytes11. In addition to cellular clearance and secretion, the lysosome mediates a range of biological processes, such as plasma membrane repair, cell homeostasis, energy metabolism and the immune response. Little is known about how lysosomal function varies in different cells, tissues,, life stages and individuals, and under different physiological conditions. However, in recent years the static view of the lysosome has progressively changed into a much broader and dynamic perspective. The ability of the lysosome to adapt to different environmental cues became evident with the discovery that lysosomal biogenesis and function are subject to global transcriptional regulation. This novel concept of lysosomal adaptation is important to our understanding of how basic biological processes, ranging from cellular clearance to the control of energy metabolism, respond to environmental cues.
In this Review we will first describe the structure of the lysosome and its established role in cellular clearance. We will then consider the emerging roles of lysosomes, including their function in plasma membrane repair and signaling, before discussing the identification of the transcription factor EB (TFEB) as a key molecule that regulates lysosomal biogenesis and autophagy12, 13. Finally, we will discuss how lysosomal dysfunction leads to human disease.
Lysosome structure
The complex series of events leading to the formation of a mature lysosome have been described in recent articles2, 14-21. The mature lysosome has an acidic lumen encircled by a cholesterol-poor membrane22 (Box 1). The main function of the lysosomal membrane is to segregate the “aggressive” acidic environment of the lumen from the rest of the cell. This is ensured by the presence of a thick glycocalyx that lines the internal perimeter to prevent the lysosomal membrane being degraded by luminal acid hydrolases. The lysosomal membrane also actively mediates the fusion of lysosomes with other cellular structures, such as late endosomes, autophagosomes and the plasma membrane, as well as the transport of metabolites, ions and soluble substrates into and out of lysosomes.
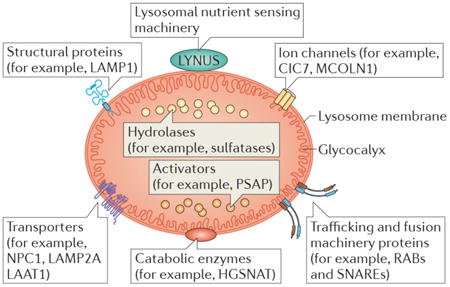
Box 1. The structure of the lysosome.
Lysosomes have a limiting membrane, which is composed of a single lipid bilayer and integral and peripheral proteins, and an acidic lumen that contains soluble hydrolytic enzymes and activators. 47, 49, 194, 195. A glycocalyx lines the internal lysosomal perimeter, protecting the membrane from the acidic environment of the lumen. Soluble enzymes are directly involved in the degradation of metabolites, while the lysosomal membrane segregates this catalytic potential and also actively participates in the maintenance of membrane integrity, the establishment of lumen acidic pH, metabolite and ion membrane transport, lysosomal trafficking and catalysis. Some key functional categories of lysosomal membrane proteins are shown (see the figure). Trafficking and fusion machinery proteins comprise SNAREs and RABs. Structural proteins include LAMP1, which is the most abundant lysosomal membrane protein accounting for 50% of this membrane's total protein. LAMP1 is mainly involved in lysosomal trafficking by mediating the attachment of lysosomes to the transport machinery2, 196. The lysosomal nutrient sensing (LYNUS) machinery includes several protein complexes that interact on the lysosomal surface, and its role is to sense the nutrient content of the lysosome and signal the information to the nucleus (see main text). An important component of the LYNUS machinery is the vacuolar ATPase (vATPase), a large multimeric channel that uses the energy derived from ATP hydrolysis to transport protons across the lysosomal membrane in order to generate the acidic pH of the lysosomal lumen197, 198. Several ion channels have been identified on the lysosomal membrane. The transient receptor potential (TRP) family member mucolipin-1 (TRPML1-MCOLN1) is a non-selective cation channel199 that is involved in calcium signalling during lysosomal fusion with other membranes, such as the plasma membrane85, 87 and autophagosomes200. A deficiency in mucolipin-1 causes mucolipidosis IV, a lysosomal storage disease201, 202. CIC-7, a chloride channel, contributes to lysosomal acidification and is involved in inherited osteopetrosis60, 61, 203. Transporters in the lysosomal membrane include LAMP2A, which mediates chaperone-mediated autophagy (CMA) by binding cytosolic protein substrates to the lysosomal membrane so that they can be internalized into lysosomes for degradation5, 204. Mutations of LAMP2A cause Danon disease, which is associated with the accumulation of autophagic vacuoles in muscle cells205. NPC1 is a lysosomal membrane protein involved in the export of cholesterol from the endo-lysosomal compartment and it is mutated in Niemann-Pick disease type C1206. The recently identified lysosomal amino acid transporter 1 (LAAT-1) is involved in the transport of lysine and arginine across the lysosomal membrane and into the lysosome and it plays a crucial role in cellular amino acid homeostasis207. Enzymes on the lysosomal membrane include HEPARAN-alpha glucosaminide N-acetyltransferase (HGSNAT). This enzyme participates in the stepwise degradation of heparan sulfate208-210 and mutation of this protein causes mucopolysaccharidosis type IIIC.
Lysosomal trafficking and fusion are mediated by specific sets of membrane-associated Rab GTPases17, 23, 24 and N-ethylmaleimide-sensitive attachment protein (SNAP) receptor (SNARE) proteins25-27. Of note, the ability of minimal ‘synthetic’ endosomes to fuse in vitro with purified early endosomes, or with each other, was reconstituted by using 17 recombinant human proteins, including specific Rab GTPase and SNAREs28. RAB5 and RAB7 are specifically involved in the tethering and docking processes during endo-lysosomal membrane trafficking pathways23, 24. Furthermore, a reduction in RAB5 levels results in a decreased number of endosomes and lysosomes and in a block of the endocytic pathways29. A specific combinatorial set of SNAREs, including VAMP7, VAMP8, VTI1B, syntaxin7 and syntaxin8, form the trans-complexes that drive lysosome–endosome fusion and the homotypic fusion between endosomes25. Interestingly, recent studies revealed that SNAREs that are involved in the fusion between autophagosomes and endo-lysosomal vesicles, such as syntaxin 17, also participate in autophagosome biogenesis30,31.
The lysosomal lumen contains approximately 60 different soluble hydrolases, which are active at acidic pH. These enzymes are the main players in the execution of multistep catabolic processes. They include members of protein families such as the sulfatases, glycosidases, peptidases, phosphatases, lipases, and nucleases, which allow the lysosome to hydrolyze a vast repertoire of biological substrates, ranging from glycosaminoglycans and sphingolipids to glycogen and proteins. The targeting of most lysosomal enzymes to lysosomes, as well as their ability to be secreted and taken up again by cells, is mediated by a mannose-6-phosphate modification that they undergo in the late Golgi compartments14, 32. The ability of cells to uptake lysosomal enzymes via the mannose-6-phosphate receptor is the basis for enzyme replacement therapy for several lysosomal storage diseases (LSDs)33. A different targeting mechanism, which is mediated in part by the lysosomal receptor LIMP-2, was recently identified for β-glucocerebrosidase34.
The selective degradation of intra-lumenal membranes and lipids within lysosomes occurs in specialized intra-lysosomal vesicles that contain a complex machinery composed of proteins that are involved in lipid degradation, such as water-soluble acid hydrolases and sphingolipid activator proteins (SAPs) 35-39. The study of patients with defects of glycosphingolipid catabolism was instrumental for the understanding of this complex process 40-42.
Importantly, a number of non-lysosomal proteins modulate the functions of lysosome-resident proteins. Prominent examples of these are the two different types of mannose 6 phosphate receptors, CI-MPR and CD-MPR, which dynamically shuttle between the trans-Golgi network (TGN) and late endosomes and are involved in the targeting of lysosomal enzymes to the lysosome32, and SUMF1, an ER-resident protein responsible for a post-translational modification, a conversion of highly conserved cysteine in the active site to alpha-formyl-glycine, which is required for the activation of all sulfatases43, 44.
A variety of methods have been used to purify lysosomes and analyze their proteome45-49. Some of these approaches are based on subcellular fractionations, while others are based on specific features of soluble lysosomal proteins, such as the mannose 6-phosphate modification of their carbohydrate moieties49. In these efforts it has been difficult to distinguish between lysosomal resident proteins, which are constituents of the lysosomal machinery, and proteins that are delivered to the lysosome for degradation. Therefore, we are still far from the identification and functional characterization of all lysosomal resident proteins. Based on current data, a little over 100 bona fide lysosomal resident proteins have been identified; approximately 70 of these are lysosomal matrix proteins and approximately 50 are lysosomal membrane proteins48. However, these numbers are likely to grow in the near future.
Lysosome functions
Lysosomal functions can be schematically divided into three main types: degradation, secretion and signaling (Figure 1).
Figure 1. Main functions of the lysosome and their relationship with key cellular processes.
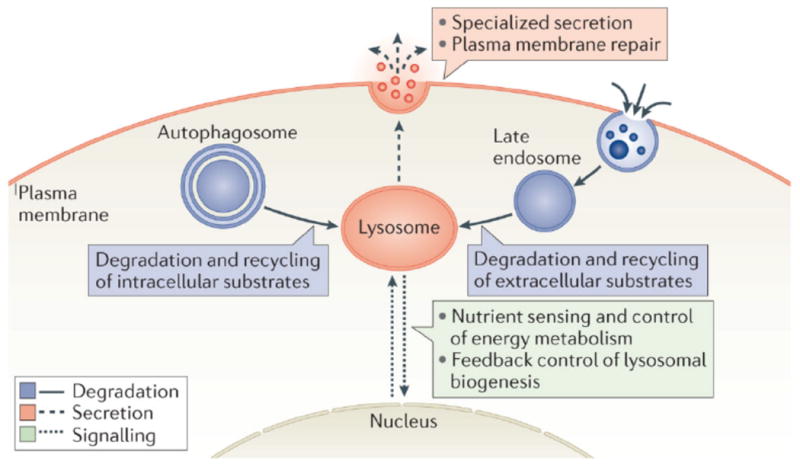
Lysosomes are involved in the degradation and recycling of extracellular material, via endocytosis, and intracellular material, via autophagy. In these processes lysosomes fuse with late endosomes and with autophagosomes, respectively. The resulting breakdown products are used to generate new cellular components and energy in response to the nutritional needs of the cell. Lysosomes also undergo Ca2+ regulated exocytosis to secrete their content into the extracellular space and to repair damaged plasma membrane. Upon plasma membrane injury, lysosomes rapidly migrate to the damaged site and fuse with the plasma membrane to allow efficient resealing. More recently, lysosomes have been identified as signaling organelles that can sense nutrient availability and activate a lysosome-to-nucleus signaling pathway that mediates the starvation response and regulates energy metabolism.
Lysosome-mediated degradation
Similar to the transport of urban waste to incinerators, the collection and transport of cellular waste to lysosomes requires complex logistics. The cell has developed different routes for transporting extracellular and intracellular waste to the lysosome. Extracellular material reaches the lysosome mainly through endocytosis. The capture of extracellular material and integral membrane proteins occurs through specific endocytosis mechanisms according to the nature of the cargo. Prominent examples of endocytosis are phagocytosis, macropinocytosis, clathrin-mediated endocytosis, caveolin-mediated endocytosis, and clathrin- and caveolin-independent endocytosis50. Signaling receptors may undergo endocytosis through clathrin-mediated endocytosis51 or clathrin-independent mechanisms52. After internalization, the receptors are routed to early endosomes53. From the endosomes, the receptors can either be recycled back to the plasma membrane to allow for repeated receptor activation, or be sorted and targeted for lysosomal degradation, resulting in the termination of receptor signaling54-56.
