

Abstract
The ICR-derived glomerulonephritis (ICGN) mouse is a unique model of nephrotic syndrome, and albuminuria becomes evident in a neonatal stage, due to a genetic mutation of tensin2. We previously provided evidence that an apparent decrease in nephrin, caused by tensin2-deficiencient states, leads to podocytopathy, albuminuria and eventually, chronic renal failure. In general, glomerular endothelial cells (ECs) function as a barrier through tight attachment of glomerular basement membrane to podocytes, while decreased ECs can worsen renal failure. Nevertheless, it is still unknown whether glomerular ECs are altered under the tensin-2-deficient states during the manifestation of chronic renal failure. Herein, we examined the changes of glomerular ECs, with focus on the expression of PECAM-1 and VE-cadherin (EC-specific markers), or of α-SMA (myofibroblast marker) in this mouse model by histological methods. Compared with the non-nephrotic (+/nep) mice, the nephrotic (nep/nep) mice exhibited the reduced expression of PECAM-1, or of VE-cadherin, in glomerular area. Notably, some glomerular ECs showed the positive stainings for both PECAM-1 and α-SMA, suggesting endothelial-to-mesenchymal transition (EndoMT) during progression of glomerular sclerosis. This is the first report showing that a decrease in glomerular ECs, at least in part, via EndoMT is involved in tensin2-deficient pathological conditions.
Keywords: ICGN mice, glomerular endothelial cells, endothelial mesenchymal transition, glomerulosclerosis, chronic kidney disease
I. Introduction
The ultrafiltration of plasma in the kidneys occurs through the capillary wall of glomerulus. The filtration barrier is composed of glomerular endothelial cells (ECs), the glomerular basement membrane (GBM), and podocytes. These three layers of the capillary wall must function on preventing the protein leakage to urine, and these layers destruction result in proteinuria. Podocyte dysfunction leads into the other slit structures. In normal kidneys, the fenestrated ECs play a major role in modulation of factors regulating vascular tone and glomerular filtration, reactive oxygen species, pro-thrombotic and anti-thrombotic factors and fibrosis [5]. EC injury and dysfunction sometimes result in cell necrosis and apoptosis as well as swelling and the detachment of ECs from the GBM [8].
So far, ICGN strain has been used as a mouse model of proteinuria and chronic kidney disease (CKD) [13, 19, 22, 26]. Genetic analysis defined that a mutation of tensin2 might cause the truncated protein, along with failure of the glomeruli to maintain expression of tensin2 [3]. Tensin2 is important for podocyte functions, and thus tensin2 deficiency may result in proteinuria, associated with the loss of slit integrity [12]. The podocyte proteins, nephrin and synaptopodin, were decreased in the glomerular tufts of nephrotic mice, especially in the central zone [12]. Taken together with recent and previous observations [12, 13, 19, 22], we have emphasized that podocytic abnormality (including nephrin loss) might cause differentiation of mesangial cells (MCs) to myofibroblasts, a key player for matrix (ECM) production and fibrogenesis.
Concerning the major components of glomeruli (i.e., GBM, podocytes, ECs), Ogura et al. firstly reported the abnormal GBM (thickness with the increase in ECM) and the loss in foot process by podocytes in the ICGN mice [26, 27]. Our group delineated the early onset of podopathy (or podocytopenia) [12]. On the other hand, emerging evidence indicate the critical role of endothelial-mesenchymal transition (EndoMT) in tissue fibrogenesis [28]. Although glomerular EC plays a key role for renal homeostasis, little is known about the behavior of glomerular ECs in the nephrotic ICGN mice, a congenital model of diffuse glomerulosclerosis (GS) [22]. In the present study, we found the decrease in glomerular ECs and this was, in part, associated with the EndoMT, as discussed later.
II. Materials and Methods
Animals
The ICGN/Oa mice were kept at 21 ± 2°C and 40 ± 5% humidity, and fed a commercial diet (MF; Oriental Yeast, Tokyo, Japan) [12, 13]. The heterozygotes (+/nep) were prepared by mating between homozygous (nep/nep) males and heterozygous (+/nep) females. To determine the natural course of glomerular morphology, the heterozygotes and homozygotes were subjected to autopsy at the ages of 5 and 20 weeks. All animal experiments were carried out according to the Guideline for Experimental Animal Care issued by the Prime Minister’s Office of Japan and approved by the Committee on Animal Experimentation of Osaka University.
PAS stain and Immunohistological analysis
Right kidney removed at each autopsy was fixed in cold 10%-buffered formaldehyde and 70% ethanol for 24 hr respectively. Transversally trimmed kidney tissues were submitted to a routine process for paraffin embedding. The renal sections were prepared, deparaffined, stained with PAS (Periodic-acid Schiff) for glomerular histology. To detect the glomerular proteins, anti-nestin IgG (1:500, BD Pharmingen, La Jolla, CA), anti-PECAM-1 IgG (1:500, BD Pharmingen), anti-VE-cadherin IgG (1:200, C-19, Santa Cruz Biotechnology, Santa Cruz, CA), and anti-α-SMA IgG (1:200, DAKO, Glostrup, Denmark) were used, respectively. For nestin staining, 10% formalin-fixed sections were used, and antigen retrieval was performed by using 1 mM EDTA at pH 8.0 at 121°C for 5 min. For PECAM, VE-cadherin, and α-SMA staining, 70% EtOH-fixed sections were used, as reported [13, 21]. The sections were dewaxed and then followed by incubation with 10% normal serum for 30 min. The sections were incubated at 4°C for overnight with primary antibodies. The sections were washed with PBS, followed by the secondary antibodies (1:600, Alexa488-conjugated or Alexa546-conjugated IgG, Invitrogen, CA) at room temperature for 20 min. The nuclear counterstain reagent TO-PRO-3 iodide (Invitrogen) was applied. The sections were mounted with crystal mount (Biomeda, CA) and observed under an LSM5 PASCAL confocal microscope (Zeiss, Germany).
For DAB staining of VE-cadherin and nestin, the sections were incubated at 4°C for overnight with the primary antibodies. The sections were washed with PBS and incubated with biotin-labeled anti-mouse and anti-goat antibody (Vector, Burlingame, CA) for 1 hr at room temperature. Avidin-biotin coupling reaction was performed on the sections using a kit (Elite®, Vector). All antigens were visualized as brown with DAB (Nacalai, Kyoto, Japan). We counted the number of glomerular ECs (i.e., VE-cadherin-positive cells), or of podocytes (i.e., nestin-positive cells), according to the previous reports [12, 24].
III. Results
Glomerular morphology and nestin localization
We examined the morphology of glomeruli in the ICGN/Oa mice by PAS staining. The glomeruli of non-nephrotic (+/nep) mice seemed to be normal (Fig. 1A, C), but nephrotic (nep/nep) mice exhibited the glomerular sclerosis at 5 weeks of age (Fig. 1B). The sclerotic lesions (in particular, hypertrophy with glomerular segmentation) became more severe at 20 weeks of age, with the thicken GBM (Fig. 1D). The number of glomerular cells was then decreased in the nephrotic (nep/nep) mice, but more than half of glomerular cells remained [12]. We then examined the expression of nestin protein in the kidney as a histological marker of podocytes [34]. In the non-nephrotic (+/nep) mice, nestin was localized in the whole area of glomerular tuft (Fig. 1E, G). In the nephrotic (nep/nep) mice, nestin was localized mainly in the marginal and peripheral areas of glomerular tuft at 5 and 20 weeks of age, respectively (Fig. 1F, H). There were significant differences in the number of nestin-positive glomerular cells between non-nephrotic and nephrotic mice in each age (5 weeks: 7.31 ± 0.77 vs. 6.18 ± 0.49, p<0.01; 20 weeks: 5.33 ± 0.38 vs. 0.76 ± 0.14, p<0.001), being similar to our report showing the “podocytopenia”, as evidenced by WT1-positive or synaptopodin-positive staining [12].
Fig. 1. .
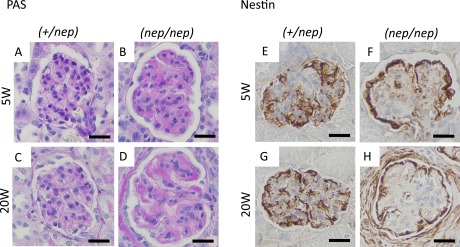
Histological images of glomeruli and podocyte-specific protein at 5 and 20 weeks of age. (A–D) PAS stain, (E–H) nestin stain. (A, E) 5W of heterozygotes, (B, F) 5W of homozygotes, (C, G) 20W of heterozygotes, (D, H) 20W of homozygotes. Bars=20 μm.