A known hallmark of endosome-to-lysosome maturation is the progressive decrease of the internal pH to around pH 5 in the mature lysosome57. This is crucial for the release of acid hydrolases from mannose-6-phosphate receptors into the endosomal lumen and the recycling of receptors back to the Golgi network15. The generation and maintenance of the lysosomal pH gradient requires the activity of a proton-pumping v-type ATPase, which uses the energy of ATP hydrolysis to pump protons into the lysosomal lumen58. Additional lysosomal membrane channels are thought to be involved in lysosomal acidification, such as the anion transporter ClC-759-61 and the cation transporters MCOLN1 and TPC262, which mediate Ca2+ release from the lysosome63-65. However, the role of each of these channels and the precise mechanisms underlying the complex regulation of lysosomal acidification and ion balance are still controversial and require further investigation.
Intracellular materials reach the lysosome through the process of autophagy, a “self-eating” catabolic pathway that is used by cells to capture their own cytoplasmic components destined for degradation and recycling. Three types of autophagy have been identified: microautophagy, chaperone-mediated autophagy (CMA) and macroautophagy. During microautophagy, cytosolic proteins are engulfed into the lysosome through the direct invagination of lysosomal or endosomal membranes6, 66, 67. In CMA, cytosolic proteins are transported into the lysosomal lumen through chaperone- and receptor-mediated internalization, which requires the unfolding of proteins and their translocation through the LAMP2a protein5, 16, 68, 69. Macroautophagy, herein referred to as autophagy, relies on the biogenesis of autophagosomes, double membrane-bound vesicles that sequester cytoplasmic material and then fuse with lysosomes. Thus, the role of all three types of autophagy in degradation and recycling processes is strictly dependent on lysosomal function.
Autophagy is activated by a broad range of cellular stress-inducing conditions and is able to degrade protein aggregates, oxidized lipids, damaged organelles, and intracellular pathogens. The resulting breakdown products are used to generate new cellular components and energy in response to the nutritional needs of the cell. The mechanisms underlying autophagy and its relevance both in health and disease have been extensively studied in the past decade and comprehensively described in recent reviews70, 71.
Lyososomal exocytosis
Lysosomes can secrete their content through a process called lysosomal exocytosis, which can be detected by the translocation of lysosomal membrane marker proteins (for example, LAMP1) to the plasma membrane7, 8, 72. In this process, lysosomes fuse with the plasma membrane through a Ca2+-regulated mechanism that leads to a bulk release of the lysosomal content into the extracellular matrix72-77. Originally lysosomal exocytosis was thought to be limited to professional secretory cells containing a subset of specialized lysosome-related organelles (LROs)75, but soon it was shown that any cell type can perform this function72. Lysosomal exocytosis mediates several physiological processes, such as degranulation in cytotoxic T lymphocytes78, bone resorption by osteoclasts10, parasite defense by mast cells and eosinophils79, 80, melanocyte function in pigmentation11, platelet function in coagulation81, and hydrolase release by spermatozoa during fertilization82.
The molecular machinery mediating Ca2+-regulated exocytosis of conventional lysosomes includes the v- SNARE VAMP-7 and synaptotagmin VII (SytVII) on lysosomes, and the t-SNARES SNAP23 and syntaxin 4 on the plasma membrane83 and several RAB proteins on the lysosomal surface8, 27, 83, 84. Another important mediator of lysosomal exocytosis is the lysosomal membrane Ca2+ channel mucolipin 1 (MCOLN1, TRPML1)85-87. It was also postulated that autophagy proteins may regulate lysosomal exocytosis. For example, lipidation of the late autophagosome marker protein LC3 is required for the secretion of lysosomal contents into the extracellular space, as this directs the lysosomes to fuse with the plasma membrane88-90. However, autophagosomes per se may not be mediating this process89.
Lysosomal exocytosis is not only responsible for the secretion of lysosomal content, but it plays a crucial role in plasma membrane repair. Plasma membrane injuries induce a rapid migration of lysosomes to the damaged site. Lysosomes then fuse to the plasma membrane and efficiently reseal the damaged sites91, 92. This process is important also in defense mechanisms against bacterial infection93, and has been implicated in a specific type of muscular dystrophy, which is characterized by a defect in muscle fiber repair94.
Lysosomal exocytosis is transcriptionally regulated by TFEB, a master gene for lysosomal biogenesis (see below). TFEB induces both the docking and fusion of lysosomes with the plasma membrane by regulating the expression of certain genes, the protein products of which increase lysosomal dynamics and cause a mucolipin 1-mediated elevation of intracellular Ca2+ 86. Interestingly, TFEB mediated regulation of lysosomal exocytosis plays an important role in osteoclast differentiation and bone resorption95.
Signaling from lysosomes
It has become evident that the lysosome plays an important role in nutrient sensing and in signaling pathways that are involved in cell metabolism and growth. Remarkably, the mammalian target of rapamycin complex 1 (mTORC1) kinase complex, a master controller of cell and organism growth96, exerts its activity on the lysosomal surface97. The lysosomal localization of mTORC1 suggests a mechanistic co-regulation between cell growth and cell catabolism. Growth factors, hormones, amino acids, glucose, stress and oxygen are the major activators of mTORC1, which in turn positively regulates protein, mRNA and lipid biosynthesis, and ATP production96,98. In this way mTORC1 regulates the balance between biosynthetic and catabolic states. When nutrients are present, mTORC1 directly phosphorylates and suppresses the activity of the kinase complex ULK1–ATG13–FIP200 (unc-51-like kinase 1/mammalian autophagy-related gene 13/focal adhesion kinase family-interacting protein of 200 kDa99-101), which is required to induce autophagosome biogenesis102, 103. The inhibition of mTORC1, by either starvation or drugs, leads to activation of ULK1–ATG13–FIP200 and autophagy. Thus, the level of cellular autophagy is inversely correlated with mTORC1 activity, and the pharmacological inhibition of mTORC1 potently stimulates autophagy.
Recent studies showed that the level of amino acids inside the lysosome lumen controls mTORC1 docking on the lysosomal surface, which is a prerequisite for its activity, and that amino acids must accumulate in the lysosomal lumen in order for mTORC1 to dock and become activated104. This observation supports the idea that mTORC1 activity is dependent on the lysosome and explains why mTORC1 is reactivated upon the lysosomal degradation of autophagic substrates that occurs during starvation105. A recent study showed that an ATP-sensitive Na+ channel, lysoNaATP, which is located on the lysosomal membrane, also interacts with mTORC1 and participates in nutrient sensing. During starvation mTORC1 is released from the lysosomal surface, and the lysoNaATP channel becomes constitutively open. Thus, lysoNaATP regulates lysosomal pH stability and amino acid homeostasis by responding to ATP levels and controlling lysosomal membrane potential106. Thus, a complex signaling machinery, which involves mTORC1 as well as additional protein complexes, is located on the lysosomal surface. This machinery, herein referred to as LYNUS (lysosome nutrient sensing), responds to lysosomal amino acid content and signals the information both to the cytoplasm and the nucleus. The main components of the LYNUS machinery are illustrated in Figure 2.
Figure 2. Model of TFEB regulation and function during starvation.
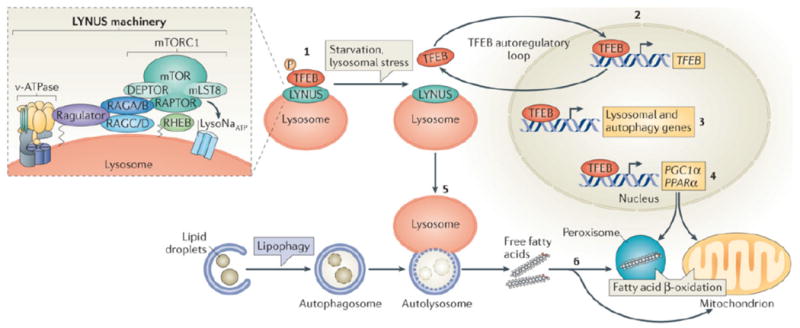
This model illustrates how transcription factor EB (TFEB) is induced by starvation and mediates the starvation response by regulating lipid catabolism. In the presence of adequate nutrition TFEB interacts with the lysosome nutrient sensing (LYNUS) machinery, which senses lysosomal nutrient levels via the vATPase complex, and is phosphorylated by mTORC1 on the lysosomal surface (1). This keeps TFEB inactive by cytosolic sequestration. During starvation mTORC1 is released from the LYNUS machinery and becomes inactive. Thus, TFEB can no longer be phosphorylated by mTORC1 and it translocates to the nucleus where it induces its own transcription (2). Therefore, starvation regulates TFEB activity through a dual mechanism that involves a post-translational modification (that is, phosphorylation) and a transcriptional autoregulatory loop. Once in the nucleus, TFEB regulates the expression of genes involved in the lysosomal–autophagy pathway (3). and of PGC1α–PPARα target genes (4). In this way TFEB controls the starvation response by activating both macrolipophagy (5) and fatty-acid oxidation (6).
The insert shows the main components of the LYNUS machinery. The mTORC1 complex, which includes regulatory proteins associated with mTOR, such as RAPTOR, LST8, and DEPTOR211, physically interacts with the RAG GTPases on the lysosomal surface and it is activated by them212. A complex known as the Ragulator mediates the activation and docking of RAGS to the lysosomal membrane97, 213 and the small GTPase RHEB is also involved in the growth factor-mediated activation of mTORC1214, 215. The vATPase complex is involved in amino acid sensing and it mediates amino acid-sensitive interactions between Rags and Ragulator, which is the initial step in lysosomal signaling104. The ATP-sensitive Na+ channel lysoNaATP, which is comprised of the subunits TPC1 and TPC2, is located on the lysosomal membrane and it has recently been shown to interact with mTORC1 and participates in nutrient sensing106 The nature of interaction between lysoNaATP and mTORC1 is unknown but seems to be independent form other components of the LYNUS machinery and the transcription factor EB (TFEB) and its interacting proteins (see text).
The involvement of the lysosome in nutrient sensing is a new concept that expands our view of this organelle from simply being an effector of cellular clearance to being a sensor and regulator of a variety of cellular functions, ranging from cell cycle progression and growth to macromolecule biosynthesis and autophagy107. The recent discovery of a starvation-induced lysosome-to-nucleus signaling mechanism (see below) further supports this concept108. Interestingly, autophagic lysosomal reformation (ALR), a recently described evolutionarily-conserved process by which nascent lysosomes are formed from autolysosomal membranes, also requires mTORC1 reactivation during prolonged starvation105, 109, 110. Furthermore, prolonged starvation also controls lysosomal reformation through the kinase activity of PI4KIIIβ and important regulator of lysosomal efflux 21
Regulation of lysosome function
The recent discovery of a “lysosomal gene network” and of its master gene TFEB, has revealed that lysosomal function can be coordinated to respond and adapt to environmental cues. Here we discuss the central role of TFEB in regulating lysosomal biogenesis, lysosome-nucleus signalling and lipid catabolism.
TFEB regulates lysosomal biogenesis and cellular clearance
Lysosome-mediated cellular clearance processes require the concerted action of hydrolases, acidification machinery and membrane proteins. The expression and activity of these components must be coordinated to allow optimal lysosomal function in different physiological and pathological conditions, such as growth, starvation, infection, and the intracellular accumulation of storage products. This concept of lysosomal adaptation has only emerged recently, as little attention was given to the study of the transcriptional regulation of the genes encoding lysosomal proteins. The recent discovery of a lysosomal gene network — the coordinated lysosomal expression and regulation (CLEAR) network — and of its master regulator TFEB, a member of the MITF subfamily of transcription factors111 that was previously implicated in a chromosomal translocation associated with renal carcinoma112, provides experimental evidence that lysosomal function is globally controlled12. The systems biology approach used to identify the CLEAR network is summarised in Box 2. Consistent with its role as a modulator of the CLEAR network TFEB positively regulates the expression of lysosomal genes, controls the number of lysosomes, and promotes the ability of cells to degrade lysosomal substrates12, 113. Further unbiased genomic and expression analyses, integrated with deep sequencing of TFEB chromatin-immunoprecipitates, provided a more detailed analysis of the CLEAR network and revealed that TFEB is a central regulator of cellular degradative pathways114. Specifically, it activates the transcription of genes that encode proteins involved in several aspects of cellular clearance, such as lysosomal biogenesis, autophagy, exocytosis, endocytosis, and additional lysosome-associated processes, such as phagocytosis, the immune response and lipid catabolism. Interestingly, many non-lysosomal proteins involved in the degradation of known autophagy substrates were also found to be members of the network114. These observations suggested that TFEB also regulates autophagy114. Indeed, TFEB overexpression in cultured cells significantly increases the number of autophagosomes, and enhances lysosome-to-autophagosome fusion and the degradation of long-lived proteins that are known autophagy substrates13. Consistently, viral-mediated overexpression of TFEB in the liver induced autophagy13. Thus, although the delivery of autophagy substrates to the lysosome and their degradation by lysosomal enzymes are distinct cellular processes, they are mechanistically linked by a common transcriptional regulation13, 115.
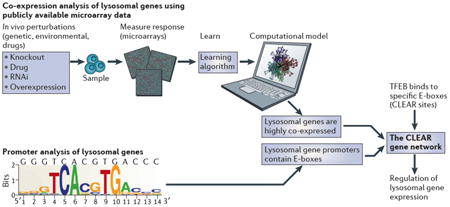
Box 2. The identification of the CLEAR gene network.
Gene networks control several aspects of cellular function and metabolism, such as the coordination of the cellular response to environmental conditions. In specialized organelles, this coordination is facilitated by compartmentalization. A systems biology approach was used to test the hypothesis that lysosomal genes are co-expressed, regulated by common factors, and able to respond to similar environmental cues (see the figure). The expression behaviour of genes encoding lysosomal proteins was analysed using publicly available microarray data. This analysis revealed that lysosomal genes have a statistically significant tendency to be co-expressed in a variety of different tissues and cell types and under different conditions12. Subsequently, pattern discovery analysis revealed the presence of a palindromic 10-base site in the promoters of known lysosomal genes. This sequence was previously identified as a specific version of a known target site for basic helix-loop-helix (bHLH) transcription factors, also known as an E-box. Thus, these two independent approaches, namely co-expression and promoter analyses, identified a new gene network which was named CLEAR (coordinated lysosomal expression and regulation). Further studies demonstrated that the transcription factor EB (TFEB), which belongs to the MITF subfamily of bHLH transcription factors, binds to CLEAR target sites in the promoters of lysosomal genes and positively regulates their expression, acting as a master gene of the CLEAR network12.
Other examples of transcription factors-regulating autophagy have been reported116-123. The FOXO transcription factor family (FOXO1, 3, 4 and 6) is negatively regulated by the insulin pathway in an AKT dependent manner. FOXOs are well conserved and have a critical role in many cellular processes, including in the regulation of autophagy120, 124. FOXO3 is activated during fasting and mediates the transcription of many genes that directly and indirectly regulate autophagy induction121, 122. FOXO3 regulation and function is very similar to that of TFEB, suggesting possible interactions between the two pathways. Indeed, FOXO3A overexpression increases cellular glutamine levels and inhibits mTORC1 activity, leading to TFEB activation and resulting in the coordinated transcriptional activation of lysosomal biogenesis and autophagy119 Another transcription factor regulating autophagy is the recently described ZKSCAN3, which belongs to the family of zinc-finger transcription factors that contain KRAB and SCAN domains and has recently been identified as a repressor of autophagy123. When ZKSCAN3 is silenced, cellular senescence and autophagy are promoted. When ZKSCAN3 is overexpressed, autophagy is suppressed in diverse cellular models. ZKSCAN3 was also shown to negatively regulate the expression of genes involved in autophagy and lysosome biogenesis and function. Interestingly, starvation induces the cytoplasmic accumulation of ZKSCAN3 and thereby inhibiting its activity. Conversely, nutrient availability promotes ZKSCAN3 nuclear translocation in an mTOR-dependent manner123. In conclusion, it appears that TFEB and ZKSCAN3 work in opposite directions to regulate lysosome biogenesis and autophagy in response to cellular needs. It will be interesting to determine whether these two transcription factors work in conjunction with each other.
TFEB conveys signals from the lysosome to the nucleus
Transcriptional mechanisms that control crucial cellular functions should respond to environmental cues. Under basal conditions, in most cell types TFEB is located in the cytoplasm. However, under specific conditions, such as starvation or lysosomal dysfunction, TFEB rapidly translocates from the cytoplasm to the nucleus12, 13. The nuclear translocation of TFEB is controlled by its phosphorylation status. Phosphorylated TFEB is located predominantly in the cytoplasm, while its dephosphorylated form is found in the nucleus13. Phosphoproteomic studies identified at least 10 different phosphorylation sites in the TFEB protein, suggesting a complex regulatory mechanism125. At least three different kinases have been shown to phosphorylate TFEB: ERK213, 126, mTORC1108, 126-129, PKCβ95. The phosphorylation of Ser142 by ERK2 and of both Ser142 and Ser211 by mTORC1, are crucial in determining the subcellular localization of TFEB. Mutation of either or both of these serines into alanines results in the constitutive nuclear localization of TFEB13, 126-129. On the other hand, during osteoclasts differentiation PKCβ-mediated phosphorylation of three serine residues located in the last 15 amino acids of TFEB stabilizes the protein and increases its activity95.
Interestingly, cytoplasmic TFEB is located both in the cytosol and on the lysosomal surface where it interacts with mTORC1 and the LYNUS machinery108, 130 (Figure 2). This observation suggests a mechanism by which the lysosome regulates its own biogenesis by controlling TFEB subcellular localization. Cellular conditions that lead to mTORC1 inactivation, such as stress, starvation, and lysosomal inhibition, induce TFEB nuclear translocation and thus activate the lysosomal system108, 127, 129. In addition, several isoforms of the 14-3-3 protein family play an important role in controlling TFEB subcellular localization by retaining phosphorylated TFEB in the cytoplasm127, 129. More recently, TFEB was shown to interact with active RAG guanosine triphosphatases (GTPases)130. This interaction promotes the lysosomal localization of TFEB and its mTORC1-dependent phosphorylation 130. Interestingly, other members of the basic-helix-loop-helix (bHLH) family of transcription factors, such as MITF and TFE3, the sequence of which is closely related to TFEB, appear to be regulated by a similar mechanism129, 130. It will be interesting to investigate whether other additional mTOR-independent mechanisms also regulate the nuclear translocation of TFEB.
Recent data indicate that cellular nutrient levels also regulate TFEB at the transcriptional level. The absence of serum and amino acids from the cell culture medium induces TFEB expression, while their re-administration turns it off. In a similar manner depriving mice of food for 24-hours induces TFEB expression in multiple tissues131. Interestingly, the transcriptional response of TFEB to nutrients is mediated by an autoregulatory feedback loop in which TFEB binds to its own promoter in a starvation-dependent manner and induces its own expression131. Thus, the regulation of TFEB activity by nutrients involves a rapid, phosphorylation-dependent, post-transcriptional switch, which is responsible for the nuclear translocation of TFEB and a transcriptional autoregulatory component, which allows for a slower, more sustained, response. This complex regulation mediates the cellular starvation response by inducing lipid catabolism (see below)131.
In conclusion, TFEB participates in a lysosome-to-nucleus signaling mechanism, which conveys information on lysosomal status to the nucleus to trigger a transcriptional response. This “cross-talk” between the lysosome and the nucleus controls cellular clearance and energy metabolism. A proposed model of TFEB regulation by nutrients is illustrated in Figure 2.
TFEB regulates lipid catabolism
Autophagy plays a central role in lipid metabolism by shuttling lipid droplets to lysosomes, where they are hydrolyzed into free fatty acids (FFAs) and glycerol. This process, called macrolipophagy132, 133, indicates the presence of a tight relationship between intracellular lipid metabolism and lysosomes. Interestingly, excessive lipid overload may in turn inhibit autophagy. This could be caused by either an alteration of the composition of the lysosomal membrane, rendering it less prone to fusion with autophagosomes134, 135, or by the down-regulation of autophagy genes136. Restoring liver autophagy ameliorates the metabolic phenotype of genetically induced obese mice (Ob/Ob), suggesting that enhancing lysosomal function may be a possible therapeutic strategy for the treatment of obesity136. Interestingly, lysosomal dysfunction was associated with an altered energy balance in murine models of LSDs137. In addition, in Wolman disease, the deficiency of lysosomal acid lipase leads to a severe intracellular fat accumulation138.
These studies suggest that the regulation of the lysosomal and autophagic pathways may impact cellular lipid metabolism. Indeed, TFEB was found to regulate liver lipid metabolism131. Transcriptome analysis in the mouse liver after viral-mediated TFEB overexpression revealed that this transcription factor positively regulates the expression of genes involved in several steps of lipid breakdown, such as lipophagy, fatty acid oxidation, and ketogenesis. Interestingly, PGC1α and PPARα, key regulators of lipid metabolism in response to starvation131, 139, are significantly induced by TFEB. In addition, TFEB was shown to directly bind to the PGC1α promoter in a starvation-sensitive manner131.
Remarkably, while liver-specific TFEB knock-out (KO) caused defective lipid degradation during starvation, TFEB overexpression enhanced liver fat catabolism and prevented diet-induced obesity131. Thus, TFEB controls the starvation response by responding to nutrient levels, and by inducing a metabolic switch that allows the organism to generate energy from stored lipids. These observations shed new light on the role of the lysosome in cellular energy metabolism, and on mechanisms underlying obesity and metabolic syndrome. A proposed model for the role of TFEB in lipid catabolism is illustrated in Figure 2.
TFEB regulation and function are evolutionarily conserved
The C. elegans genome encodes a single homologue of TFEB, HLH-30, which is a transcription factor that recognizes a DNA motif similar to the CLEAR motif and drives the transcription of metabolic genes140. HLH-30 acts in a similar manner to TFEB during C. elegans starvation. Hlh-30 mRNA progressively accumulates during starvation and rapidly decreases after the re-introduction of food, as is the case with mammalian TFEB131, 141. The HLH-30 protein also responds to starvation in a manner similar to its human counterpart as it can be detected mainly in the cytoplasm of well-fed C. elegans and predominantly in the nucleus of fasting animals141. The autoregulatory loop that regulates TFEB transcription is conserved in C. elegans131. Interestingly, HLH-30 activity is required to mobilize cytosolic lipids in fasting nematodes. Starved hlh-30 mutants failed to mobilize lipids as promptly as wild type animals131, 141, indicating that HLH-30 is required for C. elegans to efficiently use lipid stores during periods of starvation. HLH-30 is essential for the induction of lipid catabolism genes, such as lipase -2, lipase-3 and lipase-5, during fasting141 and the induction of lipid catabolism genes is greatly compromised in starved nematodes when hlh-30 is deleted131. Notably, starved hlh-30 mutants fail to mobilize their lipid stores due to a severe transcriptional response defect.