A decrease in glomerular ECs during the progression of : glomerulosclerosis
Several lines of evidence indicate that renal capillary loss is one of the most key events for accelerating CKD. We used VE-cadherin as a histological marker to detect glomerular ECs [31]. Indeed, we confirmed that this marker was localized along the capillary lumen (i.e., inside of synaptopodin-positive podocytic foot process) (not shown), and such a staining pattern was similar to the previous report [31]. In the non-nephrotic (+/nep) mice, the number of glomerular ECs (i.e., VE-cadherin-positive cells) remained unchanged between 5 and 20 weeks after birth (Fig. 2A, C). In contrast, there was a gradual decrease of the glomerular ECs in the nephrotic ICGN (nep/nep) mice (Fig. 2B, D). There was a significant difference in the number of glomerular ECs (5 and 20 weeks of age) between the normal and nephrotic groups (Fig. 2E).
Fig. 2. .
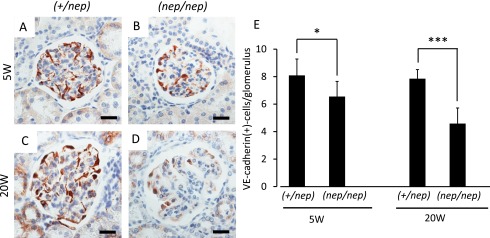
Immunohistochemical images of VE-cadherin at 5 and 20 weeks of age. (A) 5W of heterozygotes, (B) 5W of homozygotes, (C) 20W of heterozygotes, (D) 20W of homozygotes. (E) Quantification of VE-cadherin-positive cells per glomerulus. All data are expressed as mean ± SD (n=6). Statistical analysis: *p < 0.05, ***p < 0.001 as compared with age-matched heterozygous mice. Bars=20 μm.
This result was also reproduced of the fluorescence methods for detecting PECAM-1 (Fig. 3). PECAM-1 was tightly localized in the whole glomerular tuft of the non-nephrotic mice (+/nep) (Fig. 3A, C). Likewise, PECAM-1 was recognized in the whole glomerulus of nephrotic (nep/nep) mice at 5 weeks of age (Fig. 3B). However, PECAM-1 expression became sparse in the glomeruli (Fig. 3D). Of note, the PECAM-1 expression was observed at the center of glomerular tuft in nephrotic mice. These results indicate that the number of glomerular ECs gradually decreased, according to the progressions of glomerular sclerosis.
Fig. 3. .

Immunofluorescence images of PECAM-1 (green) at 5 and 20 weeks of age, and counterstained with TO-PRO-3 (blue). (A) 5W of heterozygotes, (B) 5W of homozygotes, (C) 20W of heterozygotes, (D) 20W of homozygotes. Bars=20 μm.
The possible EndoMT in the glomeruli of nephrotic ICGN/Oa mice
We predicted that EndoMT might be involved in the loss of ECs. To test our hypothesis, the double staining of PECAM-1 (i.e., EC marker) and α-SMA (i.e., myofibroblast marker) was performed for the renal tissue section. In the non-nephrotic (+/nep) mice, PECAM-1 is tightly expressed in the whole glomerular tuft, but α-SMA existed along the capillary area (Fig. 4A–C). In contrast, α-SMA was strongly localized in the whole glomerular tuft of nephrotic (nep/nep) mice (Fig. 4E), being same as the previous results obtained with an immunoperoxidase-based staining method [13, 21, 22]. Of note, some PECAM-1-positive fluorescence signals were also detectable among the α-SMA-positive cell population (Fig. 4), thereby suggesting the possible involvement of EndoMT in glomelular sclerogenesis, as discussed below.
Fig. 4. .

Fluorescence images of PECAM-1 (green) and α-SMA (red) in the heterozygotes (A–C) and homozygotes (D–I) at 20 weeks of age. Bars=20 μm (A–F), 5 μm (G–I). Note: Very few populations of glomerular ECs seem to acquire mesenchyme-like phenotype (arrowhead), while EndoMT becomes evident in the glomerulus of nephrotic mice (arrow).