In wild-type C. elegans, starvation results in lifespan extension142. However, loss of hlh-30 was shown to result in the abrogation of starvation-induced lifespan extension131, 141, suggesting an important role for HLH30 and TFEB in longevity. Consistently, worms that are mutant for daf-2, which encodes the insulin-like growth factor 1 (IGF-1) receptor in C. elegans, have an increased life-span and it has been shown that they upregulate autophagy143. In conclusion, HLH-30 and murine TFEB share evolutionarily conserved functions in the adaptation of organisms to starvation. As observed for mammalian TFEB, hlh-30 expression is autoregulated, required for lipid mobilization, and is essential for the starvation response. The C. elegans model will be very useful for studying, in more detail, the potential role of TFEB in cell survival and aging in different conditions, considering that TFEB total loss of function is embryonic lethal in mice144. The striking conservation of TFEB function in C. elegans suggests that this regulatory mechanism evolved early to facilitate organismal adaptation to challenging nutritional conditions131, 141.
Lysosomal dysfunction and human disease
Lysosomal dysfunction has been associated with several human diseases, as well as with the process of aging, which may be associated with a decline in lysosomal function and a progressive accumulation of intracellular material (for example, lipofuscin and ubiquitin)145. Indeed, enhancement of the autophagic—lysosomal pathway appears to be an important determinant of the anti-aging effect of caloric restriction146. The identification of genes that regulate lysosomal biogenesis and function, such as TFEB, should pave the way to the development of novel therapeutics for diseases in which lysosomal dysfunction is aberrant.
Lysosomal dysfunction in LSDs and neurodegenerative diseases
For more than three centuries it has been known that genetic defects in specific lysosomal components leads to the accumulation of undegraded substrates in the lysosomal lumen, followed by progressive lysosomal dysfunction in several tissues and organs. These disorders are known as lysosomal storage diseases (LSDs). The classification of LSDs, as well as their clinical features, has been reviewed in detail in several recent articles147-152. Although these diseases were among the first for which both the biochemical and the molecular basis were recognized, the mechanisms by which the storage of undegraded material in lysosomes translates into cellular and tissue dysfunction and clinical symptoms has yet to be fully elucidated. The main mechanisms that have been identified so far are summarized in Box 3. In this context, a global impairment of lysosomal function plays an important role in the pathogenesis of several LSDs because a deficiency in individual lysosomal proteins can have broad consequences on the basic functions of lysosomes147. In particular, several studies have demonstrated an impairment of the autophagic pathway in LSDs147, 153-157. This results in the secondary accumulation of autophagy substrates, such as dysfunctional mitochondria and poly-ubiquitinated proteins, which play a crucial role in disease pathogenesis157. A block of autophagy in LSDs may be caused by a defect in the fusion between lysosomes and autophagosomes, as observed in Multiple Sulfatase Deficiency (MSD) and Mucopolysaccharidosis type IIIA (MPS IIIA), which may be caused by abnormalities in membrane lipid composition and SNARE protein distribution156.
Box 3. Mechanisms of lysosomal storage diseases (LSDs).
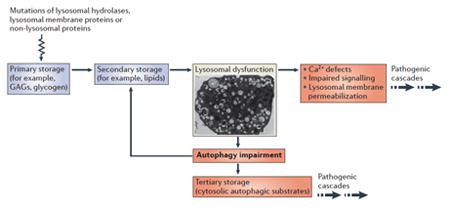
LSDs are a group of individually rare, recessively inherited, inborn errors of metabolism with an overall incidence of 1 in 5000, caused by mutations of genes encoding proteins that localize to the lysosomal lumen, lysosomal membrane, or other cellular compartments that contribute to lysosomal function. These disorders are characterized by the progressive accumulation of a variety of undegraded material in the lysosomes of most cells and tissues. Approximately 60 different types of LSDs have been recognized. Historically, LSDs have been classified based on the type of material that accumulates in the lysosomes, such as mucopolysaccharidoses, sphingolipidoses, glycoproteinoses, glycogenosis, and lipofuscinoses. LSDs often show a multisystemic phenotype associated with severe neurodegeneration, mental decline, cognitive problems, and behavioral abnormalities. Other tissues that are commonly affected are bone and muscle. Cell and tissue pathology are the result of a complex series of pathogenic cascades that occur downstream of lysosomal dysfunction. The figure illustrates the main steps underlying LSD pathogenesis (see the figure). Mutations in genes that are important for lysosomal function result in the accumulation of specific undegraded substrates in the lysosome (primary storage). This leads to a secondary accumulation of additional lysosomal substrates (secondary storage) due to a blockage in lysosomal trafficking. Excessive lysosomal storage has a broad impact on lysosomal function by causing defects in Ca2+ homeostasis, signaling abnormalities, and lysosomal membrane permeabilization. In addition, lysosomal dysfunction is associated with autophagy impairment, due to a defect in the fusion between lysosomes and autophagosomes, causing the “tertiary storage” of autophagy substrates (such as aggregate prone proteins and dysfunctional mitochondria), both of which contribute to neurodegeneration.
Current therapeutic strategies for LSDs are aimed at either restoring or replacing the activity of defective lysosomal enzymes by the use of molecular chaperones, enzyme replacement or viral-mediated gene therapy158. Inhibition of substrate synthesis is another available therapeutic option for some LSDs158. These strategies suffer from major limitations, such as the difficulty of delivering the enzyme, or the gene, to the required target sites in the body. For instance, a major hurdle for delivering therapies to the brain is crossing the blood-brain-barrier (BBB). In addition, in most cases each therapy is strictly disease-specific. This makes the overall costs of preclinical studies and clinical trials for LSDs extremely high, when one considers that LSDs include over 60 different disease entities.
Accumulating evidence indicates that lysosomal and autophagy dysfunction is one of the main mechanisms underlying common neurodegenerative diseases such as Parkinson's (PD), Alzheimer's (AD), and Huntington's (HD) diseases159, 160 (Figure 3). Mutated aggregate-prone proteins that cause neurodegenerative diseases, such as expanded huntingtin in HD and mutated α-synuclein in PD, are cleared by boosting the lysosomal-autophagic pathway161-163. In addition, aggregate-prone proteins may in turn affect the efficiency of autophagy by inhibiting cargo recognition by autophagosomes164, 165.
Figure 3. Defective cellular clearance in neurodegenerative diseases.
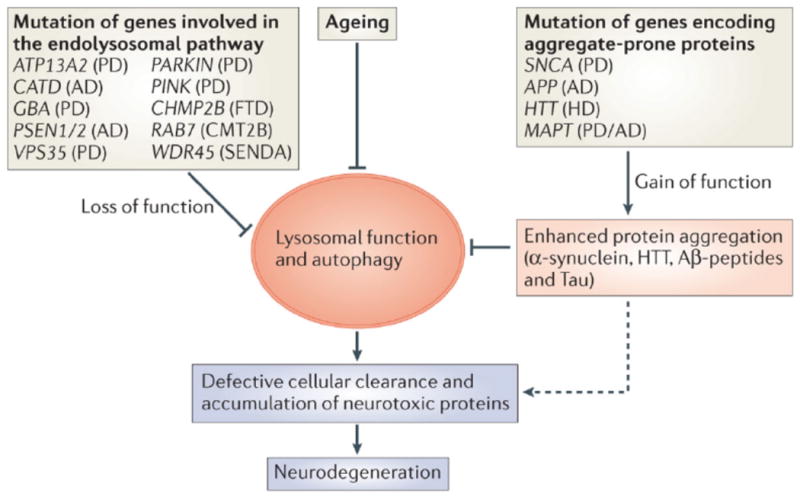
Defective cellular clearance, leading to neurodegeneration, can result from two different mechanisms. First, loss-of-function mutations of genes involved in the lysosomal–-autophagic pathway (for example, ATP13A2, CATD, GBA1, PSEN1/2, VPS35, PINK, PARK, CHMP2B, RAB7, and WDR45) can affect cellular degradation and recycling processes. Second, gain-of-function mutations of aggregate-prone proteins (for example, SNCA, APP, HTT and MAPT) may lead to enhanced protein aggregation and engulfment of lysosomal–autophagic pathways. In addition, a global decrease of lysosomal–autophagy function has been observed during ageing and may contribute to an impairment of cellular clearance. Ultimately, and regardless of the mechanism involved, defective cellular clearance leads to the accumulation of neurotoxic proteins and neuronal cell death. (PD = Parkinson's disease; AD = Alzheimer's diseases; FTD = Fronto-Temporal Dementia; CMT2b = Charcot-Marie-Tooth type 2B; SENDA = Static Encephalopathy of Childhood with Neurodegeneration in Adulthood).
Mutations in genes encoding essential components of the endolysosomal-autophagic pathway have also been described in several neurodegenerative diseases. A significant number of patients with PD, particularly among Ashkenazi Jews166, are heterozygous for mutations in the gene encoding the lysosomal enzyme β-glucocerebrosidase167. Homozygous mutations in the same gene cause Gaucher's disease, a neurodegenerative lysosomal storage disease168. It has been proposed that lower levels of β-glucocerebrosidase lead to an increased accumulation of glucosylceramide that in turn accelerates the synthesis and stabilization of soluble α-synuclein oligomers that eventually convert into amyloid fibrils. Furthermore, the accumulation of α-synuclein also blocks the trafficking of newly synthesized β-glucocerebrosidase to the lysosome and thus further amplifies glucosylceramide accumulation169. In addition, mutations in ATP13A2, a component of the lysosomal acidification machinery, were found in patients with hereditary Parkinsonism170 and are associated with lysosomal dysfunction, defective clearance of autophagosomes and accumulation of α-synuclein171. Similarly, mutations in PINK and PARK genes are associated with the defective clearance of mitochondria via an organelle-specific type of autophagy known as mitophagy, leading to Parkinson disease 172-175. PD was also observed in patients carrying mutations in the VPS35 gene, which encodes an endosomal protein involved in the retrograde transport between endosomes and the Trans-Golgi network 176, 177.
Lysosomal and autophagy dysfunction have also been identified in patients with AD carrying mutations of the presenilin 1 gene178. At least two different mechanisms, one involving a defect in lysosomal acidification machinery178 and the other a defect in lysosomal Ca+2 homeostasis179, have been proposed to explain lysosomal dysfunction in these patients. Additional examples of neurodegenerative diseases due to mutations of proteins involved in endosome and lysosome maturation include Fronto-Temporal Dementia and Charcot-Marie-Tooth type 2B, which are due to mutations in CHMP2B180 and RAB7181, respectively. Of note, a mutation in the autophagic protein WD repeat domain 45 (WDR45) has been recently associated with static encephalopathy of childhood with neurodegeneration in adulthood (SENDA), a neurodegenerative disease characterized by iron accumulation into the brain182.
TFEB activation as a potential therapy
The similarity between the mechanisms that lead to LSDs and common neurodegenerative diseases suggest that therapeutic strategies aimed at rescuing and/or enhancing lysosomal and autophagic function may impact both types of diseases. Several attempts have been made to treat animal models of neurodegenerative diseases by enhancing the lysosomal—autophagic pathway160, 183-189. An appealing therapeutic perspective, which has become available since the recent discovery of TFEB, would be to enhance cellular clearance by inducing TFEB function. Preliminary evidence showed that cells with enhanced TFEB levels displayed a faster rate of glycosaminoglycans (GAGs) clearance compared to controls12. TFEB overexpression also resulted in a striking reduction of GAGs and of cellular vacuolization in glia-differentiated neuronal stem cells (NSCs) that were isolated from mouse models of MSD and MPSIIIA, two severe types of LSD86. Similar results were obtained using this approach in cells from patients and/or from murine models other types of LSDs, including Neuronal Ceroid Lipofuscinosis type 3 (CLN3, Batten disease) and Pompe disease86. In all cases TFEB overexpression led to the clearance of the storage material within the cells. TFEB-mediated cellular clearance was also observed in vivo in murine models of MSD and Pompe disease upon TFEB viral-mediated gene transfer86, 190. TFEB overexpression in a mouse model of Pompe disease reduced glycogen load and lysosomal size, improved autophagosome processing and alleviated the accumulation of autophagic vacuoles. Interestingly, the clearance effect of TFEB was found to be dependent on the autophagy pathway, and in the muscle, TFEB was shown to induce exocytosis of autophagolysosomes (also known as autolysosomes) via their fusion with the plasma membrane190. Notably, TFEB was also used as a tool to promote cellular clearance in common neurodegenerative diseases. TFEB gene delivery in a murine model of PD ameliorated tissue pathology191. In a recent study, TFEB was identified as the main mediator of the ability of PGC1α to promote cellular clearance and to rescue neurotoxicity in a murine model of HD192. Finally, TFEB overexpression in the liver of mice carrying a mutated form of α1 anti-trypsin resulted in the clearance of the mutated protein and in the rescue of liver fibrosis193.