IV. Discussion
Glomerular ECs play key roles as an interface of vascular tissue structure and body fluid, or as a regulatory barrier for blood clearance [8, 35]. Indeed, clinical and experimental studies revealed that a decrease in glomerular ECs correlates with renal dysfunction [6, 9, 24, 25], leading to hypoxic and fibrotic events. With regard to this, recent studies revealed that EndoMT is, in part, responsible for tissue fibrosis under chronic injuries in hearts [37] and lungs [1]. Notably, EndoMT is also involved in renal fibrogenesis during CKDs. Indeed, confocal microscopy revealed that EndoMT was seen in tubulo-interstitial space, rather than glomerular area, in rodent models of CKDs, such as diabetic nephropathy [16, 18], obstructive nephropathy (UUO) or Alport syndrome [38], and adenine-induced CKD [35]. However, little is known about the onset of EndoMT in GS, possibly due to a lack in typical models of GS.
In the late 1990s, we identified the nephrotic ICGN mice as a unique model of multi-focal and global GS [19–22]. This strain manifests diffuse GS at lease from 3 weeks after birth, along with a high level of urinary albumin (>20 mg/ml). Indeed, GS index is more than 3 in 6–10 weeks of age with global myofibroblastosis [19, 22]. Using this model of severe GS, we found the decrease in glomerular ECs during CKD, and such a capillary loss seems to be linked to the progression of tubular atrophy, fibrosis and renal failure, as reported [19, 21, 23]. Considering that prevention of endothelial dysfunction can be a therapeutic target for CKD [7], the reduction in glomerular ECs may trigger the downstream events, such as tubular atrophy and renal failure [13, 19, 21–23]. Indeed, attenuation of renal capillary loss by HGF leads to producing the therapeutic effects of CKD (unpublished data), convincing a role of ECs for renal homeostasis.
One of the most important findings in this study is that EndoMT is involved not only in peri-tubular areas [18, 35, 38] but also in glomerular areas. The co-localized expression of α-SMA with PECAM-1 implies the involvement of EndoMT in GS development, as reported during arterial sclerogenesis in a rat model of CKD [35]. Among growth factors, TGF-β1 is known to induce a differentiation of ECs into myofibroblasts in vitro [17, 37] and in vivo [10, 18], possibly via Smad3 activation. In our mouse model (ICGN strain), TGF-β1 up-regulation becomes evident in the infiltrated macrophages or resident cells, especially in the sclerotic glomeruli [21, 33]. Our chronological study indicated that TGF-β1 levels were >3-fold higher in the 20-week-old nephrotic mice than in the non-nephrotic littermates (in preparation). Thus, it is likely that glomerular ECs are converted to myofibroblasts via a paracrine system of macrophage- or mesangium-released TGF-β1 (i.e., in situ differentiation).
It is important to find the decrease in the number of glomerular cells per se in our model, during the CKD progression [13, 19, 22]. This was associated with the increase in apoptotic cells, including glomerular ECs (not shown), as reported in other models of CKD [15, 29, 30]. TGF-β is critical for the induction of apoptosis in ECs in vitro [32] and in vivo [4]. In our mouse model, TGF-β expression becomes evident in sclerosing glomeruli [21, 33]. Together, we predict that mesangium-derived TGF-β may, in addition to EndoMT, be contributable to the capillary loss, partly via inducing apoptosis. PECAM-1 is known to inhibit TGF-β-mediated apoptosis via cross-talk pathways between MCs and ECs, whereby MC β8-integrin sequesters TGF-β [14]. Thus, loss in PECAM-1 may be associated with the possible enhancements of TGF-β-mediated pathological actions, including EndoMT, apoptosis and fibrosis. In our model, podocytopenia becomes evident at 20 weeks after birth, along with the TGF-β1 up-regulation. TGF-β1 is critical for conversion of podocytes to myofibroblasts [2]. The potential contribution of TGF-β1 to podocytepenia (including the conversion pathway) warrants future attentions.
A tensin2 mutation is responsible for the renal phenotypes of ICGN mice [3, 12]. Thus, we will discuss the role of tensin2-deficiency in the illness events (including ECs loss). Tensin2 is expressed in podocytes, rather than in ECs or mesangial cells, indicating the initial event in podocytes (i.e., loss in nephrin stabilization [12]). Under such a nephrin-reduced condition, albuminuria becomes evident, and then inflammatory cells (such as macrophages) infiltrates into renal interstitial areas, followed by TGF-β1 up-regulation. Collectively, we predict that macrophage-secreted TGF-β1 is crucial for CKD progression under the tensin2-deficiency as follows: (i) induction of EndoMT in ECs for further sclerogenesis; (ii) apoptosis-mediated vessel loss; and (iii) local hypoxia with a decline in glomerular filtration rate. We are now under the process of investigating the localization and roles of tensin2 in renal resident cells.
Emerging evidence shows that glomerular capillary loss or injury is a histological hallmark of advanced CKD in humans and animals [6, 9, 25, 36]. So far, resident MCs and podocytes have been believed to be a candidate to cellular origins of glomerular myofibroblasts [2, 11]. Since the nephrotic ICGN mice manifest GS even if peri-tubular fibrosis is negligible [19], EndoMT seems to be contributable for GS itself, rather than peri-tubular fibrosis. Giving that a small population of ECs undergoes EndoMT in (+/nep) mice, we cannot exclude a possibility that the differentiated cells (i.e., EC-like mesenchymal cells) may be propagated in response to fibrogenic regulators (such as PDGF or TGF-β [21]) in the nephrotic (nep/nep) mice. Although further studies are necessary for elucidating the molecular mechanism of capillary loss, we at least emphasize that ECs can be a source of glomerular myofibroblasts during CKD.
V. Acknowledgments
This work was supported by grants from the Ministry of Education, Science, Technology, Sports and Culture of Japan (No. 20590398 and 23590458 to SM, No. 26860268 to TK).
VI. References
- 1.Arciniegas E., Frid M. G., Douglas I. S. and Stenmark K. R. (2007) Perspectives on endothelial to mesenchymal transition: potential contribution to vascular remodeling in chronic pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 293; L1–8. [DOI] [PubMed] [Google Scholar]
- 2.Chen C. A., Hwang J. C., Guh J. Y., Tsai J. C. and Chen H. C. (2006) TGF-beta1 and integrin synergistically facilitate the differentiation of rat podocytes by increasing alpha-smooth muscle actin expression. Transl. Res. 148; 134–141. [DOI] [PubMed] [Google Scholar]
- 3.Cho A. R., Uchio-Yamada K., Torigai T., Miyamoto T., Miyoshi I., Matsuda J., Kurosawa T., Kon Y., Asano A., Sasaki N. and Agui T. (2006) Deficiency of the tensin2 gene in the ICGN mouse: an animal model for congenital nephrotic syndrome. Mamm. Genome 17; 407–416. [DOI] [PubMed] [Google Scholar]
- 4.Fierlbeck W., Liu A., Coyle R. and Ballermann B. J. (2003) Endothelial cell apoptosis during glomerular capillary lumen formation in vivo. J. Am. Soc. Nephrol. 14; 1349–1354. [DOI] [PubMed] [Google Scholar]
- 5.Fogo A. B. and Kon V. (2010) The glomerulus—a view from the inside—the endothelial cell. Int. J. Biochem. Cell Biol. 42; 1388–1397. [DOI] [PubMed] [Google Scholar]
- 6.Futrakul N., Panichakul T., Sirisinha S., Futrakul P. and Siriviriyakul P. (2004) Glomerular endothelial dysfunction in chronic kidney disease. Ren. Fail. 26; 259–264. [DOI] [PubMed] [Google Scholar]
- 7.Futrakul N., Butthep P., Patumraj S., Siriviriyakul P. and Futrakul P. (2006) Microvascular disease and endothelial dysfunction in chronic kidney diseases: therapeutic implication. Clin. Hemorheol. Microcirc. 34; 265–271. [PubMed] [Google Scholar]
- 8.Futrakul P., Sitprija V., Yenrudi S., Poshyachinda M., Sensirivatana R., Watana D., Singklwa V., Jungthirapanich J. and Futrakul N. (1997) Glomerular endothelial dysfunction determines disease progression: a hypothesis. Am. J. Nephrol. 17; 533–540. [DOI] [PubMed] [Google Scholar]
- 9.Haddad G., Zhu L. F., Rayner D. C. and Murray A. G. (2013) Experimental glomerular endothelial injury in vivo. PLoS One 8; e78244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He J., Xu Y., Koya D. and Kanasaki K. (2013) Role of the endothelial-to-mesenchymal transition in renal fibrosis of chronic kidney disease. Clin. Exp. Nephrol. 17; 488–497. [DOI] [PubMed] [Google Scholar]