The mechanism by which TFEB promotes the clearance of storage material needs further elucidation. TFEB induction rescues lysosomal storage in LSDs in spite of a complete deficiency of one or more lysosomal enzymes. A prevailing mechanism in this case may be the activation of lysosomal exocytosis, by which the storage material may be secreted outside the cells upon TFEB overexpression. However, in general, it is likely that TFEB-mediated cellular clearance is the result of the combined effects of lysosomal biogenesis, autophagy, and lysosomal exocytosis (Fig. 4). The possibility of pharmacologically modulating lysosomal function, for example by inhibiting TFEB phosphorylation or by increasing TFEB dephosphorylation, represents an attractive therapeutic approach to promote cellular clearance in all of the above-mentioned diseases. Therefore, drug screening approaches aimed at identifying molecules that promote TFEB nuclear translocation present an interesting path forward. Careful, long-term studies for the evaluation of potential side effects will have to be undertaken. In this context pulsatile treatments, in which one can boost TFEB activity only for limited periods of time, may be the best option for diseases in which storage material takes a long time to accumulate. At this stage it is too early to determine whether TFEB induction will end up being a viable therapeutic option for LSDs or for other diseases. However, the broad spectrum of diseases on which this therapeutic strategy may impact makes it a very appealing avenue.
Figure 4. TFEB regulates cellular clearance.
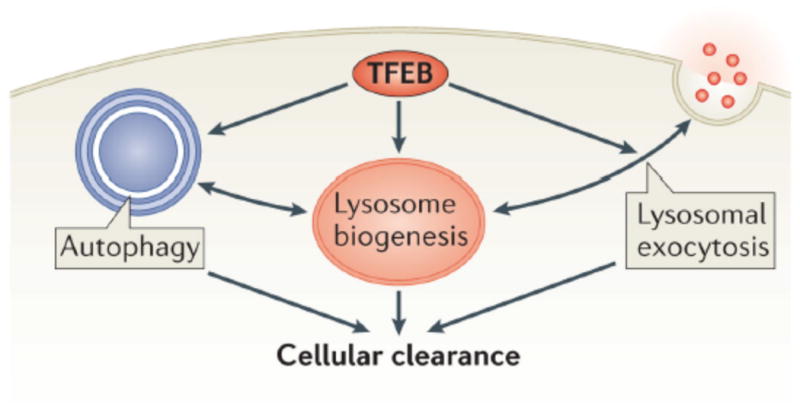
TFEB controls lysosomal biogenesis by regulating the level of lysosomal enzymes, lysosomal acidification and the number of lysosomes. TFEB also controls autophagy by regulating the number of autophagosomes and the fusion between autophagosomes and lysosomes. Finally. TFEB regulates docking and fusion of lysosomes to the plasma membrane in the process of lysosomal exocytosis. The concerted action of these three processes leads to cellular clearance. TFEB
Conclusions and future perspectives
The emerging role of the lysosome in important processes, such as nutrient sensing, signaling, and metabolism, requires further investigation - what we see today is just the tip of the iceberg. Systematic approaches, such as transcriptomics, proteomics, and metabolomics, coupled with the power of systems biology will be particularly important for identifying all of the components of the lysosome and understanding the role of the “greater lysosomal system”152. These systematic approaches should be complemented by in vivo imaging and intravital microscopy, which allow the visualization of lysosomes in the context of a living organism and in specific physiological or pathological conditions.
Interdisciplinary approaches will also allow us to answer intriguing questions such as: how does lysosome number, size, and content vary in different cell types, in different tissues, or in different individuals? How many different types of lysosomes are there with specialized roles? To what extent do environmental or pathological conditions influence the composition, function, or identity of lysosomes? What is the physiological role of lysosomal signaling and its involvement in human disease?
Besides the involvement in neurodegenerative diseases, the role of the lysosome in other pathological processes, such as abnormalities of lipid metabolism, infections, and even aging, is still largely unexplored. Transcriptomic and proteomic analysis of patient-derived tissues and whole genome and exome sequencing of patients' DNA may lead to the discovery of lysosomal “variation” as a predisposing factor for additional human diseases. Furthermore, studying lysosomal function in a variety of disease processes will have a significant impact in developing novel therapeutic strategies. In this context, the development of high content screening approaches will pave the way to the identification of novel compounds that are able to modulate lysosomal function, which could in turn be made into effective drugs to promote cellular clearance.
Acknowledgments
We thank T. Braulke, A. De Matteis, J. Irazoqui, D. Rubinsztein, D. Sabatini, P. Saftig and R Zoncu for the helpful suggestions. We thank G. Diez-Roux for helpful discussions and support in manuscript preparation. We thank E. Abrams for manuscript editing. We acknowledge the support of the Italian Telethon Foundation grant numbers TGM11CB6 (C.S., A.B); the Beyond Batten Disease Foundation (C.S. and A.B.); European Research Council Advanced Investigator grant no. 250154 (A.B.); March of Dimes #6-FY11-306 (A.B); US National Institutes of Health (R01-NS078072) (A.B).
Glossary
- Glycocalyx
The polysaccharide-based coating on the inner side of a lysosomal membrane that protects it from digestion by lysosomal enzymes
- mucolipidosis IV
Mucolipidosis IV is an autosomal recessive neurodegenerative lysosomal storage disorder due to mutations in the gene encoding mucolipin-1. It is characterized by psychomotor retardation and ophthalmologic abnormalities
- Danon disease
Danon disease is an X-linked dominant disorder due to mutations in the gene encoding lysosome-associated membrane protein-2 (LAMP2), predominantly affecting cardiac muscle
- Niemann-Pick disease type C1
an autosomal recessive lipid storage disorder caused by mutation in the NPC1 gene and characterized by progressive neurodegeneration
- Wolman disease
early-onset fulminant disorder of infancy with massive infiltration of the liver, spleen, and other organs by macrophages filled with cholesteryl esters and triglyceridess. It is caused by homozygous or compound heterozygous mutation in the LIPA gene
- Gaucher disease
an autosomal recessive lysosomal storage disorder due to deficient activity of beta-glucocerebrosidase
- Multiple Sulfatase Deficiency
an autosomal recessive inborn error of metabolism caused by homozygous or compound heterozygous mutation in the sulfatase-modifying factor-1 gene (SUMF1)
- Mucopolysaccharidosis
a group of metabolic disorders caused by the absence or malfunctioning of lysosomal enzymes needed to break down molecules
- Autophagosome
intracytoplasmic vacuole containing elements of a cell's own cytoplasm; it fuses with a lysosome and the contents are subjected to enzymatic digestion
- lysosome-related organelles
cell type-specific compartments, which include melanosomes, lytic granules, MHC class II compartments, platelet-dense granules, basophil granules, azurophil granules, and Drosophila pigment granules
- Fronto-Temporal Dementia
refers to a clinical manifestation of the pathologic finding of frontotemporal lobar degeneration
- Charcot-Marie-Tooth type 2B
autosomal dominant peripheral sensory neuropathy due to mutations in the small GTPase late endosomal protein RAB7
- Neuronal Ceroid Lipofuscinosis
neuronal ceroid lipofuscinoses (NCL; CLN) are a clinically and genetically heterogeneous group of neurodegenerative disorders characterized by the intracellular accumulation of autofluorescent lipopigment storage material
- Pompe disease
Glycogen storage disease II, an autosomal recessive disorder, is the prototypic lysosomal storage disease caused by mutation in the gene encoding acid alpha-1,4-glucosidase
References
- 1.de Duve C. The lysosome turns fifty. Nature cell biology. 2005;7:847–9. doi: 10.1038/ncb0905-847. [DOI] [PubMed] [Google Scholar]
- 2.Saftig P, Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nature reviews Molecular cell biology. 2009;10:623–35. doi: 10.1038/nrm2745. [DOI] [PubMed] [Google Scholar]
- 3.Luzio JP, Parkinson MD, Gray SR, Bright NA. The delivery of endocytosed cargo to lysosomes. Biochemical Society transactions. 2009;37:1019–21. doi: 10.1042/BST0371019. [DOI] [PubMed] [Google Scholar]
- 4.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends in cell biology. 2012;22:407–17. doi: 10.1016/j.tcb.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mijaljica D, Prescott M, Devenish RJ. Microautophagy in mammalian cells: revisiting a 40-year-old conundrum. Autophagy. 2011;7:673–82. doi: 10.4161/auto.7.7.14733. [DOI] [PubMed] [Google Scholar]
- 7.Chieregatti E, Meldolesi J. Regulated exocytosis: new organelles for non-secretory purposes. Nature reviews Molecular cell biology. 2005;6:181–7. doi: 10.1038/nrm1572. [DOI] [PubMed] [Google Scholar]
- 8.Verhage M, Toonen RF. Regulated exocytosis: merging ideas on fusing membranes. Current opinion in cell biology. 2007;19:402–8. doi: 10.1016/j.ceb.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 9.Blott EJ, Griffiths GM. Secretory lysosomes. Nature reviews Molecular cell biology. 2002;3:122–31. doi: 10.1038/nrm732. [DOI] [PubMed] [Google Scholar]
- 10.Mostov K, Werb Z. Journey across the osteoclast. Science. 1997;276:219–20. doi: 10.1126/science.276.5310.219. [DOI] [PubMed] [Google Scholar]
- 11.Stinchcombe J, Bossi G, Griffiths GM. Linking albinism and immunity: the secrets of secretory lysosomes. Science. 2004;305:55–9. doi: 10.1126/science.1095291. [DOI] [PubMed] [Google Scholar]
- 12.Sardiello M, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–7. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- 13.Settembre C, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–33. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Braulke T, Bonifacino JS. Sorting of lysosomal proteins. Biochimica et biophysica acta. 2009;1793:605–14. doi: 10.1016/j.bbamcr.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 15.Luzio JP, Pryor PR, Bright NA. Lysosomes: fusion and function. Nature reviews Molecular cell biology. 2007;8:622–32. doi: 10.1038/nrm2217. [DOI] [PubMed] [Google Scholar]
- 16.Pfeffer SR. Rab GTPases: specifying and deciphering organelle identity and function. Trends in cell biology. 2001;11:487–91. doi: 10.1016/s0962-8924(01)02147-x. [DOI] [PubMed] [Google Scholar]
- 17.Zerial M, McBride H. Rab proteins as membrane organizers. Nature reviews Molecular cell biology. 2001;2:107–17. doi: 10.1038/35052055. [DOI] [PubMed] [Google Scholar]
- 18.Rink J, Ghigo E, Kalaidzidis Y, Zerial M. Rab conversion as a mechanism of progression from early to late endosomes. Cell. 2005;122:735–49. doi: 10.1016/j.cell.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 19.Henne WM, Buchkovich NJ, Emr SD. The ESCRT pathway. Developmental cell. 2011;21:77–91. doi: 10.1016/j.devcel.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 20.Luzio JP, et al. ESCRT proteins and the regulation of endocytic delivery to lysosomes. Biochemical Society transactions. 2009;37:178–80. doi: 10.1042/BST0370178. [DOI] [PubMed] [Google Scholar]
- 21.Sridhar S, et al. The lipid kinase PI4KIIIbeta preserves lysosomal identity. The EMBO journal. 2013;32:324–39. doi: 10.1038/emboj.2012.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schulze H, Kolter T, Sandhoff K. Principles of lysosomal membrane degradation: Cellular topology and biochemistry of lysosomal lipid degradation. Biochimica et biophysica acta. 2009;1793:674–83. doi: 10.1016/j.bbamcr.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 23.Rojas R, et al. Regulation of retromer recruitment to endosomes by sequential action of Rab5 and Rab7. The Journal of cell biology. 2008;183:513–26. doi: 10.1083/jcb.200804048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang T, Ming Z, Xiaochun W, Hong W. Rab7: role of its protein interaction cascades in endo-lysosomal traffic. Cellular signalling. 2011;23:516–21. doi: 10.1016/j.cellsig.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 25.Pryor PR, et al. Combinatorial SNARE complexes with VAMP7 or VAMP8 define different late endocytic fusion events. EMBO reports. 2004;5:590–5. doi: 10.1038/sj.embor.7400150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weber T, et al. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–72. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- 27.Jahn R, Scheller RH. SNAREs--engines for membrane fusion. Nature reviews Molecular cell biology. 2006;7:631–43. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- 28.Ohya T, et al. Reconstitution of Rab- and SNARE-dependent membrane fusion by synthetic endosomes. Nature. 2009;459:1091–7. doi: 10.1038/nature08107. [DOI] [PubMed] [Google Scholar]
- 29.Zeigerer A, et al. Rab5 is necessary for the biogenesis of the endolysosomal system in vivo. Nature. 2012;485:465–70. doi: 10.1038/nature11133. [DOI] [PubMed] [Google Scholar]
- 30.Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012;151:1256–69. doi: 10.1016/j.cell.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 31.Hamasaki M, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013 doi: 10.1038/nature11910. [DOI] [PubMed] [Google Scholar]
- 32.Ghosh P, Dahms NM, Kornfeld S. Mannose 6-phosphate receptors: new twists in the tale. Nature reviews Molecular cell biology. 2003;4:202–12. doi: 10.1038/nrm1050. [DOI] [PubMed] [Google Scholar]
- 33.Neufeld EF. The uptake of enzymes into lysosomes: an overview. Birth defects original article series. 1980;16:77–84. [PubMed] [Google Scholar]
- 34.Reczek D, et al. LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase. Cell. 2007;131:770–83. doi: 10.1016/j.cell.2007.10.018. [DOI] [PubMed] [Google Scholar]
- 35.Gallala HD, Breiden B, Sandhoff K. Regulation of the NPC2 protein-mediated cholesterol trafficking by membrane lipids. Journal of neurochemistry. 2011;116:702–7. doi: 10.1111/j.1471-4159.2010.07014.x. [DOI] [PubMed] [Google Scholar]
- 36.Munford RS, Sheppard PO, O'Hara PJ. Saposin-like proteins (SAPLIP) carry out diverse functions on a common backbone structure. Journal of lipid research. 1995;36:1653–63. [PubMed] [Google Scholar]
- 37.Kolter T, Sandhoff K. Principles of lysosomal membrane digestion: stimulation of sphingolipid degradation by sphingolipid activator proteins and anionic lysosomal lipids. Annual review of cell and developmental biology. 2005;21:81–103. doi: 10.1146/annurev.cellbio.21.122303.120013. [DOI] [PubMed] [Google Scholar]
- 38.Furst W, Sandhoff K. Activator proteins and topology of lysosomal sphingolipid catabolism. Biochimica et biophysica acta. 1992;1126:1–16. doi: 10.1016/0005-2760(92)90210-m. [DOI] [PubMed] [Google Scholar]
- 39.Mobius W, Herzog V, Sandhoff K, Schwarzmann G. Intracellular distribution of a biotin-labeled ganglioside, GM1, by immunoelectron microscopy after endocytosis in fibroblasts. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 1999;47:1005–14. doi: 10.1177/002215549904700804. [DOI] [PubMed] [Google Scholar]
- 40.Burkhardt JK, et al. Accumulation of sphingolipids in SAP-precursor (prosaposin)-deficient fibroblasts occurs as intralysosomal membrane structures and can be completely reversed by treatment with human SAP-precursor. European journal of cell biology. 1997;73:10–8. [PubMed] [Google Scholar]
- 41.Bradova V, et al. Prosaposin deficiency: further characterization of the sphingolipid activator protein-deficient sibs. Multiple glycolipid elevations (including lactosylceramidosis), partial enzyme deficiencies and ultrastructure of the skin in this generalized sphingolipid storage disease. Human genetics. 1993;92:143–52. doi: 10.1007/BF00219682. [DOI] [PubMed] [Google Scholar]
- 42.Schnabel D, et al. Simultaneous deficiency of sphingolipid activator proteins 1 and 2 is caused by a mutation in the initiation codon of their common gene. The Journal of biological chemistry. 1992;267:3312–5. [PubMed] [Google Scholar]
- 43.Cosma MP, et al. The multiple sulfatase deficiency gene encodes an essential and limiting factor for the activity of sulfatases. Cell. 2003;113:445–56. doi: 10.1016/s0092-8674(03)00348-9. [DOI] [PubMed] [Google Scholar]
- 44.Dierks T, et al. Multiple sulfatase deficiency is caused by mutations in the gene encoding the human C(alpha)-formylglycine generating enzyme. Cell. 2003;113:435–44. doi: 10.1016/s0092-8674(03)00347-7. [DOI] [PubMed] [Google Scholar]
- 45.Bagshaw RD, Mahuran DJ, Callahan JW. Lysosomal membrane proteomics and biogenesis of lysosomes. Molecular neurobiology. 2005;32:27–41. doi: 10.1385/MN:32:1:027. [DOI] [PubMed] [Google Scholar]
- 46.Callahan JW, Bagshaw RD, Mahuran DJ. The integral membrane of lysosomes: its proteins and their roles in disease. Journal of proteomics. 2009;72:23–33. doi: 10.1016/j.jprot.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 47.Lubke T, Lobel P, Sleat DE. Proteomics of the lysosome. Biochimica et biophysica acta. 2009;1793:625–35. doi: 10.1016/j.bbamcr.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schroder BA, Wrocklage C, Hasilik A, Saftig P. The proteome of lysosomes. Proteomics. 2010;10:4053–76. doi: 10.1002/pmic.201000196. [DOI] [PubMed] [Google Scholar]
- 49.Sleat DE, Jadot M, Lobel P. Lysosomal proteomics and disease. Proteomics Clinical applications. 2007;1:1134–46. doi: 10.1002/prca.200700250. [DOI] [PubMed] [Google Scholar]
- 50.Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 51.Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annual review of biochemistry. 2009;78:857–902. doi: 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]
- 52.Hansen CG, Nichols BJ. Molecular mechanisms of clathrin-independent endocytosis. Journal of cell science. 2009;122:1713–21. doi: 10.1242/jcs.033951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sorkin A, von Zastrow M. Endocytosis and signalling: intertwining molecular networks. Nature reviews Molecular cell biology. 2009;10:609–22. doi: 10.1038/nrm2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Katzmann DJ, Babst M, Emr SD. Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell. 2001;106:145–55. doi: 10.1016/s0092-8674(01)00434-2. [DOI] [PubMed] [Google Scholar]
- 55.Raiborg C, Stenmark H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature. 2009;458:445–52. doi: 10.1038/nature07961. [DOI] [PubMed] [Google Scholar]
- 56.Haglund K, Dikic I. The role of ubiquitylation in receptor endocytosis and endosomal sorting. Journal of cell science. 2012;125:265–75. doi: 10.1242/jcs.091280. [DOI] [PubMed] [Google Scholar]
- 57.Ohkuma S, Poole B. Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proceedings of the National Academy of Sciences of the United States of America. 1978;75:3327–31. doi: 10.1073/pnas.75.7.3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ohkuma S, Moriyama Y, Takano T. Identification and characterization of a proton pump on lysosomes by fluorescein-isothiocyanate-dextran fluorescence. Proceedings of the National Academy of Sciences of the United States of America. 1982;79:2758–62. doi: 10.1073/pnas.79.9.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Graves AR, Curran PK, Smith CL, Mindell JA. The Cl-/H+ antiporter ClC-7 is the primary chloride permeation pathway in lysosomes. Nature. 2008;453:788–92. doi: 10.1038/nature06907. [DOI] [PubMed] [Google Scholar]
- 60.Kasper D, et al. Loss of the chloride channel ClC-7 leads to lysosomal storage disease and neurodegeneration. The EMBO journal. 2005;24:1079–91. doi: 10.1038/sj.emboj.7600576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weinert S, et al. Lysosomal pathology and osteopetrosis upon loss of H+-driven lysosomal Cl- accumulation. Science. 2010;328:1401–3. doi: 10.1126/science.1188072. [DOI] [PubMed] [Google Scholar]
- 62.Mindell JA. Lysosomal acidification mechanisms. Annual review of physiology. 2012;74:69–86. doi: 10.1146/annurev-physiol-012110-142317. [DOI] [PubMed] [Google Scholar]
- 63.Zhang F, Jin S, Yi F, Li PL. TRP-ML1 functions as a lysosomal NAADP-sensitive Ca2+ release channel in coronary arterial myocytes. Journal of cellular and molecular medicine. 2009;13:3174–85. doi: 10.1111/j.1582-4934.2008.00486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang F, Xu M, Han WQ, Li PL. Reconstitution of lysosomal NAADP-TRP-ML1 signaling pathway and its function in TRP-ML1(-/-) cells. American journal of physiology Cell physiology. 2011;301:C421–30. doi: 10.1152/ajpcell.00393.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Calcraft PJ, et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature. 2009;459:596–600. doi: 10.1038/nature08030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sahu R, et al. Microautophagy of cytosolic proteins by late endosomes. Developmental cell. 2011;20:131–9. doi: 10.1016/j.devcel.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ahlberg J, Marzella L, Glaumann H. Uptake and degradation of proteins by isolated rat liver lysosomes. Suggestion of a microautophagic pathway of proteolysis. Laboratory investigation; a journal of technical methods and pathology. 1982;47:523–32. [PubMed] [Google Scholar]
- 68.Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996;273:501–3. doi: 10.1126/science.273.5274.501. [DOI] [PubMed] [Google Scholar]
- 69.Chiang HL, Terlecky SR, Plant CP, Dice JF. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science. 1989;246:382–5. doi: 10.1126/science.2799391. [DOI] [PubMed] [Google Scholar]
- 70.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annual review of genetics. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ravikumar B, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiological reviews. 2010;90:1383–435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 72.Rodriguez A, Webster P, Ortego J, Andrews NW. Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. The Journal of cell biology. 1997;137:93–104. doi: 10.1083/jcb.137.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chavez RA, Miller SG, Moore HP. A biosynthetic regulated secretory pathway in constitutive secretory cells. The Journal of cell biology. 1996;133:1177–91. doi: 10.1083/jcb.133.6.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Coorssen JR, Schmitt H, Almers W. Ca2+ triggers massive exocytosis in Chinese hamster ovary cells. The EMBO journal. 1996;15:3787–91. [PMC free article] [PubMed] [Google Scholar]
- 75.Stinchcombe JC, Griffiths GM. Regulated secretion from hemopoietic cells. The Journal of cell biology. 1999;147:1–6. doi: 10.1083/jcb.147.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Andrews NW. Regulated secretion of conventional lysosomes. Trends in cell biology. 2000;10:316–21. doi: 10.1016/s0962-8924(00)01794-3. [DOI] [PubMed] [Google Scholar]
- 77.Jaiswal JK, Andrews NW, Simon SM. Membrane proximal lysosomes are the major vesicles responsible for calcium-dependent exocytosis in nonsecretory cells. The Journal of cell biology. 2002;159:625–35. doi: 10.1083/jcb.200208154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stinchcombe JC, Griffiths GM. Secretory mechanisms in cell-mediated cytotoxicity. Annual review of cell and developmental biology. 2007;23:495–517. doi: 10.1146/annurev.cellbio.23.090506.123521. [DOI] [PubMed] [Google Scholar]
- 79.Logan MR, Odemuyiwa SO, Moqbel R. Understanding exocytosis in immune and inflammatory cells: the molecular basis of mediator secretion. The Journal of allergy and clinical immunology. 2003;111:923–32. quiz 933. [PubMed] [Google Scholar]
- 80.Wesolowski J, Paumet F. The impact of bacterial infection on mast cell degranulation. Immunologic research. 2011;51:215–26. doi: 10.1007/s12026-011-8250-x. [DOI] [PubMed] [Google Scholar]
- 81.Ren Q, Ye S, Whiteheart SW. The platelet release reaction: just when you thought platelet secretion was simple. Current opinion in hematology. 2008;15:537–41. doi: 10.1097/MOH.0b013e328309ec74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tulsiani DR, Abou-Haila A, Loeser CR, Pereira BM. The biological and functional significance of the sperm acrosome and acrosomal enzymes in mammalian fertilization. Experimental cell research. 1998;240:151–64. doi: 10.1006/excr.1998.3943. [DOI] [PubMed] [Google Scholar]
- 83.Rao SK, Huynh C, Proux-Gillardeaux V, Galli T, Andrews NW. Identification of SNAREs involved in synaptotagmin VII-regulated lysosomal exocytosis. The Journal of biological chemistry. 2004;279:20471–9. doi: 10.1074/jbc.M400798200. [DOI] [PubMed] [Google Scholar]