- 11.Johnson R. J., Floege J., Yoshimura A., Iida H., Couser W. G. and Alpers C. E. (1992) The activated mesangial cell: a glomerular “myofibroblast”? J. Am. Soc. Nephrol. 2; S190–S197. [DOI] [PubMed] [Google Scholar]
- 12.Kato T., Mizuno S., Taketo M. M. and Kurosawa T. M. (2008) The possible involvement of tensin2 in the expression and extension of nephrin by glomerular podocytes in mice. Biomed. Res. 29; 279–287. [DOI] [PubMed] [Google Scholar]
- 13.Kato T., Kurosawa T. M. and Taketo M. M. (2009) Proteinuria-induced chronic kidney disease in the ICGN/Oa mice with a mutation of Tensin2 gene. Ren. Fail. 31; 229–238. [DOI] [PubMed] [Google Scholar]
- 14.Khan S., Lakhe-Reddy S., McCarty J. H., Sorenson C. M., Sheibani N., Reichardt L. F., Kim J. H., Wang B., Sedor J. R. and Schelling J. R. (2011) Mesangial cell integrin αvβ8 provides glomerular endothelial cell cytoprotection by sequestering TGF-β and regulating PECAM-1. Am. J. Pathol. 178; 609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kitamura H., Shimizu A., Masuda Y., Ishizaki M., Sugisaki Y. and Yamanaka N. (1998) Apoptosis in glomerular endothelial cells during the development of glomerulosclerosis in the remnant-kidney model. Exp. Nephrol. 6; 328–336. [DOI] [PubMed] [Google Scholar]
- 16.Kizu A., Medici D. and Kalluri R. (2009) Endothelial-mesenchymal transition as a novel mechanism for generating myofibroblasts during diabetic nephropathy. Am. J. Pathol. 175; 1371–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kokudo T., Suzuki Y., Yoshimatsu Y., Yamazaki T., Watabe T. and Miyazono K. (2008) Snail is required for TGFbeta-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J. Cell Sci. 121; 3317–3324. [DOI] [PubMed] [Google Scholar]
- 18.Li J., Qu X., Yao J., Caruana G., Ricardo S. D., Yamamoto Y., Yamamoto H. and Bertram J. F. (2010) Blockade of endothelial-mesenchymal transition by a Smad3 inhibitor delays the early development of streptozotocin-induced diabetic nephropathy. Diabetes 59; 2612–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mizuno S., Yue B. F., Okamoto M., Horikawa Y. and Kurosawa T. (1997) Diffuse glomerulosclerosis without tubular injury does not directly manifest renal dysfunction in nephrotic mice (ICGN strain). Exp. Nephrol. 5; 498–507. [PubMed] [Google Scholar]
- 20.Mizuno S., Horikawa Y., Okamoto M. and Kurosawa T. (1998) Preventive effect of ACE inhibitor on interstitial myofibroblast formation and matrix deposition in a nephrotic model. Ren. Fail. 20; 481–491. [DOI] [PubMed] [Google Scholar]
- 21.Mizuno S., Kurosawa T., Matsumoto K., Mizuno-Horikawa Y., Okamoto M. and Nakamura T. (1998) Hepatocyte growth factor prevents renal fibrosis and dysfunction in a mouse model of chronic renal disease. J. Clin. Invest. 101; 1827–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mizuno S., Mizuno-Horikawa Y., Yue B. F., Okamoto M. and Kurosawa T. (1999) Nephrotic mice (ICGN strain): a model of diffuse mesangial sclerosis in infantile nephrotic syndrome. Am. J. Nephrol. 19; 73–82. [DOI] [PubMed] [Google Scholar]
- 23.Mizuno S., Matsumoto K., Kurosawa T., Mizuno-Horikawa Y. and Nakamura T. (2000) Reciprocal balance of hepatocyte growth factor and transforming growth factor-beta1 in renal fibrosis in mice. Kidney Int. 57; 937–948. [DOI] [PubMed] [Google Scholar]
- 24.Mizuno S., Wen J. and Mizuno-Horikawa Y. (2004) Repeated streptozotocin injections cause early onset of glomerulosclerosis in mice. Exp. Anim. 53; 175–180. [DOI] [PubMed] [Google Scholar]