- 84.Bossi G, Griffiths GM. CTL secretory lysosomes: biogenesis and secretion of a harmful organelle. Seminars in immunology. 2005;17:87–94. doi: 10.1016/j.smim.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 85.LaPlante JM, et al. Lysosomal exocytosis is impaired in mucolipidosis type IV. Molecular genetics and metabolism. 2006;89:339–48. doi: 10.1016/j.ymgme.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 86.Medina DL, et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell. 2011;21:421–30. doi: 10.1016/j.devcel.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dong XP, et al. Activating mutations of the TRPML1 channel revealed by proline-scanning mutagenesis. The Journal of biological chemistry. 2009;284:32040–52. doi: 10.1074/jbc.M109.037184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cadwell K, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–63. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.DeSelm CJ, et al. Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Developmental cell. 2011;21:966–74. doi: 10.1016/j.devcel.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ganesan AK, et al. Genome-wide siRNA-based functional genomics of pigmentation identifies novel genes and pathways that impact melanogenesis in human cells. PLoS genetics. 2008;4:e1000298. doi: 10.1371/journal.pgen.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gerasimenko JV, Gerasimenko OV, Petersen OH. Membrane repair: Ca(2+)-elicited lysosomal exocytosis. Current biology : CB. 2001;11:R971–4. doi: 10.1016/s0960-9822(01)00577-2. [DOI] [PubMed] [Google Scholar]
- 92.Reddy A, Caler EV, Andrews NW. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell. 2001;106:157–69. doi: 10.1016/s0092-8674(01)00421-4. [DOI] [PubMed] [Google Scholar]
- 93.Roy D, et al. A process for controlling intracellular bacterial infections induced by membrane injury. Science. 2004;304:1515–8. doi: 10.1126/science.1098371. [DOI] [PubMed] [Google Scholar]
- 94.Han R, et al. Dysferlin-mediated membrane repair protects the heart from stress-induced left ventricular injury. The Journal of clinical investigation. 2007;117:1805–13. doi: 10.1172/JCI30848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ferron M, et al. A RANKL-PKCβ-TFEB Signaling Cascade is Necessary for Lysosomal Biogenesis in Osteoclasts. Genes & Development. doi: 10.1101/gad.213827.113. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sancak Y, et al. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Efeyan A, et al. Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature. 2012 doi: 10.1038/nature11745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ganley IG, et al. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. The Journal of biological chemistry. 2009;284:12297–305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hosokawa N, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Molecular biology of the cell. 2009;20:1981–91. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jung CH, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Molecular biology of the cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chan EY, Kir S, Tooze SA. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. The Journal of biological chemistry. 2007;282:25464–74. doi: 10.1074/jbc.M703663200. [DOI] [PubMed] [Google Scholar]
- 103.Hara T, et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. The Journal of cell biology. 2008;181:497–510. doi: 10.1083/jcb.200712064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zoncu R, et al. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011;334:678–83. doi: 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yu L, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465:942–6. doi: 10.1038/nature09076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cang C, et al. mTOR Regulates Lysosomal ATP-Sensitive Two-Pore Na(+) Channels to Adapt to Metabolic State. Cell. 2013;152:778–90. doi: 10.1016/j.cell.2013.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nature reviews Molecular cell biology. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Settembre C, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. The EMBO journal. 2012;31:1095–108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rong Y, et al. Spinster is required for autophagic lysosome reformation and mTOR reactivation following starvation. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:7826–31. doi: 10.1073/pnas.1013800108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rong Y, et al. Clathrin and phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome reformation. Nature cell biology. 2012;14:924–34. doi: 10.1038/ncb2557. [DOI] [PubMed] [Google Scholar]
- 111.Rehli M, Den Elzen N, Cassady AI, Ostrowski MC, Hume DA. Cloning and characterization of the murine genes for bHLH-ZIP transcription factors TFEC and TFEB reveal a common gene organization for all MiT subfamily members. Genomics. 1999;56:111–20. doi: 10.1006/geno.1998.5588. [DOI] [PubMed] [Google Scholar]
- 112.Medendorp K, et al. Molecular mechanisms underlying the MiT translocation subgroup of renal cell carcinomas. Cytogenetic and genome research. 2007;118:157–65. doi: 10.1159/000108296. [DOI] [PubMed] [Google Scholar]
- 113.Ma X, Godar RJ, Liu H, Diwan A. Enhancing lysosome biogenesis attenuates BNIP3-induced cardiomyocyte death. Autophagy. 2012;8:297–309. doi: 10.4161/auto.18658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Palmieri M, et al. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Human molecular genetics. 2011;20:3852–66. doi: 10.1093/hmg/ddr306. [DOI] [PubMed] [Google Scholar]
- 115.Cuervo AM. Cell biology. Autophagy's top chef. Science. 2011;332:1392–3. doi: 10.1126/science.1208607. [DOI] [PubMed] [Google Scholar]
- 116.Ma D, Panda S, Lin JD. Temporal orchestration of circadian autophagy rhythm by C/EBPbeta. The EMBO journal. 2011;30:4642–51. doi: 10.1038/emboj.2011.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rzymski T, et al. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene. 2010;29:4424–35. doi: 10.1038/onc.2010.191. [DOI] [PubMed] [Google Scholar]
- 118.Rouschop KM, et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. The Journal of clinical investigation. 2010;120:127–41. doi: 10.1172/JCI40027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.van der Vos KE, et al. Modulation of glutamine metabolism by the PI(3)K-PKB-FOXO network regulates autophagy. Nature cell biology. 2012;14:829–37. doi: 10.1038/ncb2536. [DOI] [PubMed] [Google Scholar]
- 120.Demontis F, Perrimon N. FOXO/4E-BP signaling in Drosophila muscles regulates organism-wide proteostasis during aging. Cell. 2010;143:813–25. doi: 10.1016/j.cell.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhao J, et al. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell metabolism. 2007;6:472–83. doi: 10.1016/j.cmet.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 122.Mammucari C, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell metabolism. 2007;6:458–71. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 123.Chauhan S, et al. ZKSCAN3 Is a Master Transcriptional Repressor of Autophagy. Molecular cell. 2013 doi: 10.1016/j.molcel.2013.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Calnan DR, Brunet A. The FoxO code. Oncogene. 2008;27:2276–88. doi: 10.1038/onc.2008.21. [DOI] [PubMed] [Google Scholar]
- 125.Dephoure N, et al. A quantitative atlas of mitotic phosphorylation. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:10762–7. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cea M, et al. Targeting NAD+ salvage pathway induces autophagy in multiple myeloma cells via mTORC1 and extracellular signal-regulated kinase (ERK1/2) inhibition. Blood. 2012 doi: 10.1182/blood-2012-03-416776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8:903–14. doi: 10.4161/auto.19653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pena-Llopis S, et al. Regulation of TFEB and V-ATPases by mTORC1. The EMBO journal. 2011;30:3242–58. doi: 10.1038/emboj.2011.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Roczniak-Ferguson A, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Science signaling. 2012;5:ra42. doi: 10.1126/scisignal.2002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Martina JA, Puertollano R. Rag GTPases mediate amino acid-dependent recruitment of TFEB and MITF to lysosomes. The Journal of cell biology. 2013;200:475–91. doi: 10.1083/jcb.201209135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Settembre C, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nature cell biology. 2013;15:647–58. doi: 10.1038/ncb2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell metabolism. 2011;13:495–504. doi: 10.1016/j.cmet.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Singh R, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–5. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rodriguez-Navarro JA, Cuervo AM. Dietary lipids and aging compromise chaperone-mediated autophagy by similar mechanisms. Autophagy. 2012;8 doi: 10.4161/auto.20649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rodriguez-Navarro JA, et al. Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E705–14. doi: 10.1073/pnas.1113036109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell metabolism. 2010;11:467–78. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Woloszynek JC, Coleman T, Semenkovich CF, Sands MS. Lysosomal dysfunction results in altered energy balance. The Journal of biological chemistry. 2007;282:35765–71. doi: 10.1074/jbc.M705124200. [DOI] [PubMed] [Google Scholar]
- 138.Du H, Duanmu M, Witte D, Grabowski GA. Targeted disruption of the mouse lysosomal acid lipase gene: long-term survival with massive cholesteryl ester and triglyceride storage. Human molecular genetics. 1998;7:1347–54. doi: 10.1093/hmg/7.9.1347. [DOI] [PubMed] [Google Scholar]
- 139.Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. The Journal of clinical investigation. 2006;116:615–22. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Grove CA, et al. A multiparameter network reveals extensive divergence between C. elegans bHLH transcription factors. Cell. 2009;138:314–27. doi: 10.1016/j.cell.2009.04.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.O'Rourke EJ, Ruvkun G. MXL-3 and HLH-30 transcriptionally link lysosomal lipolysis and autophagy to nutrient availability. Nature Cell Biology. doi: 10.1038/ncb2741. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kaeberlein TL, et al. Lifespan extension in Caenorhabditis elegans by complete removal of food. Aging cell. 2006;5:487–94. doi: 10.1111/j.1474-9726.2006.00238.x. [DOI] [PubMed] [Google Scholar]
- 143.Melendez A, et al. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science. 2003;301:1387–91. doi: 10.1126/science.1087782. [DOI] [PubMed] [Google Scholar]
- 144.Steingrimsson E, Tessarollo L, Reid SW, Jenkins NA, Copeland NG. The bHLH-Zip transcription factor Tfeb is essential for placental vascularization. Development. 1998;125:4607–16. doi: 10.1242/dev.125.23.4607. [DOI] [PubMed] [Google Scholar]
- 145.Cuervo AM, Dice JF. When lysosomes get old. Experimental gerontology. 2000;35:119–31. doi: 10.1016/s0531-5565(00)00075-9. [DOI] [PubMed] [Google Scholar]
- 146.Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–95. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 147.Ballabio A, Gieselmann V. Lysosomal disorders: from storage to cellular damage. Biochimica et biophysica acta. 2009;1793:684–96. doi: 10.1016/j.bbamcr.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 148.Cox TM, Cachon-Gonzalez MB. The cellular pathology of lysosomal diseases. The Journal of pathology. 2012;226:241–54. doi: 10.1002/path.3021. [DOI] [PubMed] [Google Scholar]
- 149.Futerman AH, van Meer G. The cell biology of lysosomal storage disorders. Nature reviews Molecular cell biology. 2004;5:554–65. doi: 10.1038/nrm1423. [DOI] [PubMed] [Google Scholar]
- 150.Schultz ML, Tecedor L, Chang M, Davidson BL. Clarifying lysosomal storage diseases. Trends in neurosciences. 2011;34:401–10. doi: 10.1016/j.tins.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Vitner EB, Platt FM, Futerman AH. Common and uncommon pathogenic cascades in lysosomal storage diseases. The Journal of biological chemistry. 2010;285:20423–7. doi: 10.1074/jbc.R110.134452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Walkley SU. Pathogenic cascades in lysosomal disease-Why so complex? Journal of inherited metabolic disease. 2009;32:181–9. doi: 10.1007/s10545-008-1040-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lieberman AP, et al. Autophagy in lysosomal storage disorders. Autophagy. 2012;8:719–30. doi: 10.4161/auto.19469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.de Pablo-Latorre R, et al. Impaired parkin-mediated mitochondrial targeting to autophagosomes differentially contributes to tissue pathology in lysosomal storage diseases. Human molecular genetics. 2012;21:1770–81. doi: 10.1093/hmg/ddr610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Di Malta C, Fryer JD, Settembre C, Ballabio A. Astrocyte dysfunction triggers neurodegeneration in a lysosomal storage disorder. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E2334–42. doi: 10.1073/pnas.1209577109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Fraldi A, et al. Lysosomal fusion and SNARE function are impaired by cholesterol accumulation in lysosomal storage disorders. The EMBO journal. 2010;29:3607–20. doi: 10.1038/emboj.2010.237. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 157.Settembre C, et al. A block of autophagy in lysosomal storage disorders. Human molecular genetics. 2008;17:119–29. doi: 10.1093/hmg/ddm289. [DOI] [PubMed] [Google Scholar]
- 158.Cox TM. In: Lysoosmal Stroage Disorders. A Practical Guide. Mehta Atul, W B., editors. Wiley-Blackwell; West Sussex: 2012. pp. 153–165. [Google Scholar]
- 159.Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nature neuroscience. 2010;13:805–11. doi: 10.1038/nn.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Harris H, Rubinsztein DC. Control of autophagy as a therapy for neurodegenerative disease. Nature reviews Neurology. 2012;8:108–17. doi: 10.1038/nrneurol.2011.200. [DOI] [PubMed] [Google Scholar]
- 161.Jeong H, et al. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell. 2009;137:60–72. doi: 10.1016/j.cell.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–5. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 163.Winslow AR, et al. alpha-Synuclein impairs macroautophagy: implications for Parkinson's disease. The Journal of cell biology. 2010;190:1023–37. doi: 10.1083/jcb.201003122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Orenstein SJ, et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nature neuroscience. 2013 doi: 10.1038/nn.3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Martinez-Vicente M, et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nature neuroscience. 2010;13:567–76. doi: 10.1038/nn.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R. Mutations in the glucocerebrosidase gene and Parkinson's disease in Ashkenazi Jews. The New England journal of medicine. 2004;351:1972–7. doi: 10.1056/NEJMoa033277. [DOI] [PubMed] [Google Scholar]
- 167.Sidransky E, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. The New England journal of medicine. 2009;361:1651–61. doi: 10.1056/NEJMoa0901281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Brady RO, Kanfer JN, Shapiro D. Metabolism of Glucocerebrosides. Ii. Evidence of an Enzymatic Deficiency in Gaucher's Disease. Biochemical and biophysical research communications. 1965;18:221–5. doi: 10.1016/0006-291x(65)90743-6. [DOI] [PubMed] [Google Scholar]
- 169.Mazzulli JR, et al. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146:37–52. doi: 10.1016/j.cell.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ramirez A, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nature genetics. 2006;38:1184–91. doi: 10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- 171.Usenovic M, Krainc D. Lysosomal dysfunction in neurodegeneration: The role of ATP13A2/PARK9. Autophagy. 2012;8:987–8. doi: 10.4161/auto.20256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Valente EM, et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304:1158–60. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 173.Geisler S, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nature cell biology. 2010;12:119–31. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 174.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. The Journal of cell biology. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kitada T, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–8. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 176.Zimprich A, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. American journal of human genetics. 2011;89:168–75. doi: 10.1016/j.ajhg.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Chartier-Harlin MC, et al. Translation initiator EIF4G1 mutations in familial Parkinson disease. American journal of human genetics. 2011;89:398–406. doi: 10.1016/j.ajhg.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Lee JH, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–58. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Coen K, et al. Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. The Journal of cell biology. 2012;198:23–35. doi: 10.1083/jcb.201201076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Skibinski G, et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nature genetics. 2005;37:806–8. doi: 10.1038/ng1609. [DOI] [PubMed] [Google Scholar]
- 181.Verhoeven K, et al. Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. American journal of human genetics. 2003;72:722–7. doi: 10.1086/367847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Saitsu H, et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nature genetics. 2013 doi: 10.1038/ng.2562. [DOI] [PubMed] [Google Scholar]
- 183.Yang DS, et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer's disease ameliorates amyloid pathologies and memory deficits. Brain : a journal of neurology. 2011;134:258–77. doi: 10.1093/brain/awq341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Mueller-Steiner S, et al. Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer's disease. Neuron. 2006;51:703–14. doi: 10.1016/j.neuron.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 185.Sun B, et al. Cystatin C-cathepsin B axis regulates amyloid beta levels and associated neuronal deficits in an animal model of Alzheimer's disease. Neuron. 2008;60:247–57. doi: 10.1016/j.neuron.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ravikumar B, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nature genetics. 2004;36:585–95. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 187.Menzies FM, et al. Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain : a journal of neurology. 2010;133:93–104. doi: 10.1093/brain/awp292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Rose C, et al. Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington's disease. Human molecular genetics. 2010;19:2144–53. doi: 10.1093/hmg/ddq093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Tanaka M, et al. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nature medicine. 2004;10:148–54. doi: 10.1038/nm985. [DOI] [PubMed] [Google Scholar]
- 190.Spampanato C, et al. Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Molecular Medicine. 2013;5:691–706. doi: 10.1002/emmm.201202176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Dehay B, et al. Pathogenic lysosomal depletion in Parkinson's disease. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:12535–44. doi: 10.1523/JNEUROSCI.1920-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Tsunemi T, et al. PGC-1alpha rescues Huntington's disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Science translational medicine. 2012;4:142ra97. doi: 10.1126/scitranslmed.3003799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Pastore N, et al. Gene transfer of master autophagy regulator TFEB results in clearance of toxic protein and correction of hepatic disease in alpha-1-anti-trypsin deficiency. EMBO Molecular Medicine. 2013 doi: 10.1002/emmm.201202046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Bagshaw RD, Mahuran DJ, Callahan JW. A proteomic analysis of lysosomal integral membrane proteins reveals the diverse composition of the organelle. Molecular & cellular proteomics : MCP. 2005;4:133–43. doi: 10.1074/mcp.M400128-MCP200. [DOI] [PubMed] [Google Scholar]
- 195.Kobayashi T, et al. Separation and characterization of late endosomal membrane domains. The Journal of biological chemistry. 2002;277:32157–64. doi: 10.1074/jbc.M202838200. [DOI] [PubMed] [Google Scholar]
- 196.Andrejewski N, et al. Normal lysosomal morphology and function in LAMP-1-deficient mice. The Journal of biological chemistry. 1999;274:12692–701. doi: 10.1074/jbc.274.18.12692. [DOI] [PubMed] [Google Scholar]
- 197.Nishi T, Forgac M. The vacuolar (H+)-ATPases--nature's most versatile proton pumps. Nature reviews Molecular cell biology. 2002;3:94–103. doi: 10.1038/nrm729. [DOI] [PubMed] [Google Scholar]
- 198.Marshansky V, Futai M. The V-type H+-ATPase in vesicular trafficking: targeting, regulation and function. Current opinion in cell biology. 2008;20:415–26. doi: 10.1016/j.ceb.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Dong XP, et al. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature. 2008;455:992–6. doi: 10.1038/nature07311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Wong CO, Li R, Montell C, Venkatachalam K. Drosophila TRPML Is Required for TORC1 Activation. Current biology : CB. 2012;22:1616–21. doi: 10.1016/j.cub.2012.06.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Bargal R, et al. Identification of the gene causing mucolipidosis type IV. Nature genetics. 2000;26:118–23. doi: 10.1038/79095. [DOI] [PubMed] [Google Scholar]
- 202.Bassi MT, et al. Cloning of the gene encoding a novel integral membrane protein, mucolipidin-and identification of the two major founder mutations causing mucolipidosis type IV. American journal of human genetics. 2000;67:1110–20. doi: 10.1016/s0002-9297(07)62941-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Jentsch TJ, Poet M, Fuhrmann JC, Zdebik AA. Physiological functions of CLC Cl- channels gleaned from human genetic disease and mouse models. Annual review of physiology. 2005;67:779–807. doi: 10.1146/annurev.physiol.67.032003.153245. [DOI] [PubMed] [Google Scholar]
- 204.Cuervo AM, Gomes AV, Barnes JA, Dice JF. Selective degradation of annexins by chaperone-mediated autophagy. The Journal of biological chemistry. 2000;275:33329–35. doi: 10.1074/jbc.M005655200. [DOI] [PubMed] [Google Scholar]
- 205.Nishino I, et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease) Nature. 2000;406:906–10. doi: 10.1038/35022604. [DOI] [PubMed] [Google Scholar]
- 206.Lloyd-Evans E, et al. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nature medicine. 2008;14:1247–55. doi: 10.1038/nm.1876. [DOI] [PubMed] [Google Scholar]
- 207.Liu B, Du H, Rutkowski R, Gartner A, Wang X. LAAT-1 is the lysosomal lysine/arginine transporter that maintains amino acid homeostasis. Science. 2012;337:351–4. doi: 10.1126/science.1220281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Hrebicek M, et al. Mutations in TMEM76* cause mucopolysaccharidosis IIIC (Sanfilippo C syndrome) American journal of human genetics. 2006;79:807–19. doi: 10.1086/508294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Fan X, et al. Identification of the gene encoding the enzyme deficient in mucopolysaccharidosis IIIC (Sanfilippo disease type C) American journal of human genetics. 2006;79:738–44. doi: 10.1086/508068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Durand S, Feldhammer M, Bonneil E, Thibault P, Pshezhetsky AV. Analysis of the biogenesis of heparan sulfate acetyl-CoA:alpha-glucosaminide N-acetyltransferase provides insights into the mechanism underlying its complete deficiency in mucopolysaccharidosis IIIC. The Journal of biological chemistry. 2010;285:31233–42. doi: 10.1074/jbc.M110.141150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Efeyan A, Zoncu R, Sabatini DM. Amino acids and mTORC1: from lysosomes to disease. Trends in molecular medicine. 2012 doi: 10.1016/j.molmed.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Sancak Y, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM. Ragulator Is a GEF for the Rag GTPases that Signal Amino Acid Levels to mTORC1. Cell. 2012;150:1196–208. doi: 10.1016/j.cell.2012.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes & development. 2003;17:1829–34. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Current biology : CB. 2003;13:1259–68. doi: 10.1016/s0960-9822(03)00506-2. [DOI] [PubMed] [Google Scholar]