- 25.Ochodnický P., Henning R. H., Buikema H. J., de Zeeuw D., Provoost A. P. and van Dokkum R. P. (2010) Renal vascular dysfunction precedes the development of renal damage in the hypertensive Fawn-Hooded rat. Am. J. Physiol. Renal Physiol. 298; F625–633. [DOI] [PubMed] [Google Scholar]
- 26.Ogura A., Asano T., Matsuda J., Takano K., Nakagawa M. and Fukui M. (1989) Characteristics of mutant mice (ICGN) with spontaneous renal lesions: a new model for human nephrotic syndrome. Lab. Anim. 23; 169–174. [DOI] [PubMed] [Google Scholar]
- 27.Ogura A., Fujimura H., Asano T., Koura M., Naito I. and Kobayashi Y. (1995) Early ultrastructural glomerular alterations in neonatal nephrotic mice (ICGN strain). Vet. Pathol. 32; 321–323. [DOI] [PubMed] [Google Scholar]
- 28.Piera-Velazquez S., Li Z. and Jimenez S. A. (2011) Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am. J. Pathol. 179; 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shimizu A., Kitamura H., Masuda Y., Ishizaki M., Sugisaki Y. and Yamanaka N. (1997) Rare glomerular capillary regeneration and subsequent capillary regression with endothelial cell apoptosis in progressive glomerulonephritis. Am. J. Pathol. 151; 1231–1239. [PMC free article] [PubMed] [Google Scholar]
- 30.Sun Y. B., Qu X., Zhang X., Caruana G., Bertram J. F. and Li J. (2013) Glomerular endothelial cell injury and damage precedes that of podocytes in adriamycin-induced nephropathy. PLoS One. 8; e55027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takata S., Sawa Y., Uchiyama T. and Ishikawa H. (2013) Expression of toll-like receptor4 in glomerular endothelial cells under diabetic conditions. Acta Histochem. Cytochem. 46; 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsukada T., Eguchi K., Migita K., Kawabe Y., Kawakami A., Matsuoka N., Takashima H., Mizokami A. and Nagataki S. (1995) Transforming growth factor beta-1 induces apoptotic cell death in cultured human umbilical vein endothelial cells with down-regulated expression of bcl-2. Biochem. Biophys. Res. Commun. 210; 1076–1082. [DOI] [PubMed] [Google Scholar]
- 33.Uchio K., Manabe N., Kinoshita A., Tamura K., Miyamoto M., Ogura A., Yamamoto Y. and Miyamoto H. (1999) Abnormalities of extracellular matrices and transforming growth factor beta1 localization in the kidney of the hereditary nephrotic mice (ICGN strain). J. Vet. Med. Sci. 61; 769–776. [DOI] [PubMed] [Google Scholar]
- 34.Wagner N., Wagner K. D., Scholz H., Kirschner K. M. and Schedl A. (2006) Intermediate filament protein nestin is expressed in developing kidney and heart and might be regulated by the Wilms’ tumor suppressor Wt1. Am. J. Physiol. Regul. Integr. Comp. Physiol. 291; R779–R787. [DOI] [PubMed] [Google Scholar]
- 35.Wu M., Tang R. N., Liu H., Xu M., Pan M. M. and Liu B. C. (2013) Cinacalcet attenuates the renal endothelial-to-mesenchymal transition in rats with adenine-induced renal failure. Am. J. Physiol. Renal. Physiol. 306; F138–146. [DOI] [PubMed] [Google Scholar]
- 36.Yamanaka N. and Shimizu A. (1999) Role of glomerular endothelial damage in progressive renal disease. Kidney Blood Press. Res. 22; 13–20. [DOI] [PubMed] [Google Scholar]
- 37.Zeisberg E. M., Tarnavski O., Zeisberg M., Dorfman A. L., McMullen J. R., Gustafsson E., Chandraker A., Yuan X., Pu W. T., Roberts A. B., Neilson E. G., Sayegh M. H., Izumo S. and Kalluri R. (2007) Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 13; 952–961. [DOI] [PubMed] [Google Scholar]
- 38.Zeisberg E. M., Potenta S. E., Sugimoto H., Zeisberg M. and Kalluri R. (2008) Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J. Am. Soc. Nephrol. 19; 2282–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]